

Abstract
Concerns have been raised about whether preclinical models sufficiently mimic molecular disease processes observed in nonalcoholic steatohepatitis (NASH) patients, bringing into question their translational value in studies of therapeutic interventions in the process of NASH/fibrosis. We investigated the representation of molecular disease patterns characteristic for human NASH in high‐fat diet (HFD)‐fed Ldlr‐/‐.Leiden mice and studied the effects of obeticholic acid (OCA) on these disease profiles. Multiplatform serum metabolomic profiles and genome‐wide liver transcriptome from HFD‐fed Ldlr‐/‐.Leiden mice were compared with those of NASH patients. Mice were profiled at the stage of mild (24 weeks HFD) and severe (34 weeks HFD) fibrosis, and after OCA intervention (24‐34 weeks; 10 mg/kg/day). Effects of OCA were analyzed histologically, biochemically, by immunohistochemistry, using deuterated water technology (de novo collagen formation), and by its effect on the human‐based transcriptomics and metabolomics signatures. The transcriptomics and metabolomics profile of Ldlr‐/‐.Leiden mice largely reflected the molecular signature of NASH patients. OCA modulated the expression of these molecular profiles and quenched specific proinflammatory‐profibrotic pathways. OCA attenuated specific facets of cellular inflammation in liver (F4/80‐positive cells) and reduced crown‐like structures in adipose tissue. OCA reduced de novo collagen formation and attenuated further progression of liver fibrosis, but did not reduce fibrosis below the level before intervention. Conclusion: HFD‐fed Ldlr‐/‐.Leiden mice recapitulate molecular transcriptomic and metabolomic profiles of NASH patients, and these signatures are modulated by OCA. Intervention with OCA in developing fibrosis reduces collagen deposition and de novo synthesis but does not resolve already manifest fibrosis in the period studied. These data show that human molecular signatures can be used to evaluate the translational character of preclinical models for NASH.
Abbreviations
- ALT
alanine aminotransferase
- CD
clusters of differentiation
- CLS
crown‐like structures
- eWAT
epididymal white adipose tissue
- FXR
farnesoid X receptor
- HFD
high‐fat diet
- LC‐MS
OCA, IL, interleukin
- mRNA
messenger RNA
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- OCA
obeticholic acid
- SHG
second harmonic generation
- TGF
transforming growth factor
- WAT
white adipose tissue
Nonalcoholic steatohepatitis (NASH) is a chronic progressive liver disease with a multifactorial etiology that is characterized by a metabolic and an inflammatory component.1, 2 The disease is closely associated with central obesity, insulin resistance, and hyperlipidemia, and has been linked to diets rich in energy‐dense foods with high saturated fatty acid and carbohydrate content.3, 4 NASH can progress to liver fibrosis, which is considered the most important predictor of nonalcoholic fatty liver disease (NAFLD)‐related mortality.5, 6
A prolonged disturbance of metabolic homeostasis in the liver is believed to evoke a chronic inflammatory response (“metabolic inflammation”), which is a driver of disease progression toward liver fibrosis.1, 7 At a histological level, liver inflammation in NASH patients is characterized by the presence of lobular inflammatory aggregates (i.e., clusters of activated immune cells containing macrophages, neutrophils, and T cells).2, 8 At a molecular level, livers of high‐risk patients are characterized by the activation of distinct proinflammatory pathways (e.g., hepatic stellate cell activation, interleukin [IL]‐8 signaling) and their upstream regulators (e.g., transforming growth factor [TGF]‐β, tumor necrosis factor [TNF]‐α). These pathways are also partly reflected in molecular gene expression signatures that distinguish patients at different disease stages.9, 10 Recent serum profiling studies in NAFLD/NASH patients have shown that advanced metabolomics technologies allow patients to be categorized into major subtypes, thereby supporting the view that the NASH patient population is heterogeneous.11, 12
Considerable efforts are being made to develop pharmacological therapies that normalize the metabolic‐inflammatory disturbances in the liver and thereby attenuate the development of NASH and fibrosis.1 The farnesoid X receptor (FXR) agonist obeticholic acid (OCA) is one of the most promising candidate drugs, based on preclinical studies in acute models of inflammation and fibrosis13, 14 as well as on published results from clinical studies.15, 16 However, the mechanisms by which OCA can attenuate metabolically induced inflammation and associated pathways leading to fibrosis remain largely unknown. One of the reasons for this is a lack of appropriate translational preclinical models that exhibit the metabolic risk factors and phenotypic characteristics of patients as well as the chronic nature of the pathogenesis in a sufficiently translational way.10
Herein we describe a preclinical model of NASH in obesity, Ldlr‐/‐.Leiden mice that develop pronounced liver fibrosis in response to energy‐dense high‐fat diets (HFDs) with a macronutrient composition resembling that of human diets (not requiring amino‐acid deficiency or cholesterol supplementation). The model displays phenotypical and metabolic features of high‐risk patients, including insulin resistance.17, 18 Combined transcriptomics (liver) and metabolomics (serum) profiling revealed that Ldlr‐/‐.Leiden mice recapitulate many of the molecular pathways of human disease including specific fingerprint genes9, 10 identified in patients with progressive NASH, as well as lipidome/metabolome signatures11 of NASH patients. In this model, we evaluated the effects of intervention with OCA in the ongoing disease process (i.e., when mice had developed lobular inflammation with early fibrosis). The effects of OCA on metabolically‐induced inflammatory and profibrotic processes were investigated at the histopathological, cellular, molecular, and gene expression level. Specific emphasis was placed on the effects on key regulators of inflammatory and profibrotic pathways, and a potential resolution of fibrosis. The effects of OCA on de novo synthesis of major collagen types were quantified by in vivo deuterated‐water labeling and liquid chromatography–mass spectrometry (LC‐MS) analysis of deuterium incorporation into several types of collagen.
Materials and Methods
Animal Studies
Animal experiments were approved by an independent Committee on the Ethics of Animal Experiments (DEC‐Zeist, the Netherlands) and were performed in compliance with European Community specifications regarding use of laboratory animals. Male Ldlr‐/‐.Leiden mice (TNO Metabolic Health Research, Leiden, the Netherlands) were used. This strain is a substrain of the commercially available Ldlr‐/‐ mouse (The Jackson Laboratory) and has a 94% C57BL/6J background and 6% 129S1/SvImJ background. At 12 weeks of age, 42 mice were started on a NASH‐inducing HFD containing 45 kcal% fat from lard and 35 kcal% from carbohydrates (primarily sucrose) (D12451, Research Diets, New Brunswick, NJ). This diet was not supplemented with cholesterol but does contain 0.01% cholesterol (wt/wt) based on the natural cholesterol content of the lard in the diet. Eighteen mice were fed standard chow (Ssniff R/M‐H, Soest, Germany). After 24 weeks of diet feeding, n = 14 HFD‐fed mice and n = 9 chow‐fed mice were sacrificed to determine liver pathology as a start‐of‐intervention reference. The remaining HFD‐fed mice were matched into two groups (n = 14/group) based on body weight and plasma cholesterol, triglycerides, and blood glucose. One group continued on the HFD (HFD) and the other continued on HFD with 10 mg/kg/day OCA (HFD+OCA) (Bio‐Connect, Huissen, the Netherlands, provided as dietary admixture) for an additional 10 weeks. Mice were group‐housed (3‐5 mice per cage) and received food and water ad libitum. Food intake (cage level, expressed as grams/mouse/day) and body weight were monitored throughout the study. Blood samples for ethylene diamine tetraacetic acid plasma were taken from the tail vein after a 5‐hour fast. Animals were sacrificed by gradual‐fill CO2 asphyxiation in week 34. Terminal blood was collected through cardiac puncture to prepare serum (unfasted), and livers and epididymal white adipose tissue (eWAT) were isolated. A part of the left lobe was formalin‐fixed and paraffin‐embedded for histological analyses. The remaining part of the left lobe and the other liver lobes were flash‐frozen in N2 and subsequently stored at −80°C for biochemical and gene expression analyses. The eWAT was formalin‐fixed and paraffin‐embedded for histological analysis.
Plasma Measurements
Plasma analyses were performed using standardized protocols and tools.19, 20 Briefly, blood glucose was measured at the time of blood sampling using a hand‐held glucometer (Freestyle Disectronic, Vianen, the Netherlands). Plasma cholesterol and triglyceride levels were determined using enzymatic assays (CHOD‐PAP and GPO‐PAP, respectively; Roche Diagnostics, Almere, the Netherlands). Plasma insulin was analyzed by ELISA (Mercodia, Uppsala, Sweden). Plasma alanine aminotransferase (ALT) levels were measuring using a spectrophotometric activity assay (Reflotron‐Plus, Roche).
Histological Analysis of White Adipose Tissue Inflammation and Hypertrophy
White adipose tissue (WAT) hypertrophy and inflammation were assessed in 5‐µm sections of the eWAT. Sections were stained with hematoxylin phloxine saffron and adipose tissue morphometry was analyzed using Adiposoft,21 an open‐source automated plugin for the image processing package Fiji22 for imageJ.23 Crown‐like structures (CLS) were counted as a measure of adipose tissue inflammation in the same fields used for the morphometry analysis and expressed as number of CLS/1,000 adipocytes.
Liver Histopathology
Development of NAFLD was analyzed in 3‐µm liver sections that were stained with hematoxylin and eosin and analyzed blindly by two board‐certified pathologists using a well‐established adapted scoring method for human NAFLD.24 Data shown herein represents the average of these two independent assessments. Briefly, steatosis was expressed as the percentage of the total liver section affected by microvesicular or macrovesicular steatosis. Hepatic inflammation was analyzed by counting the number of inflammatory foci per field at ×100 magnification (view size 2.3 mm2), in five nonoverlapping fields per specimen. In addition, NASH was scored using the NAFLD activity score (NAS).25 Liver cross‐sections were assessed for macrophage content using a rat antimouse F4/80 monoclonal antibody (clone BM8, 14‐4801; eBioscience [Thermo Fisher], Landsmeer, the Netherlands). F4/80‐positive crown‐like structures26 were counted by a pathologist in five nonoverlapping fields (×100 magnification). Clusters of differentiation (CD) 4–positive cells were identified using a rat antimouse CD4 monoclonal antibody (clone 4SM95, 14‐9766; eBioscience) and CD8‐cells were identified using a rat anti‐mouse CD8 monoclonal antibody (clone 4SM15, 14‐0808; eBioscience). Quantitative analysis of the number of CD4+ and CD8+ cells was performed using an automated macro in ImageJ (National Institutes of Health, Bethesda, MD23) that counts positively stained cells in an entire cross‐section, and expressed as the number of cells per square millimeter nucleus area.
Histological analysis of fibrosis development was performed in picrosirius red–stained liver sections by a liver pathologist (Amsterdam Medical Center, Netherlands) and expressed as the percentage of perihepatocellular fibrosis relative to the total perisinusoidal area. Multiphoton and second harmonic generation (SHG) imaging of hepatic collagen was performed using a Genesis 200 imaging system and subsequent computer‐assisted data analysis (HistoIndex, Singapore). SHG is a nonlinear optical process that is highly sensitive to non‐centrosymmetric structures such as collagen fibrils and fibers.27 Images of SHG collagen were segmented into two categories based on a size and intensity Otsu threshold28: the “thin fibers” category contains all of the interstitial and fibrotic collagen, below said threshold. The thick bundles of collagen fibers typically found around the sample of blood vessels are labeled as “aggregated fibers.” The area occupied by collagen relative to the total area of the sample is expressed as the “collagen area ratio.” After the collagen area was defined, a skeletonization of the collagen was applied, giving a schematic representation of the collagen fibers and their intersection points. The “collagen reticulation index” is the total number of intersection points in the given collagen area, which informs on the structure and interconnection of collagen fibers and provides an estimation of how reticulated the collagen fiber network is. These two features were measured for both the thin and aggregated collagen fibers.
Biochemical Liver Analyses
Liver lipids were extracted from freshly prepared liver homogenates using methanol and chloroform following the Bligh & Dyer method29 and separated by high‐performance thin‐layer chromatography as described previously.17 Briefly, lipids were stained with a color reagent (5 g of MnCl24H2O, 32 mL of 95%‐97% H2SO4 added to 960 mL of CH3OH/H2O = 1:1 [vol/vol]) and triglycerides, cholesteryl esters, and free cholesterol were quantified (TINA version 2.09 software, Raytest, Straubenhardt, Germany).
Hepatic total collagen content was determined biochemically after acid hydrolysis of liver biopsies for 20 hours at 95°C. Hydroxyproline was measured in the freshly prepared hydrolysate using a total collagen assay relative to a collagen standard per manufacturer’s instructions (Quickzyme, Leiden, the Netherlands).
De Novo Collagen Analysis
De novo collagen formation during the 2 weeks before sacrifice was assessed using stable isotope labeling and LC‐MS‐based mass isotopomer analysis as previously described.30, 31 Mice received a bolus intraperitoneal injection of 100% 2H2O to bring the total body water enrichment to about 5%, followed by 8% 2H2O in drinking water to maintain this level of body water enrichment for the remainder of the labeling period. After sacrifice, frozen liver tissue was subjected to sequential protein extraction to fractionate guanidine‐soluble collagens and residual insoluble collagens, and the fraction of newly synthesized collagen was determined by measuring the incorporation of deuterium (2H) from heavy water (2H2O) into the stable C‐H bonds of hydroxyproline in the newly synthesized collagen, as described previously.32
Metabolomics
Terminal serum samples were analyzed by a combined metabolomics/lipidomics approach with dedicated platforms to analyze, among others, (1) fatty acyls, bile acids, steroids, and lysoglycerophospholipids; (2) glycerolipids, glycerophospholipids, sterol lipids, and sphingolipids; and (3) amino acids and derivatives. This combined analysis was established for human and rodent serum by OWL Metabolomics (Derio, Spain). Briefly, serum samples were used for metabolite extraction using methanol and chloroform or methanol alone. Extracts were subjected to mass spectrometry coupled with ultrahigh‐performance liquid chromatography–based analytical platforms as described in detail by Alonso et al.11 Metabolomics data were preprocessed using the TargetLynx application manager for MassLynx 4.1 (Waters Corp., Milford, MA). Intrabatch and interbatch normalization followed the procedure described.33 A cohort of 535 patients who underwent liver biopsy analysis (353 with a diagnosis of simple steatosis and 182 with a diagnosis of NASH) was used to identify NAFLD subtypes based on Ldlr‐/‐.Leiden mouse serum metabolic signature. Details of the inclusion criteria and clinical data of this cohort have been described previously.11
Gene Expression and Pathway Analysis
The RNA‐Bee Total‐RNA Isolation Kit (Bio‐Connect, Huissen, the Netherlands) was used for RNA extraction from snap‐frozen liver tissue (left lobe). RNA concentration was determined spectrophotometrically using Nanodrop 1000 (Isogen Life Science, De Meern, Netherlands), and RNA quality was assessed using the 2100 Bioanalyzer (Agilent Technologies, Amstelveen, Netherlands). RNA was then used to generate strand‐specific messenger RNA (mRNA)–seq libraries for next generation sequencing at GenomeScan B.V. (Leiden, the Netherlands). Libraries were multiplexed, clustered, and sequenced on an Illumina NextSeq 500 with a single‐read 75‐cycles sequencing protocol, 13 million reads per sample, and indexing. Differentially expressed genes were determined for HFD versus chow at t = 24 weeks, HFD versus chow at t = 34 weeks, and HFD+OCA versus HFD at t = 34 weeks, using the DESeq‐method34 with statistical cut‐off false discovery rate < 0.05.
To evaluate the representation of human pathophysiological pathways in HFD‐fed Ldlr‐/‐.Leiden mice, murine hepatic gene expression profiles were compared with published data on hepatic gene expression profiles in human NASH: a disease signature for NASH versus control10 that was downloaded from the Gene Expression Omnibus (GEO) with accession number GSE48452 and a gene profile that differentiates severe “progressive” NASH patients (fibrosis stage 3 or 4) from mild NASH patients (fibrosis stage 0 or 1)9 (GEO accession number GSE31803).
The effects of OCA on hepatic gene expression were analyzed by gene enrichment analysis across pathways and biological processes using Ingenuity Pathway Analysis (IPA) suite (www.ingenuity.com, accessed 2016) as described previously.19, 35 The upstream regulator analysis tool of IPA was used to assess the activity of transcription factors as well as other upstream regulators (e.g., receptors, enzymes, metabolites) essentially as reported.36 Gene expression data were used to predict activation or deactivation of upstream regulators (e.g., of TGF‐β or the insulin receptor). A negative z‐score of less than −2 indicates significantly reduced transcriptional activity based on the direction of gene expression changes of target genes. A positive z‐score of greater than 2 indicates significant activation of the upstream regulator. In addition, the effects of OCA on the gene expression profiles for human NASH9, 10 were investigated.
Statistical Analysis
All data are shown as mean ± SD. Statistical significance between the OCA‐treated group and the HFD control group was assessed by Student t test for normally distributed parameters (with Welch correction in the case of unequal variances) or Mann‐Whitney U test for nonparametric parameters. For plasma parameters, gene expression data and metabolomics data, we tested the null hypothesis that these did not differ between HFD and HFD+OCA (using two‐sided tests). For the histopathological and liver biochemical parameters, we tested the null hypothesis that OCA did not reduce development of NASH (using one‐sided tests). Hierarchical clustering analysis of the NAFLD patients based on serum metabolome was performed with the cluster R package, including Ward’s minimum variance method as the agglomeration method. Validation of the classification procedure was described previously.11
Results
High‐Fat Diet–Treated Ldlr‐/‐.Leiden Mice Display Similarities to Human Disease at Multiple Levels
Ldlr‐/‐.Leiden mice fed a HFD for 24 weeks developed pronounced obesity compared with littermates fed a low‐fat reference diet (chow) (Table 1). In response to HFD feeding, mice developed stable hypertriglyceridemia and hypercholesterolemia. Fasting plasma insulin levels were strongly and significantly increased by HFD, which resulted in maintenance of blood glucose levels comparable to those in chow‐fed mice. Furthermore, HFD feeding led to significantly increased levels of plasma ALT, indicative of liver injury in this group. Histopathological analysis of livers revealed significant HFD‐induced increases in macrovesicular steatosis, microvesicular steatosis, and lobular inflammation (Table 1). Perihepatocellular fibrosis was not significantly increased in HFD‐fed animals (P = 0.15 relative to chow), indicating that the t = 24‐week time point reflects the early onset of liver fibrosis.
Table 1.
Metabolic Parameters and Liver Histology After 24 Weeks of Chow or HFD
| Chow | HFD | |
|---|---|---|
| Metabolic parameters | ||
| Body weight (g) | 43.3 ± 4.5 | 52.3 ± 2.3 * |
| Plasma triglycerides (mM) | 2.6 ± 1.1 | 4.8 ± 2.1† |
| Plasma cholesterol (mM) | 11.5 ± 3.2 | 35.3 ± 7.3 * |
| Blood glucose (mM) | 7.6 ± 1.2 | 7.1 ± 0.6 |
| Plasma insulin (ng/ml) | 5.9 ± 4.7 | 33.6 ± 25.9 * |
| ALT (U/L at 37ºC) | 92.4 ± 59.5 | 350.5 ± 111.8 * |
| Liver histology | ||
| Macrovesicular steatosis (% of hepatocytes) | 10.6 ± 9.3 | 28.0 ± 5.6 * |
| Microvesicular steatosis (% of hepatocytes) | 33.1 ± 27.2 | 46.2 ± 6.8‡ |
| Hepatic inflammation (number of inflammatory clusters/5 fields at ×100) | 12.7 ± 25.5 | 24.2 ± 15.0† |
| Hepatic fibrosis (% of perihepatocellular area) | 2.8 ± 0.8 | 5.2 ± 5.2§ |
Data shown are mean ± SD.
P < 0.001.
P < 0.01.
P = 0.10.
P = 0.15 relative to chow.
To evaluate the representation of molecular patterns and processes that are characteristic for human NASH in HFD‐fed Ldlr‐/‐.Leiden mice, we compared murine serum metabolomic profiles and hepatic gene expression profiles to those observed in patients. First, the HFD‐induced alterations in serum metabolome in Ldlr‐/‐.Leiden mice (Fig. 1A) were compared with the serum metabolome of 535 patients with biopsy‐proven NAFLD. This analysis revealed that the 50 most significantly regulated metabolites in the Ldlr‐/‐.Leiden mice (specified in Supporting Table S1) were reflected in a substantial proportion of NAFLD/NASH patients (Fig. 1B). Next, murine hepatic gene expression profiles were compared with published human hepatic gene expression profiles,10 to investigate representation of pathophysiological pathways at the gene expression level. HFD‐fed Ldlr‐/‐.Leiden mice displayed a gene expression profile that was similar to the reported disease signature that distinguishes NASH patients from normal controls (Fig. 2A). Furthermore, a set of genes that differentiates progressive NASH patients (fibrosis stage 3 or 4) from patients with mild NASH (fibrosis stage 0 or 1)9 was also induced by HFD feeding in Ldlr‐/‐.Leiden mice (Fig. 2B). Genome‐wide pathway analysis of all differentially expressed genes showed that the experimental conditions in the model reflected most of the NASH‐related disease processes and pathways enriched in human NASH livers (Fig. 2C). Altogether these data indicate that the diet‐inducible Ldlr‐/‐.Leiden model recapitulated many phenotypical and molecular facets of human disease, including similarities at the level of single genes, disease pathways, and a characteristic metabolome signature. This allowed us to assess the effects of OCA on further progression of NASH toward fibrosis under translational experimental conditions.
Figure 1.
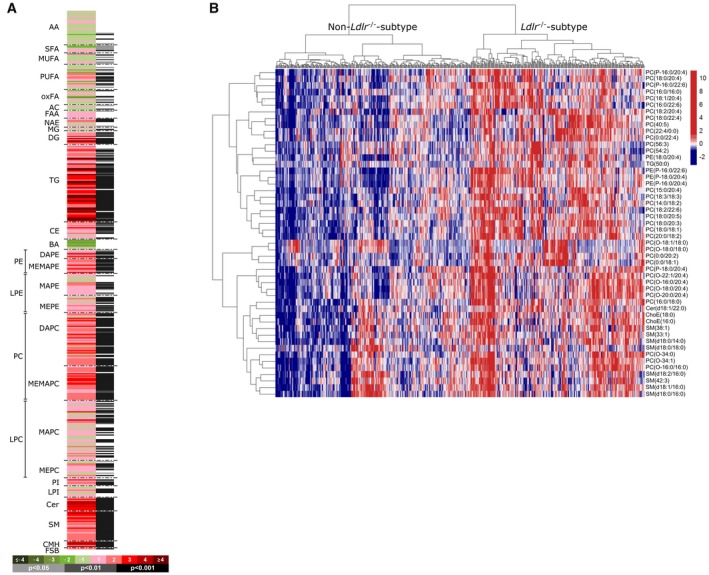
Representation of human metabolome in 24‐week HFD‐fed Ldlr‐/‐.Leiden mice. (A) Heatmap showing HFD‐induced alterations in serum metabolome in Ldlr‐/‐.Leiden mice as compared with chow‐fed Ldlr‐/‐.Leiden mice. Green indicates down‐regulation; red indicates up‐regulation. (B) Heatmap representation of the serum metabolomic profile from 535 biopsy‐confirmed NAFLD/NASH patients. Each data point corresponds to the relative ion abundance of a given metabolite (horizontal axis) in an individual patient’s serum (vertical axis), showing the top 50 metabolites that differentiated HFD‐fed Ldlr‐/‐.Leiden mice from chow. Blue indicates down‐regulation and red indicates up‐regulation relative to the level in the rest of the study population (mean value: 0). Hierarchical clustering based on the optimum average silhouette width divided the patients into two groups: the Ldlr‐/‐.Leiden subtype (serum metabolome resembles that observed in HFD‐fed Ldlr‐/‐.Leiden mice; right‐hand cluster) and the non‐Ldlr‐/‐.Leiden subtype (patients that have a different serum metabolome; left‐hand cluster). Abbreviations: AA, amino acid; AC, acyl carnitine; BA, bile acid; CE, cholesteryl ester; Cer, ceramide; CMH, monohexosylceramide; DG, diglyceride; FAA, fatty acyl amide; FSB, free sphingoid base; LPC, lysophosphatidylcholine; LPE, lysophosphatidylethanolamine; LPI, lysophosphatidylinositol; MG, monoglyceride; MUFA, monounsaturated fatty acid; NAE, N‐acylethanolamine; oxFA, oxidized fatty acid; PC, phosphatidylcholine; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PUFA, polyunsaturated fatty acid; SFA, saturated fatty acid; SM, sphingomyelin; and TG, triglyceride.
Figure 2.
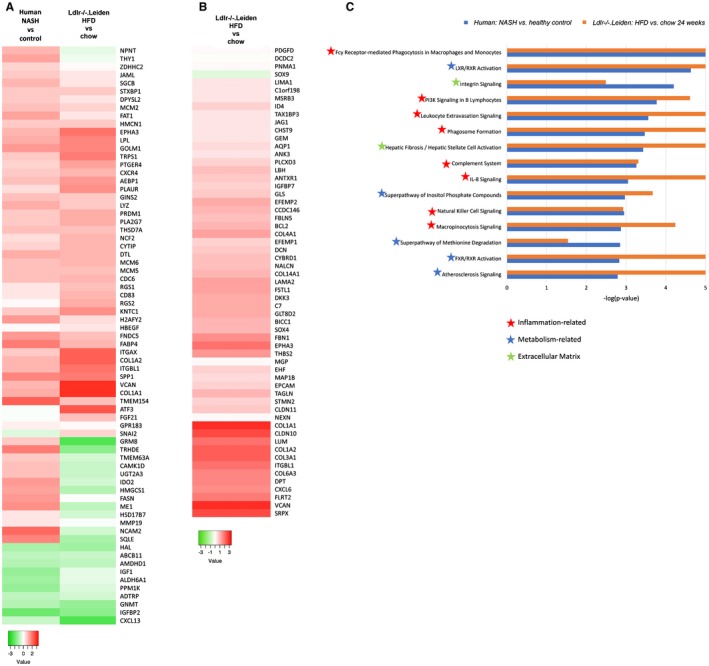
Representation of human pathophysiological processes in 24‐week HFD‐fed Ldlr‐/‐.Leiden mice. (A) Heatmap showing significantly regulated genes in human NASH livers versus normal controls10 are largely represented in 24‐week HFD‐fed Ldlr‐/‐.Leiden mice. (B) Heatmap diagram showing recapitulation of hepatic gene expression profile that differentiates mild (fibrosis stage 0‐1) from severe (fibrosis stage 3‐4) NASH patients9 in 24‐week HFD‐fed Ldlr‐/‐.Leiden mice. Green indicates down‐regulation; red indicates up‐regulation. (C) Enrichment of the top 15 human NASH‐related processes10 in 24‐week HFD‐fed Ldlr‐/‐.Leiden mice. P values equal to or smaller than −log(P value) of 5 are shown as −log(P value) of 5.
Intervention With OCA Attenuates Progression of Inflammation and Liver Fibrosis
After 24 weeks of HFD feeding, mice received HFD supplemented with a relatively low dose of OCA (10 mg/kg/day; HFD+OCA group) or control HFD (HFD group) for an additional 10 weeks. The intervention had no effect on body weight (53.5 ± 5.6 g in HFD, 54.7 ± 5.2 g in HFD+OCA) or food intake (average during treatment period: 2.7 ± 0.2 g/mouse/day in HFD, 2.7 ± 0.3 g/mouse/day in HFD+OCA). OCA strongly lowered plasma triglyceride levels (4.1 ± 1.5 mM in HFD, 1.7 ± 0.7 mM in HFD+OCA; P < 0.001) and tended to lower total cholesterol levels (33.4 ± 12.4 mM in HFD, 25.2 ± 7.4 mM in HFD + OCA; P = 0.06). OCA treatment tended to increase plasma insulin levels (11.4 ± 5.1 ng/mL in HFD, 18.8 ± 12.7 mM in HFD+OCA, P = 0.08), but did not affect blood glucose (7.3 ± 1.1 mM in HFD, 8.1 ± 1.1 mM in HFD+OCA).
We then investigated whether OCA may affect WAT hypertrophy and inflammation, both of which are associated with adipose tissue insulin resistance.37, 38 OCA significantly reduced the average adipocyte size in eWAT (Fig. 3A). This was attributable to a shift toward an increase in the percentage of smaller adipocytes (< 2,000 µm2, 2,000‐4,000 µm2) and a reduction in the percentage of larger adipocytes (6,000‐8,000 µm2, > 8,000 µm2) (Fig. 3B). OCA significantly reduced adipose tissue inflammation, as assessed by the number of crown‐like structures present (Fig. 3C).
Figure 3.
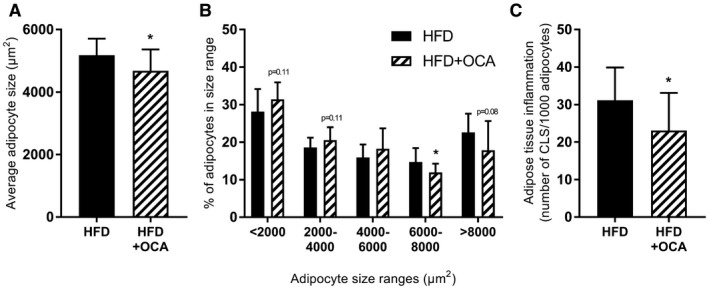
OCA treatment reduces adipocyte hypertrophy and adipose tissue inflammation in the epidydimal depot. Mice were treated with OCA (10 mg/kg/day) from t = 24 weeks to t = 34 weeks. (A) OCA treatment reduced the average adipocyte size in the eWAT, reducing the presence of large adipocytes while tending to increase the presence of smaller adipocytes. (B) Adipose tissue inflammation, expressed as the number of CLS/1,000 adipocytes, was significantly reduced by OCA. Data are mean ± SD. *P < 0.05 HFD at 34 weeks.
Next, we investigated whether OCA may affect the development of NASH and fibrosis. Plasma ALT was significantly reduced by OCA (342.1 ± 138.7 U/L at 37ºC in HFD, 221.0 ± 88.9 U/L at 37ºC in HFD+OCA; P = 0.018), indicating an effect on liver pathology. Indeed, OCA tended to reduce macrovesicular steatosis (28.4% ± 12.8% of hepatocytes in HFD, 22.3% ± 9.8% in HFD+OCA; P = 0.06), whereas microvesicular steatosis was not reduced (28.4% ± 8.0% of hepatocytes in HFD, 33.3% ± 9.3% in HFD+OCA) (Fig. 4A). Biochemical analysis of liver lipids showed significant reductions in hepatic triglycerides and cholesteryl esters in OCA‐treated mice, while free cholesterol levels remained unchanged (Fig. 4B).
Figure 4.
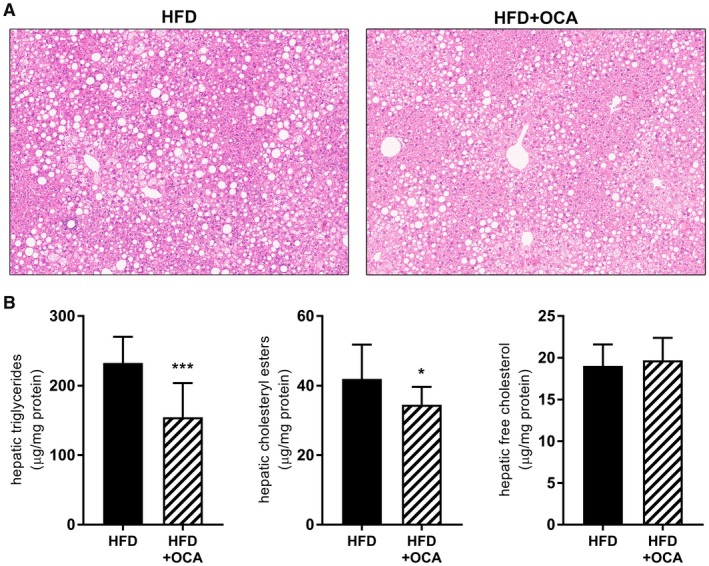
OCA treatment reduces hepatic steatosis in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice. Mice were treated with OCA (10 mg/kg/day) from t = 24 weeks to t = 34 weeks. (A) Representative images of hematoxylin and eosin–stained liver cross‐sections showing reduced hepatic steatosis in OCA‐treated mice. (B) Biochemically determined liver lipid content revealed a reduction in hepatic triglycerides and hepatic cholesteryl esters in OCA‐treated animals, whereas free cholesterol levels were not affected. Data are mean ± SD. *P < 0.05; ***P < 0.001 versus HFD at 34 weeks.
Quantitative analysis of inflammatory aggregates revealed a trend to reduced counts in OCA‐treated mice (Fig. 5A). In comparison to the number of inflammatory aggregates at the start of the OCA treatment, OCA fully halted further progression of the inflammation‐inducing effect of HFD. A refined quantitative immunohistochemical analysis of cells that stained positive for F4/80 (macrophages), CD4 (T helper cells) and CD8 (cytotoxic T cells) showed that OCA fully prevented further HFD‐induced recruitment of macrophages (Fig. 5B) with a significant reduction relative to HFD control. In addition to this reduction in the quantity of macrophages in the liver, treatment with OCA also resulted in an increase in the ratio of Arg1:CD68 gene expression (Fig. 5C) and tended to reduce the ratio of CD11c:CD68 (Fig. 5D) and CCR2:CD68 (Fig. 5E) expression, indicating a shift toward a more anti‐inflammatory macrophage phenotype after OCA treatment, in line with previously described effects of OCA in adipose tissue.39 The number of other inflammatory cell types (CD4+ [Fig. 5F] and CD8+ [Fig. 5G]) decreased over time during HFD feeding (from t = 24 weeks to t = 34 weeks), indicating a different dynamic of these cells during NASH pathogenesis. OCA tended to enhance their disappearance. Overall, the NAS tended to be reduced in mice treated with OCA (4.18 ± 1.19 in HFD, 3.50 ± 0.80 in HFD+OCA; P = 0.09). In line with what was observed using the adapted scoring system for murine NASH24 described previously, this was attributable to a tendency toward a reduction in the inflammation score (1.83 ± 0.94 in HFD, 1.33 ± 0.78 in HFD+OCA; P = 0.10). The steatosis score was not affected by OCA (2.33 ± 0.49 in HFD, 2.18 ± 0.0.39 in HFD+OCA; non‐significant).
Figure 5.
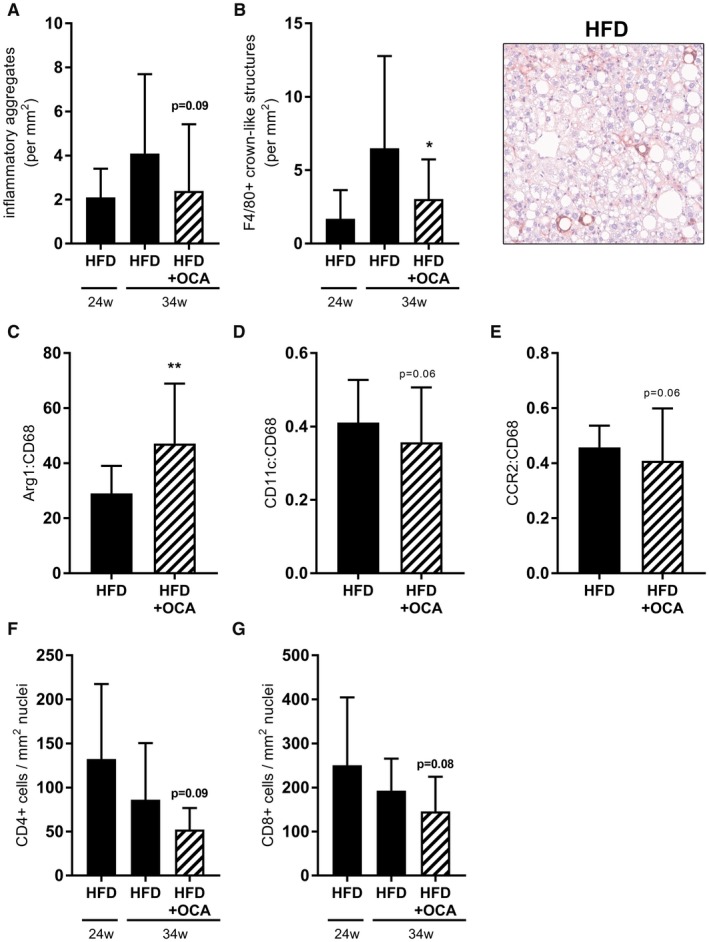
OCA treatment reduces hepatic inflammation in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice. Mice were treated with OCA (10 mg/kg/day) from t = 24 weeks to t = 34 weeks. (A) OCA‐treatment tended to reduce the number of inflammatory clusters in liver. (B) The number of F4/80+ CLS was reduced by OCA. (C) OCA significantly increased the ratio of Arg1 to CD68 mRNA expression. The ratio of CD11c to CD68 mRNA expression (D) and CCR2 to CD68 mRNA expression (E) tended to be reduced in HFD+OCA relative to HFD. OCA tended to enhance the disappearance of CD4+ (F) and CD8+ (G) cells. Data are mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001 versus HFD at 34 weeks.
Histochemical staining of hepatic collagen by picrosirius red (Fig. 6A) followed by a pathologist’s analysis of perihepatocellular fibrosis revealed a significant reduction in fibrosis development in OCA‐treated mice (Fig. 6B), with levels remaining similar to those at the start of treatment (t = 24 weeks). This effect on liver fibrosis was confirmed by biochemical analysis of hepatic collagen content, which revealed a significant reduction of hepatic collagen in HFD+OCA relative to HFD (Fig. 6C). Collagen levels in the HFD+OCA group remained at the level observed at the start of the treatment period (t = 24 weeks). More refined analysis of hepatic fibrosis by multiphoton microscopy and SHG imaging—which provides additional information on individual collagen fibers and their physical attributes (e.g., diameter for differentiation between “thin” and thicker “aggregated” collagen fibers, cross‐linkage for the degree of reticulation of fibers)—revealed a pronounced effect of OCA on the total area of thin‐fiber collagen as well as the degree of reticulation of these thin fibers (Fig. 6D). Similarly, although less markedly, OCA reduced the area of aggregated collagen fibers together with their degree of reticulation (Fig. 6E).
Figure 6.
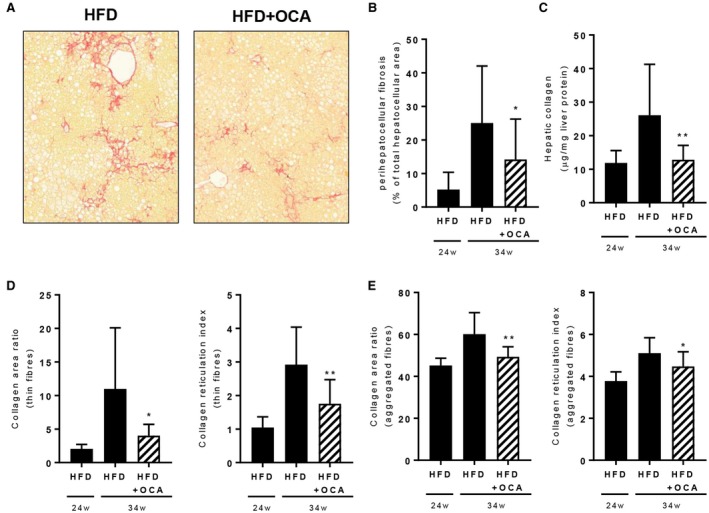
OCA treatment reduces hepatic fibrosis in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice. Mice were treated with OCA (10 mg/kg/day) from t = 24 weeks to t = 34 weeks. (A) Picrosirius red–stained liver cross‐sections showing reduced fibrosis in OCA‐treated mice. (B) Pathologist’s assessment of perihepatocellular fibrosis revealed reduced progression of fibrosis in HFD+OCA. (C) Biochemically determined hepatic collagen was reduced in HFD+OCA. OCA reduced the collagen area and degree of reticulation of thin‐fiber collagen (D) and aggregated‐fiber collagen (E) (multiphoton and SHG imaging). Data are mean ± SD. *P < 0.05; **P < 0.01 versus HFD at 34 weeks.
Analysis of de novo collagen formation using stable isotope labeling followed by LC‐MS isotopomer analysis revealed a significant attenuating effect of OCA on the percentage of newly synthesized collagen during the 14 days prior to sacrifice (Fig. 7A), indicating that OCA reduces the deposition of new collagen. Further investigation of the fractional synthesis rate of several specific types of collagen showed a significant reduction in formation of the guanidine‐insoluble fraction of collagen α‐1(I) chain, collagen α‐2(I) chain, and collagen α‐1(III) chain (Fig. 7B) as well as the guanidine‐soluble fraction of collagen α‐1(I) chain and collagen α‐2(I) chain (Fig. 7C). Overall, these data show that OCA attenuates further progression of fibrosis, at least in part by reducing the deposition of newly formed collagen.
Figure 7.
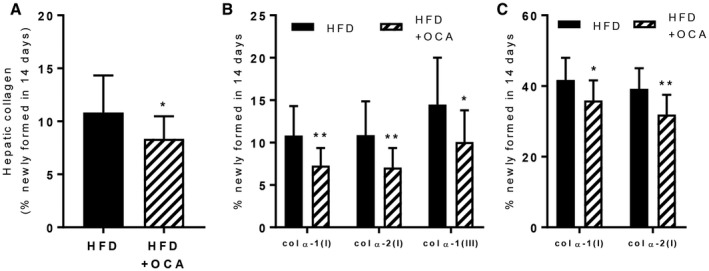
OCA treatment reduces de novo collagen synthesis in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice. Mice were treated with OCA (10 mg/kg/day) from t = 24 weeks to t = 34 weeks. (A) OCA reduced de novo total collagen synthesis assessed by stable‐isotope labeling and in parallel reduced de novo synthesis of individual collagen chains in the guanidine‐insoluble (B) and guanidine‐soluble (C) fractions. Data are mean ± SD. *P < 0.05; **P < 0.01 versus HFD at 34 weeks.
OCA Treatment Normalizes Metabolic and Inflammatory Gene Expression
To gain insight into the molecular processes affected by OCA, livers were subjected to gene profiling analysis using RNA sequencing. Gene expression analysis was followed by upstream regulator analysis, which predicts the activation state of a protein, enzyme, or transcription factor based on the expression pattern of the genes downstream of this factor. This analysis showed that in HFD‐fed mice, there was a global activation of inflammatory pathways that involved activation of chemokines, cytokines, and toll‐like receptors as well as transcriptional regulators of inflammation (Table 2). Treatment with OCA had pronounced anti‐inflammatory effects on some, but not all of these pathways. For instance, OCA quenched the inflammatory pathways downstream of TNF‐α (z = −2.4, P = 2.0‐13) and interferon‐γ (z = −3.7, P = 9.5‐11), and activated anti‐inflammatory IL‐10 signaling (IL‐10 receptor subunit alpha; z = 3.1, P = 8.5‐15). With regard to fibrosis, OCA significantly attenuated TGF‐β signaling (z = −2.7, P = 2.3‐19), thereby corroborating the histological and biochemical effects on collagen formation.
Table 2.
Effects of OCA on Hepatic Gene Expression
| Upstream Regulator | HFD Versus Chow | HFD+OCA Versus HFD | ||
|---|---|---|---|---|
| Upstream Regulator | Activation z Score | P Value | Upstream Regulator | |
| Chemokines | ||||
| CCR2 | 2.1 | 4.7E‐04 | n/a | n/a |
| CCL5 | 2.1 | 5.2E‐09 | n/a | n/a |
| CCL6 | 2.2 | 8.5E‐03 | n/a | 3.4E‐02 |
| CXCL8 | 3.2 | 1.1E‐02 | n/a | n/a |
| CXCR4 | 2.7 | 7.1E‐03 | n/a | n/a |
| CXCL10 | 2.2 | 4.2E‐02 | n/a | n/a |
| CXCL12 | 4.1 | 1.0E‐05 | −2.4 | 9.1E‐02 |
| Cytokines | ||||
| IL1B | 7.0 | 7.4E‐35 | −0.9 | 2.1E‐06 |
| IL2 | 4.4 | 1.3E‐18 | −1.7 | 1.6E‐04 |
| IL3 | 2.9 | 4.2E‐14 | −1.7 | 6.8E‐06 |
| IL4 | 2.9 | 5.0E‐27 | −0.8 | 9.0E‐12 |
| IL5 | 3.9 | 3.9E‐16 | n/a | n/a |
| IL6 | 4.8 | 7.1E‐34 | −1.7 | 3.1E‐09 |
| IL7 | 1.4 | 1.1E‐02 | n/a | n/a |
| IL9 | 2.0 | 1.0E+00 | n/a | n/a |
| IL10 | 0.7 | 1.3E‐14 | −1.6 | 7.0E‐10 |
| IL10RA | −3.9 | 9.5E‐25 | 3.1 | 8.5E‐15 |
| IL11 | 1.7 | 2.1E‐04 | n/a | n/a |
| IL11RA | 2.4 | 4.8E‐02 | n/a | n/a |
| IL12 (complex) | 2.9 | 6.8E‐07 | −2.0 | 1.4E‐05 |
| IL13 | 2.4 | 1.5E‐27 | −2.1 | 4.2E‐06 |
| IL13RA2 | 2.0 | 3.5E‐02 | n/a | n/a |
| IL15 | 3.6 | 3.8E‐07 | n/a | n/a |
| IL17A | 3.4 | 5.8E‐10 | n/a | n/a |
| IL17C | 2.4 | 3.4E‐01 | n/a | n/a |
| IL17D | 2.0 | 1.9E‐01 | n/a | n/a |
| IL18 | 4.3 | 1.5E‐02 | −1.2 | 4.1E‐02 |
| IL21 | 3.3 | 5.2E‐05 | n/a | n/a |
| IL27 | 3.9 | 6.2E‐04 | −1.1 | 1.1E‐02 |
| IL32 | 0.7 | 2.8E‐02 | n/a | n/a |
| CSF1 | 4.1 | 2.4E‐13 | −2.5 | 1.5E‐04 |
| CSF2 | 7.1 | 3.1E‐21 | −4.0 | 5.3E‐08 |
| TNF | 9.2 | 5.4E‐59 | −2.4 | 2.0E‐13 |
| OSM | 3.8 | 4.0E‐15 | −2.0 | 2.2E‐01 |
| MIF | 2.1 | 5.1E‐04 | n/a | n/a |
| IFN Beta | 3.7 | 1.8E‐04 | n/a | n/a |
| IFNG | 7.3 | 4.9E‐54 | −3.7 | 9.5E‐11 |
| Inflammatory regulators | ||||
| NFkB (complex) | 6.6 | 1.2E‐19 | −1.8 | 4.3E‐02 |
| NFKB1 | 4.3 | 1.7E‐09 | n/a | n/a |
| NFKB2 | 0.8 | 4.0E‐02 | n/a | n/a |
| NfkB1‐RelA | 2.2 | 4.3E‐01 | n/a | n/a |
| NFKBIA | 3.7 | 7.3E‐24 | −2.3 | 5.7E‐03 |
| STAT1 | 5.8 | 7.8E‐14 | −2.7 | 5.6E‐05 |
| STAT3 | 3.1 | 8.1E‐24 | −0.5 | 2.7E‐04 |
| STAT4 | 3.3 | 6.5E‐10 | −2.7 | 6.8E‐03 |
| STAT5a/b | 1.9 | 5.4E‐05 | −2.0 | 3.1E‐01 |
| Jnk | 5.2 | 1.1E‐08 | −2.3 | 1.1E‐01 |
| ITK | 3.4 | 1.9E‐05 | −3.0 | 1.5E‐03 |
| Ap1 | 3.8 | 7.4E‐05 | n/a | n/a |
| NFATC2 | 3.5 | 2.0E‐02 | n/a | n/a |
| TLR signaling | ||||
| TLR2 | 1.9 | 1.0E‐01 | n/a | 3.3E‐02 |
| TLR3 | 4.6 | 2.7E‐05 | n/a | n/a |
| TLR4 | 5.7 | 6.8E‐12 | −1.5 | 3.8E‐02 |
| TLR5 | 2.4 | 1.4E‐01 | n/a | n/a |
| TLR7 | 4.8 | 4.9E‐03 | −2.1 | 9.0E‐02 |
| TLR8 | 2.0 | 1.0E+00 | n/a | n/a |
| TLR9 | 4.5 | 8.5E‐08 | −0.1 | 1.2E‐02 |
| MYD88 | 5.9 | 6.1E‐11 | −2.5 | 1.9E‐01 |
| Growth factors | ||||
| TGFB1 | 7.2 | 2.0E‐66 | −2.8 | 2.3E‐19 |
| VEGFA | 2.5 | 1.3E‐15 | −2.8 | 1.3E‐01 |
| Oxidative stress (response) | ||||
| NOS2 | 3.7 | 4.7E‐06 | n/a | n/a |
| CAT | −2.5 | 2.8E‐04 | n/a | n/a |
| SOD1 | −2.2 | 2.8E‐10 | n/a | 1.6E‐02 |
| NRF1 | −2.1 | 2.0E‐02 | n/a | n/a |
| NRF2 | −2.8 | 4.0E‐22 | 1.7 | 8.5E‐04 |
| Metabolism‐related | ||||
| PPARA | 0.8 | 9.9E‐42 | −0.2 | 1.6E‐18 |
| PPARD | −0.2 | 2.2E‐10 | 0.8 | 4.0E‐04 |
| PPARG | −3.3 | 1.8E‐15 | 0.7 | 1.2E‐03 |
| PPARGC1A | −3.8 | 3.4E‐11 | 1.2 | 3.6E‐05 |
| PPARGC1B | −3.7 | 1.5E‐04 | n/a | n/a |
| ACOX1 | −6.7 | 2.3E‐38 | 3.1 | 4.4E‐10 |
| SREBF1 | −3.7 | 4.1E‐18 | −0.1 | 1.0E‐03 |
| SREBF2 | −4.5 | 1.6E‐19 | n/a | n/a |
| FXR | n/a | −1.0E+00 | 3.2 | 8.8E‐05 |
| INSIG1 | −0.1 | 1.6E‐16 | 2.3 | 7.2E‐06 |
| INSIG2 | 2.8 | 1.2E‐03 | n/a | n/a |
| CYP51A1 | 2.7 | 1.3E‐07 | n/a | n/a |
| ChREBP | −2.4 | 2.4E‐01 | n/a | n/a |
| INSR | −5.8 | 4.2E‐19 | 1.6 | 4.5E‐03 |
| CNR1 | 2.2 | 3.3E‐02 | −2.1 | 7.6E‐03 |
| ESRRA | −1.6 | 1.1E‐03 | 2.0 | 1.5E‐01 |
| CAR | n/a | n/a | 2.2 | 1.0E‐09 |
The z score indicates the predicted activation state of a transcription factor or key regulator: z ≤ −2 indicates relevant inhibition (shown in green); z ≥ 2 indicates relevant activation (shown in red). The P value indicates significant enrichment of the genes downstream of a regulator (P < 0.01 was considered statistically significant, indicated in gray). n/a indicates an insufficient number of differentially expressed genes to link gene effects to an upstream regulator.
In addition to these anti‐inflammatory and antifibrotic effects, OCA also affected a number of metabolic processes. OCA‐treated mice showed significantly enhanced transcriptional activity of FXR after OCA treatment as expected (z = 3.2, P = 8.8‐05). In line with the observed reduction in hepatic lipids, OCA enhanced fatty acid catabolism (involving activation of ACOX1, z = 3.1, P = 4.4‐10) and attenuated sterol biosynthesis (involving INSIG1 activation; z = 2.3, P = 7.2‐06). Because OCA tended to increase plasma insulin levels, we performed a more granular analysis of hepatic insulin signaling and metabolites associated with insulin resistance. Upstream regulator analysis predicted a significant deactivation of the insulin receptor in HFD‐fed mice relative to chow‐fed mice (z score: −5.8; P = 4.4‐19). OCA did not affect the activation state of this receptor or other regulators linked to insulin signaling and resistance (e.g., Akt2, PI3K, PKC, JNK1). Furthermore, the serum levels of diacylglycerols and ceramides—two lipid species that have been associated with hepatic insulin resistance40—were increased in HFD‐fed mice relative to chow (diacylglycerols: fold change 1.95, P < 0.01; ceramides: fold change 1.49, P < 0.05). Treatment with OCA significantly lowered the serum level of diacylglycerols, and non‐significantly lowered the level of ceramides relative to HFD controls (diacylglycerols: fold change 0.64, P < 0.05; ceramides: fold change 0.82, not significant).
Effects of OCA on Human‐Based Omics Signatures
We next investigated whether treatment with OCA would affect, and potentially normalize, the metabolomics and transcriptomics signatures characteristic for NASH patients (shown in Figures 1 and 2). Of the 205 serum metabolites that were altered by HFD feeding (Supporting Table S2, OCA had an effect on 104 metabolites (Supporting Table S3), of which 44 were a counteraction of the HFD‐induced increase or reduction (Fig. 8A and Supporting Table S3). Among these 44 metabolites were several lipid species that have been implicated in the pathogenesis of NASH, such as monoglycerides, diglycerides, ceramides, and several sphingomyelins that were increased by HFD and attenuated by OCA treatment (Supporting Table S3). Hepatic gene expression profiling showed that OCA modulated the expression of specific genes that characterize NASH patients (gene profile described by Teufel et al.10; Fig. 8B). Furthermore, OCA reverted the expression of most genes that characterize progressive NASH patients (gene set of Moylan et al.9; Fig. 8C).
Figure 8.
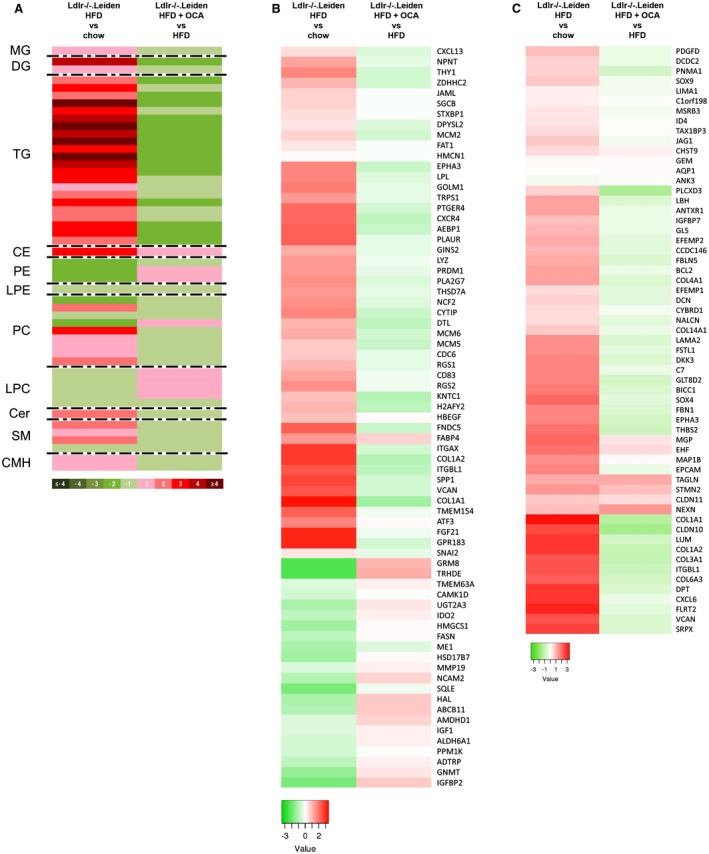
OCA treatment modulates human‐based omics signatures. (A) Heatmap showing HFD‐induced alterations in Ldlr‐/‐.Leiden metabolome at 34 weeks (relative to chow) and effects of OCA thereupon. (B) Heatmap showing expression of genes differentially regulated in human NASH livers versus normal controls10 in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice relative to chow and OCA‐treated mice relative to HFD. (C) Heatmap showing recapitulation of hepatic gene expression profile that differentiates mild (fibrosis stage 0‐1) from severe (fibrosis stage 3‐4) NASH patients9 in 34‐week HFD‐fed Ldlr‐/‐.Leiden mice relative to chow and modulation of this profile in OCA‐treated mice. Green indicates down‐regulation; red indicates up‐regulation. Abbreviations: CE, cholesteryl ester; Cer, ceramide; CMH, monohexosylceramide; DG, diglyceride; LPC, lyso‐phosphatidylcholine; LPE, lysophosphatidylethanolamine; MG, monoglyceride; PC, phosphatidylcholine; PE, phosphatidyl‐ethanolamine; SM, sphingomyelin; and TG, triglyceride.
Discussion
Recent advances in transcriptomics and metabolomics technologies allow comprehensive profiling of human disease phenotypes at the molecular level. We show that such technologies are valuable tools to determine the translational aspects of disease models as well as for studying the effects of therapeutic interventions on processes and plasma metabolites derived from human studies. Obese Ldlr‐/‐.Leiden mice fed a high‐caloric diet mimic NASH patients in two complementary aspects: the disease‐associated hepatic gene expression profile and the disease‐associated serum metabolite profile that integrates multiple organ activities.
In the present study these translational profiling techniques (metabolomics, transcriptomics) are used to study the effects of OCA, which is currently in a phase III clinical trial for the treatment of NASH. However, little is known about the effects of OCA at the molecular pathophysiological level, and a detailed investigation of the anti‐inflammatory and anti‐fibrotic effects of OCA has not been performed to date. Here, we investigated the effects of OCA on hepatic inflammatory and profibrotic processes in the HFD‐fed Ldlr‐/‐.Leiden mouse, which recapitulates many phenotypical and molecular facets of NASH in humans.18, 41 Previously available data on the effects of OCA are based largely on preclinical studies using experimental conditions that appear less relevant to the clinical situation, such as the use of toxic chemicals to induce liver injury or the use of extreme diets (lacking essential amino acids or containing supra‐physiological concentrations of saturated fat, fructose, or cholesterol). These conditions carry the risk of overemphasizing one aspect of disease at the expense of others. Indeed, a recent comparison of the most discriminating gene expression changes in patient liver tissue and several mouse models of NASH10 has raised concerns about whether preclinical models adequately mimic these molecular disease profiles. Such doubts bring into question the translational value of these models in studies of therapeutic interventions in the process of NASH/fibrosis. Here we compared the gene expression profile of Ldlr‐/‐.Leiden mice fed an HFD (without added cholesterol) with the same human liver gene expression data set described by Teufel et al.10 and found a much greater representation of human pathophysiological processes than reported in that study for other rodent models. However, it is unlikely that a single model would allow investigation of all pathogenic processes seen in patients. In‐depth molecular characterization of other preclinical models may help to define a combination of experimental conditions that reflects disease diversity in humans.
Possible explanations for the differences between experimental models and patients include (1) the type of inducer (dietary versus nondietary), how metabolic overload is induced in organs that control metabolic homeostasis, and (2) whether a model mimics the multifactorial nature of the disease, including crosstalk between the liver and other organs.42 For example, Ldlr‐/‐.Leiden mice develop NASH in the context of obesity, adipose tissue inflammation and insulin resistance, and factors released by WAT under comparable experimental conditions used herein have been shown to contribute to NASH pathology.43 Models with nonobese phenotypes such as methionine‐choline deficient diet feeding, STZ treatment, or Pten deficiency44 are typically more rapid, extreme, and liver‐centric, and run the risk of underestimating the contributions of other organs. However, such models can be of value in studying specific aspects of pathology, provided that the investigators have a clear understanding of the pathology they wish to study and choose the animal model that best suits their research questions.45
Next to comparison at the gene expression level, we also investigated the representation of the serum metabolome of NASH patients in the HFD‐fed Ldlr‐/‐.Leiden mouse, using an approach that is inherently different from that applied in the gene expression comparisons. Although gene expression analyses are based on the comparison of the average gene expression in one group (NASH patients, regardless of their differences in histological presentation) relative to that of another group (controls), and therefore reveal those genes that are on average most strongly regulated across all patients, the large number of patients included in metabolomic data sets (n = 535 versus the typical n = 30‐40 in gene expression studies) allow identification of subtypes of NASH patients based on their individual metabolomic profile. In addition, the serum metabolome has been shown to reflect the liver metabolome in two experimental NASH models.11, 12 The serum metabolome also comprises metabolites processed and released by other organs, and therefore may provide clues to the contributions of other organs to NASH progression (such as the adipose tissue and the gut).46, 47 Serum metabolomic profiles therefore form a comprehensive systemic characterization of subjects with NASH that may also allow more holistic treatment of systemic metabolic dysfunction. Although metabolomic profiling appears promising, it should be noted that much is unknown about the dynamics of metabolome fluxes. Because metabolomic studies represent a snapshot at the time of blood collection, commonalities and differences in metabolome should be interpreted with caution. Here we show that the HFD‐fed Ldlr‐/‐.Leiden mouse metabolome reflects that of a subset of NASH patients. Whether the observed degree of overlap between the Ldlr‐/‐.Leiden mouse metabolome and the human NASH metabolome can be considered as adequate or even good for a preclinical NASH model cannot be determined until many different models have been characterized in a similar way, thereby allowing comparison of the different models. As it is assumed that human NASH has various disease etiologies, it is unlikely that a single mouse model would reflect all possible etiologies. Support for this belief is provided by previous comparisons of the same human metabolome data set with two different mouse models11, 12 (i.e., MAT1A‐KO mice and 0.1MCD‐fed mice, both marked by a reduced hepatic S‐adenosylmethionine [SAMe] content). A major subset of the studied human cohort was reflected by these models (classified as M+ subtype), indicating that a subset of NASH patients can be characterized by an impaired synthesis of SAMe. Interestingly, the classification based on the Ldlr‐/‐.Leiden mouse described herein revealed that of the patients classified as non‐M+ subtype in those two studies, most (80%) were found to reflect the Ldlr‐/‐.Leiden subtype, indicating that the Ldlr‐/‐.Leiden model is complementary to these models and may reflect a distinct subset of patients.
Results from the phase II FLINT trial showed that plasma insulin levels were increased in OCA‐treated patients.16 The reasons for this are unknown. In the present study, plasma insulin concentrations tended to be increased after treatment with OCA. Adipose tissue and liver were therefore analyzed in more detail. Adipose tissue hypertrophy and inflammation have been associated with insulin resistance,37, 38 and inflamed eWAT has been causally linked to the development of NAFLD/NASH in mice.43 In the present study, OCA significantly improved hypertrophy and inflammation in eWAT. This is in line with earlier findings in wild‐type mice fed an atherogenic (cholesterol‐containing) diet for 24 weeks, treated with the same dose of OCA as used in this study (10 mg/kg) from 16 to 24 weeks of atherogenic diet feeding.39 We also found no indications of a detrimental effect of OCA on insulin signaling in the liver, and serum metabolites linked to insulin resistance (such as diacylglycerides40) were lowered by OCA. Altogether, these data indicate that the observed elevation of plasma insulin is most likely not attributable to effects in eWAT or liver. Other tissues that may explain effects on plasma insulin such as skeletal muscle and pancreas were not isolated in this study. Because FXR is not expressed by skeletal muscle tissue,48 it is unlikely that OCA had a direct effect on the insulin sensitivity of this tissue, although we cannot exclude the possibility that OCA could have had an indirect effect on muscle insulin sensitivity. As FXR activation in the pancreas has been shown to stimulate insulin secretion,49 it is possible that the observed insulin‐elevating effects are caused by an effect on insulin secretion per se.
In line with the observations in the FLINT trial,16 we observed an improvement in hepatic fibrosis after OCA treatment in the present study. Although the reported improvement in the FLINT trial was based on improvement of the fibrosis severity score (a qualitative measure), we investigated the antifibrotic effects of OCA in a quantitative manner, combining several methods of analysis to provide a comprehensive description of the effects of OCA. We show that OCA quantitatively reduces hepatic fibrosis as measured by traditional picrosirius red staining, as well as biochemically measured collagen levels. This is in line with the findings of others, who report reduced hepatic collagen content after OCA treatment in chemically induced liver fibrosis13, 14, 50 and in AMLN diet (high trans‐fat and fructose diet supplemented with 2% cholesterol)–fed ob/ob mice.51, 52 In addition to the observed reduction in the level of collagen in the liver in the present study, OCA also lowered the degree of reticulation of collagen (of both thin and aggregated collagen fibers) as assessed by multiphoton imaging, a technology that allows the dissection of fibrosis stages in human biopsies.53 Because hepatic fibrosis is the result of an imbalance between the synthesis and degradation of extracellular matrix proteins,54 any effect on hepatic fibrosis can be explained by an effect on either (or both) of these processes. Whether fibrotic changes in the liver are completely reversible remains a matter of debate, although it is increasingly thought that resolution of fibrosis is achievable, at least to a certain extent.55 Here we show that the antifibrotic effects of OCA are at least in part attributable to a reduction in the deposition of new collagen (as evidenced by reduced rates of de novo collagen synthesis). Our results do not indicate an effect on the breakdown of already existing collagen, as the levels of hepatic fibrosis were not reduced beyond the level observed in the start‐of‐treatment reference group. However, we cannot exclude the possibility that resolution would have occurred if the treatment period had been lengthened or the dose of OCA had been increased.
Overall, we show that HFD‐fed Ldlr‐/‐.Leiden mice recapitulate specific molecular metabolomic and transcriptomic signatures of NASH patients. Treatment with a relatively low dose of OCA modulates these human disease profiles and reduces fibrosis progression, but does not resolve already‐manifest fibrosis in the period studied (10 weeks). These data indicate that human molecular signatures may be used to estimate the translational value of preclinical models for NASH and assess the effects of interventions in a comprehensive manner.
Potential conflict of interest
Dr. Alonso and Dr. Martinez‐Arranz are employed by OWL. Dr. Mato advises, consults, and owns stock in OWL; he advises and consults for Abbott, and consults for Galmed.
Author contributions
Conception and design of the study: R.K., R.H., and P.Y.W.; experiments/data acquisition: M.C.M., V.L., S.K., J.V., A.M., M.I.L., L.G., W.Y., S.T., M.P.M.C., E.J.P., and C.A.; analysis and interpretation of data: M.C.M., M.I.L., I.M.A., R.S., A.vK., A.M.vdH., J.M.M., and C.A.; manuscript writing: M.C.M. and R.K.; and critical revision of drafted manuscript and approval of final version: all authors.
Supporting information
Supported by Netherlands Organisation for Applied Scientific Research (research program Personalized Health Technologies), National Institutes of Health (RO1AT001576 to J.M.M.), Agencia Estatal de Investigación (MINECO SAF2014‐52097R, ISCiii PIE14/00031, and CIBERehd‐ISCiii), and Severo Ochoa Excellence Accreditation (SEV‐2016‐0644 to J.M.M.).
References
Author names in bold designate shared co‐first authorship.
- 1. Musso G, Cassader M, Gambino R. Non‐alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov 2016;15:249‐274. [DOI] [PubMed] [Google Scholar]
- 2. Tiniakos DG, Vos MB, Brunt EM. Nonalcoholic fatty liver disease: pathology and pathogenesis. Annu Rev Pathol 2010;5:145‐171. [DOI] [PubMed] [Google Scholar]
- 3. Anderson N, Borlak J. Molecular mechanisms and therapeutic targets in steatosis and steatohepatitis. Pharmacol Rev 2008;60:311‐357. [DOI] [PubMed] [Google Scholar]
- 4. Loomba R, Sanyal AJ. The global NAFLD epidemic. Nat Rev Gastroenterol Hepatol 2013;10:686‐690. [DOI] [PubMed] [Google Scholar]
- 5. Angulo P, Kleiner DE, Dam‐Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, et al. Liver fibrosis, but no other histologic features, is associated with long‐term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389‐397.e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ekstedt M, Hagström H, Nasr P, Fredrikson M, Stål P, Kechagias S, et al. Fibrosis stage is the strongest predictor for disease‐specific mortality in NAFLD after up to 33 years of follow‐up. Hepatology 2015;61:1547‐1554. [DOI] [PubMed] [Google Scholar]
- 7. Haas JT, Francque S, Staels B. Pathophysiology and mechanisms of nonalcoholic fatty liver disease. Annu Rev Physiol 2016;78:181‐205. [DOI] [PubMed] [Google Scholar]
- 8. Heymann F, Tacke F. Immunology in the liver—from homeostasis to disease. Nat Rev Gastroenterol Hepatol 2016;13:88‐110. [DOI] [PubMed] [Google Scholar]
- 9. Moylan CA, Pang H, Dellinger A, Suzuki A, Garrett ME, Guy CD, et al. Hepatic gene expression profiles differentiate presymptomatic patients with mild versus severe nonalcoholic fatty liver disease. Hepatology 2014;59:471‐482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Teufel A, Itzel T, Erhart W, Brosch M, Wang XY, Kim YO, et al. Comparison of gene expression patterns between mouse models of nonalcoholic fatty liver disease and liver tissues from patients. Gastroenterology 2016;151:513‐525, e510. [DOI] [PubMed] [Google Scholar]
- 11. Alonso C, Fernández‐Ramos D, Varela‐Rey M, Martínez‐Arranz I, Navasa N, Van Liempd SM, et al. Metabolomic identification of subtypes of nonalcoholic steatohepatitis. gastroenterology 2017;152:1449‐1461, e1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iruarrizaga‐Lejarreta M, Varela‐Rey M, Fernández‐Ramos D, Martínez‐Arranz I, Delgado TC, Simon J, et al. Role of aramchol in steatohepatitis and fibrosis in mice. Hepatology Communications 2017;1:911‐927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verbeke L, Mannaerts I, Schierwagen R, Govaere O, Klein S, Vander Elst I, et al. FXR agonist obeticholic acid reduces hepatic inflammation and fibrosis in a rat model of toxic cirrhosis. Sci Rep 2016;6:33453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang D‐G, Zhang C, Wang J‐X, Wang B‐W, Wang H, Zhang Z‐H, et al. Obeticholic acid protects against carbon tetrachloride‐induced acute liver injury and inflammation. Toxicol Appl Pharmacol 2017;314:39‐47. [DOI] [PubMed] [Google Scholar]
- 15. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013;145:574‐582, e571. [DOI] [PubMed] [Google Scholar]
- 16. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al. Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Morrison MC, Mulder P, Salic K, Verheij J, Liang W, van Duyvenvoorde W, et al. Intervention with a caspase‐1 inhibitor reduces obesity‐associated hyperinsulinemia, non‐alcoholic steatohepatitis and hepatic fibrosis in LDLR‐/‐.Leiden mice. Int. J Obesity 2016;40:1416‐1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. van Koppen A, Verschuren L, van den Hoek AM, Verheij J, Morrison MC, Li K, et al. Uncovering a predictive molecular signature for the onset of NASH‐related fibrosis in a translational NASH mouse model. Cell Mol Gastroenterol Hepatol 2018;5:83‐98, e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Salic K, Morrison MC, Verschuren L, Wielinga PY, Wu L, Kleemann R, et al. Resolvin E1 attenuates atherosclerosis in absence of cholesterol‐lowering effects and on top of atorvastatin. Atherosclerosis 2016;250:158‐165. [DOI] [PubMed] [Google Scholar]
- 20. Mulder P, Morrison MC, Verschuren L, Liang W, van Bockel JH, Kooistra T, et al. Reduction of obesity‐associated white adipose tissue inflammation by rosiglitazone is associated with reduced non‐alcoholic fatty liver disease in LDLr‐deficient mice. Sci Rep 2016;6:31542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Galarraga M, Campión J, Muñoz‐Barrutia A, Boqué N, Moreno H, Martínez JA, et al. Adiposoft: automated software for the analysis of white adipose tissue cellularity in histological sections. J Lipid Res 2012;53:2791‐2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Meth 2012;9:676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schneider CA, Rasband WS, Eliceiri KW. NIH image to ImageJ: 25 years of image analysis. Nat Meth 2012;9:671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liang W, Menke AL, Driessen A, Koek GH, Lindeman JH, Stoop R, et al. Establishment of a general NAFLD scoring system for rodent models and comparison to human liver pathology. PLoS ONE 2014;9:e115922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kleiner DE, Brunt EM, Natta MV, Behling C, Contos MJ, Cummings OW, et al. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 26. Itoh M, Kato H, Suganami T, Konuma K, Marumoto Y, Terai S, et al. Hepatic crown‐like structure: a unique histological feature in non‐alcoholic steatohepatitis in mice and humans. PLoS One 2013;8:e82163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sun W, Chang S, Tai DC, Tan N, Xiao G, Tang H, et al. Nonlinear optical microscopy: use of second harmonic generation and two‐photon microscopy for automated quantitative liver fibrosis studies. J Biomed Opt 2008;13:064010. [DOI] [PubMed] [Google Scholar]
- 28. Otsu N. A Threshold selection method from gray‐level histograms. IEEE Trans Syst Man Cybern 1979;9:62‐66. [Google Scholar]
- 29. Bligh EG, Dyer WJ. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 1959;37:911‐917. [DOI] [PubMed] [Google Scholar]
- 30. Decaris ML, Gatmaitan M, FlorCruz S, Luo F, Li K, Holmes WE, et al. Proteomic analysis of altered extracellular matrix turnover in bleomycin‐induced pulmonary fibrosis. Mol Cell Proteomics 2014;13:1741‐1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Didangelos A, Yin X, Mandal K, Baumert M, Jahangiri M, Mayr M. Proteomics characterization of extracellular space components in the human aorta. Mol Cell Proteomics 2010;9:2048‐2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Price JC, Holmes WE, Li KW, Floreani NA, Neese RA, Turner SM, et al. Measurement of human plasma proteome dynamics with 2H2O and liquid chromatography tandem mass spectrometry. Anal Biochem 2012;420:73‐83. [DOI] [PubMed] [Google Scholar]
- 33. Martínez‐Arranz I, Mayo R, Pérez‐Cormenzana M, Mincholé I, Salazar L, Alonso C, et al. Enhancing metabolomics research through data mining. J Proteomics 2015;127:275‐288. [DOI] [PubMed] [Google Scholar]
- 34. Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol 2010;11:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morrison MC, Liang W, Mulder P, Verschuren L, Pieterman E, Toet K, et al. Mirtoselect, an anthocyanin‐rich bilberry extract, attenuates non‐alcoholic steatohepatitis and associated fibrosis in ApoE *3Leiden mice. J Hepatol 2015;62:1180‐1186. [DOI] [PubMed] [Google Scholar]
- 36. Liang W, Tonini G, Mulder P, Kelder T, van Erk M, van den Hoek AM, et al. Coordinated and interactive expression of genes of lipid metabolism and inflammation in adipose tissue and liver during metabolic overload. PLoS One 2013;8:e75290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kim JI, Huh JY, Sohn JH, Choe SS, Lee YS, Lim CY, et al. Lipid‐overloaded enlarged adipocytes provoke insulin resistance independent of inflammation. Mol Cell Biol 2015;35:1686‐1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McArdle M, Finucane O, Connaughton R, McMorrow A, Roche H. Mechanisms of obesity‐induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol 2013;4:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Haczeyni F, Poekes L, Wang H, Mridha AR, Barn V, Haigh WG, et al. Obeticholic acid improves adipose morphometry and inflammation and reduces steatosis in dietary but not metabolic obesity in mice. Obesity 2017;25:155‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Samuel Varman T, Shulman Gerald I. Mechanisms for insulin resistance: common threads and missing links. Cell 2012;148:852‐871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Morrison MC, Kleemann R, vanKoppen A, Hanemaaijer R, Verschuren L. Key inflammatory processes in human NASH are reflected in Ldlr−/−.Leiden mice: a translational gene profiling study. Front Physiol 2018;9:132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Byrne CD, Targher G. NAFLD: A multisystem disease. J Hepatol 2015;62:S47‐S64. [DOI] [PubMed] [Google Scholar]
- 43. Mulder P, Morrison MC, Wielinga PY, van Duyvenvoorde W, Kooistra T, Kleemann R. Surgical removal of inflamed epididymal white adipose tissue attenuates the development of non‐alcoholic steatohepatitis in obesity. Int J Obes 2016;40:675‐684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takahashi Y, Soejima Y, Fukusato T. Animal models of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J Gastroenterol 2012;18:2300‐2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Castro RE, Diehl AM. Towards a definite mouse model of NAFLD. J Hepatol 2018;69:272‐274. [DOI] [PubMed] [Google Scholar]
- 46. Zhang X, Ji X, Wang Q, Li JZ. New insight into inter‐organ crosstalk contributing to the pathogenesis of non‐alcoholic fatty liver disease (NAFLD). Protein Cell 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Targeting the gut‐liver axis in liver disease. J. Hepatol 2018;9:164‐177. [DOI] [PubMed] [Google Scholar]
- 48. Cariou B, van Harmelen K, Duran‐Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, et al. The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice. J Biol Chem 2006;281:11039‐11049. [DOI] [PubMed] [Google Scholar]
- 49. Düfer M, Hörth K, Krippeit‐Drews P, Drews G. The significance of the nuclear farnesoid X receptor (FXR) in β cell function. Islets 2012;4:333‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Úbeda M, Lario M, Muñoz L, Borrero M‐J, Rodríguez‐Serrano M, Sánchez‐Díaz A‐M, et al. Obeticholic acid reduces bacterial translocation and inhibits intestinal inflammation in cirrhotic rats. J Hepatol 2016;64:1049‐1057. [DOI] [PubMed] [Google Scholar]
- 51. Roth JD, Feigh M, Veidal SS, Fensholdt LK, Rigbolt KT, Hansen HH, et al. INT‐767 improves histopathological features in a diet‐induced ob/ob mouse model of biopsy‐confirmed non‐alcoholic steatohepatitis. World J Gastroenterol 2018;24:195‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Tolbol KS, Kristiansen MN, Hansen HH, Veidal SS, Rigbolt KT, Gillum MP, et al. Metabolic and hepatic effects of liraglutide, obeticholic acid and elafibranor in diet‐induced obese mouse models of biopsy‐confirmed nonalcoholic steatohepatitis. World J Gastroenterol 2018;24:179‐194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wang Y, Vincent R, Yang J, Asgharpour A, Liang X, Idowu MO, et al. Dual‐photon microscopy‐based quantitation of fibrosis‐related parameters (q‐FP) to model disease progression in steatohepatitis. Hepatology 2017;65:1891‐1903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Huang Y, Deng X, Liang J. Modulation of hepatic stellate cells and reversibility of hepatic fibrosis. Exp Cell Res 2017;352:420‐426. [DOI] [PubMed] [Google Scholar]
- 55. Zoubek ME, Trautwein C, Strnad P. Reversal of liver fibrosis: from fiction to reality. Best Pract Res Clin Gastroenterol 2017;31:129‐141. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials