

Abstract
Prohibitin1 (PHB1) is a mitochondrial chaperone with diverse functions that include cell proliferation, apoptosis, and mitochondrial homoeostasis. Liver‐specific Phb1 knockout (KO) mice develop spontaneous injury and hepatocellular carcinoma (HCC). Our previous work demonstrated that PHB1 negatively regulates the H19‐insulin‐like growth factor 2 (IGF2)‐H19‐IGF2 axis signaling pathway and E‐box activity in hepatocytes and HCC cells. Phb1 KO livers exhibited increased expression of multiple wingless/integrated (WNT) target genes compared to control littermates. Therefore, we hypothesized that PHB1 is a negative regulator of WNT‐beta‐catenin signaling in the liver. Analysis of livers from Phb1 KO mice demonstrated an activation of the WNT‐beta‐catenin pathway as determined by phosphorylation of glycogen synthase kinase 3 (GSK3)betaserine [Ser]9 and protein kinase B (AKT)Ser473. Phb1 KO livers showed increased messenger RNA (mRNA) levels of multiple WNT ligands, with Wnt7a (79‐fold), Wnt10a (12‐fold), and Wnt16 (48‐fold) being most highly overexpressed compared to control littermates. Subcellular fractionation of liver cells from Phb1 KO mice indicated that hepatocytes are the main source of WNT ligands. Immunostaining and cellular colocalization analysis of Phb1 KO livers demonstrated expression of WNT7a, WNT10a, and WNT16 in hepatocytes. Chromatin immunoprecipitation revealed increased binding of transcription factor E2F1 (E2F1) to the Wnt10a promoter in Phb1 KO livers and WNT9A in HepG2 cells. PHB1 silencing in HepG2 cells activated WNT signaling, whereas its overexpression caused inactivation of this pathway. PHB1 silencing in HepG2 cells induced the expression of multiple WNT ligands of which WNT9A induction was partly regulated through E2F1. Conclusion: PHB1 acts as a negative regulator of WNT signaling, and its down‐regulation causes the induction of multiple WNT ligands and downstream activation of canonical WNT‐beta‐catenin signaling in murine liver and human HCC cells, in part through E2F1.
Abbreviations
- AKT
protein kinase B
- CCA
cholangiocarcinoma
- Ccnd1/Ccne1
cyclin D1/E1
- ChIP
chromatin immunoprecipitation
- c‐Myc
Myc proto‐oncogene
- E2F1
transcription factor E2F1
- EMT
epithelial‐mesenchymal transition
- EV
empty vector
- GEO
Gene Expression Omnibus
- GSK3
glycogen synthase kinase 3
- HCC
hepatocellular carcinoma
- HNF4
hepatocyte nuclear factor 4
- IGF2
insulin‐like growth factor 2
- IgG
immunoglobulin G
- KO
knockout
- LEF
lymphoid enhancer‐binding factor 1
- LRP 5/6
low density lipoprotein receptor‐related protein 5/6
- mRNA
messenger RNA
- NC
negative control siRNA
- NIH
National Institutes of Health
- OE
overexpressing
- p‐
phosphorylated
- PHB1
prohibitin 1
- pos
positive
- qPCR
quantitative polymerase chain reaction
- Rb
retinoblastoma protein
- Ser
serine
- si
small interfering
- TCF
T‐cell‐specific transcription factor
- WNT
wingless/integrated
- WT
wild type
PHB1 is an evolutionarily conserved mitochondrial chaperone protein proposed to play a role in cellular proliferation,1 transcriptional regulation,2, 3 mitochondrial homeostasis,4 and cellular signaling.5 It was first identified in the regenerating rat liver where its expression was down‐regulated and consequently thought to act as a negative regulator of cell proliferation.1 The diverse functions of PHB1 are controversial and determined by cell type and cellular localization, such as at the plasma membrane, nucleus, and mitochondria, in addition to its posttranslational modifications.5, 6, 7 Our previous study demonstrated that liver‐specific deletion of Phb1 in mice causes chronic liver injury, bile duct metaplasia, cell proliferation, and spontaneous development of HCC.8 PHB1 negatively regulates the proliferation of hepatocytes and human HCC cells, in part through suppression of the H19‐IGF2 signaling axis.9 Importantly, PHB1 expression has been shown to be down‐regulated in human HCC and cholangiocarcinoma (CCA) and also negatively regulates E‐box activity in human HCC cells.10
WNT‐beta‐catenin signaling is a highly conserved and essential pathway for normal development and tissue regeneration of various organs, including liver.11, 12 Deregulated WNT‐beta‐catenin signaling has been shown to correlate with tumorigenesis.12, 13 The WNT family consists of 19 secreted ligands, and each one is differentially regulated at the transcriptional and posttranscriptional levels.14 WNT signaling activation initiates when a ligand binds to its transmembrane receptors Frizzled and low‐density lipoprotein receptor‐related protein (LRP)5/6 and is followed by cascades of protein phosphorylation that lead to increased expression of WNT target genes. WNT signaling consists of beta‐catenin‐dependent (canonical) and beta‐catenin‐independent (noncanonical) pathways. Canonical WNT signaling is primarily regulated by the transcriptional co‐activator beta‐catenin through T‐cell‐specific transcription factor (TCF)/lymphoid enhancer‐binding factor 1 (LEF) transcription factors. In the absence of WNT, cytoplasmic beta‐catenin is degraded by the action of the destruction complex composed of the scaffolding protein axin, the tumor suppressor adenomatous polyposis coli gene product, casein kinase 1 (CK1), and glycogen synthase kinase 3 (GSK3) beta. CK1 and GSK3beta sequentially phosphorylate the amino terminal region of beta‐catenin, resulting in its ubiquitination. Following WNT ligand interaction with coreceptors Frizzled/LRP5/6, the beta‐catenin destruction complex gets inactivated. GSK3beta is a negative regulator of canonical WNT‐beta‐catenin signaling. Phosphorylation of GSK3beta on Ser9 by kinases, such as AKT, leads to its inactivation and results in stabilization and increased nuclear translocation of beta‐catenin and transcriptional activation of WNT target genes.13
The WNT‐beta‐catenin pathway plays an important role in liver development and regeneration.12, 15 On the other hand, overactive WNT‐beta‐catenin signaling positively correlates with human HCC and mouse models of HCC.15 Because Phb1 KO mice livers exhibit an extensive regenerative response at an early age and develop HCC later in life, we hypothesized that WNT signaling is hyperactivated in these mice livers and that this could drive regeneration and later tumorigenesis. Our data for Phb1 KO mice livers and in vitro gene silencing/overexpression in HepG2 cells demonstrate that PHB1 negatively regulates WNT signaling in these systems. PHB1 suppresses the expression of multiple WNT ligands partly in an E2F1‐dependent manner. In summary, our data demonstrate for the first time a novel role for PHB1 in regulating one of the major oncogenic pathways in liver and identify yet another mechanism of how PHB1 acts as a tumor suppressor in murine liver and human liver cancer cells.
Materials and Methods
Materials and Reagents
All general reagents used were analytical grade purchased from Sigma‐Aldrich (St. Louis, MO) unless specified.
Human Liver Tissues
Human HCC and CCA tissues and adjacent nontumor tissues collected during liver resection were used in this study, which was approved by institutional review boards of Cedars‐Sinai Medical Center and Keck School of Medicine, University of Southern California. All human materials were obtained with patients’ informed consent. Both tumor and nontumor adjacent tissues were histologically verified by pathologists at the respective institutes. All tissues samples were de‐identified and then frozen in liquid nitrogen for long‐term storage.
Animal Experiments
Animals were bred, maintained, and cared for as per National Institutes of Health (NIH) guidelines, and protocols were approved by the Institutional Animal Care and Use Committee of Cedars‐Sinai Medical Center, Los Angeles, CA. Liver‐specific Phb1 KO mice were generated as described.8 Liver tissues from 3‐week‐old male and female Phb1flox/flox;AlbuminCre (hereafter, Phb1 KO) mice and Flox wild‐type control (WT) littermates were used for various analyses described in this study.
Cell Culture
Human hepatoblastoma cell line HepG2 was purchased from the American Type Culture Collection. HepG2, Huh7, and Hep3B cell lines were cultured as described.9, 16
Quantitative Real‐Time Polymerase Chain Reaction
Total RNA was isolated using the column‐based purification method according to the manufacturer's protocol (Quick‐RNA MiniPrep; Zymo Research, Irvine, CA). Quantitative real‐time polymerase chain reaction (qPCR) was performed as described.9 Glyceraldehyde 3‐phosphate dehydrogenase (Gapdh; mouse) or hypoxanthine phospho ribosyl transferase 1 (HPRT1; human) were used for normalizing gene expression. Gene‐specific primers were designed by the Roche assay design method and purchased from Eurofins MWG Operon USA (Louisville, KY). The probes and primers used in this study are listed in Supporting Tables S1 and S2.
Gene Silencing and Overexpression In Vitro
We seeded 1.5 × 105 cells in six‐well plates for PHB1 or E2F1 overexpression or silencing as described.9 E2F1 plasmid was purchased from Addgene (Cambridge, MA). For gene knockdown, prevalidated Silencer Select small interfering (si)RNAs against human PHB1, E2F1, or universal negative siRNA control (NC) (Thermo Scientific, Waltham, MA) were reverse transfected into cells at a dose of 20 nM in six‐well plates by using Lipofectamine RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA). Primary mouse hepatocytes were isolated from C57B6J mice (Jackson Laboratory, Bar Harbor, ME) as described below and were plated on collagen precoated tissue culture plates. Cells were forward transfected with siRNA against mouse Phb1 or NC for 24 hours.
Cell Isolation From Mouse Liver
Hepatocytes and nonparenchymal cells were isolated by following published protocols17 with minor modifications. Mouse liver was perfused with collagenase for 15 minutes, and hepatocytes were isolated by centrifugation at 50g for 1 minute at 4°C. Total nonparenchymal cells were isolated after three serial centrifugations at 50g for 1 minute to remove hepatocytes, and finally nonparenchymal cell fractions were pelleted at 200g for 7 minutes at 4°C. Freshly isolated mouse hepatocytes were used for in vitro Phb1 silencing as described previously.
Immunofluorescence Staining
WT and Phb1 KO mice livers were fixed in 4% paraformaldehyde (Poly Sciences, Warrington, PA) and processed for histology analysis. A 5‐μm‐thick paraffin‐embedded liver tissue section was used for immunofluorescence staining as described.18 Primary antibodies used in this study are listed in Supporting Table S3. Fluorescence signals were detected by using Cy3 (Jackson Immuno Research Laboratories, West Grove, PA) or Alexa Fluor488 (Abcam, Cambridge, United Kingdom) conjugated secondary antibodies. Images were acquired with a KEYENCE BZ‐X710 inverted fluorescent microscope (KEYENCE Corporation of America, Itasca, IL).
Chromatin Immunoprecipitation Assay
Putative E2F1 binding sites were identified by ALGGEN PROMO version 3.0.2 prediction software. E2F1 binding to mouse Wnt10a and human WNT9A promoters was examined by chromatin immunoprecipitation (ChIP) assay using the EpiTect ChIP OneDay kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions. Briefly, cross‐linked and precleared chromatin was immunoprecipitated with 2 μg E2F1 antibody for 16 hours. Immunoprecipitated chromatin was washed and reverse cross‐linked followed by genomic DNA isolation. qPCR was performed using ChIP primers (provided in Supporting Table S2). Relative target occupancy was determined as described.9
Western Blot Analysis
Total tissue or cell lysates were prepared by using modified radioimmunoprecipitation assay buffer with a protease and phosphatase inhibitor cocktail (Sigma‐Aldrich) and performed western blotting as described.9, 18 Densitometry analyses and quantification of western blots were determined using ImageJ software version 1.50i (NIH).
TCF Promoter Activity Assay
TCF promoter activity was measured by the TOPFlash and FOPFlash assay methods.19 After 24 hours of PHB1 knockdown or overexpression in HepG2 cells, cells were transfected with Renilla plasmid (0.1 μg) and TOPFlash or FOPFlash plasmids (1 μg) (Addgene), which specifically measures β‐catenin/TCF transcriptional activity. Cells were grown for an additional 24 hours. Twenty‐four hours after luciferase plasmid transfection in control HepG2 cells in experiments with conditioned media, media was changed to conditioned media collected from siPHB1 or NC siRNA‐transfected HepG2 cells for an additional 24 hours. Luminescence produced by Firefly and Renilla luciferase was measured with a Tecan GENios spectrophotometer (Tecan, Männedorf, Switzerland) using the DualGlo Luciferase assay system (Promega, Madison, WI). Relative luciferase activity was reported as fold induction after normalization to Renilla luciferase values.
Statistical Analysis
Data are expressed as mean ± SEM values. Statistical analysis was performed using analysis of variance and Fisher's exact test. Expression levels of genes and proteins were normalized to respective housekeeping genes and proteins, and fold change was represented. Significance was defined by P < 0.05.
Results
WNT‐Beta‐Catenin Signaling is Activated in Phb1 KO Mice Livers
Previously we demonstrated that Phb1 KO mice exhibit severe liver injury and develop HCC by 7‐8 months of age.8 Analysis of 3‐week‐old Phb1 KO livers showed increased proliferation of hepatocytes as determined by a large increase in the number of Ki67positive [pos] hepatocyte nuclear factor 4 (HNF4)alphapos cells in KO livers compared to WT livers (Fig. 1A ). Moreover, there was a 3‐fold to 5‐fold increase in the expression of the WNT target genes cyclin E1 (Ccne1) and Myc proto‐oncogene (c‐Myc) in Phb1 KO livers compared to WT controls (Fig. 1B). We also previously showed a significant increase in cyclin D1 (Ccnd1) expression in Phb1 KO livers compared to WT littermates.8 Based on these observations, we tested the hypothesis that WNT‐beta‐catenin signaling is activated in Phb1 KO livers and that this activation might drive the extensive regenerative response and tumorigenesis in Phb1 KO livers. GSK3beta is a critical regulator of canonical WNT‐beta‐catenin signaling, and its activity is negatively regulated by its phosphorylation on Ser9 by protein kinases, such as AKT.20, 21 We found that GSK3betaSer9 phosphorylation but not its total content increased 2.5‐fold in Phb1 KO livers compared to WT controls (Fig. 1C). Next, we investigated whether the upstream kinase AKT is activated in Phb1 KO livers. AKT activity is regulated by multiple amino acid residue phosphorylation events for which Ser473 phosphorylation is required for full AKT activity.22 AKTSer473 phosphorylation increased by approximately 50% in Phb1 KO livers compared to WT controls (Fig. 1D).
Figure 1.
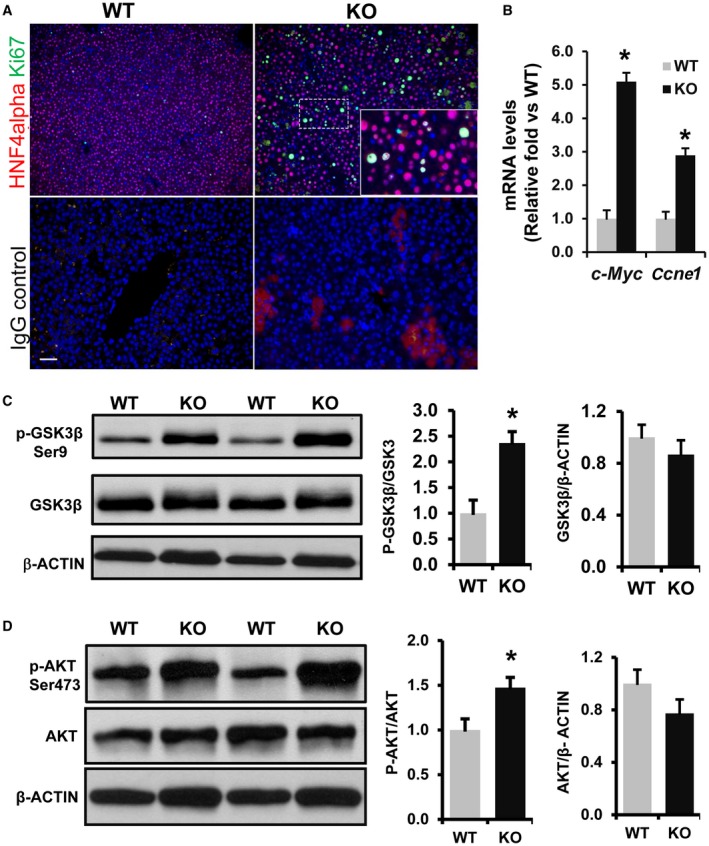
Increased cell proliferation and activation of WNT signaling in 3‐week‐old Phb1 KO livers. (A) Paraffin‐embedded liver sections from 3‐week‐old Phb1 KO mice and WT controls were subjected to co‐immunofluorescence staining for proliferation marker Ki67 (green) with hepatocyte marker HNF4alpha (red) and control IgG (staining control). Nuclei are stained with DAPI (blue). Images represent three independent staining on four different KO and WT livers. Scale bar, 25 μm. (B) Increased expression of WNT target genes in Phb1 KO liver. Total RNA was prepared from 3‐week‐old WT and Phb1KO livers and subjected to qPCR as described in Materials and Methods. Relative expressions of c‐Myc and Ccne1 were compared with WT controls. Results represent mean ± SEM from n = 7 mice in each group; *P < 0.01 versus WT. Total protein was extracted from 3‐week‐old Phb1 KO and WT livers and immunoblotted for (C) p‐GSK3betaSer9, total GSK3beta, and beta‐ACTIN and (D) p‐AKTSer473, total AKT, and beta‐ACTIN. Representative blots are shown. Protein band intensity was quantified by densitometry analysis by ImageJ software (NIH). The ratio between normalized phospho/total protein band intensities was calculated, then mean activation fold was represented over WT. Results represent mean ± SEM from n = 7 mice in each group; * P < 0.05 versus WT. Abbreviations: c‐Myc, Myc Proto‐oncogene; DAPI, 4´,6‐diamidino‐2‐phenylindole.
Multiple WNT Ligands are Induced in Phb1 KO Livers
WNT signaling initiates when WNT ligands bind to Frizzled/LRP5/6, the transmembrane receptors complex. To test whether activation of WNT signaling in Phb1 KO livers is associated with increased expression of WNT ligands, qPCR was performed on RNA extracted from total livers of KO and WT mice. We found Wnt7a (79‐fold), Wnt10a (12‐fold), and Wnt16 (48‐fold) were significantly increased in Phb1 KO livers compared to WT controls (Fig. 2A ). Wnt10b (4.6‐fold) and Wnt4 (2.5‐fold) were also increased to a lesser extent in KO livers (Fig. 2A). WNT ligands and Frizzled receptors that were not detected/changed in KO livers are shown in Supporting Fig. S1A,B. Analysis of Phb1 KO livers also revealed increased expression of epithelial‐mesenchymal transition (EMT) markers, such as Vimentin and Snail family zinc finger 1 (Snai1) (Supporting Fig. S2A).
Figure 2.
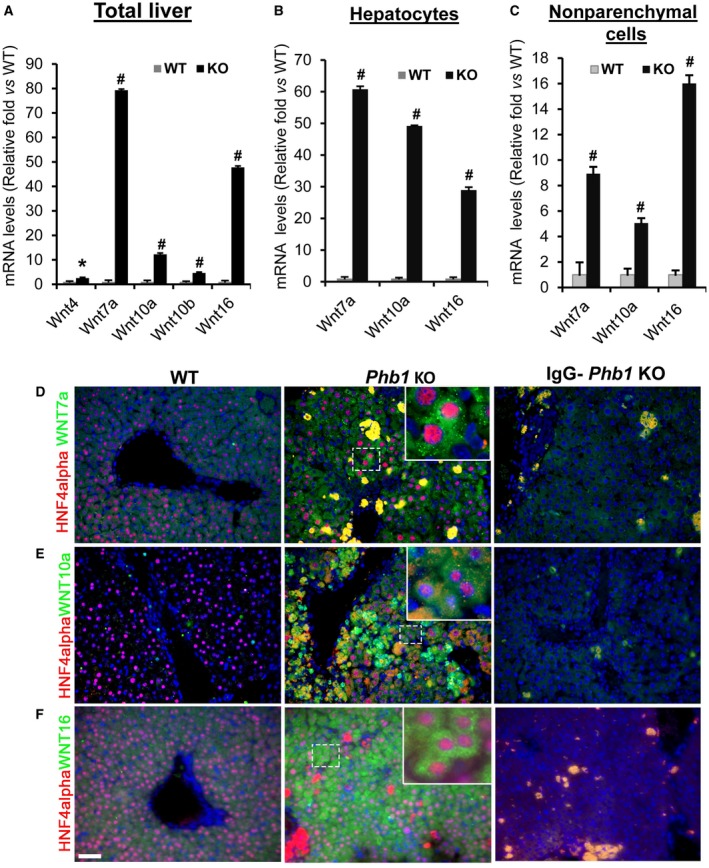
Induction of multiple WNT ligands in 3‐week‐old Phb1 KO livers. (A) Total RNA was isolated from 3‐week‐old Phb1 KO and WT control livers, and qPCR was performed as described in Materials and Methods. Relative expression levels of Wnt4, Wnt7a, Wnt10a, Wnt10b, and Wnt16 were compared to WT control livers. Results represent mean ± SEM from n = 7 mice per group; #P < 0.001, *P < 0.05 versus WT. (B) Total RNA was prepared from hepatocytes isolated from 3‐week‐old Phb1 KO and WT livers and subjected to qPCR. Relative expression of Wnt7a, Wnt10, and Wnt16 was compared to hepatocytes from WT control livers; n = 4 mice in each group; #P < 0.001 versus WT. (C) Total RNA was prepared from nonparenchymal cells isolated from Phb1 KO and WT control livers, and qPCR was performed as described in Materials and Methods. Relative expression of Wnt7a, Wnt10a, and Wnt16 was compared to the nonparenchymal fraction from WT controls; n = 4 per group; #P < 0.001 versus WT. (D‐F) Paraffin‐embedded liver sections from 3‐week‐old Phb1 KO and WT mice were subjected to co‐immunofluorescence staining for hepatocyte marker HNF4alpha (red) with (D) WNT7a, (E) WNT10a, and (F) WNT16. IgG staining was performed in parallel to determine nonspecific staining for each antibody. Nuclei are stained with DAPI (blue). Images represent three to four independent stainings on four different KO and WT livers. Scale bar, 25 μm. Stained areas seen in the IgG control of Phb1 KO livers are nonspecific staining of the necrotic areas in the liver. Inset is a representation of marked area just to show localization (~6x of the marked area with dashed white lines) Abbreviation: DAPI, 4´,6‐diamidino‐2‐phenylindole.
WNT Ligands Are Induced In Hepatocytes And Nonparenchymal Cells in Phb1 KO Livers
To determine the cellular origin of WNT ligands in Phb1 KO livers, hepatocytes and nonparenchymal cells were fractionated from WT and KO livers. qPCR data demonstrated that WNT ligands are mainly derived from the hepatocyte fraction. There was a 50‐fold to 60‐fold increase in the mRNA levels of Wnt7a and Wnt10a and about a 30‐fold increase in Wnt16 in KO hepatocytes compared to hepatocytes from WT controls (Fig. 2B). All three Wnt ligands were also significantly increased in the nonparenchymal cell fraction but to a much lower extent (Fig. 2C). To confirm that hepatocytes express WNT ligands, co‐immunofluorescence was performed on paraffin‐embedded liver sections prepared from Phb1 KO and WT mice. Hepatocyte marker HNF4alpha colocalization with WNT7a, WNT10a, and WNT16 further confirmed hepatocyte expression of WNT ligands in Phb1 KO liver (Fig. 2D,E,F). In KO livers we found several small nonhepatocyte cells that also stained positive for WNT10a. Immunostaining with mesenchymal marker colocalization was not conclusive due to nonspecific staining; hence, we could not determine the nonparenchymal cell types that expressed WNT ligands. To further validate whether Phb1 deletion directly influenced the expression of WNT ligands in hepatocytes, Phb1 silencing was performed using in vitro cultured mouse primary hepatocytes for 24 hours. Both Wnt7a and Wnt10a mRNA levels increased by 30%‐70% after ~75% silencing of Phb1 (Fig. 3A,B). Wnt16 expression was undetectable in primary hepatocytes after 24 hours of silencing Phb1 in vitro compared to NC.
Figure 3.
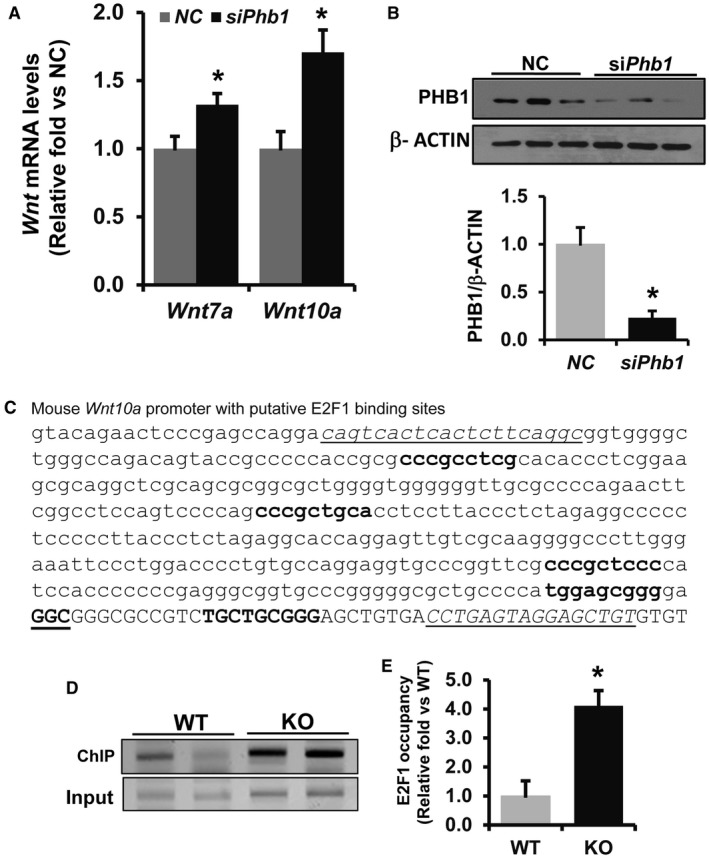
Increased binding of E2F1 to the Wnt10a promoter in Phb1 KO livers. (A,B) Phb1 silencing in mouse primary hepatocytes induced Wnt7a and Wnt10a. Total RNA and protein were prepared and subjected to qPCR and western blot, respectively. Relative expression was represented as fold induction compared to NC; n = 3; *P < 0.05 versus NC. (C) Predicted E2F1 binding sites in the mouse Wnt10a (Accession ID, U61969.1) sequence as determined by ALGGEN PROMO Transcription Factor bindings prediction software. Transcription start site is shown as bold with GGC underlined. Underlined sequences represent primer sequences used for ChIP PCR assay. (D,E) E2F1 binding to the promoter was determined by ChIP assay of chromatin prepared from 3‐week‐old Phb1 KO and WT mice livers. ChIP assay was performed as described in the Materials and Methods. Relative target site occupancy is represented as fold over WT control. Results represent mean ± SEM from six WT and KO livers in duplicate experiments; *P < 0.05 versus WT.
Increased E2F1 Binding To WNT10A Promoter In Phb1 KO Livers
Studies have demonstrated that PHB1 directly interacts with the retinoblastoma protein (Rb) and negatively regulates E2F1 function.3, 23, 24 Because Wnt10a and Wnt7a were induced both in vivo and in vitro in normal mouse hepatocytes when Phb1 was silenced, we examined whether their induction in Phb1 KO hepatocytes is mediated by E2F1. Putative E2F1 binding sites were identified in the Wnt7a and Wnt10a promoter sequence using ALGGEN PROMO prediction software. The putative E2F1 binding sites in the Wnt10a promoter sequence are shown in Fig. 3C. ChIP assay followed by qPCR showed a 4‐fold increase in E2F1 binding to the Wnt10a promoter in Phb1 KO livers tissues compared to WT controls (Fig. 3D); however, we were not able to detect clear E2F1 binding to the Wnt7a promoter between the KO and WT livers by ChIP assay (data not shown).
PHB1 Negatively Regulates Canonical WNT Signaling In HepG2 Cells
Next, we examined whether PHB1 regulates WNT signaling in HepG2 cells, a human liver cancer cell line. PHB1 silencing by 70% resulted in a 3‐fold increase in TCF promoter activity when compared to the NC as determined by TOPFlash assay (Fig. 4A,B). PHB1 silencing in HepG2 cells increased the phosphorylation of GSK3betaSer9 (inactive) by 3‐fold and AKT phosphorylation at Ser473 (active) by 50% (Fig. 4C,D). On the other hand, overexpression of PHB1 by 50% in HepG2 cells resulted in a 60% decrease in TCF promoter activity compared to empty vector transfected cells (Fig. 5A,B). Western blot analysis further showed that phosphorylation of AKTSer473 and GSK3betaSer9 was reduced by 40%‐60% in PHB1 overexpressing HepG2 cells compared to controls (Fig. 5C,D).
Figure 4.
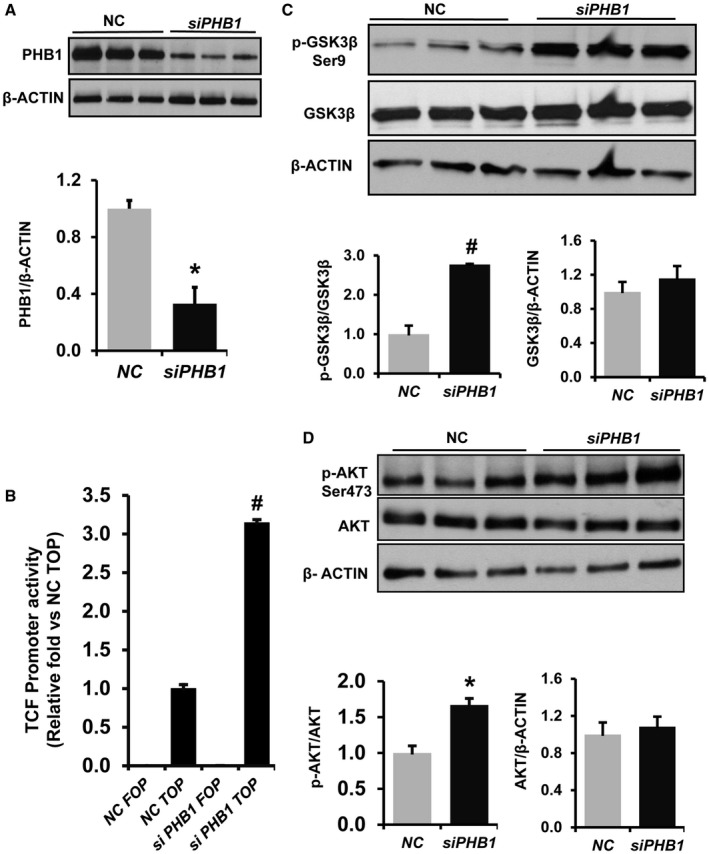
Effect of PHB1 silencing on WNT signaling in HepG2 cells. HepG2 cells were forward transfected with PHB1 siRNA or NC. (A) Total protein was prepared from transfected cells, and western blot was performed for PHB1 and beta‐ACTIN. The graph below represents the densitometry quantification of PHB1 expression level normalized to beta‐ACTIN. Results represent mean ± SEM; n = 3; *P < 0.05 versus NC. (B) PHB1 was silenced in HepG2 cells for 24 hours and cotransfected with a TCF TOPFlash reporter or its mutant FOPFlash plasmid for an additional 24 hours. Luciferase activity was measured using the Promega luciferase assay kit and normalized to Renilla luciferase activity; fold increase in promoter activity is shown. Results represent mean ± SEM from three independent experiments performed in duplicates. #P < 0.005 versus NC TOPFlash. (C) Western blot analysis of pGSK3betaSer9, total GSK3beta, and beta‐ACTIN and (D) pAKTSer473, total AKT, and beta‐ACTIN in PHB1‐silenced HepG2 cells. Densitometry quantification of the protein band intensity was performed using ImageJ software (NIH). Ratios were calculated to determine fold induction and activation. Results represent mean ± SEM from three independent experiments; *P < 0.05 versus NC.
Figure 5.
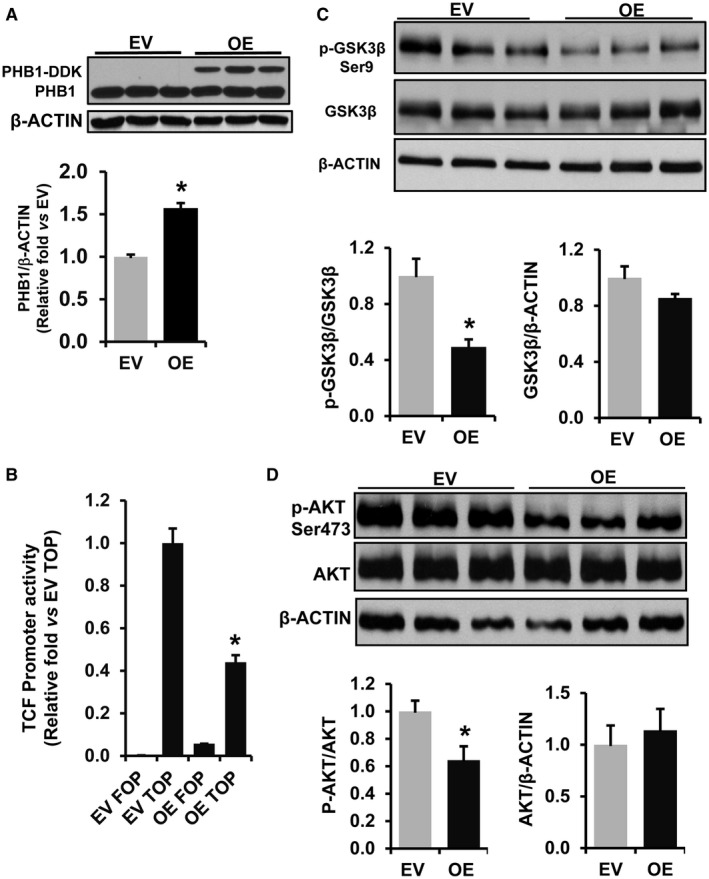
PHB1 overexpression down‐regulates WNT signaling in HepG2 cells. (A) HepG2 cells were transfected with EV or PHB1 OE vector in DDK tag, and western blot was performed. The graph represents densitometry quantification of PHB1 expression normalized to beta‐ACTIN. Results represent mean ± SEM; n = 3; *P < 0.05 versus EV. (B) HepG2 cells were transfected with EV or PHB1 OE vector. After 24 hours, cells were cotransfected with TCF‐TOPFlash reporter or its mutant plasmid FOPFlash. Luciferase activity was measured and normalized to Renilla luciferase activity; fold change in promoter activity is shown. Results represent mean ± SEM from three independent experiments performed in duplicates. #P < 0.005 versus TOPFlash EV. (C) Western blot analysis of pGSK3betaSer9, total GSK3beta, and beta‐ACTIN and (D) pAKTSer473, total AKT, and beta‐ACTIN in PHB1 OE HepG2 cells. Densitometry quantification of protein band intensity was performed using ImageJ (NIH), and ratios were calculated to determine fold change and activation. Results represent mean ± SEM from three independent experiments; *P < 0.05 versus EV.
PHB1 Silencing Causes Induction Of Multiple WNT Ligands In HCC Cells
An examination of WNT ligands that are increased in PHB1‐silenced HepG2 cells by real‐time (RT)2‐WNT‐PCR array (data not shown) (Qiagen Inc., Germantown, MD) followed by qPCR revealed a multiple WNT ligand induction profile, which was slightly different from Phb1 KO livers. Like Phb1 KO livers, HepG2 cells exhibited an induction of WNT10A following PHB1 silencing (Fig. 6A). However, unlike Phb1 KO livers, PHB1 silencing for 48 hours induced WNT11 and WNT9A at much higher levels compared to the NCs (Fig. 6A), whereas WNT7A and WNT16 were undetectable in HepG2 cells. A similar trend of multiple WNT ligand induction with PHB1 silencing was also observed in two other HCC cell lines, Huh7 and Hep3B (Supporting Fig. S3). Similar to Phb1 KO livers, PHB1 silencing in HepG2 cells also resulted in the induction of the EMT markers VIMENTIN and zinc finger E‐box binding homeobox 2 (ZEB2), but no change in SNAI1 level was observed (Supporting Fig. S2B). To determine whether WNT ligands induced in the absence of PHB1 silencing are secreted as functional proteins to induce downstream signaling activation in a paracrine fashion, conditioned media from PHB1‐silenced cells were tested for WNT signaling activation in HepG2 cells by TOPFlash assay. siPHB1‐conditioned media caused a 50% increase in TCF promoter activity compared to control siRNA‐conditioned media (Supporting Fig. S4).
Figure 6.
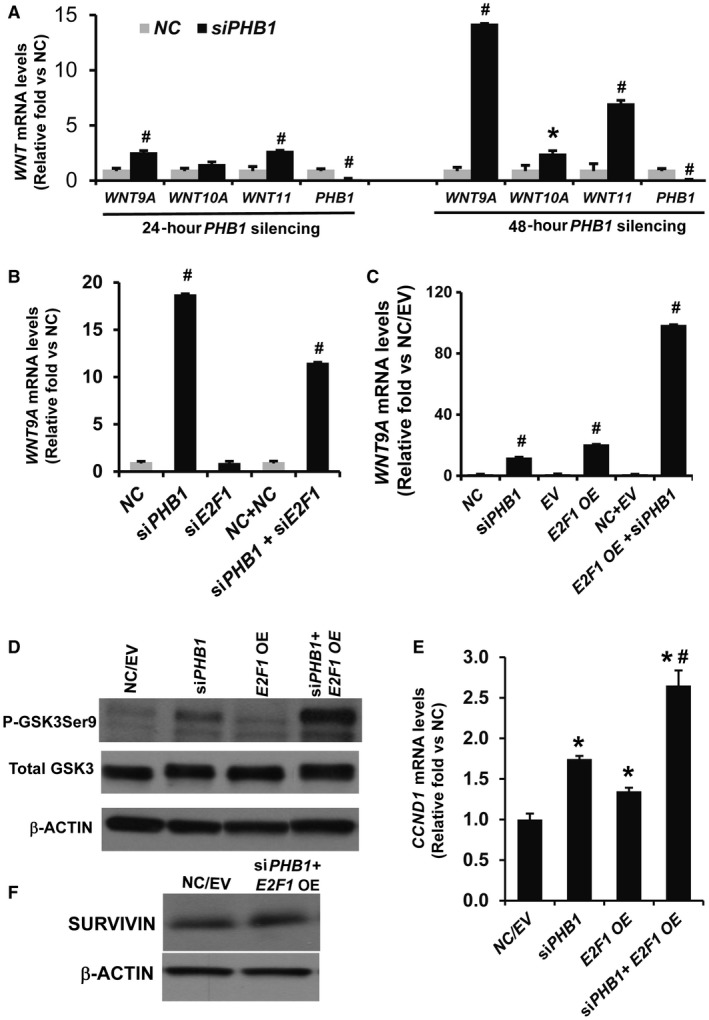
WNT9A induction by PHB1 silencing is E2F1 dependent. (A) Effect of PHB1 silencing on WNT ligand induction in HepG2 cells. HepG2 cells were forward transfected with NC or PHB1 siRNA for 24 hours and 48 hours. Relative expression of WNT9A, WNT10A, WNT16, and PHB1 was compared to the NC. Results represent mean ± SEM from three independent experiments; #P < 0.001, *P < 0.05 versus NC. (B) Effect of PHB1 and E2F1 cosilencing for 48 hours on WNT9A expression in HepG2 cells. Results represent mean ± SEM from three independent experiments performed in duplicates; #P < 0.001 for siPHB1 versus NC, siPHB1 versus siPHB1+siE2F1. (C) Effect of PHB1 cosilencing with E2F1 overexpression on WNT9A levels in HepG2 cells. Results represent mean ± SEM from three independent experiments performed in duplicates. #P < 0.001 for siPHB1 versus NC, EV versus E2F1 OE, siPHB1 versus siPHB1+E2F1 OE, E2F1 OE versus siPHB1+E2F1 OE. (D) Effect of E2F1 OE for 48 hours in conjunction with 72 hours of PHB1 silencing on phosphorylation of GSK3betaSer9. Data represent two independent experiments. (E) Effect of E2F1 OE for 48 hours in conjunction with 72 hours of PHB1 silencing on CCND1 mRNA levels. Data represent three independent experiments; *P < 0.05 versus NC/EV, #P < 0.05 versus siPHB1 and E2F1 OE. (F) Effect of PHB1 silencing and E2F1 overexpression as in (E) on the expression of anti‐apoptotic protein SURVIVIN in HepG2 cells. Data represent two independent experiments.
Because we found increased binding of E2F1 to Wnt10a promoter in Phb1 KO livers, we examined whether E2F1 has any regulatory role in WNT9A (a ligand that is induced at the highest level) induction in response to PHB1 silencing in HepG2 cells. E2F1 was cosilenced with PHB1 for 48 hours in HepG2 cells, and the level of WNT9A mRNA was measured. E2F1 silencing alone did not change the expression of WNT9A compared to the control. However, cosilencing with PHB1 caused about a 40% decrease in WNT9A induction compared to PHB1 silencing alone (Fig. 6B). Conversely, E2F1 overexpression for 48 hours in conjunction with 72 hours PHB1 silencing caused an ~8‐fold increase in WNT9A mRNA expression compared to PHB1 silencing alone and ~5‐fold increase compared to E2F1 overexpression alone (Fig. 6C). PHB1 silencing also caused down‐regulation of protein and mRNA levels of E2F1 by 40%‐60%, respectively (Supporting Fig. S5). Increased WNT ligand induction by E2F1 in conjunction with PHB1 silencing caused an activation of WNT signaling in HepG2 cells as determined by increased GSK3betaSer9 phosphorylation and expression of WNT target gene CCND1 (Fig. 6D,E). Because E2F1 can induce both cell proliferation and apoptosis, we checked the levels of SURVIVIN, an anti‐apoptotic protein. E2F1 overexpression for 48 hours along with PHB1 silencing for 72 hours did not change SURVIVIN protein levels in HepG2 cells (Fig. 6F).
To determine whether E2F1 regulates WNT9A transcription in HepG2 cells, we examined the promoter sequence of WNT9A for putative E2F1 binding sites. Following analysis of the human WNT9A promoter sequence by PROMO prediction software, we found that WNT9A promoter has several putative E2F1 binding sites (Fig. 7A). We then performed ChIP assays to investigate the functional role of E2F1 binding to the WNT9A promoter on its induction in the absence of PHB1. We found nearly a 3.3‐fold increase in E2F1 binding to the WNT9A promoter in PHB1‐silenced HepG2 cells compared to the NC (Fig. 7B). Next, we performed ChIP assays to determine whether E2F1 overexpression in a setting of PHB1 silencing resulted in increased WNT9A promoter binding that potentially correlated with increased levels of WNT9A induction. Our results showed that E2F1 binding to the WNT9A promoter was significantly increased under a PHB1‐silenced condition when compared to E2F1 overexpression in the presence of endogenous PHB1 (Fig. 7C).
Figure 7.
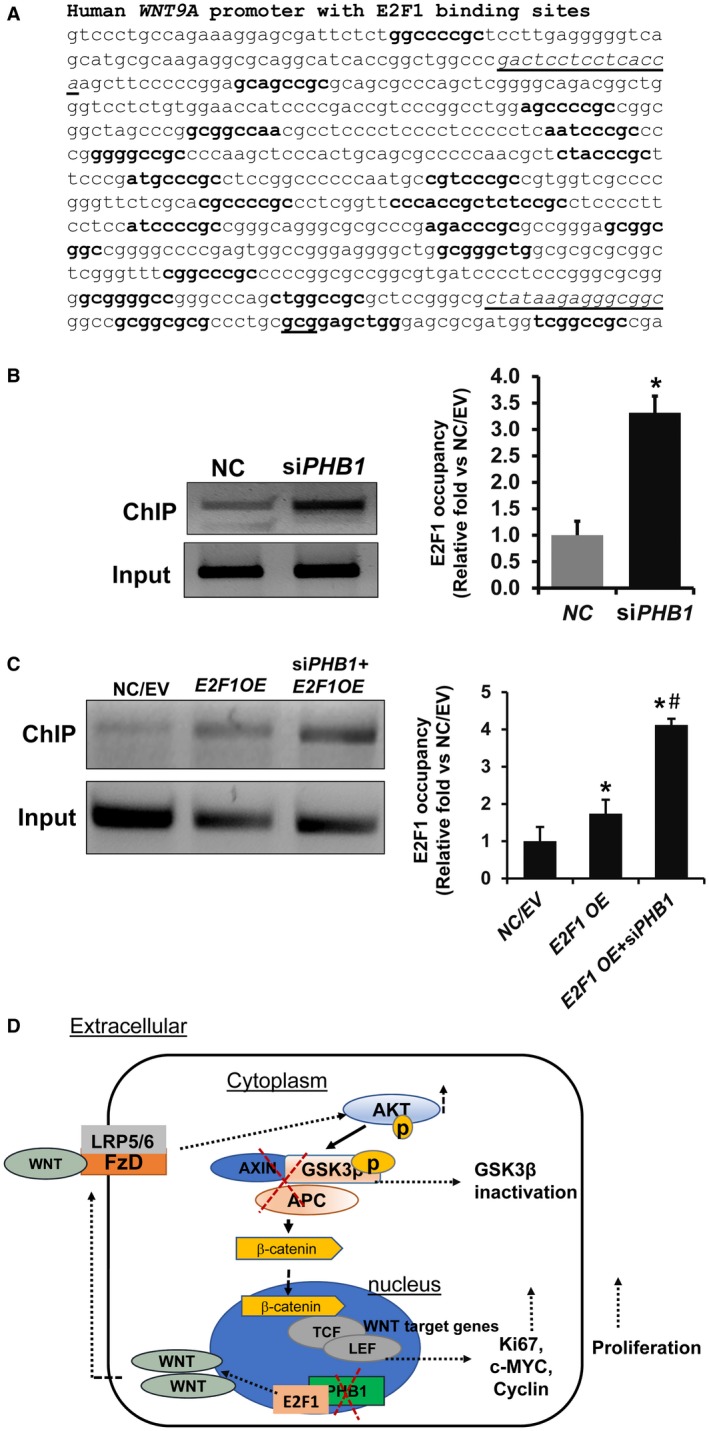
PHB1 silencing leads to increased E2F1 binding to the WNT9A promoter in HepG2 cells. (A) Predicted E2F1 binding sites (in bold) in the human WNT9A (Accession ID, HM015601.1) sequence was determined by ALGGEN PROMO transcription factor bindings prediction software. Transcription start site is gcg underlined in bold. Sequences that are underlined are primer sequences used for the ChIP assay. (B) E2F1 binding to the WNT9A promoter was determined by ChIP assay. Chromatin was prepared from NC and PHB1‐silenced HepG2 cells, and ChIP assay was performed as described in Materials and Methods. Relative target site occupancy is represented as fold over NC. Results represent mean ± SEM from three independent experiments in duplicates; *P < 0.05 versus NC. (C) Effect of E2F1 OE for 48 hours in conjunction with 72 hours of PHB1 silencing on E2F1 binding to the WNT9A promoter in HepG2 cells. Results represent mean ± SEM from three to six independent experiments in duplicates; *P < 0.05 versus NC/EV, #P < 0.05 E2F1 OE versus E2F1 OE+siPHB1. (D) Summary demonstrating the potential role of PHB1 in modulating WNT signaling in Phb1 KO livers and HepG2 cells. PHB1 deletion leads to induction of WNT ligands in an E2F1‐dependent manner. WNT ligands induce downstream activation of WNT‐beta‐catenin signaling through the AKT‐GSK3beta signaling pathway. This results in increased TCF transcriptional activity and cell proliferation. Abbreviations: APC, adenomatous polyposis coli; c‐MYC, Myc Proto‐Oncogene; FzD, Frizzled.
Expression Pattern Of WNT Ligands With Decreased PHB1 Expression In Human Liver Cancers
Our previous study demonstrated that the expression of PHB1 is significantly down‐regulated in human HCC and CCA tissues compared to nontumor adjacent tissues.10 To determine whether PHB1 down‐regulation is associated with induction of WNT ligands in HCC, we analyzed CCA tissues and the publicly available Gene Expression Omnibus (GEO) database. These analyses showed that in many patients where PHB1 expression was down‐regulated, there was increased expression of at least one of the WNT ligands (WNT9A, WNT10A, or WNT7A) that we identified either in Phb1 KO liver or PHB1 silenced HepG2 cells in up to 45%‐49% of the cases (Table 1). The values that are underlined in Table 1 depict the number/percentages of cases with decreased PHB1 expression that exhibited increased WNT ligand expression compared to controls. Additional GEO data sets with up‐regulated WNT ligands in human HCC tissues were also identified; however, expression data for PHB1 were not available for the same data sets and therefore were not included (data not shown). This analysis showed a high‐level heterogeneous and dynamic expression pattern of WNT ligands and PHB1 in human HCC liver tissues.
Table 1.
Expression Pattern of WNT Ligands and PHB1 in Human HCC/CCA
| GSE Serial # | Description | Total Cases | Expression Ratio: HCC/Adjacent Nontumor Control | |||
|---|---|---|---|---|---|---|
| PHB1 | WNT9A | WNT10A | WNT7A | |||
| Down | Up | Up | Up | |||
| GSE76427 | Microarray expression data for tumor and adjacent nontumor tissues from patients with HCC | 114 | 57 (50%) | 67 (58%) 28 (49%) | 39 (34%) 27 (47%) | 29 (25%) 18 (32%) |
| GSE6764 | Early HCC versus normal liver tissue | 18 | 14 (78%) | 7 (39%) 5 (36%) | 2 (11%) 1 (7%) | 2 (11%) 1 (7%) |
| GSE14323 | RNA expression data for liver samples from subjects with HCC or normal liver | 38 | 18 (47%) | n/a | n/a | 13 (34%) 5 (28%) |
| GSE60502 | Gene expression profiling of HCC and adjacent nontumorous liver tissue | 18 | 11 (61%) | n/a | n/a | 9 (50%) 5 (45%) |
| GSE22058 | mRNAs from paired tumor and adjacent nontumor tissues from patients with HCC | 100 | 55 (55%) | 42 (42%) 22 (40%) | 13 (13%) 10 (40%) | 16 (16%) 5 (9%) |
| HCC tissues | mRNA levels in paired tumor/adjacent nontumor tissue and by qPCR assay | 18 pairs | 9 (50%) | 8 (44%) 4 (44%) | 8 (44%) 5 (56%) | 2 (11%) 1 (11%) |
| CCA tissues | mRNA levels in paired tumor/adjacent nontumor tissue and by qPCR assay | 7 pairs | 5 (71%) | 5 (71%) 3 (60%) | 5 (71%) 5 (100%) | 6 (86%) 4 (86%) |
Publicly available National Center for Biotechnology Information–GEO data sets as well as HCC/CCA tissues were analyzed to determine the pattern of PHB1 and WNT ligand expression. Upper row in each WNT category is the number (percentage) of patients with respective up‐regulated WNT ligand expression compared to the total number of cases. Values that are underlined are the number/percentage of patients with both WNT up‐regulation and PHB1 down‐regulation.
Abbreviation: GSE, gene series.
Discussion
PHB1 is a ubiquitously expressed chaperone protein localized to various cellular compartments, including the nucleus, plasma membrane, cytoplasm, and mitochondria, with distinct functions.7 PHB1 was originally discovered to be down‐regulated in regenerating rat liver and as a consequence was thought to be a tumor suppressor.1 However, conflicting studies showing the role of PHB1 as an oncogene have also been reported.25 Previous studies from our laboratory have reported that liver‐specific Phb1 gene deletion in mice causes spontaneous injury, inflammation, bile duct metaplasia, and HCC.8 Deregulated regenerative response and inflammation often predispose tissues to tumorigenesis, and therefore timely regulation of these signaling pathways is critical for tissue homeostasis to prevent malignant transformation.15 Although chronic inflammation in the liver‐specific Phb1 KO mouse could have contributed to HCC formation, our recent studies show PHB1 acts directly as a tumor suppressor in the liver by negatively regulating the H19‐IGF2 axis9 and the E‐box element as a heterodimer with MYC‐associated factor X (MAX).10 In this study, we investigated the link between PHB1 and one of the major tissue regenerative and oncogenic pathways, WNT‐beta‐catenin signaling. Our data show that PHB1 acts as a negative regulator of WNT signaling by suppressing the mRNA levels of multiple WNT ligands, in part through an E2F1‐dependent mechanism. PHB1 deletion resulted in the activation of AKT and inactivation of GSK3beta (phosphorylated form), a critical negative regulator of canonical WNT signaling both in vivo in Phb1 KO livers and in PHB1‐silenced HepG2 cells, with increased TCF promoter activity. Conversely PHB1 overexpression in HepG2 cells resulted in the reverse effect. Therefore, both in vivo and in vitro findings from our study unequivocally show that PHB1 negatively regulates canonical WNT signaling in murine liver and HepG2 cells. Activation of AKT and inactivation of GSK3beta leads to stabilization of beta‐catenin followed by its nuclear translocation and transcriptional activation of WNT target genes. GSK3beta is a negative regulator of WNT signaling and acts as a tumor suppressor by promoting beta‐catenin ubiquitination.26
Although PHB1 was originally described as a mitochondrial chaperone protein, it has diverse functions.7, 27, 28 In addition to serving as a negative regulator of H19‐IGF2 signaling and E‐box‐driven promoter activity in hepatocytes,9, 10 PHB1 has also been shown to increase the transcriptional activity of p53 in breast and prostate cancer cells.29 Despite these reports, the role of PHB1 as a tumor suppressor is controversial, and its expression is increased in many types of cancer where it has been shown to promote cell proliferation. PHB1 was found to be required for Ras‐mediated Raf activation and potentially acts as a scaffold protein in the plasma membrane of HeLa cells,25 whereas PHB1 expression was associated with increased drug resistance in uterine and lung cancer cells.30 Cumulatively, these findings suggest that PHB1 function is greatly influenced by the cell type, cellular localization, and potentially interacting proteins.
Phb1 deletion in mice as well as in vitro PHB1 silencing in HepG2 cells induced expression of multiple WNT ligands at variable levels. Of the 19 WNT ligands, Wnt7a exhibited the highest expression compared to the other ligands in Phb1 KO livers. Although the expression of Wnt10a is much lower compared to Wnt7a in total liver, KO hepatocytes expressed similarly high levels of Wnt7a and Wnt10a compared to the nonparenchymal cell fraction isolated from KO livers. Because liver‐specific Phb1 knockdown is mediated through the Albumin;Cre promoter, in vivo Phb1 deletion occurs only in hepatocytes and cholangiocytes compared to other liver cell types. Therefore, the high level of Wnt7a and Wnt10a induction in Phb1 KO hepatocytes is a direct effect of PHB1 knockdown. This was further confirmed by in vitro Phb1 silencing in mouse primary hepatocytes and WNT7a/WNT10a co‐immunofluorescence staining with hepatocyte marker HNF4alpha. Overall induction of Wnt10a and Wnt7a after 24 hours of in vitro Phb1 silencing was lower in mouse primary hepatocytes compared to Phb1 KO livers. The expression pattern may be time dependent and could explain why Wnt16 was below detection level in primary hepatocytes. In addition to hepatocytes, the nonparenchymal cell fraction also expressed higher levels of Wnt7a, Wnt10a, and Wnt16 mRNA in Phb1 KO livers compared to WT controls. Phb1 KO livers are characterized by extensive injury, inflammation, and regenerative response.8 Nonparenchymal cells are known sources of cytokines and growth factors during liver regeneration. Therefore, it is likely that nonparenchymal cells are also an indirect source of WNT ligands in Phb1 KO livers. Studies have shown that macrophages are the main source of WNT in experimental models of liver and intestinal injury and regeneration.31, 32 Increased WNT signaling in inflammatory macrophages has been associated with CCA growth in experimental models.33 WNT signaling has been shown to play a critical role in hepatobiliary repair during cholestatic liver injury models.34 Therefore, increased WNT ligand expression from hepatocytes and nonparenchymal cells and downstream activation of this pathway could be one of the mechanisms that is responsible for increased cellular proliferation, regenerative response, and development of HCC and CCA in Phb1 KO mice livers.
The pattern of WNT ligand induction in PHB1‐silenced HCC cells is different from Phb1 KO livers in that these cells did not exhibit WNT7A expression; instead, other ligands were induced, such as WNT9A, as well as WNT11 and WNT10A to a lesser extent. The difference could be attributed to the overall genetic/epigenetic differences between HCC cells and normal mouse hepatocytes. Moreover, the Phb1 KO liver represents an in vivo system where the normal regenerative response is highly deregulated in addition to extensive liver necrosis and inflammation, and this could affect gene expression patterns in hepatocytes as well. Signaling crosstalk between hepatocytes and nonhepatocytes in the liver during injury and regeneration could also influence the overall gene expression profile in the mouse liver. Regardless of the specific WNT ligands, these secreted proteins can cause similar cellular and physiological effects by activating downstream WNT‐beta‐catenin signaling, as we observed in Phb1 KO livers and in HepG2 cells. Our recent study showed that PHB1 is significantly down‐regulated in human HCC and CCA.10 The GEO database search found a trend toward decreased PHB1 expression and up‐regulation of multiple WNT ligands in HCC and CCA tissues. However, we did not a find a significant inverse correlation between WNT ligands and PHB1 expression in these data sets, possibly due to high‐level heterogeneity in WNT ligand expression levels among patients. Nevertheless, this suggests a potential association between activated WNT signaling and reduced PHB1 expression in many HCC cases. These findings are clinically relevant and could be partly correlated with the underlying downstream molecular mechanism of liver tumorigenesis in patients with low levels of hepatic PHB1 expression.
An extensive expression pattern of Wnt ligands in mouse liver as well as in individual liver cell types has been reported.35 According to this study, majority of Wnt ligands are expressed in embryonic liver compared to adult mouse liver except Wnt4, Wnt5a, and Wnt5b. Many Wnt ligands were either not detected or expressed at low levels in 3‐week‐old WT control mice livers, which is comparable to the previous study.35 Importantly, Wnt7a, Wnt10a, and Wnt16 are highly induced in 3‐week‐old Phb1 KO livers. Therefore, the downstream WNT signaling activation observed in KO livers could be the cumulative effect of induction of multiple WNT ligands in hepatocytes and nonparenchymal cells. Wnt2, Wnt2b, Wnt5b, Wnt7b, and Wnt9a were detected in both WT and Phb1 KO livers. Expression of these WNT ligands in WT control livers is likely due to the difference in mouse age and the more sensitive assay method we used in this study compared to the previous report.35
Multiple pathways, including nuclear factor kappa B, transforming growth factor beta, notch, signal transducer and activator of transcription 3, and interleukin 6, have been proposed to regulate human WNT5A and WNT2B at the transcriptional level. However, detailed molecular studies are lacking, possibly because of the dynamic expression pattern and multiplicity of these proteins.36, 37 PHB1 interacts with E2F1 and represses its transcriptional activity.3, 38 We found that E2F1 binding to the Wnt10a and WNT9A promoters was significantly increased in Phb1 KO livers and in PHB1‐silenced HepG2 cells, respectively. E2F1 gene silencing in HepG2 cells significantly reduced PHB1 knockdown‐mediated WNT9A induction compared to PHB1 silencing alone, whereas its overexpression had the opposite effect. ChIP data further indicated that in the setting of PHB1 deficiency, E2F1 exhibits enhanced WNT9A promoter binding activity, potentially leading to WNT9A induction. This suggests that PHB1 might act as a docking factor to negatively regulate E2F1‐mediated WNT9A induction in HepG2 cells. Increased GSK3Ser9 phosphorylation and expression of CCND1 as well as unchanged levels of SURVIVIN, an anti‐apoptotic protein in E2F1 overexpressing cells in a setting of PHB1 silencing, suggests that the E2F1‐induced WNT signaling axis in PHB1‐deficient cells promotes cell proliferation and may surpass any apoptotic signal induced by E2F1 under these experimental conditions in HepG2 cells. E2F1 transcriptional activity is highly dependent on the cell cycle.39 Both oncogenic and tumor suppressive functions have been reported for E2F1 in murine liver,40 whereas its expression is increased in human HCC.41, 42, 43 Taken together, our results demonstrate for the first time that PHB1 acts as a tumor suppressor, in part through suppressing the canonical WNT signaling in murine liver and HCC cells. The molecular mechanism involves serving as a negative regulator of multiple WNT ligands, some of which may be transcriptional targets of E2F1. These WNT ligands can activate canonical WNT signaling in both paracrine and autocrine manners. Studies in breast and prostate cancer cells have demonstrated that PHB1 negatively regulates the transcriptional activity of E2F1 through its direct interaction with the Rb protein,3, 24, 27 whereas its interaction with p53 increases its transcriptional activity.29 PHB1 interacts with the Rb protein and inhibits E2F activity through a mechanism that involves recruitment of histone deacetylase 1 and nuclear receptor corepressor 1.2 A previous study from our laboratory demonstrated that reduced PHB1 expression in an immortalized normal mouse hepatocyte cell line resulted in increased E2F1 binding to the Ccnd1 promoter and increased expression of Ccnd1.8 These findings strengthen the tumor‐suppressive roles of PHB1 in liver, breast, and prostate cancer cells. Conversely, increased expression of PHB1 has been reported in lung, cervical, and hematologic cancers.25, 44, 45 PHB1 function is thought to be determined by various factors, such as its cellular localization, cell types, and posttranslational modifications, that could result in differential protein–protein interactions and functional outcomes. Therefore, in‐depth studies are required to uncover what determines PHB1 function at a molecular level in various cell types and cancers.
In summary, we have demonstrated a novel repressive role for PHB1 in the regulation of WNT‐beta‐catenin signaling, one of the major regenerative and oncogenic pathways in mammalian cells (Fig. 7D). PHB1 silencing resulted in the up‐regulation of multiple WNT ligands both in vivo and in HCC cells, in part through increased E2F1 transactivating activity.
Potential conflict of interest
Dr. Mato advises, consults for, and owns stock in Owl; he advises and consults for Abbott and consults for Galmed. The other authors have nothing to report.
Supporting information
Supported by the National Institutes of Health (R01CA172086 to S.C.L., J.M.M.), Plan Nacional de I+D SAF (2014‐52097R to J.M.M.), and Departmento de Educación del Gobierno Vasco (to J.M.M.); liver sections prepared by the Histology Core of the University of Southern California Research Center for Liver Diseases (P30DK48522).
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Contributor Information
Nirmala Mavila, Email: nirmala.mavila@cshs.org.
Shelly C. Lu, Email: shelly.lu@cshs.org
References
Author names in bold designate shared co‐first authorship.
- 1. McClung JK, Danner DB, Stewart DA, Smith JR, Schneider EL, Lumpkin CK, et al. Isolation of a cDNA that hybrid selects antiproliferative mRNA from rat liver. Biochem Biophys Res Commun 1989;164:1316‐1322. [DOI] [PubMed] [Google Scholar]
- 2. Wang S, Fusaro G, Padmanabhan J, Chellappan SP. Prohibitin co‐localizes with Rb in the nucleus and recruits N‐CoR and HDAC1 for transcriptional repression. Oncogene 2002;21:8388‐8396. [DOI] [PubMed] [Google Scholar]
- 3. Wang S, Nath N, Adlam M, Chellappan S. Prohibitin, a potential tumor suppressor, interacts with RB and regulates E2F function. Oncogene 1999;18:3501‐3510. [DOI] [PubMed] [Google Scholar]
- 4. Kasashima K, Sumitani M, Satoh M, Endo H. Human prohibitin 1 maintains the organization and stability of the mitochondrial nucleoids. Exp Cell Res 2008;314:988‐996. [DOI] [PubMed] [Google Scholar]
- 5. Mishra S, Ande SR, Nyomba BL. The role of prohibitin in cell signaling. FEBS J 2010;277:3937‐3946. [DOI] [PubMed] [Google Scholar]
- 6. Osman C, Merkwirth C, Langer T. Prohibitins and the functional compartmentalization of mitochondrial membranes. J Cell Sci 2009;122:3823‐3830. [DOI] [PubMed] [Google Scholar]
- 7. Mishra S, Murphy LC, Murphy LJ. The prohibitins: emerging roles in diverse functions. J Cell Mol Med 2006;10:353‐363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ko KS, Tomasi ML, Iglesias‐Ara A, French BA, French SW, Ramani K, et al. Liver‐specific deletion of prohibitin 1 results in spontaneous liver injury, fibrosis, and hepatocellular carcinoma in mice. Hepatology 2010;52:2096‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ramani K, Mavila N, Ko KS, Mato JM, Lu SC. Prohibitin 1 regulates the H19‐Igf2 axis and proliferation in hepatocytes. J Biol Chem 2016;291:24148‐24159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fan W, Yang H, Liu T, Wang J, Li TW, Mavila N, et al. Prohibitin 1 suppresses liver cancer tumorigenesis in mice and human hepatocellular and cholangiocarcinoma cells. Hepatology 2017;65:1249‐1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Clevers H, Loh KM, Nusse R. Stem cell signaling. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014;346:1248012. [DOI] [PubMed] [Google Scholar]
- 12. Monga SP. beta‐Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148:1294‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clevers H, Nusse R. Wnt/beta‐catenin signaling and disease. Cell 2012;149:1192‐1205. [DOI] [PubMed] [Google Scholar]
- 14. Willert K, Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol 2012;4:a007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nejak‐Bowen KN, Monga SP. Beta‐catenin signaling, liver regeneration and hepatocellular cancer: sorting the good from the bad. Semin Cancer Biol 2011;21:44‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Liu T, Yang H, Fan W, Tu J, Li TWH, Wang J, et al. Mechanisms of MAFG dysregulation in cholestatic liver injury and development of liver cancer. Gastroenterology 2018;155:557‐571.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rountree CB, Senadheera S, Mato JM, Crooks GM, Lu SC. Expansion of liver cancer stem cells during aging in methionine adenosyltransferase 1A‐deficient mice. Hepatology 2008;47:1288‐1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mavila N, James D, Shivakumar P, Nguyen MV, Utley S, Mak K, et al. Expansion of prominin‐1‐expressing cells in association with fibrosis of biliary atresia. Hepatology 2014;60:941‐953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li TW, Peng H, Yang H, Kurniawidjaja S, Panthaki P, Zheng Y, et al. S‐Adenosylmethionine and methylthioadenosine inhibit β‐catenin signaling by multiple mechanisms in liver and colon cancer. Mol Pharmacol. 2015;87:77‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase‐3 by insulin mediated by protein kinase B. Nature 1995;378:785‐789. [DOI] [PubMed] [Google Scholar]
- 21. Manning BD, Toker A. AKT/PKB signaling: navigating the network. Cell 2017;169:381‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Scheid MP, Woodgett JR. Unravelling the activation mechanisms of protein kinase B/Akt. FEBS Lett 2003;546:108‐112. [DOI] [PubMed] [Google Scholar]
- 23. Schneider M, Schambony A, Wedlich D. Prohibitin1 acts as a neural crest specifier in Xenopus development by repressing the transcription factor E2F1. Development 2010;137:4073‐4081. [DOI] [PubMed] [Google Scholar]
- 24. Wang S, Nath N, Fusaro G, Chellappan S. Rb and prohibitin target distinct regions of E2F1 for repression and respond to different upstream signals. Mol Cell Biol 1999;19:7447‐7460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rajalingam K, Wunder C, Brinkmann V, Churin Y, Hekman M, Sievers C, et al. Prohibitin is required for Ras‐induced Raf‐MEK‐ERK activation and epithelial cell migration. Nat Cell Biol 2005;7:837‐843. [DOI] [PubMed] [Google Scholar]
- 26. McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Sokolosky M, Abrams SL, et al. GSK‐3 as potential target for therapeutic intervention in cancer. Oncotarget 2014;5:2881‐2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Theiss AL, Sitaraman SV. The role and therapeutic potential of prohibitin in disease. Biochim Biophys Acta 2011;1813:1137‐1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Peng YT, Chen P, Ouyang RY, Song L. Multifaceted role of prohibitin in cell survival and apoptosis. Apoptosis 2015;20:1135‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fusaro G, Dasgupta P, Rastogi S, Joshi B, Chellappan S. Prohibitin induces the transcriptional activity of p53 and is exported from the nucleus upon apoptotic signaling. J Biol Chem 2003;278:47853‐47861. [DOI] [PubMed] [Google Scholar]
- 30. Patel N, Chatterjee SK, Vrbanac V, Chung I, Mu CJ, Olsen RR, et al. Rescue of paclitaxel sensitivity by repression of prohibitin1 in drug‐resistant cancer cells. Proc Natl Acad Sci USA 2010;107:2503‐2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Saha S, Aranda E, Hayakawa Y, Bhanja P, Atay S, Brodin NP, et al. Macrophage‐derived extracellular vesicle‐packaged WNTs rescue intestinal stem cells and enhance survival after radiation injury. Nat Commun 2016;7:13096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boulter L, Govaere O, Bird TG, Radulescu S, Ramachandran P, Pellicoro A, et al. Macrophage‐derived Wnt opposes Notch signaling to specify hepatic progenitor cell fate in chronic liver disease. Nat Med 2012;18:572‐579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Boulter L, Guest RV, Kendall TJ, Wilson DH, Wojtacha D, Robson AJ, et al. WNT signaling drives cholangiocarcinoma growth and can be pharmacologically inhibited. J Clin Invest 2015;125:1269‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Okabe H, Yang J, Sylakowski K, Yovchev M, Miyagawa Y, Nagarajan S, et al. Wnt signaling regulates hepatobiliary repair following cholestatic liver injury in mice. Hepatology 2016;64:1652‐1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zeng G, Awan F, Otruba W, Muller P, Apte U, Tan X, et al. Wnt'er in liver: expression of Wnt and frizzled genes in mouse. Hepatology 2007;45:195‐204. [DOI] [PubMed] [Google Scholar]
- 36. Katoh M, Katoh M. Transcriptional mechanisms of WNT5A based on NF‐kappaB, Hedgehog, TGFbeta, and Notch signaling cascades. Int J Mol Med 2009;23:763‐769. [DOI] [PubMed] [Google Scholar]
- 37. Katoh M, Katoh M. Transcriptional regulation of WNT2B based on the balance of Hedgehog, Notch, BMP and WNT signals. Int J Oncol 2009;34:1411‐1415. [PubMed] [Google Scholar]
- 38. Joshi B, Ko D, Ordonez‐Ercan D, Chellappan SP. A putative coiled‐coil domain of prohibitin is sufficient to repress E2F1‐mediated transcription and induce apoptosis. Biochem Biophys Res Commun 2003;312:459‐466. [DOI] [PubMed] [Google Scholar]
- 39. Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 1993;365:349‐352. [DOI] [PubMed] [Google Scholar]
- 40. Conner EA, Lemmer ER, Omori M, Wirth PJ, Factor VM, Thorgeirsson SS. Dual functions of E2F–1 in a transgenic mouse model of liver carcinogenesis. Oncogene 2000;19:5054‐5062. [DOI] [PubMed] [Google Scholar]
- 41. Malz M, Pinna F, Schirmacher P, Breuhahn K. Transcriptional regulators in hepatocarcinogenesis–key integrators of malignant transformation. J Hepatol 2012;57:186‐195. [DOI] [PubMed] [Google Scholar]
- 42. Kent LN, Bae S, Tsai SY, Tang X, Srivastava A, Koivisto C, et al. Dosage‐dependent copy number gains in E2f1 and E2f3 drive hepatocellular carcinoma. J Clin Invest 2017;127:830‐842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Farra R, Grassi G, Tonon F, Abrami M, Grassi M, Pozzato G, et al. The role of the transcription factor E2F1 in hepatocellular carcinoma. Curr Drug Deliv 2017;14:272‐281. [DOI] [PubMed] [Google Scholar]
- 44. Chiu CF, Ho MY, Peng JM, Hung SW, Lee WH, Liang CM, et al. Raf activation by Ras and promotion of cellular metastasis require phosphorylation of prohibitin in the raft domain of the plasma membrane. Oncogene 2013;32:777‐787. [DOI] [PubMed] [Google Scholar]
- 45. Ross JA, Robles‐Escajeda E, Oaxaca DM, Padilla DL, Kirken RA. The prohibitin protein complex promotes mitochondrial stabilization and cell survival in hematologic malignancies. Oncotarget 2017;8:65445‐65456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials