

Abstract
Plasminogen activator inhibitor 1 (PAI‐1), an essential regulator of fibrinolysis, is increasingly implicated in the pathogenesis of metabolic disorders, such as obesity and nonalcoholic fatty liver disease (NAFLD). Pharmacologic inhibition of PAI‐1 is emerging as a highly promising therapeutic strategy for obesity and its sequelae. Given the well‐established profibrotic function of PAI‐1, we considered whether PAI‐1 may serve as a target for antifibrotic therapy in nonalcoholic steatohepatitis (NASH). We therefore determined the effect of genetic Pai‐1 deletion and pharmacologic PAI‐1 inhibition on the development of NASH‐related fibrosis in mice. Pai‐1 knockout (Pai‐1 –/–) and wild‐type control (Pai‐1 +/+) mice were fed a high‐fat/high‐cholesterol high‐sugar (HFHS) diet or a methionine‐ and choline‐deficient (MCD) diet to induce steatohepatitis with fibrosis. PAI‐1 was pharmacologically inhibited using the small molecule inhibitor TM5441 in wild‐type C57BL/6 mice fed an HFHS or MCD diet. Either genetic deletion of Pai‐1 or pharmacologic inhibition of PAI‐1 attenuated MCD diet‐induced hepatic steatosis but did not prevent hepatic inflammation or fibrosis. Targeted inhibition of PAI‐1 conferred transient protection from HFHS diet‐induced obesity and hepatic steatosis, an effect that was lost with prolonged exposure to the obesigenic diet. Neither genetic deletion of Pai‐1 nor pharmacologic inhibition of PAI‐1 prevented HFHS diet‐induced hepatic inflammation or fibrosis. Conclusion: Pai‐1 regulates hepatic lipid accumulation but does not promote NASH progression. The PAI‐1 inhibitor TM5441 effectively attenuates diet‐induced obesity and hepatic steatosis but does not prevent NASH‐related fibrosis in mice.
Abbreviations
- Acc
acetyl‐coenzye A carboxylase
- Aco
acyl‐coenzyme A oxidase
- ALT
alanine aminotransferase
- Col1α1
collagen 1α1
- Cpt1α
carnitine palmitoyltransferase 1 alpha
- ECM
extracellular matrix
- Fas
fatty acid synthase
- H&E
hematoxylin and eosin
- HFHS
high‐fat/high‐cholesterol, high‐sugar
- hpf
high‐power field
- MCD
methionine‐ and choline‐deficient
- Mcp‐1
monocyte chemoattractant protein 1
- MCS
methionine‐ and choline‐sufficient
- Mip1α
macrophage inflammatory protein 1α
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PAI‐1
plasminogen activator inhibitor 1
- PCR
polymerase chain reaction
- Pgc1α
peroxisome proliferator‐activated receptor γ coactivator 1α
- Pparα
peroxisome proliferator‐activated receptor α
- SFD
standard fat control diet
- Srebp1c
sterol regulatory element binding protein 1c
- Timp‐1
tissue inhibitor of metalloproteinase 1
- Tnfα
tumor necrosis factor α
- tPA
tissue plasminogen activator
NAFLD, the hepatic manifestation of metabolic syndrome, is rapidly becoming a major public health crisis, yet there is no definitive pharmacologic therapy to prevent or treat this disease. In patients with NASH, the degree of hepatic fibrosis is considered to be the best predictor of mortality.1, 2 Furthermore, recent data indicate that improvement in hepatic fibrosis, even reversal of cirrhosis, is possible in NASH.3, 4 As such, antifibrotic agents are actively being investigated as potential novel therapies to prevent or reverse NASH‐related hepatic fibrosis.
PAI‐1 is an essential regulator of fibrinolysis, with a known profibrotic function in numerous tissue types.5, 6 It is now well established that patients with NAFLD have elevated plasma levels and hepatic expression of PAI‐1.7, 8, 9, 10 Furthermore, the degree of elevation in serum PAI‐1 has been shown to correlate with the degree of fibrosis on liver biopsies in patients with NASH.11 These data have raised speculation that PAI‐1 may promote NASH‐related fibrosis.
Although best known for its roles in regulating fibrinolysis and fibrosis, PAI‐1 is increasingly recognized as a mediator of metabolic diseases, including obesity, diabetes, and NAFLD.7, 10, 12, 13 Heterozygosity of a null PAI‐1 mutation in humans is associated with lower fasting insulin levels and a lower prevalence of diabetes.14 Furthermore, a novel small molecule inhibitor of PAI‐1, TM5441, has recently been shown to attenuate high‐fat diet‐induced obesity and hepatic steatosis in mice.15, 16 Given the potential role of PAI‐1 in promoting metabolic disease combined with its established profibrotic properties, we hypothesized that PAI‐1 may serve as an ideal therapeutic target for NASH‐related fibrosis. As such, we determined the effects of genetic deletion of Pai‐1 and pharmacologic inhibition of PAI‐1 on the development of steatohepatitis with fibrosis, using two well‐established murine dietary models of progressive NASH.
Materials and Methods
Animals and Diets
Male mice bearing a global deletion of Pai‐1 (Pai‐1 –/–) and wild‐type (Pai‐1 +/+) littermate controls, 8 weeks of age, in a C57BL/6 background were fed an MCD or a methionine‐ and choline‐sufficient (MCS) diet (MP Biomedical, Solon, OH) for 8 weeks or an HFHS diet or standard fat control diet (SFD) for 16 weeks. The HFHS diet had 40% of energy as fat (milk fat, 12% saturated) with 2% cholesterol (AIN‐76 Western Diet, TestDiet) and included drinking water supplemented with 42 g/L of 55% fructose/45% glucose by weight. To investigate the effects of pharmacologic PAI‐1 inhibition, wild‐type male C57BL/6J mice (Jackson Laboratories, Bar Harbor, ME) were fed an MCD or MCS diet supplemented with TM5441 (a small molecule inhibitor of PAI‐1) for 8 weeks or an HFHS diet or SFD supplemented with TM5441 for up to 16 weeks. TM5441 was mixed in the diet to achieve a final dosing of 20 mg/kg/day. Details regarding the development and validation of TM5441 and related chemical analogues have been reported.17, 18 Mice underwent 14/10‐hour light/dark cycling and were given free access to food and water. Mice were euthanized by CO2 inhalation. Blood was collected by cardiac puncture and centrifuged to collect the plasma. The livers were rapidly excised, weighed, and flushed with ice‐cold saline, and an aliquot was fixed in 10% formalin for histologic analysis. The remainder of the liver was sectioned and snap frozen in liquid nitrogen. All animal protocols were approved by the Northwestern University Institutional Animal Care and Use Committee. All animals received humane care according to criteria outlined in the Guide for the Care and Use of Laboratory Animals prepared by the National Academy of Sciences and published by the National Institutes of Health.
Histologic Analysis
Liver sections were stained with hematoxylin and eosin (H&E), F4/80, and Masson’s trichrome stain at the Northwestern University Mouse Histology and Phenotyping Laboratory (Chicago, IL). Slides were blindly scored for grade of steatosis and inflammation. Steatosis grade was as follows: grade 0, <5% of liver parenchyma; grade 1, 5%‐25% of liver parenchyma; grade 2, 26%‐50% of liver parenchyma; grade 3, 51%‐75% of liver parenchyma; grade 4, >75% of liver parenchyma. Inflammation was scored by the number of inflammatory foci per 20× field. F4/80 staining was quantified by the number of positively stained cells per 100 hepatocytes. Quantification of trichrome staining (percentage of area) was performed using ImageJ software.
Liver and Plasma Chemistries
Liver samples were homogenized in Dulbecco’s phosphate‐buffered saline for hepatic lipid analysis (100 mg liver tissue/1 mL). Triglyceride levels were measured in liver homogenate using an Infinity spectrophotometric assay per the manufacturer’s protocol (Thermo Electron Corporation, Melbourne, Australia). Plasma alanine aminotransferase (ALT) was measured using a spectrophotometric assay per the manufacturer’s protocol (Teco Diagnostics, Anaheim, CA). Plasma levels of total and active PAI‐1 were measured using enzyme‐linked immunosorbent assay (ELISA) kits for total and active PAI‐1 (Molecular Innovations, Novi, MI). Levels of active tissue plasminogen activator (tPA) were measured in liver homogenate with a Mouse tPA Activity ELISA kit (Molecular Innovations).
Analysis of Gene Expression by Real‐Time Quantitative Polymerase Chain Reaction
Total RNA from frozen liver samples was isolated using TRIzol reagent (Invitrogen, Carlsbad, CA). Two micrograms of total RNA was used for reverse‐transcription polymerase chain reaction (PCR) using a qScript complementary DNA synthesis kit (Quanta BioSciences, Gaithersburg, MD). Real‐time quantitative PCR was performed using Quantitect SYBR Green PCR Mastermix (Qiagen, Valencia, CA) along with primers specific for the gene of interest. Glyceraldehyde 3‐phosphate dehydrogenase was used as a housekeeping gene. Amplification was performed on an ABI 7300 sequence detector (Applied Biosystems, Foster City, CA). Gene expression was calculated relative to respective age‐ and sex‐matched controls, using the comparative threshold cycle method.
Hydroxyproline Assay
The hydroxyproline content in liver samples was measured as described.19 Briefly, 500 μL of 6 N HCl was added to 50 mg of liver tissue and incubated at 100°C for 24 hours, followed by neutralization with 500 μL 6 N NaOH. The sample was then centrifuged at 13,000g for 12 minutes. This was followed by incubating 120 μL of sample with 75 μL of chloramines T solution for 10 minutes and then adding 450 μL of Ehrlich’s solution (perchloric acid/ para‐dimethylaminobenzaldehyde/isopropranol). Samples were incubated for 30 minutes at 65°C and read at an absorbance of 561 nm.
Statistical Analysis
Data are presented as mean ± SD. Comparisons between groups were performed using Student t test analysis.
Results
Pai‐1 Deletion Attenuates MCD Diet‐Induced Hepatic Steatosis but does not Prevent Hepatic Inflammation or Fibrosis
To determine the role of Pai‐1 in the development of murine steatohepatitis and fibrosis, we fed Pai‐1 –/– and Pai‐1 +/+ control mice an MCD or MCS (control) diet for 8 weeks. MCD feeding increased the plasma PAI‐1 level and induced hepatic Pai‐1 expression (Fig. 1A,B). As expected, 8 weeks of MCD feeding induced significant hepatic steatosis in wild‐type (Pai‐1 +/+) mice (Fig. 2A). Pai‐1 –/– mice showed a significant reduction in diet‐induced hepatic steatosis and hepatic triglyceride accumulation compared to Pai‐1 +/+ controls (Fig. 2A‐C). MCD and MCS diets contain a higher sucrose content than standard murine chow diets, and as a result, wild‐type mice fed an MCS control diet for 8 weeks developed a mild increase in hepatic triglyceride content relative to chow‐fed mice. Pai‐1 –/– mice were protected from the mild increase in hepatic triglyceride content induced by prolonged MCS feeding (Fig. 2).
Figure 1.
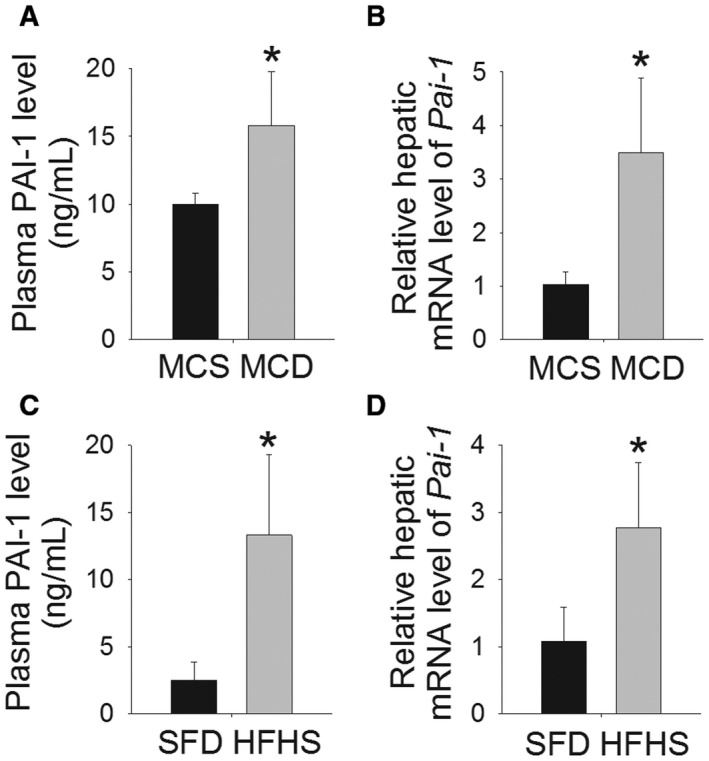
PAI‐1 is induced in murine dietary models of NASH. (A) Plasma level of PAI‐1 (ng/mL) and (B) hepatic mRNA level of Pai‐1 in wild‐type Pai‐1+/+ mice fed an MCD diet or MCS diet for 8 weeks. (C) Plasma level of PAI‐1 (ng/mL) and (D) hepatic mRNA level of Pai‐1 in wild‐type Pai‐1+/+ mice fed an HFHS diet or SFD for 16 weeks. Values are expressed as mean ± SD; n = 8; *P < 0.05 versus respective control diet. Abbreviation: mRNA, messenger RNA.
Figure 2.
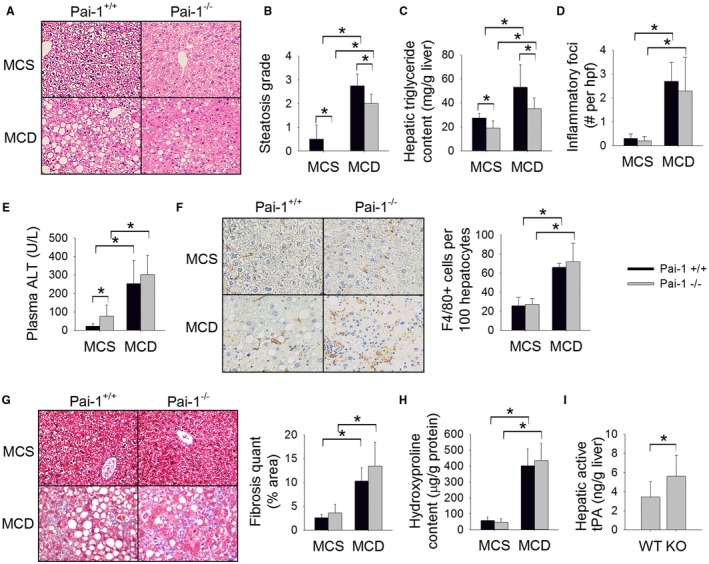
Pai‐1 deletion attenuates MCD diet‐induced hepatic steatosis but does not prevent hepatic inflammation or fibrosis. (A) H&E‐stained liver sections, (B) steatosis grade, (C) hepatic triglyceride content (mg/g liver), (D) number of inflammatory foci on H&E sections per hpf, (E) plasma ALT (U/L), (F) F4/80‐stained liver sections with quantification (# cells/100 hepatocytes), (G) Masson’s trichrome‐stained liver sections with quantification of trichrome staining, (H) hepatic hydroxyproline content (µg/g protein), and (I) hepatic level of active tPA (ng/g liver) in Pai‐1 –/– and Pai‐1+/+ mice fed an MCD or MCS (control) diet for 8 weeks. Values are expressed as mean ± SD; n = 7‐8; *P < 0.05. Abbreviations: KO, knockout; WT, wild type.
We next measured the expression of key genes regulating hepatic lipid metabolism among Pai‐1–/– and Pai‐1 +/+ controls fed an MCD diet (Table 1). Pai‐1–/– mice showed an increase in expression of peroxisome proliferator‐activated receptor α (Pparα) and its target gene peroxisome proliferator‐activated receptor γ coactivator 1α (Pgc1α), relative to Pai‐1+/+ mice in response to MCD feeding, suggesting that the attenuated hepatic steatosis in this model may be due in part to enhanced fatty acid oxidation. There was no significant effect of Pai‐1 deletion on the expression of fatty acid synthesis genes.
Table 1.
Hepatic Gene Expression in PAI‐1–/– and PAI‐1+/+ Mice Fed an MCD or MCS Diet for 8 Weeks
| MCS | MCD | ||||
|---|---|---|---|---|---|
| Pai‐1 +/+ | Pai‐1 –/– | Pai‐1 +/+ | Pai‐1 –/– | ||
| Triglyceride synthesis | Srebp‐1c | 1.1 ± 0.5 | 1.2 ± 0.7 | 1.0 ± 0.5 | 1.1 ± 0.7 |
| Fas | 1.0 ± 0.4 | 1.0 ± 0.3 | 0.4* ± 0.3 | 0.4* ± 0.2 | |
| Acc | 1.1 ± 0.5 | 0.6 ± 0.3 | 1.1 ± 0.5 | 0.8 ± 0.1 | |
| Fatty acid oxidation | Pparα | 1.0 ± 0.4 | 1.5 ± 1.0 | 0.9 ± 0.4 | 2.7*, † ± 1.5 |
| Pgc1α | 1.0 ± 0.2 | 1.2 ± 0.2 | 2.1* ± 0.5 | 4.5*, † ± 1.5 | |
| Aco | 1.1 ± 0.6 | 1.6 ± 0.5 | 0.9 ± 0.3 | 1.2 ± 0.5 | |
| Cpt1α | 1.0 ± 0.2 | 1.0 ± 0.2 | 0.8 ± 0.1 | 1.1 ± 0.4 | |
| Hepatic inflammation | Tnfα | 1.1 ± 0.4 | 1.1 ± 0.4 | 4.6* ± 1.3 | 3.6* ± 1.5 |
| Mcp‐1 | 1.1 ± 0.7 | 1.5 ± 0.4 | 9.1* ± 4.4 | 19.0* ± 10.0 | |
| Mip1α | 1.1 ± 0.6 | 1.3 ± 0.6 | 13.8* ± 6.1 | 19.6* ± 10.2 | |
| Hepatic fibrosis | Timp‐1 | 1.1 ± 0.8 | 1.7 ± 1.0 | 14.3* ± 5.0 | 14.9* ± 4.8 |
| Col1α1 | 1.5 ± 1.0 | 1.5 ± 0.8 | 6.8* ± 2.7 | 7.0* ± 2.8 | |
Relative expression, mean ± SD.
P < 0.05 versus MCS‐fed mice of the same genotype;
P < 0.05 versus Pai‐1 +/+ mice fed the same diet.
The MCD diet has been shown to promote inflammation and early stage fibrosis with prolonged feeding.20, 21, 22 As expected, MCD feeding induced hepatic inflammation as evidenced by an increased number of inflammatory foci on H&E‐stained liver sections, marked elevation in plasma ALT level, increased hepatic macrophage infiltration as quantified by hepatic F4/80 staining, and induction of hepatic inflammatory genes, including tumor necrosis factor α (Tnfα), monocyte chemoattractant protein 1 (Mcp‐1), and macrophage inflammatory protein 1α (Mip1α) (Fig. 2D‐F; Table 1). Deletion of Pai‐1 did not prevent the development of hepatic inflammation as evidenced by an equivalent number of inflammatory foci, a similar degree of ALT elevation, equivalent F4/80 staining, and similar induction of hepatic proinflammatory genes among Pai‐1 –/– and Pai‐1 +/+ mice.
MCD‐fed Pai‐1 +/+ mice showed early stage hepatic fibrosis as quantified on Masson’s trichrome‐stained liver sections (Fig. 2G). The degree of fibrosis was not attenuated by Pai‐1 deletion. Likewise, Pai‐1 –/– and Pai‐1 +/+ mice showed an equivalent increase in hepatic hydroxyproline content in response to MCD feeding (Fig. 2H). Furthermore, MCD‐fed Pai‐1 –/– and Pai‐1 +/+ mice showed a similar induction of hepatic genes associated with fibrosis, including tissue inhibitor of metalloproteinase 1 (Timp‐1) and collagen 1α1 (Col1α1) (Table 1). Deletion of Pai‐1 has been shown to be protective against hepatic fibrosis induced by bile duct ligation, an effect that has been attributed to enhanced activation of hepatic tPA.23 MCD‐fed Pai‐1–/– mice showed an expected increase in hepatic active tPA level, indicating that the absence of protection from fibrosis among Pai‐1–/– mice is not due to failed activation of hepatic tPA (Fig. 2K).
The MCD diet induces histologic features of NASH; however, there are numerous well‐established metabolic sequelae of MCD feeding that do not parallel human NASH, including weight loss and improved insulin sensitivity.24, 25 Notably, deletion of Pai‐1 had no impact on these MCD diet‐induced phenotypic features; MCD feeding resulted in an approximately 40% reduction in body weight during the feeding protocol, which was equivalent among Pai‐1 –/– and Pai‐1 +/+ mice (body weight reduction, –41.2% ± 2.6% versus –39.8% ± 2.2%, respectively; not significant). MCD feeding also caused a similar reduction in fasting blood glucose among Pai‐1 –/– and Pai‐1 +/+ mice (80 ± 8 versus 90 ± 17 mg/dL, respectively; not significant).
Pai‐1 Deletion does not Prevent HFHS Diet‐Induced Hepatic Fibrosis
The HFHS (“fast food diet”) is rapidly becoming the preferred murine dietary model of NASH. This diet has been shown to induce marked hepatic steatosis, inflammation, and early fibrosis with prolonged feeding, yet contrary to the MCD diet, the HFHS diet induces a metabolic phenotype similar to human NAFLD.26, 27, 28 Therefore, we next determined the effect of Pai‐1 deletion on HFHS diet‐induced NAFLD. Pai‐1 –/– and Pai‐1 +/+ control mice were fed an HFHS diet for 16 weeks. The development of obesity was associated with a rise in plasma levels of PAI‐1 and increased hepatic expression of Pai‐1 among wild‐type mice (Fig. 1C,D). Pai‐1 –/– mice showed early transient protection from weight gain relative to Pai‐1 +/+ mice fed the HFHS diet (Fig. 3A). In weeks 8 through 16 of HFHS feeding, however, weight gain among Pai‐1 –/– mice was not statistically different than that of Pai‐1 +/+ littermates. Likewise, Pai‐1 –/– and Pai‐1 +/+ mice showed a similar degree of hepatic steatosis and an equivalent increase in hepatic triglyceride content by week 16 of HFHS feeding (Fig. 3B‐D). Consistent with equivalent hepatic lipid accumulation, hepatic expression of lipogenesis genes was similar among Pai‐1 –/– and Pai‐1 +/+ mice (Table 2). Similar to the pattern observed among MCD‐fed mice, Pai‐1 –/– mice showed increased hepatic expression of Pparα relative to Pai‐1 +/+ mice at week 16 of HFHS feeding.
Figure 3.
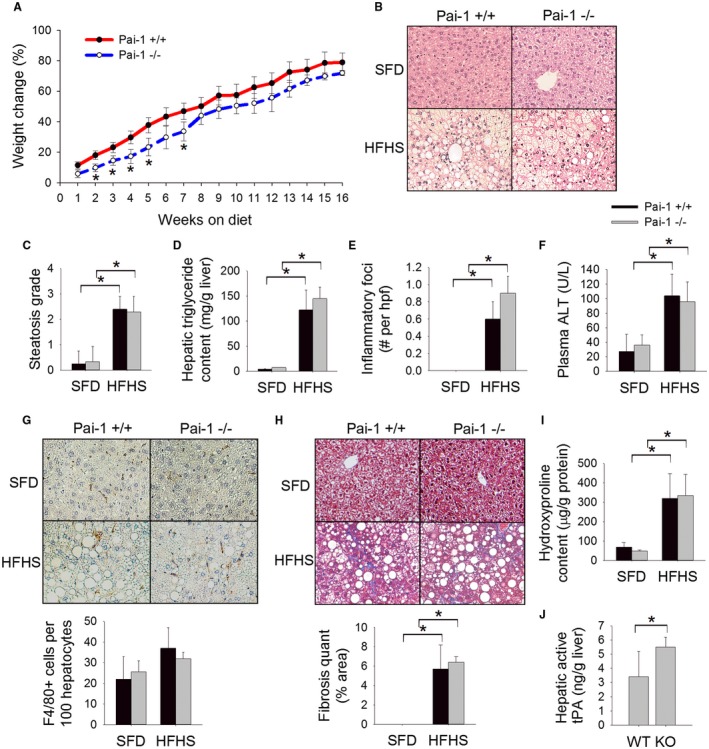
Pai‐1 deletion does not prevent HFHS diet‐induced hepatic inflammation or fibrosis. (A) Body weight change (%), (B) H&E‐stained liver sections, (C) steatosis grade, (D) hepatic triglyceride content (mg/g liver), (E) number of inflammatory foci on H&E sections per hpf, (F) plasma ALT (U/L), (G) F4/80‐stained liver sections with quantification (# cells/100 hepatocytes), (H) Masson’s trichrome‐stained liver sections with quantification of trichrome staining, (I) hepatic hydroxyproline content (µg/g protein), and (J) hepatic level of active tPA (ng/g liver) in Pai‐1 –/– and Pai‐1+/+ mice fed an HFHS diet or SFD for 16 weeks. Values are expressed as mean ± SD; n = 6‐9; *P < 0.05. Abbreviations: KO, knockout; WT, wild type.
Table 2.
Hepatic Gene Expression in PAI‐1–/– and PAI‐1+/+ Mice Fed an HFHS or SFD for 16 Weeks
| SFD | HFHS | ||||
|---|---|---|---|---|---|
| Pai‐1 +/+ | Pai‐1 –/– | Pai‐1 +/+ | Pai‐1 –/– | ||
| Triglyceride synthesis | Srebp‐1c | 1.1 ± 0.4 | 1.3 ± 0.3 | 2.3* ± 1.1 | 2.3* ± 0.7 |
| Fas | 1.2 ± 0.8 | 0.6 † ± 0.3 | 1.1 ± 0.6 | 0.6 † ± 0.1 | |
| Acc | 1.0 ± 0.1 | 0.7 † ± 0.1 | 0.8 ± 0.2 | 0.7 ± 0.1 | |
| Fatty acid oxidation | Pparα | 1.0 ± 0.2 | 0.9 ± 0.7 | 0.3* ± 0.2 | 1.0 † ± 0.7 |
| Pgc1α | 1.0 ± 0.3 | 0.5 † ± 0.1 | 0.2* ± 0.1 | 0.2* ± 0.1 | |
| Aco | 1.1 ± 0.5 | 0.9 ± 0.8 | 1.4 ± 0.4 | 1.9* ± 0.7 | |
| Cpt1α | 1.0 ± 0.1 | 1.7 † ± 0.2 | 1.1 ± 0.3 | 1.3 ± 0.1 | |
| Hepatic inflammation | Tnfα | 1.0 ± 0.3 | 1.1 ± 0.4 | 1.9* ± 0.7 | 3.3* ± 1.6 |
| Mcp‐1 | 1.3 ± 0.9 | 2.3 † ± 0.8 | 2.3* ± 1.7 | 3.9* ± 1.8 | |
| Mip1α | 1.1 ± 0.5 | 1.8 ± 0.6 | 2.5* ± 0.9 | 5.5*, † ± 3.8 | |
| Hepatic fibrosis | Timp‐1 | 1.2 ± 0.8 | 0.7 † ± 0.2 | 3.4* ± 1.5 | 4.5* ± 1.9 |
| Col1α1 | 1.1 ± 0.5 | 0.4 † ± 0.1 | 3.8* ± 1.8 | 3.7* ± 1.2 | |
Relative expression, mean ± SD.
P < 0.05 versus SFD‐fed mice of the same genotype;
P < 0.05 versus Pai‐1 +/+ mice fed the same diet.
Deletion of Pai‐1 did not prevent the development of hepatic inflammation as evidenced by an equivalent number of inflammatory foci in liver sections, a similar degree of plasma ALT elevation, equivalent hepatic macrophage infiltration, and equal induction of proinflammatory genes among Pai‐1 –/– and Pai‐1 +/+ mice (Fig. 3E‐G; Table 2). As was observed with MCD feeding, the degree of fibrosis induced by an HFHS diet was not attenuated by Pai‐1 deletion as evidenced by equivalent liver trichrome staining, equivalent hepatic hydroxyproline content, and similar induction of hepatic fibrosis genes (Fig. 3H,I; Table 2). Pai‐1 –/– mice showed an appropriate increase in hepatic active tPA relative to Pai‐1 +/+ mice (Fig. 3J).
Pharmacologic Inhibition of Pai‐1 Attenuates MCD Diet‐Induced Hepatic Steatosis but does not Prevent Hepatic Fibrosis
With the ultimate goal of identifying a pharmacologic agent to treat NASH‐induced fibrosis, we next explored the effect of TM5441, an orally active highly specific small molecule inhibitor of PAI‐1, on diet‐induced NASH.17 Wild‐type C57BL/6 mice were fed an MCD or MCS diet with or without TM5441 for 8 weeks. Among MCD‐fed mice, TM5441 resulted in a 57% reduction in the plasma level of active PAI‐1 (Fig. 4A). Similar to genetic deletion of Pai‐1, pharmacologic inhibition of PAI‐1 caused a modest reduction in MCD diet‐induced hepatic steatosis (Fig. 4B,C). Quantification of the hepatic triglyceride content demonstrated a trend toward reduced hepatic triglyceride content among mice treated with TM5441 (Fig. 4D). Given the modest effect of TM5441 on MCD diet‐induced hepatic triglyceride accumulation, we considered whether a dose–response relationship exists. We therefore fed an additional cohort of mice the MCD diet with a higher dose of TM5441 (40 mg/kg/day) for 8 weeks. Increasing the dose of TM5441 did not result in enhanced reduction in hepatic steatosis (Supporting Fig. S1). As was seen among Pai‐1 –/– mice fed an MCD diet, pharmacologic inhibition of PAI‐1 resulted in enhanced induction of Pparα and Pgc1α in response to MCD feeding (Table 3).
Figure 4.
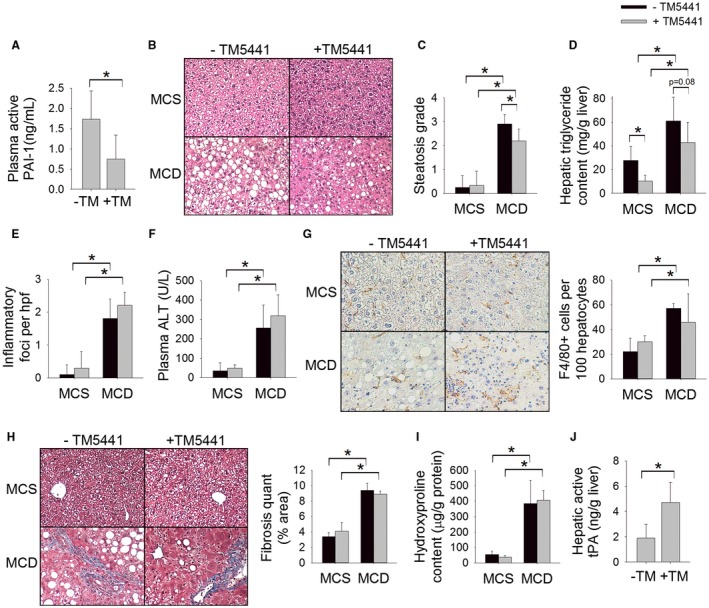
Pharmacologic inhibition of PAI‐1 attenuates MCD diet‐induced hepatic steatosis but does not prevent hepatic fibrosis. (A) Plasma level of active PAI‐1 (ng/mL), (B) H&E‐stained liver sections, (C) steatosis grade, (D) hepatic triglyceride content (mg/g liver), (E) number of inflammatory foci on H&E sections per hpf, (F) plasma ALT (U/L), (G) F4/80‐stained liver sections with quantification (# cells/100 hepatocytes), (H) Masson’s trichrome‐stained liver sections with quantification of trichrome staining, (I) hepatic hydroxyproline content (µg/g protein), and (J) hepatic level of active tPA (ng/g liver) in C57BL/6J mice fed an MCD or MCS diet with or without TM5441 for 8 weeks. Values are expressed as mean ± SD; n = 8; *P < 0.05.
Table 3.
Hepatic Gene Expression in C57BL/6J Mice Fed an MCD or MCS Diet With or Without TM5441 for 8 Weeks
| MCS | MCD | ||||
|---|---|---|---|---|---|
| –TM5441 | +TM5441 | –TM5441 | +TM5441 | ||
| Triglyceride synthesis | Srebp‐1c | 1.1 ± 0.4 | 1.6 ± 1.3 | 1.3 ± 0.6 | 2.3 ± 0.7 |
| Fas | 1.0 ± 0.3 | 1.1 ± 0.5 | 0.4* ± 0.2 | 0.3* ± 0.1 | |
| Acc | 1.0 ± 0.2 | 0.8 ± 0.1 | 0.7 ± 0.2 | 0.7 ± 0.2 | |
| Fatty acid oxidation | Pparα | 1.0 ± 0.2 | 1.0 ± 0.2 | 1.0 ± 0.6 | 2.3*, † ± 1.2 |
| Pgc1α | 1.0 ± 0.2 | 1.5 ± 0.7 | 2.8* ± 1.7 | 2.9* ± 1.6 | |
| Aco | 1.0 ± 0.3 | 1.5 ± 0.7 | 0.7 ± 0.3 | 0.9 ± 0.3 | |
| Cpt1α | 1.0 ± 0.3 | 1.0 ± 0.0 | 0.9 ± 0.1 | 1.7 ± 1.2 | |
| Hepatic inflammation | Tnfα | 1.1 ± 0.7 | 1.1 ± 0.4 | 5.6* ± 3.0 | 7.0* ± 2.7 |
| Mcp‐1 | 1.1 ± 0.6 | 1.1 ± 0.6 | 5.3* ± 2.8 | 6.3* ± 6.2 | |
| Mip1α | 1.1 ± 0.6 | 1.2 ± 0.4 | 11.4* ± 6.7 | 9.5* ± 5.5 | |
| Hepatic fibrosis | Timp‐1 | 1.1 ± 0.5 | 1.3 ± 0.6 | 15.0* ± 9.0 | 18.1* ± 7.1 |
| Col1α1 | 1.1 ± 0.7 | 2.0 ± 1.0 | 6.8* ± 2.7 | 10.1* ± 2.8 | |
Relative expression, mean ± SD.
P < 0.05 versus MCS‐fed mice with the same drug exposure (+/–TM5441);
P < 0.05 versus nondrug‐treated mice (–TM5441) fed the same diet.
Similar to genetic Pai‐1 deletion, pharmacologic inhibition of PAI‐1 did not attenuate markers of hepatic inflammation or hepatic fibrosis (Fig. 4E‐I; Table 3). Pharmacologic inhibition of PAI‐1 appropriately increased the hepatic level of active tPA (Fig. 4J).
Inhibition of Pai‐1 Attenuates HFHS Diet‐Induced Obesity and Hepatic Steatosis but does not Prevent Hepatic Fibrosis
We next examined the effect of PAI‐1‐inhibition on HFHS diet‐induced NAFLD. Wild‐type C57BL/6J mice were treated with TM5441 in conjunction with an HFHS diet or SFD for up to 16 weeks. Among mice fed an HFHS diet, TM5441 reduced the plasma level of active PAI‐1 by 53% (Fig. 5A). It has been reported that TM5441 attenuates high‐fat diet‐induced weight gain in mice.15 Similarly, we found that inhibition of PAI‐1 significantly attenuated HFHS diet‐induced weight gain beginning as early as 2 weeks into the experimental protocol (Fig. 5B). With prolonged HFHS feeding, however, the weight curves began to converge as the mice approached maximal gain. Moreover, in weeks 11 through 16, the weight gain among mice fed an HFHS diet supplemented with TM5441 was not significantly different compared to mice fed the HFHS diet alone.
Figure 5.
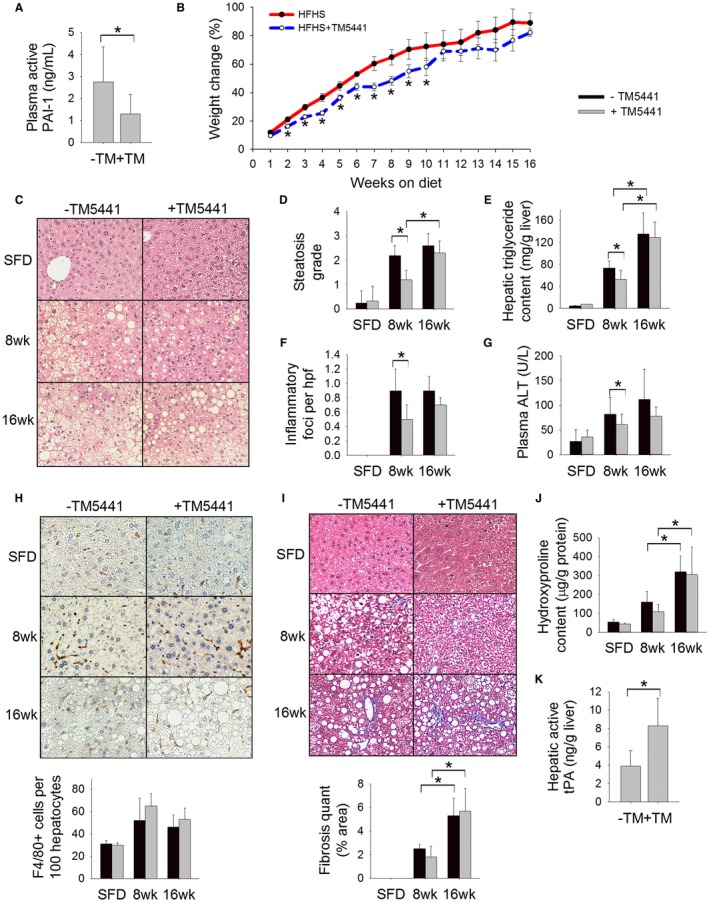
Inhibition of PAI‐1 attenuates HFHS diet‐induced obesity and hepatic steatosis but does not prevent hepatic fibrosis. (A) Plasma level of active PAI‐1 (ng/mL), (B) body weight change (%), (C) H&E‐stained liver sections, (D) steatosis grade, (E) hepatic triglyceride content (mg/g liver), (F) number of inflammatory foci on H&E sections per hpf, (G) plasma ALT (U/L), (H) F4/80‐stained liver sections with quantification (# cells/100 hepatocytes), (I) Masson’s trichrome‐stained liver sections with quantification of trichrome staining, (J) hepatic hydroxyproline content (µg/g protein), and (K) hepatic level of active tPA (ng/g liver) in C57BL/6J mice fed an HFHS diet or SFD with or without TM5441 for 8 weeks or 16 weeks. Values are expressed as mean ± SD; n = 8; *P < 0.05. SFD was statistically different (P < 0.05) than mice fed an HFHS diet for 8 or 16 weeks for all parameters measured.
It was recently shown that TM5441 attenuates high‐fat diet‐induced hepatic steatosis in mice.16 Consistent with these findings, mice fed an HFHS diet supplemented with TM5441 showed reduced hepatic steatosis and a reduced hepatic triglyceride content at week 8 compared to mice fed an HFHS diet without TM5441 (Fig. 5C‐E). However, by week 16, there was a similar degree of hepatic steatosis among mice treated with TM5441 compared to HFHS alone. HFHS feeding was associated with induction of hepatic sterol regulatory element binding protein 1c (Srebp1c) expression, consistent with the known paradoxical effect of hepatic steatosis on hepatic lipogenesis.29 Mice treated with TM5441 showed no induction of Srebp1c at 8 weeks, reflecting the decreased steatosis at this time point. By week 16, TM5441‐treated mice showed equal induction of hepatic Srebp1c compared to mice fed the HFHS diet alone.
Paralleling the reduction in steatosis at week 8, TM5441 also resulted in a reduction in plasma ALT, hepatic Tnfα expression, and the number of inflammatory foci on H&E‐stained liver sections at week 8 (Fig. 5F,G; Table 4); this effect was lost by week 16 when body weight and degree of hepatic steatosis equalized between the two cohorts. TM5441 did not significantly affect hepatic macrophage infiltration or hepatic expression of Mcp1 or Mip1α at either 8 or 16 weeks of HFHS feeding (Fig. 5H; Table 4). There was attenuated expression of fibrosis‐related genes at week 8 among mice treated with TM5441 (Table 4) yet no significant difference in hepatic trichrome staining or hepatic hydroxyproline content (Fig. 5I,J). By week 16, there was no difference in any fibrosis markers among mice fed an HFHS diet with TM5441 versus HFHS alone (Fig. 5I,J; Table 4). As expected, treatment with TM5441 increased the hepatic level of active tPA (Fig. 5K). Overall, these data indicate that PAI‐1 inhibition delays but does not prevent the development of NASH in mice.
Table 4.
Hepatic Gene Expression in C57BL/6J Mice Fed an HFHS Diet or SFD With or Without TM5441 for 8 or 16 Weeks
| SFD | HFHS × 8 Weeks | HFHS × 16 Weeks | |||||
|---|---|---|---|---|---|---|---|
| –TM5441 | +TM5441 | –TM5441 | +TM5441 | –TM5441 | +TM5441 | ||
| Triglyceride synthesis | Srebp‐1c | 1.1 ± 0.4 | 0.5 † ± 0.4 | 2.5* ± 0.6 | 0.9 † ± 0.7 | 2.8* ± 0.8 | 2.5* ± 0.8 |
| Fas | 1.2 ± 0.8 | 0.5 † ± 0.2 | 0.8 ± 0.2 | 0.3 † ± 0.2 | 1.3 ± 0.5 | 1.0 ± 0.2 | |
| Acc | 1.0 ± 0.3 | 0.6 † ± 0.1 | 0.6 ± 0.2 | 0.6 ± 0.1 | 0.8 ± 0.1 | 0.8 ± 0.1 | |
| Fatty acid oxidation | Pparα | 1.0 ± 0.2 | 1.6 ± 0.4 | 2.0* ± 0.8 | 0.9 ± 0.5 | 0.8 ± 0.2 | 3.5*, † ± 2.1 |
| Pgc1α | 1.1 ± 0.5 | 0.9 ± 0.1 | 0.5* ± 0.1 | 0.4* ± 0.3 | 0.3* ± 0.2 | 0.5* ± 0.2 | |
| Aco | 1.0 ± 0.2 | 1.6 † ± 0.3 | 1.0 ± 0.4 | 1.1 ± 0.8 | 0.6* ± 0.2 | 0.9 ± 0.3 | |
| Cpt1α | 1.0 ± 0.1 | 1.6 † ± 0.2 | 1.2 ± 0.2 | 1.1 ± 0.5 | 1.8 ± 0.6 | 1.0 ± 0.1 | |
| Hepatic inflammation | Tnfα | 1.0 ± 0.3 | 1.2 ± 0.6 | 1.7* ± 0.7 | 0.6 ± 0.5 | 0.6 ± 0.2 | 1.0 ± 0.2 |
| Mcp‐1 | 1.0 ± 0.4 | 2.1 ± 1.8 | 2.6* ± 0.6 | 5.3*, † ± 4.1 | 4.6* ± 3.9 | 2.6* ± 1.5 | |
| Mip1α | 1.1 ± 0.5 | 1.8 ± 0.6 | 1.1 ± 0.2 | 1.2 ± 0.3 | 3.2* ± 2.3 | 2.1* ± 1.3 | |
| Hepatic fibrosis | Timp‐1 | 1.2 ± 1.0 | 0.7 ± 0.2 | 5.4* ± 2.0 | 2.6*, † ± 0.8 | 5.3* ± 1.9 | 4.4* ± 0.8 |
| Col1α1 | 1.1 ± 0.5 | 0.3 † ± 0.1 | 2.1* ± 0.8 | 0.7 † ± 0.5 | 1.8* ± 1.0 | 2.5* ± 1.1 | |
Relative expression, mean ± SD.
P < 0.05 versus SFD‐fed mice with the same drug exposure (+/–TM5441);
P < 0.05 versus nondrug‐treated mice (–TM5441) fed the same diet.
Discussion
PAI‐1 is increasingly recognized as a mediator of metabolic diseases, a function distinct from its canonical role in the plasminogen activator (i.e., fibrinolytic) system.12, 13 Although initially conceptualized as a treatment for human thrombotic disease, PAI‐1 antagonism is emerging as a highly promising therapeutic strategy for metabolic disorders, including obesity and NAFLD.15, 16 Given the well‐established profibrotic function of PAI‐1, we explored whether PAI‐1 may serve as a target for antifibrotic therapy in NASH. We demonstrated that pharmacologic inhibition of PAI‐1 confers transient protection from hepatic steatosis but ultimately does not protect against hepatic fibrosis in murine models of NASH.
The improvement in hepatic steatosis among mice fed an HFHS diet supplemented with TM5441 corroborates the recent findings of Lee et al.16 who demonstrated that TM5441 protects against high‐fat diet‐induced steatosis. Contrary to our findings, however, those authors also reported that TM5441 attenuated hepatic fibrosis. The degree of inflammation and fibrosis observed by Lee et al.16 after feeding C57BL/6J mice a purely high‐fat diet for 10 weeks was remarkable given that mice are relatively resistant to the development of NASH with fibrosis. Moreover, it has been widely reported that even with prolonged high‐fat feeding, C57BL/6J mice develop scant or no hepatic fibrosis.30, 31, 32, 33, 34, 35 As such, with the goal of identifying PAI‐1 antagonism as an antifibrotic therapy in NASH, we employed two distinct dietary models of murine NASH with fibrosis. Surprisingly, neither genetic deletion of Pai‐1 nor pharmacologic inhibition of PAI‐1 protected against the development of NASH‐related fibrosis.
Although PAI‐1 has been implicated in the development of fibrosis in numerous tissues, the role of PAI‐1 in the development of hepatic fibrosis is controversial. Moreover, studies in vitro and in vivo indicate that PAI‐1 may have both profibrotic and antifibrotic effects.23, 36, 37, 38, 39 PAI‐1 blocks the conversion of plasminogen to plasmin, which can degrade the extracellular matrix (ECM) directly through degradation of ECM proteins (type IV collagen, fibrin, fibronectin) and indirectly by activation of matrix metalloproteinases.40, 41 Studies in isolated and cultured stellate cells suggest that the antifibrotic effects of PAI‐1 may be mediated by inhibition of interstitial collagenases during the initiation of fibrosis.38 Numerous studies in models of renal disease indicate that PAI‐1 deficiency is protective against renal fibrosis.5, 42 Conversely, Pai‐1 deletion has been shown to promote cardiac fibrosis.43 Pai‐1 –/– mice exposed to CCl4, a widely used model of liver fibrosis, demonstrated enhanced liver fibrosis attributed to enhanced fibrogenesis and impaired ECM degradation.36 Clearly, whether PAI‐1 exhibits a profibrotic or antifibrotic effect is highly dependent on the tissue type and experimental conditions.
Increasingly, PAI‐1 is being implicated as a driver of obesity.13, 15, 44 It has been shown that the novel small molecule inhibitor of PAI‐1, TM5441, attenuates high‐fat diet‐induced weight gain in mice.15 Our data corroborate this finding using an alternative obesigenic diet. We found, however, that the beneficial effects of TM5441 on obesity are lost with prolonged exposure to the HFHS diet when weight gain begins to plateau. Moreover, TM5441 slows rather than prevents weight gain, but ultimately, and not unexpectedly, the weight gain curves converge as the mice approach maximal weight gain. Importantly, we find that the protection from hepatic steatosis observed with TM5441 treatment is lost when weight gain reaches that of mice fed the HFHS diet without TM5441. These data suggest that the beneficial effects of TM5441 on HFHS diet‐induced hepatic steatosis may be a direct consequence of attenuated weight gain. Additionally, however, hepatic expression of Pparα was consistently induced in our models of PAI‐1 inhibition or deletion, suggesting that enhanced fatty acid oxidation may contribute to the observed attenuation of steatosis. PPARα has been shown to induce Pai‐1 gene expression; however, the mechanism for compensatory induction of Pparα in the absence of Pai‐1 is unknown.45 An additional argument against attenuated weight gain being the sole mechanism of protection against steatosis is the observation that genetic deletion of Pai‐1 and pharmacologic inhibition of PAI‐1 conferred modest protection from MCD diet‐induced hepatic steatosis in the absence of differences in body weight. Caution must be taken, however, in extrapolating mechanisms of MCD diet‐induced hepatic steatosis in mice to human hepatic steatosis. The MCD diet was chosen for this study to specifically examine the effect of PAI‐1 on hepatic fibrosis. Moreover, the MCD diet produces histologic findings that largely recapitulate human NASH with fibrosis; however, the metabolic sequelae of MCD feeding are entirely contrary to the human disease. As such, we assert that the pathogenesis of MCD diet‐induced hepatic lipid accumulation provides limited insight into the mechanism of human hepatic steatosis.
It is a well‐established observation that steatohepatitis induces hepatic PAI‐1 expression and increases circulating PAI‐1 levels in humans and murine models of NASH; yet it was previously unknown whether PAI‐1 induction drives the hepatic inflammatory response that differentiates steatohepatitis from simple steatosis. We found that disruption of PAI‐1 did not protect against hepatic inflammation, suggesting that PAI‐1 induction in NASH models is a consequence rather than a cause of the hepatic inflammatory response. Moreover, inflammation is a well‐established inducer of hepatic Pai‐1 expression.46 Furthermore, proinflammatory mediators linked to NASH, such as TNFα, have been shown to directly induce Pai‐1 expression.46, 47 These data raise the possibility that PAI‐1 may mediate sequelae of advanced NASH rather than underlie its pathogenesis. Given the known function of PAI‐1 in promoting thrombosis and cancer, it is an intriguing speculation that PAI‐1 may mediate the increased risk of cardiovascular disease, thrombosis, and/or hepatocellular carcinoma in patients with advanced stages of NASH. Further studies are warranted to determine whether enhanced PAI‐1 expression in the setting of progressive NASH contributes to the development of these and other well‐established sequelae of NASH.
The prevalence of NAFLD is reaching epidemic proportions, yet effective pharmacotherapy is lacking. Our findings do not support a role for PAI‐1 antagonism as an antifibrotic therapy in NASH; however, these data do support a potential role for PAI‐1 inhibitors in the management of obesity and hepatic steatosis. Furthermore, if PAI‐1 inhibition were to have proven benefit in human obesity, we assert that there may be therapeutic benefit to concomitantly reducing steatosis even in the absence of an anti‐inflammatory or antifibrotic effect on the liver. Moreover, it has become evident that hepatic steatosis is associated with severe comorbidities even in the absence of advanced liver disease. Most notably, NAFLD has been identified as an independent risk factor for cardiovascular disease and confers increased mortality largely attributable to malignancy and cardiovascular complications.48, 49, 50 Future studies are warranted to determine the role of PAI‐1 inhibition in preventing, or ideally reversing, human metabolic diseases.
Potential conflict of interest
Dr. Miyata owns stock in Renascience. The other authors have nothing to report.
Supporting information
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases (National Institutes of Health [NIH] award K08DK095992 to A.S.H.), the National Heart, Lung, and Blood Institute (NIH award R01HL051387 to D.E.V.), and a U.S. Department of Veterans Affairs VA Merit Award (BX‐003854‐01 to A.S.H.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH or Department of Veterans Affairs.
References
- 1. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Dulai PS, Singh S, Patel J, Soni M, Prokop LJ, Younossi Z, et al. Increased risk of mortality by fibrosis stage in nonalcoholic fatty liver disease: systematic review and meta‐analysis. Hepatology 2017;65:1557‐1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Singh S, Khera R, Allen AM, Murad MH, Loomba R. Comparative effectiveness of pharmacological interventions for nonalcoholic steatohepatitis: a systematic review and network meta‐analysis. Hepatology 2015;62:1417‐1432. [DOI] [PubMed] [Google Scholar]
- 4. Boettcher E, Csako G, Pucino F, Wesley R, Loomba R. Meta‐analysis: pioglitazone improves liver histology and fibrosis in patients with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther 2012;35:66‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Huang Y, Haraguchi M, Lawrence DA, Border WA, Yu L, Noble NA. A mutant, noninhibitory plasminogen activator inhibitor type 1 decreases matrix accumulation in experimental glomerulonephritis. J Clin Invest 2003;112:379‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kaikita K, Fogo AB, Ma L, Schoenhard JA, Brown NJ, Vaughan DE. Plasminogen activator inhibitor‐1 deficiency prevents hypertension and vascular fibrosis in response to long‐term nitric oxide synthase inhibition. Circulation 2001;104:839‐844. [DOI] [PubMed] [Google Scholar]
- 7. Targher G, Bertolini L, Scala L, Zenari L, Lippi G, Franchini M, et al. Plasma PAI‐1 levels are increased in patients with nonalcoholic steatohepatitis. Diabetes Care 2007;30:e31‐e32. [DOI] [PubMed] [Google Scholar]
- 8. Targher G, Bertolini L, Rodella S, Lippi G, Franchini M, Zoppini G. NASH predicts plasma inflammatory biomarkers independently of visceral fat in men. Obesity (Silver Spring) 2008;16:1394‐1399. [DOI] [PubMed] [Google Scholar]
- 9. Sookoian S, Castano GO, Burgueno AL, Rosselli MS, Gianotti TF, Mallardi P, et al. Circulating levels and hepatic expression of molecular mediators of atherosclerosis in nonalcoholic fatty liver disease. Atherosclerosis 2010;209:585‐591. [DOI] [PubMed] [Google Scholar]
- 10. Thuy S, Ladurner R, Volynets V, Wagner S, Strahl S, Konigsrainer A, et al. Nonalcoholic fatty liver disease in humans is associated with increased plasma endotoxin and plasminogen activator inhibitor 1 concentrations and with fructose intake. J Nutr 2008;138:1452‐1455. [DOI] [PubMed] [Google Scholar]
- 11. Verrijken A, Francque S, Mertens I, Prawitt J, Caron S, Hubens G, et al. Prothrombotic factors in histologically proven nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology 2014;59:121‐129. [DOI] [PubMed] [Google Scholar]
- 12. Vaughan DE. PAI‐1 and atherothrombosis. J Thromb Haemost 2005;3:1879‐1883. [DOI] [PubMed] [Google Scholar]
- 13. De Taeye B, Smith LH, Vaughan DE. Plasminogen activator inhibitor‐1: a common denominator in obesity, diabetes and cardiovascular disease. Curr Opin Pharmacol 2005;5:149‐154. [DOI] [PubMed] [Google Scholar]
- 14. Khan SS, Shah SJ, Klyachko E, Baldridge AS, Eren M, Place AT, et al. A null mutation in SERPINE1 protects against biological aging in humans. Sci Adv 2017;3:eaao1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Piao L, Jung I, Huh JY, Miyata T, Ha H. A novel plasminogen activator inhibitor‐1 inhibitor, TM5441, protects against high‐fat diet‐induced obesity and adipocyte injury in mice. Br J Pharmacol 2016;173:2622‐2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SM, Dorotea D, Jung I, Nakabayashi T, Miyata T, Ha H. TM5441, a plasminogen activator inhibitor‐1 inhibitor, protects against high fat diet‐induced non‐alcoholic fatty liver disease. Oncotarget 2017;8:89746‐89760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boe AE, Eren M, Murphy SB, Kamide CE, Ichimura A, Terry D, et al. Plasminogen activator inhibitor‐1 antagonist TM5441 attenuates Nomega‐nitro‐L‐arginine methyl ester‐induced hypertension and vascular senescence. Circulation 2013;128:2318‐2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Eren M, Boe AE, Murphy SB, Place AT, Nagpal V, Morales‐Nebreda L, et al. PAI‐1‐regulated extracellular proteolysis governs senescence and survival in Klotho mice. Proc Natl Acad Sci U S A 2014;111:7090‐7095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Reddy GK, Enwemeka CS. A simplified method for the analysis of hydroxyproline in biological tissues. Clin Biochem 1996;29:225‐229. [DOI] [PubMed] [Google Scholar]
- 20. Yamaguchi K, Yang L, McCall S, Huang J, Yu XX, Pandey SK, et al. Inhibiting triglyceride synthesis improves hepatic steatosis but exacerbates liver damage and fibrosis in obese mice with nonalcoholic steatohepatitis. Hepatology 2007;45:1366‐1374. [DOI] [PubMed] [Google Scholar]
- 21. Ip E, Farrell GC, Robertson G, Hall P, Kirsch R, Leclercq I. Central role of PPARalpha‐dependent hepatic lipid turnover in dietary steatohepatitis in mice. Hepatology 2003;38:123‐132. [DOI] [PubMed] [Google Scholar]
- 22. Rinella ME, Elias MS, Smolak RR, Fu T, Borensztajn J, Green RM. Mechanisms of hepatic steatosis in mice fed a lipogenic methionine choline‐deficient diet. J Lipid Res 2008;49:1068‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wang H, Zhang Y, Heuckeroth RO. PAI‐1 deficiency reduces liver fibrosis after bile duct ligation in mice through activation of tPA. FEBS Lett 2007;581:3098‐3104. [DOI] [PubMed] [Google Scholar]
- 24. Rizki G, Arnaboldi L, Gabrielli B, Yan J, Lee GS, Ng RK, et al. Mice fed a lipogenic methionine‐choline‐deficient diet develop hypermetabolism coincident with hepatic suppression of SCD‐1. J Lipid Res 2006;47:2280‐2290. [DOI] [PubMed] [Google Scholar]
- 25. Rinella ME, Green RM. The methionine‐choline deficient dietary model of steatohepatitis does not exhibit insulin resistance. J Hepatol 2004;40:47‐51. [DOI] [PubMed] [Google Scholar]
- 26. Maher JJ. New insights from rodent models of fatty liver disease. Antioxid Redox Signal 2011;15:535‐550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Charlton M, Krishnan A, Viker K, Sanderson S, Cazanave S, McConico A, et al. Fast food diet mouse: novel small animal model of NASH with ballooning, progressive fibrosis, and high physiological fidelity to the human condition. Am J Physiol Gastrointest Liver Physiol 2011;301:G825‐G834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ganz M, Bukong TN, Csak T, Saha B, Park JK, Ambade A, et al. Progression of non‐alcoholic steatosis to steatohepatitis and fibrosis parallels cumulative accumulation of danger signals that promote inflammation and liver tumors in a high fat‐cholesterol‐sugar diet model in mice. J Transl Med 2015;13:193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferre P, Foufelle F. Hepatic steatosis: a role for de novo lipogenesis and the transcription factor SREBP‐1c. Diabetes Obes Metab 2010;12(Suppl. 2):83‐92. [DOI] [PubMed] [Google Scholar]
- 30. Ito M, Suzuki J, Tsujioka S, Sasaki M, Gomori A, Shirakura T, et al. Longitudinal analysis of murine steatohepatitis model induced by chronic exposure to high‐fat diet. Hepatol Res 2007;37:50‐57. [DOI] [PubMed] [Google Scholar]
- 31. Savard C, Tartaglione EV, Kuver R, Haigh WG, Farrell GC, Subramanian S, et al. Synergistic interaction of dietary cholesterol and dietary fat in inducing experimental steatohepatitis. Hepatology 2013;57:81‐92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ogasawara M, Hirose A, Ono M, Aritake K, Nozaki Y, Takahashi M, et al. A novel and comprehensive mouse model of human non‐alcoholic steatohepatitis with the full range of dysmetabolic and histological abnormalities induced by gold thioglucose and a high‐fat diet. Liver Int 2011;31:542‐551. [DOI] [PubMed] [Google Scholar]
- 33. Gaemers IC, Stallen JM, Kunne C, Wallner C, van Werven J, Nederveen A, et al. Lipotoxicity and steatohepatitis in an overfed mouse model for non‐alcoholic fatty liver disease. Biochim Biophys Acta 2011;1812:447‐458. [DOI] [PubMed] [Google Scholar]
- 34. Lo L, McLennan SV, Williams PF, Bonner J, Chowdhury S, McCaughan GW, et al. Diabetes is a progression factor for hepatic fibrosis in a high fat fed mouse obesity model of non‐alcoholic steatohepatitis. J Hepatol 2011;55:435‐444. [DOI] [PubMed] [Google Scholar]
- 35. Kubota N, Kado S, Kano M, Masuoka N, Nagata Y, Kobayashi T, et al. A high‐fat diet and multiple administration of carbon tetrachloride induces liver injury and pathological features associated with non‐alcoholic steatohepatitis in mice. Clin Exp Pharmacol Physiol 2013;40:422‐430. [DOI] [PubMed] [Google Scholar]
- 36. von Montfort C, Beier JI, Kaiser JP, Guo L, Joshi‐Barve S, Pritchard MT, et al. PAI‐1 plays a protective role in CCl4‐induced hepatic fibrosis in mice: role of hepatocyte division. Am J Physiol Gastrointest Liver Physiol 2010;298:G657‐G666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bergheim I, Guo L, Davis MA, Duveau I, Arteel GE. Critical role of plasminogen activator inhibitor‐1 in cholestatic liver injury and fibrosis. J Pharmacol Exp Ther 2006;316:592‐600. [DOI] [PubMed] [Google Scholar]
- 38. Leyland H, Gentry J, Arthur MJ, Benyon RC. The plasminogen‐activating system in hepatic stellate cells. Hepatology 1996;24:1172‐1178. [DOI] [PubMed] [Google Scholar]
- 39. Ng VL, Sabla GE, Melin‐Aldana H, Kelley‐Loughnane N, Degen JL, Bezerra JA. Plasminogen deficiency results in poor clearance of non‐fibrin matrix and persistent activation of hepatic stellate cells after an acute injury. J Hepatol 2001;35:781‐789. [DOI] [PubMed] [Google Scholar]
- 40. Liotta LA, Goldfarb RH, Brundage R, Siegal GP, Terranova V, Garbisa S. Effect of plasminogen activator (urokinase), plasmin, and thrombin on glycoprotein and collagenous components of basement membrane. Cancer Res 1981;41:4629‐4636. [PubMed] [Google Scholar]
- 41. Mackay AR, Corbitt RH, Hartzler JL, Thorgeirsson UP. Basement membrane type IV collagen degradation: evidence for the involvement of a proteolytic cascade independent of metalloproteinases. Cancer Res 1990;50:5997‐6001. [PubMed] [Google Scholar]
- 42. Oda T, Jung YO, Kim HS, Cai X, Lopez‐Guisa JM, Ikeda Y, et al. PAI‐1 deficiency attenuates the fibrogenic response to ureteral obstruction. Kidney Int 2001;60:587‐596. [DOI] [PubMed] [Google Scholar]
- 43. Ghosh AK, Bradham WS, Gleaves LA, De Taeye B, Murphy SB, Covington JW, et al. Genetic deficiency of plasminogen activator inhibitor‐1 promotes cardiac fibrosis in aged mice: involvement of constitutive transforming growth factor‐beta signaling and endothelial‐to‐mesenchymal transition. Circulation 2010;122:1200‐1209. [DOI] [PubMed] [Google Scholar]
- 44. Ma LJ, Mao SL, Taylor KL, Kanjanabuch T, Guan Y, Zhang Y, et al. Prevention of obesity and insulin resistance in mice lacking plasminogen activator inhibitor 1. Diabetes 2004;53:336‐346. [DOI] [PubMed] [Google Scholar]
- 45. Banfi C, Auwerx J, Poma F, Tremoli E, Mussoni L. Induction of plasminogen activator inhibitor I by the PPARalpha ligand, Wy‐14,643, is dependent on ERK1/2 signaling pathway. Thromb Haemost 2003;90:611‐619. [DOI] [PubMed] [Google Scholar]
- 46. Kruithof EK. Regulation of plasminogen activator inhibitor type 1 gene expression by inflammatory mediators and statins. Thromb Haemost 2008;100:969‐975. [PubMed] [Google Scholar]
- 47. Hou B, Eren M, Painter CA, Covington JW, Dixon JD, Schoenhard JA, et al. Tumor necrosis factor alpha activates the human plasminogen activator inhibitor‐1 gene through a distal nuclear factor kappaB site. J Biol Chem 2004;279:18127‐18136. [DOI] [PubMed] [Google Scholar]
- 48. Stepanova M, Younossi ZM. Independent association between nonalcoholic fatty liver disease and cardiovascular disease in the US population. Clin Gastroenterol Hepatol 2012;10:646‐650. [DOI] [PubMed] [Google Scholar]
- 49. Lazo M, Hernaez R, Bonekamp S, Kamel IR, Brancati FL, Guallar E, et al. Non‐alcoholic fatty liver disease and mortality among US adults: prospective cohort study. BMJ 2011;343:d6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dunn W, Xu R, Wingard DL, Rogers C, Angulo P, Younossi ZM, et al. Suspected nonalcoholic fatty liver disease and mortality risk in a population‐based cohort study. Am J Gastroenterol 2008;103:2263‐2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials