

Abstract
Fatty liver disease is one of the most prevalent forms of chronic liver disease that encompasses both alcoholic liver disease (ALD) and nonalcoholic fatty liver disease (NAFLD). Alcoholic steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH) are intermediate stages of ALD and NAFLD, which can progress to more advanced forms, including cirrhosis and hepatocellular carcinoma. Oxidative stress and particularly alterations in mitochondrial function are thought to play a significant role in both ASH and NASH and recognized to contribute to the generation of reactive oxygen species (ROS), as documented in experimental models. Despite the evidence of ROS generation, the therapeutic efficacy of treatment with antioxidants in patients with fatty liver disease has yielded poor results. Although oxidative stress is considered to be the disequilibrium between ROS and antioxidants, there is evidence that a subtle balance among antioxidants, particularly in mitochondria, is necessary to avoid the generation of ROS and hence oxidative stress. Conclusion: As mitochondria are a major source of ROS, the present review summarizes the role of mitochondrial oxidative stress in ASH and NASH and presents emerging data indicating the need to preserve mitochondrial antioxidant balance as a potential approach for the treatment of human fatty liver disease, which may pave the way for the design of future trials to test the therapeutic role of antioxidants in fatty liver disease.
Abbreviations
- AIF
apoptosis‐inducing factor
- ALD
alcoholic liver disease
- ALT
alanine aminotransferase
- ASH
alcoholic steatohepatitis
- ATP
adenosine triphosphate
- CPT1
carnitinepalmitoyl transferase 1
- FAO
fatty acid oxidation
- Gpx
GSH peroxidases
- Grx
glutaredoxins
- GSH
reduced glutathione
- GSHEE
GSH ethyl ester
- GSSG
oxidized GSH
- MCD
methionine and choline diet
- mGSH
mitochondrial GSH
- MnP
meso‐tetrakis (N‐ethylpyridinium‐2‐yl) porphyrin
- MnTBAP
Mn(III)tetrakis(4‐benzoic acid)porphyrin chloride
- MRC
mitochondrial respiratory chain
- mtDNA
mitochondrial DNA
- NAC
N‐acetylcysteine
- NADPH
nicotinamide adenine dinucleotide phosphate, reduced form
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NO
nitric oxide
- OXPHOS
oxidative phosphorylation
- Prx
peroxiredoxins
- RNS
reactive nitrogen species
- ROS
reactive oxygen species
- SAM
S‐adenosyl‐methionine
- SH
steatohepatitis
- SOD2
manganese superoxide dismutase
- Trx
thioredoxin
Fatty liver disease constitutes a spectrum of liver disorders that begin with steatosis, which can progress to more advanced stages, including steatohepatitis (SH), cirrhosis, and hepatocellular carcinoma. SH encompasses both alcoholic steatohepatitis (ASH) and nonalcoholic steatohepatitis (NASH), and although the predominant etiology of ASH and NASH is different—involving chronic alcohol drinking and insulin resistance/type 2 diabetes, respectively—both diseases share common biochemical features, including steatosis, inflammation, hepatocellular death, and fibrosis.1, 2, 3, 4 SH, particularly NASH, is one of the most prevalent forms of chronic liver disease worldwide due to its association with obesity and type 2 diabetes. Despite intense research, the pathogenesis of ASH and NASH is still incompletely understood.
Mitochondrial dysfunction and subsequent onset of oxidative stress are considered critical players in NASH and ASH, underlying the second hit in the “two‐hit” scenario of SH.5, 6 Indeed, although other potential mechanisms contribute to disease progression (e.g., endoplasmic reticulum [ER] stress, autophagy impairment), NASH has been considered a mitochondrial disease.6 Mitochondria are the primary intracellular sites of oxygen consumption, which takes place in the mitochondrial respiratory chain (MRC), and therefore are a major source of reactive oxygen species (ROS) generation.7 Despite evidence indicating defective MRC activity and oxidative phosphorylation (OXPHOS) in nonalcoholic fatty liver disease (NAFLD) and alcoholic liver disease (ALD),8, 9 the contribution of this functional defect to the overall progression to ASH and NASH remains to be fully understood, especially in light of data dissociating defective MRC and OXPHOS with NASH and ASH progression (see “Mitochondrial Dysfunction in NAFLD/ALD: An Ongoing Conundrum” section). Because superoxide anion is the first ROS generated in mitochondria by the transfer of electrons from MRC to molecular oxygen and the source of other ROS and reactive nitrogen species (RNS), the dismutation of superoxide anion may be a critical approach to prevent oxidative stress and the consequences in inactivating mitochondrial components that contribute to mitochondrial dysfunction and potential impact in disease pathogenesis. However, superoxide anion dismutation generates hydrogen peroxide; therefore, targeting the former requires an adaptive capacity to detoxify the latter to prevent accumulation of unwanted reactive species (ROS/RNS), which can further damage mitochondrial components and contribute to disease progression. This scenario thus defines a critical balance among antioxidants that may affect the design of future trials in testing the role of antioxidant therapy in human SH. Although mitochondria are not the only source of ROS in cells, they are important ROS generators. Thus, in the present review, we focus on mitochondrial oxidative stress and summarize the concept of oxidative stress beyond the classical view of an imbalance between oxidants and antioxidants and the emerging evidence that targeting just a single ROS species may be insufficient to prevent SH progression, which may underlie the limited therapeutic benefits of clinical trials using a particular antioxidant for the treatment of SH.
Oxidative Stress: Concept, Sources, and Defenses
Although the pathophysiology of NAFLD and ALD is complex and involves a close interaction between host genetics and environmental factors, growing evidence supports a key role for oxidative stress caused by the generation of ROS in the progression of NAFLD and ALD. As the contribution of oxidative stress in NAFLD and ALD pathogenesis has been the subject of several reviews,10, 11, 12, 13, 14 here we will briefly present the concept of ROS and oxidants, as well as the sources and strategies of defense.
Concept
As defined more than three decades ago, oxidative stress was considered an imbalance between the generation of ROS and oxidants and the counteracting activity of antioxidants.15 This concept implied that either the overgeneration of free radicals and ROS and/or the limitation or impairment in the action of antioxidants can result in the net accumulation of ROS, which can exert deleterious effects in cell function, ultimately contributing to aging and major diseases, including fatty liver disease.
Free radicals are molecules or atoms characterized by the presence of unpaired electrons; this property determines their extreme reactivity in chemical reactions for electrons exchange. Free radicals are commonly generated by homolytic bond cleavage between two atoms of similar electronegativity (e.g., O‐O or O‐N bonds) or by a single‐electron transfer to an atom or molecule (e.g., superoxide anion). Moreover, although two‐electron oxidants are not strictly radicals, they arise from the metabolism or scavenging of one‐electron radicals, as best illustrated in the generation of hydrogen peroxide from superoxide anion. The reactivity or the oxidizing potential of free radicals/oxidants can be estimated by the one‐electron reduction potential, which reflects the affinity of a molecule or atom for electrons compared with that of hydrogen (which is set at 0), with the reduction potential of superoxide anion being intermediate between hydrogen peroxide and hydroxyl radical.16, 17 Thus, among biologically relevant ROS and free radicals, hydrogen peroxide has the lowest reactivity and the highest stability and intracellular concentration, and therefore it is highly regulated in cells to avoid its overgeneration.
Sources
Although ROS and oxidants derive from different sources as subproducts of cellular metabolism, because of the consumption of molecular oxygen within the MRC, mitochondria are important ROS generators. In addition, free radicals and ROS can be generated by various enzymes in the cytosol, such as amino acids oxidases, cyclooxygenase, lipoxygenase, nitric oxide (NO) synthase, and xanthine oxidase, which generate superoxide anion and other derived ROS. These enzymes link the generation of ROS with specific signaling pathways involved in particular pathological processes.18, 19 For instance, cyclooxygenase and lipoxygenase link superoxide anion generation to arachidonic acid metabolism and inflammation, with important implications in cancer, whereas xanthine oxidase has been involved in ischemia/reperfusion injury and liver transplantation.20, 21 Oxidants also are generated by sulfhydryl oxidase in the ER during protein folding and disulfide bond formation necessary for the assembly and secretory pathway for proteins22 as well as in peroxisomes by peroxisomal oxidase. Of particular relevance for liver pathology is the burst of superoxide anion formed by nicotinamide adenine dinucleotide phosphate, reduced form (NADPH) oxidase due to the transfer of one electron from NADPH to molecular oxygen. Although NADPH oxidase was first described in phagocytes of the innate immune system (e.g., neutrophils and macrophages) and considered a first line of defense against ingested pathogens,23 NADPH oxidase has been involved in many pathologic processes, including cardiovascular disorders and liver diseases.24, 25
In addition to the extramitochondrial source of superoxide anion, its mitochondrial generation is a collateral consequence of respiration in the MRC.26, 27 (Fig. 1) The specific site of superoxide anion generation within mitochondria is not completely understood, but increasing evidence points to the involvement of the flavin mononucleotide group of complex I through reverse electron transfer,28 which is consistent with the ability of diphenyleneiodonium to inhibit succinate‐supported ROS generation without affecting the flavin group of complex II. Moreover, complex III of respiration is also known to be an important source of ROS generation through the ubiquinone‐reactive sites Q0 and Qi.29, 30 Moreover, the redox activity of p66Shc within mitochondria has been shown to directly generate hydrogen peroxide through oxidation of cytochrome c without intermediate formation of superoxide anion,31 whereas apoptosis‐inducing factor (AIF), which exhibits an NADH oxidase activity, generates superoxide anion and subsequent hydrogen peroxide.32, 33 Increased mitochondrial generation of superoxide has been shown in ASH and NASH models.34, 35 In addition to its own damaging reactivity with iron‐sulfur centers, the superoxide anion can be the source of further ROS and RNS. Superoxide anion can interact with NO to produce the highly reactive product peroxynitrite. Peroxynitrite has a short half‐life and is responsible for many damaging modifications of tyrosine residues in mitochondrial target proteins,36 which has been shown to mediate some of the features associated with mitochondrial dysfunction in NAFLD (see “Mitochondrial Dysfunction in NAFLD/ALD: An Ongoing Conundrum” section).
Figure 1.
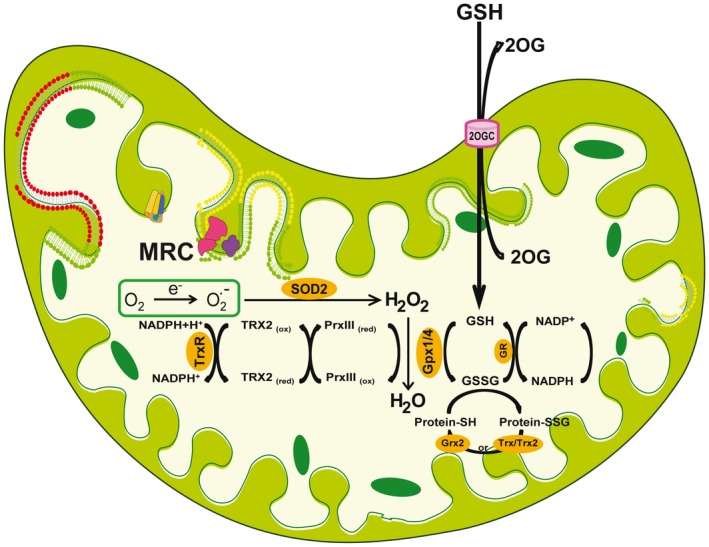
Mitochondrial ROS generation and antioxidant strategies. Oxygen consumption is a major driving force for ATP generation in the MRC. However, a collateral consequence of the OXPHOS is the transfer of electrons to molecular oxygen, resulting in the formation of superoxide anion. Because a superoxide anion is a highly reactive free radical, its down‐regulation is ensured by the presence of SOD2, which dismutates superoxide anion into hydrogen peroxide. The elimination of this oxidant is essential to prevent the generation of more reactive species by the Haber–Weiss/Fenton reactions. Hydrogen peroxide can be eliminated by the presence of several strategies, the mGSH/Gpx and the PrxIII/Trx2 couples, thus denoting the relevance of its catabolism (see “Oxidative Stress: Concept, Sources, and Defenses” section). Essential for appropriate GSH redox function is the availability of mGSH, determined from its import from cytosol by a specific process mediated by the 2‐oxoglutarate carrier. Abbreviation: 2‐OG, 2‐oxoglutarate carrier.
Defenses
Cells have developed a range of antioxidant strategies to cope with the constant generation of free radicals and reactive species, including enzymatic and nonenzymatic antioxidants.35, 37 However, because of the relevance of mitochondria as a major source of ROS generation and their role in fatty liver disease, we will focus on the strategies to control the generation of mitochondrial ROS formation (Fig. 1). The elimination of mitochondrial superoxide by manganese superoxide dismutase (MnSOD, also known as SOD2) is essential in the defense against oxidative stress and the maintenance of cell function, as illustrated by the fact that mice with global SOD2 deficiency experience neonatal death. As mentioned previously, the dismutation of superoxide generates hydrogen peroxide, which despite not strictly being an ROS, is a potent oxidant. The degradation of mitochondrial hydrogen peroxide is carried out by several antioxidant enzymes, most predominantly by peroxiredoxins (Prx), particularly PrxIII due to its mitochondrial localization, and the reduced glutathione (GSH) peroxidases (Gpx), including Gpx1, and more importantly by the mitochondrial Gpx4, which are dependent and require reduced GSH for their activity. The detoxification of hydrogen peroxide by PrxIII results in its oxidation, whose reduction occurs predominantly by thioredoxin‐2 (Trx2). In addition, the presence of glutaredoxins (Grx), in particular Grx2, which is located in the mitochondrial matrix, catalyzes the reduction of thiol‐disulfide exchange between protein thiols and oxidized GSH (GSSG) or between a glutathionylated protein and GSH; hence, Grx are critical to ensuring optimal protein activity in mitochondria.38, 39 Because most mitochondria lack catalase, the mitochondrial GSH (mGSH) pool plays a critical role in the elimination of hydrogen peroxide, implying that its source and maintenance of its reduced form is critical for appropriate detoxication of hydrogen peroxide. In addition to detoxifying hydrogen peroxide, mGSH is also key in the catabolism and detoxification of hydroperoxides present on phospholipids and other lipid sources due to the presence of GSTs in mitochondria, which also require GSH as a cofactor.38, 39 An interesting issue centers on the relative importance between the mitochondrial redox couple (mGSH/Gpx) versus the PrxIII/Trx2 system on hydrogen peroxide defense. The relevance of Gpx, particularly the mitochondrial‐targeted Gpx4, against hydrogen peroxide has been illustrated by the embryonic lethality of global deletion of Gpx4 in mice,40 whereas recent studies in HeLa cells identified PrxIII as a key defense against mitochondrial hydrogen peroxide.41 Moreover, mGSH depletion has been shown to sensitize hepatocytes against oxidant cell death.35, 37 Although specific kinetic features (e.g., Km for hydrogen peroxide and catalytic efficiency) between Gpx and PrxIII may account for the relative importance of the antioxidant systems mGSH/Gpx and PrxIII/Trx2 against hydrogen peroxide, differences in abundance and posttranslational regulation of these antioxidant components in different cells types (e.g., HeLa or hepatocytes) may also play a role in their relative importance in hydrogen peroxide defense. Interestingly, it has been shown that both antioxidant systems are interconnected. For instance, depletion of mGSH results in Trx2 oxidation,42 whereas studies in hypercholesterolemic pigs indicated that the selective depletion of mGSH in heart mitochondria led to profound decreases in the levels of the mitochondria‐specific antioxidant enzymes, such as SOD2, Trx2, and PrxIII.43 Given the relevance of mGSH in maintaining the optimal antioxidant defense strategy in mitochondria, its availability within mitochondrial matrix is essential for mitochondrial function and cell survival, as discussed in the “Antioxidants Therapy: Critical Mitochondrial Antioxidants Balance” section.
Oxidative Stress: More Than an Imbalance Between Oxidants and Antioxidants
The original conception of oxidative stress as an imbalance between oxidants and antioxidants fostered the idea that the down‐regulation of ROS by supplemented antioxidants (e.g., vitamin E, N‐acetylcysteine [NAC]) would be efficient in the treatment of diseases. However, large‐scale interventional studies with antioxidants in humans, based on this classical concept of oxidative stress, have been largely disappointing and inconsistent in demonstrating health benefits in the treatment of human‐relevant diseases, including cancer and liver diseases.44, 45, 46, 47 Although many unknown factors may have determined these unanticipated results, these findings opened new lines of thinking, including the concept that oxidative stress extends beyond the predicated imbalance between prooxidants and antioxidants and encompasses alterations in specific and discrete redox signaling pathways within cells.48 This outcome may reflect the dissociation between the reduced and oxidant condition of molecules that represent the redox status of cells. For instance, although the GSH/GSSG and cysteine/cystine redox potential have been considered a useful estimation of the balance between oxidative reactions and endogenous antioxidant defenses, data from human plasma indicated that the concentration of GSH correlated with that of cysteine; however, no correlation was found between GSSG and cystine.49 Furthermore, the values for the redox potential for plasma GSH/GSSG couple were more oxidized than the values reported in cells but less oxidized than the values of the redox potential for cysteine/cystine, indicating a lack of equilibrium of GSH/GSSG and cysteine/cystine pools between plasma and tissues, suggesting that these plasma redox levels do not necessarily reflect the complexity of oxidative stress as an imbalance between prooxidants and antioxidant systems.49 These findings thus provided a rationale for the reformulation of the concept that oxidative stress may reflect the disruption of thiol redox states rather than the reflection of a net increase in reactive species,48 underlying the so‐called redox switch that involves phosphorylation/dephosphorylation reactions. This outcome is determined by the differential sensitivity of critical cysteinyl residues between kinases and phosphatases to oxidation by oxidative stress. Thus, in addition to targeting key cell components (e.g., DNA, protein, or lipids), the oxidation of cysteinyl residues by ROS can activate specific pathways such as stress kinases (e.g., ASK1) or inactivate phosphatases (e.g., JNK phosphatases) that further contribute to sustained activation of stress kinases, which ultimately lead to cell and tissue damage.
Moreover, in addition to the control of ROS generation by antioxidants, a critical balance among antioxidant enzymes is required to avoid the overgeneration of oxidants, such as hydrogen peroxide. This has been well documented in the case of SOD and Gpx, which modulate the conversion of superoxide anion to molecular oxygen and water in a two‐step enzymatic process, involving first the dismutation of superoxide anion to hydrogen peroxide by SOD, followed by the conversion of hydrogen peroxide to molecular oxygen and water by Gpx. Overexpression of SOD activity relative to that of Gpx activity may lead to a net increase in hydrogen peroxide, which can result in the harmful generation of the hydroxyl radical by the Haber–Weiss/Fenton reactions, indicating that high levels of SOD have deleterious effects in cell function and life span. This concept has been well illustrated in experimental models, such as in bacteria that overexpress FeSOD or SOD2, which exhibit increased sensitivity to free‐radical generators such as paraquat50 or in Drosophila melanogaster in which the overexpression of Cu/ZnSOD increases mortality during the development of the mature insect.51 More importantly, fibroblasts from patients with Down syndrome, which carry three copies of the Cu/ZnSOD gene in chromosome 21 and only two copies of Gpx in chromosome 3, display increased activity of Cu/ZnSOD relative to GSHPx, resulting in increased basal levels of hydrogen peroxide.52 This scenario will be specifically developed regarding the role of mGSH as a critical determinant of the beneficial effects of superoxide anion dismutation in experimental models of NASH and ASH (see “Antioxidants Therapy: Critical Mitochondrial Antioxidants Balance” section).
Mitochondrial Dysfunction in NAFLD/ALD: An Ongoing Conundrum
In addition to being the main culprits in the generation of ROS in the MRC, mitochondria play a key role in the metabolism of nutrients to provide the energy required for multiple cell functions, lending support to the concept that mitochondrial dysfunction can contribute to NAFLD/ALD progression. However, although this association appears to be intuitive, emerging experimental data have provided counterintuitive evidence that mitochondrial dysfunction prevents rather than promotes NASH and ASH.
NAFLD
Whereas oxidation of fatty acids in mitochondria is a source of energy, the impairment in fatty acid oxidation (FAO) rates in mitochondria is thought to contribute to fat accumulation and hepatic steatosis. Previous work reported mitochondrial abnormalities in patients with NASH as well as in patients with drug‐induced NASH symptoms. In this regard, these patients exhibited enlarged and swollen mitochondria with loss of cristae and paracrystalline inclusion bodies.53, 54 The reported rate of mitochondrial DNA (mtDNA) deletions in the NASH population was lower than in patients with ALD with active drinking.55, 56 Moreover, decreased activity of hepatic MRC enzyme complexes has been reported in patients with NASH.8 Specifically, this defect involved mitochondrial (complexes I, III, IV, and V) and nuclear (complex II) DNA‐encoded subunits of mitochondrial complexes, which was not due to a lower presence of hepatic mitochondrial mass between patients and controls. Furthermore, the defects in MRC activity correlated with body mass index, plasma tumor necrosis factor levels, and homeostasis model assessment of insulin resistance index, indicative of insulin resistance, and were strongly correlated with the fibrosis staging of patients.8 In line with these findings in human NASH, similar defects in mitochondrial function have been described in experimental models of NAFLD. Thus, ob/ob mice exhibited the myristylation of specific components of MRC, in particular the ND4 subunit of complex I and cytochrome c.56 Moreover, in a nutritional NASH model, it has been described that feeding a methionine and choline diet (MCD) to rats for 4 weeks impaired carnitine palmitoyl transferase 1 (CPT1) activity, and therefore FAO, both in isolated mitochondria and hepatocytes.57 In line with this notion, it has been reported that the overexpression of an active form of CPT1 (CPT1AM), which is insensitive to malonyl‐coenzyme A (CoA) and therefore leads to a sustained increase in the rate of FAO independently of the glucose‐derived malonyl‐CoA levels, reduced obesity‐induced hepatic steatosis, weight gain, inflammation, diabetes, and insulin resistance in diet‐induced obesity in mice.58 Further genetic models provided a link between impaired mitochondrial function and hepatic steatosis and NASH. For instance, mice with deletion of histidine triad nucleotide‐binding 2 (HINT2), a member of the histidine triad superfamily of nucleotide hydrolase and transferase enzymes, exhibited decreased mitochondrial respiration and increased ROS generation and displayed morphologically deformed and fused mitochondria.59 This mitochondrial phenotype correlated with the development of fatty liver, decreased glucose tolerance, delayed recovery from insulin‐induced hypoglycemia, and increased levels of plasma insulin, leptin, adiponectin, and cholesterol.59
Despite these findings indicating the association of mitochondrial dysfunction with progressive NAFLD, recent findings using genetic mouse models of mitochondrial OXPHOS defects, as well as pharmacological approaches to target β‐oxidation, provided strong evidence that defects in mitochondrial respiratory capacity or decreased FAO rates prevent NASH and stimulate insulin sensitivity. For instance, deletion of AIF, an inner‐mitochondrial membrane‐associated protein that was originally identified as an important player in cell death, caused a progressive loss of MRC activity.60 Mice with specific deletion of AIF in liver or muscle exhibited decreased OXPHOS gene expression similar to that observed in insulin‐resistant patients. Moreover, complexes I and IV of the respiratory chain were functionally affected by loss of AIF expression in muscle, whereas liver‐specific deletion of AIF was manifested primarily as a combined deficiency of complex I and complex V activity. These defects were accompanied by increased NADH/NAD+ ratio, decreased adenosine triphosphate (ATP) and cyclic adenosine monophosphate canaliculus levels, both in muscle and liver from AIF knockout mice, as well as increased plasma lactate content, a marker of anaerobic metabolism. Quite intriguingly, however, there was no evidence for increased ROS generation or inflammation in either tissue of the mutant mice, and despite decreased mitochondrial OXPHOS, targeted deletion of AIF improved glucose tolerance and insulin sensitivity in muscle, whereas AIF deletion in liver protected against diet‐induced hepatosteatosis and liver insulin resistance.60 Thus, these data provide compelling evidence that defects in mitochondrial function due to impairment of mitochondrial complexes I, IV, and V in liver and muscle induce a lean and insulin‐sensitive metabolic state, conferring resistance to the adipogenic and diabetogenic effects of hypercaloric feeding. These intriguing findings are in line with the phenotype reported in mice with specific deletion of the mitochondrial transcription factor A (TFAM) in muscle or adipose tissue, which despite defective complex I, complexes I‐III, and complex IV activities of the MRC, exhibited increased insulin sensitivity in both tissues.61, 62 Thus, these findings challenge the view that loss of mitochondrial function is a cause of NAFLD, but rather imply that mitochondrial dysfunction may be a secondary consequence of disease progression.
ALD
In addition to these findings in NAFLD, mitochondrial dysfunction is considered a hallmark of ALD. Pioneering work described structural abnormalities of hepatic mitochondria from patients with ALD and the presence of megamitochondria, which are usually associated with a milder form of the disease.63, 64 Moreover, alcohol‐related liver disease with active drinking is linked to the expression of an mtDNA deletion and fragmentation in human ALD.53, 55 However, how these morphological alterations affect mitochondrial function in human ALD remains poorly understood. Indirect metabolic determinations using noninvasive breath tests have been considered an index of impaired mitochondrial function.65, 66 The decarboxylation of ketoisocaproate is a specific mitochondrial reaction that results in the exhalation of CO2 from 2‐keto‐isocaproic acid.65 Peak exhalation of 13CO2 from 2‐keto[1‐13C]isocaproic acid as well as the fraction of the administered dose decarboxylated for 120 minutes were both significantly reduced in patients with alcoholism compared with healthy controls, whereas aminopyrine breath test and galactose elimination capacity were not altered, indicating that the impaired mitochondrial ketoisocaproate decarboxylation is not a consequence of decreased functional hepatic mass. Moreover, the level of circulating 2‐keto[1‐13C]isocaproic acid was similar between patients with alcoholism and controls, discarding decreased bioavailability or increased dilution of labeled 2‐keto[1‐13C]isocaproic acid in patients. Importantly, the mitochondrial impairment in patients with alcoholism may be specific for decarboxylation reactions without involvement of other mitochondrial enzymes located in the inner or outer mitochondrial membranes.66 Thus, although further work is needed to critically examine the functional status of mitochondria from patients with ALD, it is conceivable that this relationship may be dependent on the stage of ALD progression and the status of active drinking.
In experimental models, however, alcohol‐mediated impairment in mitochondrial respiration appears to be species‐dependent, with opposing results reported between rats and mice. For example, several studies in rats fed alcohol orally with Lieber–DeCarli liquid diet reported decreased mitochondrial respiration (state III) and lower respiratory control ratio (state III/state IV) in isolated liver mitochondria.67, 68, 69, 70 In line with these findings, chronic alcohol intake impaired hepatic mitochondrial OXPHOS by suppressing the synthesis of protein subunits that are encoded by mitochondrial DNA, including subunits of the main respiratory complexes, NADH dehydrogenase (complex I), cytochrome b‐c1 (complex III), and cytochrome oxidase (complex IV), as well as the ATP synthase complex (complex V).71, 72 In contrast to these findings in rats, recent evidence in mice indicated increased mitochondrial respiration and higher level of complex I components in liver mitochondria.73, 74 Moreover, mice fed alcohol orally in pair‐fed liquid diet or given intragastrically exhibited increased state III respiration in isolated liver mitochondria, associated with enhanced levels of complexes I, IV, and V incorporated into the respiratory chain.75 Compared with oral alcohol intake, the magnitude of these changes was higher when alcohol was administered intragastrically and correlated with the severity of liver injury. The increase in components of mitochondrial complexes following intragastric alcohol consumption was associated with increased PGC‐1α protein expression but not of the TFAM. Furthermore, because alcohol intake has been shown to induce injury predominantly in a zonal‐dependent manner, recent studies examined whether the changes in mitochondrial function following chronic alcohol intake exhibit an acinar distribution. Interestingly, the increase in mitochondrial function from alcohol‐fed mice was predominantly observed in perivenous hepatocytes compared with periportal hepatocytes76 (and manuscript in preparation), which correlate with the area most susceptible to alcohol‐induced liver damage. Remarkably, recent findings in rats fed alcohol intragastrically reported opposing findings with respect to those described with the administration of alcohol orally, as outlined previously, exhibiting increased glutamate‐malate and acetaldehyde‐linked respiration and a mild decrease in succinate‐driven respiration,77 suggesting site‐specific changes within the MRC elicited by alcohol consumption. Moreover, of relevance for human ALD, recent findings in chimeric mice xenotransplanted with human adult hepatocytes indicated an increased mitochondrial respiration after chronic and acute alcohol administration.78 Thus, these data define a complex scenario with an apparent paradoxical role of alterations in mitochondrial function in fatty liver disease, requiring further investigation to establish whether mitochondrial dysfunction is a cause or consequence of NASH/ASH. Whether this complexity relates to magnitude and/or specificity of ROS generated or to their potential role as signaling entities remains to be further pursued. Moreover, the gender effect on mitochondrial (dys)function and subsequent ROS generation in NASH and ASH will need to be investigated in detail.
Mitochondrial Antioxidants Status in NAFLD/ALD
Given the role of mitochondria as a key player in ROS generation and the initiation of oxidative stress, and because the mitochondrial antioxidant network is important in the regulation of mitochondrial ROS generation (as described previously), we will focus on the status of mitochondrial antioxidants in NASH and ASH. Although increased superoxide anion in obese models of NAFLD is a consistent finding, the status of SOD2 is not well established, with studies indicating either decreased or increased expression.79, 80 Quite intriguingly, ob/ob mice exhibited increased levels of superoxide anion that was accompanied by a paradoxical increase in SOD2 activity as well a decrease in mitochondrial GPx. Interestingly, using nutritional and genetic NASH models, an increased expression in the levels of SOD2 has been reported; however, this was accompanied by a significant impairment in SOD2 activity, which may reflect sensitivity to oxidative/nitrosative stress.81 In this regard, SOD2 contains tyrosine residues that are susceptible to nitrosilation or oxidation by peroxynitrite, leading to enzymatic inactivation.82, 83 Of relevance, the polymorphism C47T of the SOD2 gene is associated with a reduced SOD2 activity, which results in an increased ROS production and a high susceptibility to develop progressive NASH with advanced fibrosis in pediatric patients.84 Similar to the status of SOD2 in NAFLD, the role of alcohol on SOD2 regulation is controversial, with findings indicating increased expression,85 no change,86 or decreased expression.87 Moreover, recent findings underscore that chronic ethanol metabolism inhibits hepatic SOD2 through lysine acetylation, without change in SOD2 expression.88 Interestingly, the lysine acetylation–mediated inactivation of SOD2 was preferentially seen in zone 3 of the liver, where most of the ethanol metabolism takes place. Although the mechanism of hyperacetylation by alcohol was not investigated, recent findings indicated that alcohol intake impairs SIRT3, a mitochondrial member of sirtuin family involved in protein deacetylation, by a 4‐hydroxynonenal‐mediated SIRT3 carbonylation.89 Quite intriguingly, genetic studies examining the homozygosity for alanine in the mitochondrial targeting sequence of SOD2 gene, which is associated with increased mitochondrial localization, indicated that the frequency of the alanine‐encoding allele and the percentage of alanine homozygotes correlated with the severity of ALD.83 However, homozygosity for alanine does not modify alcohol consumption and the risk of macrovacuolar steatosis in patients with alcoholism, but emerged as a risk factor for severe ALD in humans.90
In addition to these findings regarding the status of the first line of defense against mitochondrial superoxide generation, the mGSH redox cycle is a key mechanism for the metabolism of hydrogen peroxide and other hydroperoxides. This redox cycle requires reduced mGSH levels, and its depletion can impair the detoxification of peroxides. There has been evidence that mGSH status is limited in both NAFLD and ALD. In addition to the availability of NADPH to ensure the reduction of GSSG to mGSH by GRx, the homeostasis of mGSH is determined by its import from cytosol by a carrier‐mediated process that is sensitive to changes in mitochondrial physico‐chemical properties determined by alterations in the phospholipid/cholesterol molar ratio.37, 39 In the case of NAFLD, nutritional and genetic models of hepatic steatosis characterized by accumulation of mitochondrial cholesterol reported depleted mGSH levels and sensitization to inflammatory cytokines.91 Moreover, it has been shown that deficiency of microsomal triglyceride transfer protein resulted in liver steatosis and insulin resistance, and its inhibition by western diet feeding increased hepatic‐free cholesterol level and subsequent accumulation in mitochondria.92, 93 In addition, feeding a methionine‐deficient and choline‐sufficient diet, which induces NASH in mice, has been shown to deplete the phosphatidylcholine/phosphatidylethanolamine ratio in mitochondrial membranes, resulting in decreased membrane fluidity and mGSH depletion.94 In line with these findings, although the content of hepatic mitochondrial cholesterol in patients with NASH has not been reported, the overexpression of StARD1, a mitochondrial cholesterol transporting polypeptide involved in the trafficking of cholesterol to mitochondrial inner membrane, suggests enhanced mitochondrial cholesterol accumulation in human NASH,95, 96 consistent with the reported mGSH depletion seen in human NASH.97 In line with the preceding findings in NAFLD, mGSH has been shown to be depleted by alcohol feeding both in rats and mice,98, 99, 100, 101 with a preferential effect seen in perivenous hepatocytes.99, 100 The depletion of mGSH by alcohol feeding is rapidly reversed following alcohol withdrawal102, 103 and occurs in mice fed alcohol intragastrically.98 Moreover, recent evidence in rats fed an ethanol‐polyunsaturated fatty acid treatment confirmed the mitochondrial cholesterol accumulation, resulting in mGSH depletion and ASH, and these effects were prevented by betaine treatment.104 Interestingly, mGSH depletion by chronic alcohol intake has also been reported in alveolar type II cells,105 indicating that the effect of alcohol in depleting mGSH is not an exclusive feature of the liver. Overall, this cumulative evidence points to a depression in the mitochondrial antioxidant defense in both NAFLD and ALD, which lends support that increased generation of ROS and prooxidants may be culprits for disease progression, highlighting the potential use of antioxidants in the treatment of fatty liver disease.
Antioxidants Therapy: Critical Mitochondrial Antioxidants Balance
The imbalance between ROS production and antioxidant defense in favor of the former has paved the way for the rationale behind the use of antioxidants as a potential therapy for the treatment of human fatty liver disease. As a consequence, there have been a number of trials testing the role of antioxidant therapy in NAFLD and ALD. For instance, Sanyal et al. reported that vitamin E was effective in the nondiabetic adult with NASH in reducing serum alanine aminotransferase (ALT) and inflammation, although it was ineffective in improving fibrosis.47 However, the efficacy of vitamin E in NAFLD remains to be confirmed in independent studies.106 In this regard, a meta‐analysis of clinical trials did not provide clear‐cut evidence for a beneficial effect of vitamin E in normalizing serum ALT in patients with NAFLD.107 Likewise, vitamin E was also ineffective in improving serum ALT levels and NAFLD in pediatric NAFLD.108 Therefore, the responses, long‐term tolerance, and efficacy of vitamin E in patients with NAFLD with diabetes or NASH‐related cirrhosis remain to be elucidated.106
Similar to the findings in NAFLD, whether the use of antioxidants alone is able to improve the survival in patients with alcoholic hepatitis (AH), a severe form of ALD, is controversial.46, 109 In this regard, administration of an antioxidant cocktail, including vitamin E and NAC, along with nutritional supplements, to patients with AH failed to improve survival in this cohort,46 whereas Stewart et al., using a similar approach, reported increased short‐term outcome.109 Interestingly, however, the replenishment of hepatic GSH levels with either NAC or S‐adenosyl‐methionine (SAM) has been shown to increase the efficacy of prednisolone in patients with advanced ALD.110, 111 Although SAM acts as a GSH precursor, it is a methyl donor and therefore regulates multiple targets in hepatocytes.112 Despite this broad‐range effect, SAM therapy has been shown to improve survival in patients with less advanced alcoholic liver cirrhosis, such as patients with Child A and B.113
Besides these findings and given the role of superoxide anion in cell fate and oxidative stress, targeting mitochondrial superoxide anion may be of potential relevance in NAFLD and ALD. To overcome the problems associated with the use of the native SOD enzymes, such as their inaccessibility to intracellular compartments and adverse effects due to immunogenic responses, chemical SOD mimetics have been developed and characterized for SOD activity.114, 115 In this regard, it has been shown that the manganese (III) meso‐tetrakis (N‐ethylpyridinium‐2‐yl) porphyrin (MnP) protects against HFD‐induced obesity and liver steatosis.116 Although MnP is a first‐in‐class oxidoreductase, it not only scavenges superoxide anion but also possesses anti‐inflammatory properties by targeting nuclear factor kappa B, arguing that the beneficial effect of MnP may be independent of scavenging superoxide anion. Moreover, Mn(III)tetrakis(4‐benzoic acid)porphyrin chloride (MnTBAP), an anionic porphyrin SOD mimetic, has been shown to worsen nutritional (MCD) and genetic (methionine adenosyltransferase 1A) models of NAFLD despite superoxide anion scavenging and decreasing peroxynitrate generation.81 Although it has been argued that MnTBAP scavenges peroxynitrite as well, MnTBAP has been shown to functionally replace SOD2 deficiency in Sod2tm1Cje null mice, resulting in prevention of systemic pathology, hepatic lipid accumulation, and extended life span.117 Because these models of NAFLD cause mGSH depletion, the scavenging of superoxide anion by MnTBAP resulted in the overproduction of hydrogen peroxide and hydroxyl radical, and the worsening effect of MnTBAP therapy was rescued by increasing mGSH levels with GSH ethyl ester (GSHEE), a permeable form of GSH.81 Interestingly, the beneficial effects of recovering mGSH by GSHEE was recapitulated by MitoQ. Thus, these findings demonstrate that the beneficial effect of scavenging superoxide anion is determined by the availability of mGSH and illustrate the need to maintain a subtle antioxidant balance to prevent oxidative stress and NAFLD progression (Fig. 2). Moreover, SOD2 transgenic mice exhibit increased susceptibility to MCD‐induced NASH despite decreased superoxide anion and peroxynitrite generation, demonstrating that overexpression of SOD2 in the face of mGSH depletion sensitizes to nutritional NASH.
Figure 2.
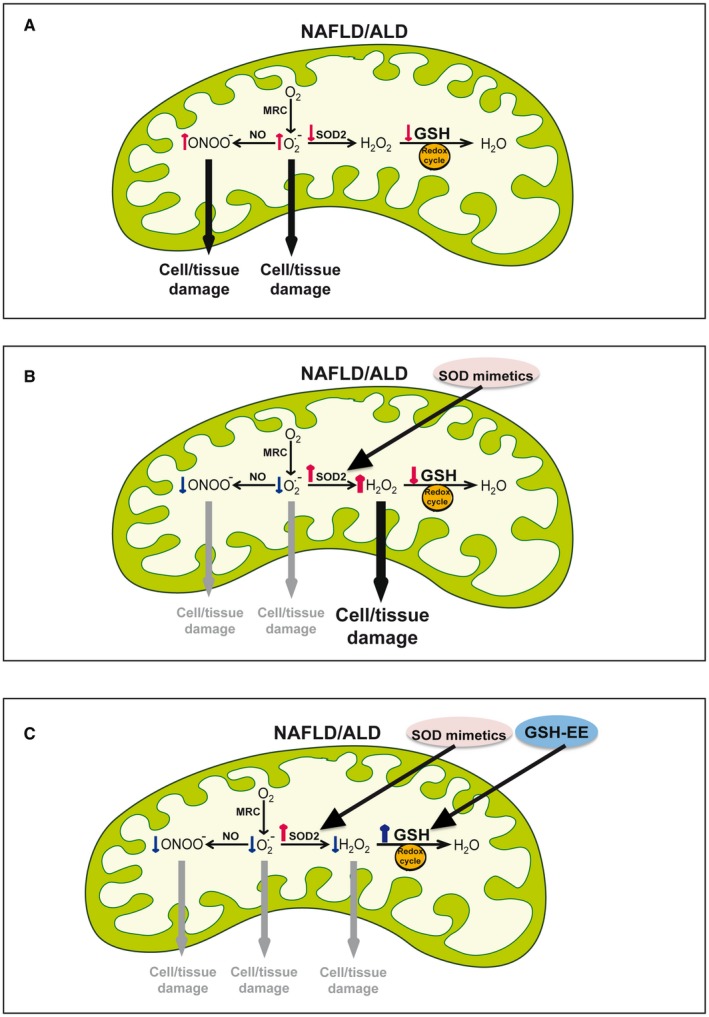
Critical antioxidant balance to prevent ROS generation and oxidative stress in fatty liver disease. (A) ROS generation and antioxidant defense status in fatty liver disease. Increased generation of superoxide anion within the MRC, which can be exacerbated by decreased levels/activity of SOD2. The increased generation of superoxide anion facilitates the formation of peroxynitrite from NO. In addition to impaired SOD2 defense, mGSH depletion contributes to limit defense against hydrogen peroxide detoxification. (B) The decreased status of SOD2 provides a rationale for the use of SOD mimetics, which decreases superoxide anion formation as well as the generation of peroxynitrite. However, because of the limitation of mGSH, the SOD mimetics results in the accumulation of hydrogen peroxide. (C) The use of both SOD mimetics with the replenishment of mGSH by the GSHEE, a permeable form of GSH prodrugs, results in the net decrease of superoxide anion, peroxynitrite, and hydrogen peroxide.
In line with these findings, it has been shown that chronic alcohol consumption induced lipid peroxidation, respiratory complex I protein carbonylation, mtDNA depletion, and respiratory complex dysfunction in transgenic mice overexpressing SOD2 (TgSOD2) but not in wild‐type mice.34, 118 Because mGSH is depleted in ALD (see “Mitochondrial Antioxidants Status in NAFLD/ALD” section), these findings suggest that the therapeutic role of SOD mimetics in ALD may also depend on the status of mGSH. Consistent with this possibility, it has been shown that acute alcohol binging exacerbated mtDNA depletion in SOD2‐deficient mice.118, 119
Very interestingly, recent findings have shown a promising effect of redox nanoparticles for the treatment of NASH.120 Redox polymers exhibiting antioxidant activity against nitroxide radicals in the hydrophobic core of the particle and amine linkages to allow the disintegration of the particles under acidic conditions due to the protonation of amino groups, were efficient in reducing hepatic ROS levels and ameliorating inflammation and fibrosis in murine NASH without changes in liver steatosis or liver weight. Although these findings underscore the potential of redox nanoparticles as a novel potential therapy for NASH, further work is needed to ascertain whether this approach targeted mitochondrial function and antioxidant defenses.
Conclusions and Future Perspectives
NAFLD and ALD are complex metabolic diseases that lack current effective treatment, likely reflecting our incomplete understanding of the molecular mechanisms underlying their pathogenesis. Although mitochondria are key in the maintenance of a myriad of cell functions, they are also the main source of ROS, which, due to their reactivity, can target multiple components, including DNA, proteins, and lipids. Thus, although counteracting ROS generation by antioxidants may have beneficial effects, this approach unfortunately has not been translated yet in human disease. Although the burst of ROS production has been viewed as an important culprit in cell pathophysiology and disease, current data suggest that increased ROS production may serve as a “signaling” mechanism to allow up‐regulation of antioxidant defenses and long‐term protection of significant relevance in diseases and aging, underlying a redox paradox and the so‐called mitohormesis theory.121 Thus, the dual function of ROS/oxidative stress as cell death triggers or players in signaling pathways may determine the final role of mitochondrial dysfunction as a cause or consequence of disease progression. In addition to this complex role of mitochondrial (dys)function in liver diseases, targeting a single antioxidant arm may be more deleterious than beneficial. Shown in both NAFLD and ALD, increasing the dismutation of mitochondrial superoxide anion with SOD mimetics in experimental models failed to protect against disease progression despite decreased superoxide anion and peroxynitrite formation. This strategy, in combination with the limitation of another key mitochondrial antioxidant defense, such as mGSH, which is key for appropriate defense of GSH redox cycle, results in the accumulation of hydrogen peroxide, which, as a potent oxidant, can lead to the generation of more harmful and reactive ROS such as hydroxyl radical. Furthermore, the use of GSH precursors to replenish mGSH levels in fatty liver disease is also of interest. Although NAC is a GSH precursor by supplying the rate‐limiting amino acid cysteine, it is unable to replenish mGSH in the face of mitochondrial cholesterol loading and hence will not be an efficient choice in NASH/ASH. In contrast, GSHEE, permeable GSH form, freely diffuses through membrane bilayer and has been shown to restore mGSH bypassing the block due to changes in mitochondrial membrane fluidity. In addition to maintaining a critical mitochondrial antioxidant balance to avoid oxidative stress, recent findings have provided evidence that therapy with redox nanoparticles counteracting nitrosative radicals may be of promise in the treatment of NASH. Further research would be needed to test whether this approach in combination with SOD mimetics and mGSH precursor could be effective for NAFLD and ALD.
Potential conflict of interest
Nothing to report.
Supported by the National Institute on Alcohol Abuse and Alcoholism/National Institutes of Health (P50AA011999). We acknlowledge the support from grants SAF2014‐57674‐R, SAF2015‐69944R and SAF2017‐85877R from Plan Nacionald de I+D, by the CIBEREHD from the Instituto de Salud Carlos III, by the AGAUR of Generalitat de Catalunya SGR‐2017‐1112, by the FUNDACION BBVA and by the European Cooperation in Science and Technology (COST) ACTION 17112 Prospective European Drug‐Induced Liver Injury Network.
References
- 1. Bedogni G, Miglioli L, Masutti F, Tiribelli C, Marchesini G, Bellentani S. Prevalence of and risk factors for nonalcoholic fatty liver disease: the Dionysos nutrition and liver study. Hepatology 2005;42:44‐52. [DOI] [PubMed] [Google Scholar]
- 2. Masarone M, Rosato V, Dallio M, Abenavoli L, Federico A, Loguercio C, et al. Epidemiology and natural history of alcoholic liver disease. Rev Recent Clin Trials 2016;11:167‐174. [DOI] [PubMed] [Google Scholar]
- 3. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Nath B, Szabo G. Alcohol‐induced modulation of signaling pathways in liver parenchymal and nonparenchymal cells: implications for immunity. Sem Liver Dis 2009;29:166‐177. [DOI] [PubMed] [Google Scholar]
- 5. Day CP, James OF. Steatohepatitis: a tale of two hits? Gastroenterology 1998;114:842‐845. [DOI] [PubMed] [Google Scholar]
- 6. Pessayre D, Fromenty B. NASH: a mitochondrial disease. J Hepatol 2005;42:928‐940. [DOI] [PubMed] [Google Scholar]
- 7. Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell 2005;120:483‐495. [DOI] [PubMed] [Google Scholar]
- 8. Perez‐Carreras M, Del Hoyo P, Martín MA, Rubio JC, Martín A, Castellano G, et al. Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology 2003;38:999‐1007. [DOI] [PubMed] [Google Scholar]
- 9. Garcia‐Ruiz C, Kaplowitz N, Fernandez‐Checa JC. Role of mitochondria in alcoholic liver disease. Curr Pathobiol Report 2013;1:159‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoek GH, Liedorp PR, Bast A. The role of oxidative stress in nonoalcoholic steatohepatitis. Clin Chim Acta 2011;412:1297‐1305. [DOI] [PubMed] [Google Scholar]
- 11. Sun J, Fu J, Li L, Chen C, Wang H, Hou Y, et al. Nrf2 in alcoholic liver disease. Toxicol Appl Pharmacol 2018;357:62‐67. [DOI] [PubMed] [Google Scholar]
- 12. Seitz HK, Bataller R, Cortez‐Pinto H, Gao B, Gual A, Lackner C, et al. Alcoholic liver diseaser. Nat Rev Dis Primers 2018;4:16. [DOI] [PubMed] [Google Scholar]
- 13. Schuppan D, Schatternberg JM. Nonalcoholic steatohepatitis: pathogenesis and novel therapeutic approaches. J Gastroenterol Hepatol 2013;(28 Suppl. 1):68‐76. [DOI] [PubMed] [Google Scholar]
- 14. Masarone M, Rosato V, Dallio M, Gravina AG, Aglitti A, Loguercio C, et al. Role of oxidative stress in pathophysiology of nonalcoholic fatty liver disease. Oxid Med Cell Longev 2018;2018:9547613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sies H. Oxidative stress: introductory remarks In: Sies H, ed. Oxidative Stress. London: Academic Press; 1985:1‐8. [Google Scholar]
- 16. Rao PS, Hayon E. Redox potentials of free radicals. IV. Superoxide and hydroperoxy radicals. J Phys Chem 1975;79:397‐402. [Google Scholar]
- 17. Hayyan M, Hashim MA, AlNashef IM. Superoxide ion: generation and chemical implications. Chem Rev 2016;116:3029‐3085. [DOI] [PubMed] [Google Scholar]
- 18. Xu L, Stevens J, Hilton MB, Seaman S, Conrads TP, Veenstra TD, et al. COX‐2 inhibition potentiates antiangiogenic cancer therapy and prevents metastasis in preclinical models. Sci Transl Med 2014;6:242ra84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goos JA, Hiemstra AC, Coupé VM, Diosdado B, Kooijman W, Delis‐Van Diemen PM, et al. Epidermal growth factor receptor (EGFR) and prostaglandin‐endoperoxide synthase 2 (PTGS2) are prognostic biomarkers for patients with resected colorectal cancer liver metastases. Br J Cancer 2014;111:749‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Peglow S, Toledo AH, Anaya‐Prado R, Lopez Neblina F, Toledo‐Pereyra AH. Allopurinol and xanthine oxidase inhibition in liver ischemia reperfusion. J Hepatobiliary Pancreat Sci 2011;18:137‐146. [DOI] [PubMed] [Google Scholar]
- 21. Taha MO, Simões MJ, Noguerol EC, Mendonça FP, Pascoalick HM, Alves RA, et al. Effects of allopurinol on ischemia and reperfusion in rabbit livers. Transplant Proc 2009;1:820‐823. [DOI] [PubMed] [Google Scholar]
- 22. Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992;257:1496‐1502. [DOI] [PubMed] [Google Scholar]
- 23. Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 2005;109:217‐226. [DOI] [PubMed] [Google Scholar]
- 24. Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antiox Redox Signal 2006;8:692‐714. [DOI] [PubMed] [Google Scholar]
- 25. De Minicis S, Bataller R, Brenner DA. NADPH oxidase in the liver: defensive, offensive, or fibrogenic? Gastroenterology 2006;131:272‐275. [DOI] [PubMed] [Google Scholar]
- 26. Chouchani ET, Pell VR, James AM, Work LM, Saeb‐Parsy K, Frezza C, et al. A unifying mechanism for mitochondrial superoxide production during ischemia‐reperfusion injury. Cell Metab 2016;23:254‐263. [DOI] [PubMed] [Google Scholar]
- 27. Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, et al. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 2018;293:9869‐9879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu Y, Fiskum G, Schubert D. Generation of reactive oxygen species by the mitochondrial electron transport chain. J Neurochem 2002;80:780‐787. [DOI] [PubMed] [Google Scholar]
- 29. Boveris A, Cadenas E, Stoppani A. Role of ubiquinone in the mitochondrial generation of hydrogen peroxide. Biochem J 1976;156:435‐444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Garcia‐Ruiz C, Colell A, Mari M, Morales A, Fernandez‐Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem 1997;272:11369‐11377. [DOI] [PubMed] [Google Scholar]
- 31. Giorgio M, Trinei M, Migliaccio E, Pelicci PG. Hydrogen peroxide: a metabolic by‐product or a common mediator of ageing signals? Nat Rev Mol Cell Biol 2007;8:722‐728. [DOI] [PubMed] [Google Scholar]
- 32. Delettre C, Yuste VJ, Moubarak RS, Bras M, Robert N, Susin SA. Identification and characterization of AIFsh2, a mitochondrial apoptosis‐inducing factor (AIF) isoform with NADH oxidase activity. J Biol Chem 2006;281:18507‐18518. [DOI] [PubMed] [Google Scholar]
- 33. Miramar MD, Costantini P, Ravagnan L, Saraiva LM, Haouzi D, Brothers G, et al. NADH oxidase activity of mitochondrial apoptosis‐inducing factor. J Biol Chem 2001;276:16391‐16398. [DOI] [PubMed] [Google Scholar]
- 34. Larosche I, Choumar A, Fromenty B, Lettéron P, Abbey‐Toby A, Van Remmen H. Prolonged ethanol administration depletes mitochondrial DNA in MnSOD‐overexpressing transgenic mice, but not in their wild type littermates. Toxicol Appl Pharmacol 2009;234:326‐338. [DOI] [PubMed] [Google Scholar]
- 35. Mari M, Colell A, Morales A, VonMontfort C, Garcia‐Ruiz,Fernández‐Checa JC. Redox control of liver function in health and disease. Antioxid Redox Signal 1010;12:1295‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. García‐Ruiz I, Fernández‐Moreira D, Solís‐Muñoz P, Rodríguez‐Juan C, Díaz‐Sanjuán T, Muñoz‐Yagüe T, et al. Mitochondrial complex I subunits are decreased in murine nonalcoholic fatty liver disease: implication of peroxynitrite. J Proteom Res 2010;9:2450‐2459. [DOI] [PubMed] [Google Scholar]
- 37. Marí M, Morales A, Colell A, García‐Ruiz C, Fernández‐Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal 2009;11:2685‐2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Marí M, Morales A, Colell A, García‐Ruiz C, Kaplowitz N, Fernández‐Checa JC. Mitochondrial glutathione: features, regulation and role in disease. Biochim Biophys Acta 2013;1830:3317‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ribas V, García‐Ruiz C, Fernández‐Checa JC. Glutathione and mitochondria. Frontiers Pharmacol 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cole‐Ezea P, Swan D, Shanley D, Hesketh J. Glutathione peroxidase 4 has a major role in protecting mitochondria from oxidative damage and maintaining oxidative phosphorylation complexes in gut epithelias cells. Free Rad Biol Med 121;53:488‐497. [DOI] [PubMed] [Google Scholar]
- 41. Chang TS, Cho CS, Park S, Yu S, Kang SW, Rhee SG. Peroxoredoxin III, a mitochondrion specific peroxidase, regulates apoptotic signaling by mitochondria. J Biol Chem 2004;279:41975‐41984. [DOI] [PubMed] [Google Scholar]
- 42. Zhang H, Go YM, Jones DP. Mitochondrial thioredoxin‐2/peroxiredoxin‐3 system functions in parallel with mitochondrial GSH system in protection against oxidative stress. Arch Biochem Biophys 2007;465:119‐126. [DOI] [PubMed] [Google Scholar]
- 43. McCommis KS, McGee AM, Laughlin MH, Bowles DK, Baines CP. Hypercholesterolemia increases mitochondrial oxidative stress and enhances the MPT response in the porcine myocardium: beneficial effects of chronic exercise. Am J Physiol Regul Integr Comp Physiol 2011;301:R1250‐R1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lippman SM, Klein EA, Goodman PJ, Lucia MS, Thompson IM, Ford LG, et al. Effect of selenium and vitamin E on risk of prostate cancer and other cancers: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009;301:39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsavachidou D, McDonnell TJ, Wen S, Wang X, Vakar‐Lopez F, Pisters LL, et al. Selenium vitamin E: cell type‐ and intervention‐specific tissue effects in prostate cancer. J Natl Cancer Inst 2009;101:306‐320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Phillips M, Curtis H, Portmann B, Donaldson N, Bomford A, O’Grady J. Antioxidants versus corticosteroids in the treatment of severe alcoholic hepatitis—a randomised clinical trial. J Hepatol 2006;44:784‐790. [DOI] [PubMed] [Google Scholar]
- 47. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. New Engl J Med 2010;362:1675‐1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sies H. Oxidative stress: a concept in redox biology and medicine. Redox Biol 2015;4:180‐183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Jones DP, Carlson JL, Mody VC, Cai J, Lynn MJ, Sternberg P. Redox state of glutathione in human plasma. Free Radic Biol Med 2000;28:625‐663. [DOI] [PubMed] [Google Scholar]
- 50. Block CA, Ausubel FM. Paraquat‐mediated selection for mutations in the manganeso‐superoxide dismutase gene sodA. J Bacteriol 1986;168:795‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Orr WC, Sohal RS. Effects of Cu, Zn superoxide dismutase overexpression of life span and resistance to oxidative stress in transgenic Drosophila melanogaster. Arch Biochem Biophys 1993;301:34‐40. [DOI] [PubMed] [Google Scholar]
- 52. De Haan JB, Francesca C, Ianello R, Bladier C, Kelner MJ, Kola I. Elevation in the ratio of Cu/Zn‐superoxide dismutase to glutathione peroxidase activity induces features of cellular senescence and this effect is mediated by hydrogen peroxide. Hum Mol Genet 1996;5:283‐291. [DOI] [PubMed] [Google Scholar]
- 53. Caldwell SH, Swerdlow RH, Khan EM, Iezzoni JC, Hespenheide EE, Parks JK, et al. Mitochondrial abnormalities in non‐alcoholic steatohepatitis. J Hepatol 1999;31:430‐434. [DOI] [PubMed] [Google Scholar]
- 54. Sanyal AJ, Campbell‐Sargent C, Mirshahi F, Rizzo WB, Contos MJ, Sterling RK, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001;120:1183‐1192. [DOI] [PubMed] [Google Scholar]
- 55. Fromenty B, Grimbert S, Mansouri A, Beaugrand M, Erlinger S, Rötig A, et al. Hepatic mitochondrial DNA deletion in alcoholics: association with microvesicular steatosis. Gastroenterology 1995;108:193‐200. [DOI] [PubMed] [Google Scholar]
- 56. Garcia‐Ruiz I, Rodríguez‐Juan C, Díaz‐Sanjuan T, del Hoyo P, Colina F, Muñoz‐Yagüe T, et al. Uric acid and anti‐TNF antibody improve mitochondrial dysfunction in ob/ob mice. Hepatology 2006;44:581‐591. [DOI] [PubMed] [Google Scholar]
- 57. Serviddio G, Giudetti AM, Bellanti F, Priore P, Rollo T, Tamborra R, et al. Oxidation of hepatic carnitine palmitoyl transferase‐I (CPT‐I) impairs fatty acid beta‐oxidation in rats fed a methionine‐choline deficient diet. PLoS ONE 2011;6:e24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Orellana‐Gavaldà JM, Herrero L, Malandrino MI, Pañeda A, Sol Rodríguez‐Peña M, Petry H, et al. Molecular therapy for obesity and diabetes based on a long‐term increase in hepatic fatty‐acid oxidation. Hepatology 2011;53:821‐832. [DOI] [PubMed] [Google Scholar]
- 59. Martin J, Maurhofer O, Bellance N, Benard G, Graber F, Hahn D, et al. Disruption of the histidine triad nucleotide‐binding hint2 gene in mice affects glycemic control and mitochondrial function. Hepatology 2013;57:2037‐2048. [DOI] [PubMed] [Google Scholar]
- 60. Pospisilik JA, Knauf C, Joza N, Benit P, Orthofer M, Cani PD, et al. Targeted deletion of AIF decreases mitochondrial oxidative phosphorylation and protects from obesity and diabetes. Cell 2007;131:476‐491. [DOI] [PubMed] [Google Scholar]
- 61. Wredenberg A, Wibom R, Wilhelmsson H, Graff C, Wiener HH, Burden SJ, et al. Increased mitochondrial mass in mitochondrial myopathy mice. Proc Natl Acad Sci 2002;99:15066‐15071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Vernochet C, Mourier A, Bezy O, Macotela Y, Boucher J, Rardin MJ, et al. Adipose‐specific deletion of TFAM increases mitochondrial oxidation and protects mice against obesity and insulin resistance. Cell Metab 2012;16:765‐776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bruguera M, Bertran A, Bombi JA, Rodes J. Giant mitochondria in hepatocytes: a diagnostic hint for alcoholic liver disease. Gastroenterology 1977;73:1383‐1387. [PubMed] [Google Scholar]
- 64. Chedid A, Mendenhall CL, Tosch T, Chen T, Rabin L, Garcia‐Pont P, et al. Significance of megamitochondria in alcoholic liver disease. Gastroenterology 1986;90:1858‐1864. [DOI] [PubMed] [Google Scholar]
- 65. Witschi A, Mossi S, Meyer B, Junker E, Lauterburg BH. Mitochondrial function reflected by the decarboxylation of [13C]ketoisocaproate is impaired in alcoholics. Alcohol Clin Exp Res 1994;18:951‐955. [DOI] [PubMed] [Google Scholar]
- 66. Jenkins WJ, Peters TJ. Mitochondrial enzyme activities in liver biopsies from patients with alcoholic liver disease. Gut 1978;19:341‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bernstein JD, Penniall R. Effects of chronic ethanol treatment upon rat liver mitochondria. Biochem Pharmacol 1978;27:2337‐2342. [DOI] [PubMed] [Google Scholar]
- 68. Cederbaum AI, Lieber CS, Rubin E. Effects of chronic ethanol treatment of mitochondrial functions damage to coupling site I. Arch Biochem Biophys 1974;165:560‐569. [DOI] [PubMed] [Google Scholar]
- 69. Spach PI, Cunningham CC. Control of state 3 respiration in liver mitochondria from rats subjected to chronic ethanol consumption. Biochim Biophys Acta 1987;894:460‐467. [DOI] [PubMed] [Google Scholar]
- 70. Chacko BK, Srivastava A, Johnson MS, Benavides GA, Chang MJ, Ye Y, et al. Mitochondria‐targeted ubiquinone (MitoQ) decreases ethanol‐dependent micro and macro hepatosteatosis. Hepatology 2011;54:153‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cunningham CC, Coleman WB, Spach PI. The effects of chronic ethanol consumption on hepatic mitochondrial energy metabolism. Alcohol Alcohol 1990;25:127‐136. [DOI] [PubMed] [Google Scholar]
- 72. Venkatraman A, Landar A, Davis AJ, Chamlee L, Sanderson T, Kim H, et al. Modification of the mitochondrial proteome in response to the stress of ethanol‐dependent hepatotoxicity. J Biol Chem 2004;279:22092‐22101. [DOI] [PubMed] [Google Scholar]
- 73. Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, et al. The role of iNOS in alcohol‐dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology 2004;40:565‐573. [DOI] [PubMed] [Google Scholar]
- 74. Zhang X, Tachibana S, Wang H, Hisada M, Williams GM, Gao B, et al. Interleukin‐6 is an important mediator for mitochondrial DNA repair after alcoholic liver injury in mice. Hepatology 2010;52:2137‐2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Han D, Ybanez MD, Johnson HS, McDonald JN, Mesropyan L, Sancheti H, et al. Dynamic adaptation of liver mitochondria to chronic alcohol feeding in mice: biogenesis, remodeling, and functional alterations. J Biol Chem 2012;287:42165‐42179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fucho R, Vallejo C, Alarcon‐Vila C, Garcia‐Ruiz C, Fernandez‐Checa JC. Ethanol feeding preferentially increases Steroidogenic acute regulatory protein, mitochondrial respiration and oxidative stress in perivenous mouse hepatocytes. J Hepatol 2017;66:S115. [Google Scholar]
- 77. Han D, Johnson HS, Rao MP, Martin G, Sancheti H, Silkwood KH, et al. Mitochondrial remodeling in the liver following chronic alcohol feeding to rats. Free Radic Biol Med 2017;102:100‐110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Insausti N, Alarcon‐Vila C, Solsona‐Vilarrasa E, Fucho R, Garcia‐Ruiz C, Fernandez‐Checa JC. Chronic alcohol feeding increases mitochondrial respiration in FRG mice with humanized liver. Hepatology 2018;68:799A. [Google Scholar]
- 79. Laurent A, Nicco C, Tran Van Nhieu J, Borderie D, Chéreau C, Conti F, et al. Pivotal role of superoxide anion and beneficial effect of antioxidant molecules in murine steatohepatitis. Hepatology 2004;39:1277‐1285. [DOI] [PubMed] [Google Scholar]
- 80. Yang S, Zhu H, Li Y, Lin H, Gabrielson K, Trush MA, et al. Mitochondrial adaptations to obesity‐related oxidant stress. Arch Biochem Biophys 2000;378:259‐268. [DOI] [PubMed] [Google Scholar]
- 81. von Montfort C, Matias N, Fernandez A, Fucho R, Conde de la Rosa L. Martinez‐Chantar ML. Mitochondrial GSH determines the toxic or therapeutic potential of superoxide scavenging in steatohepatitis. J Hepatol 2012;57:852‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Demicheli V, Quijano C, Alvarez B, Radi R. Inactivation and nitration of human superoxide dismutase (SOD) by fluxes of nitric oxide and superoxide. Free Rad Biol Med 2007;42:1359‐1368. [DOI] [PubMed] [Google Scholar]
- 83. MacMillan‐Crow LA, Cruthirds DL. Invited review: manganese superoxide dismutase in disease. Free Rad Res 2011;34:325‐336. [DOI] [PubMed] [Google Scholar]
- 84. Nobili V, Donati B, Panera N, Vongsakulyanon A, Alisi A, Dallapiccola B, et al. A 4‐polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2014;58:632‐636. [DOI] [PubMed] [Google Scholar]
- 85. Koch OR, De Leo ME, Borrello S, Palombini G, Galeotti T. Ethanol treatment up‐regulates the expression of mitochondrial manganese superoxide dismutase in rat liver. Biochem Biophys Res Commun 1994;201:1356‐1365. [DOI] [PubMed] [Google Scholar]
- 86. Wheeler MD, Nakagami M, Bradford BU, Uesugi T, Mason RP, Connor HD, et al. Overexpression of manganese superoxide dismutase prevents alcohol‐induced liver injury in the rat. J Biol Chem 2001;276:36664‐36672. [DOI] [PubMed] [Google Scholar]
- 87. Nanji AA, Griniuviene B, Sadrzadeh SM, Levitsky S, McCully JD. Effect of type of dietary fat and ethanol on antioxidant enzyme mRNA induction in rat liver. J Lipid Res 1995;36:736‐744. [PubMed] [Google Scholar]
- 88. Assiri MA, Roy SR, Harris PS, Ali H, Liang Y, Shearn CT, et al. Chronic ethanol metabolism inhibits hepatic mitochondrial superoxide dismutase via lysine acetylation. Alcohol Clin Exp Res 2017;41:1705‐1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Fritz KS, Galligan JJ, Smathers RL, Roede JR, Shearn CT, Reigan P, et al. 4‐Hydroxy‐nonenal inhibits SIRT3 via thiol‐specific modification. Chem Res Toxicol 2011;24:651‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Degoul F, Sutton A, Mansouri A, Cepanec C, Degott C, Fromenty B, et al. Homozygosity for alanine in the mitochondrial targeting sequence of superoxide dismutase and risk for severe alcoholic liver disease. Gastroenterology 2001;120:1468‐1474. [DOI] [PubMed] [Google Scholar]
- 91. Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, et al. Mitochondrial free cholesterol loading sensitizes to TNF‐ and Fas‐mediated steatohepatitis. Cell Metab 2006;4:185‐198. [DOI] [PubMed] [Google Scholar]
- 92. Ibdah JA, Perlegas P, Zhao Y, Angdisen J, Borgerink H, Shadoan MK, et al. Mice heterozygous for a defect in mitochondrial trifunctional protein develop hepatic steatosis and insulin resistance. Gastroenterology 2005;128:1381‐1390. [DOI] [PubMed] [Google Scholar]
- 93. Josekutty J, Iqbal J, Iwawaki T, Kohno K, Hussain MM. MTP inhibition induces ER stress and increases gene transcription via Ire1α/cJun to enhance plasma ALT/AST. J Biol Chem 2013;288:14372‐14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Caballero F, Fernández A, Matías N, Martínez L, Fucho R, Elena M, et al. Specific contribution of methionine and choline in nutritional nonalcoholic steatohepatitis: impact on mitochondrial S‐adenosyl‐L‐methionine and glutathione. J Biol Chem 2010;285:18528‐18536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Caballero F, Fernández A, De Lacy AM, Fernández‐Checa JC, Caballería J, García‐Ruiz C. Enhanced free cholesterol, SREBP‐2 and StAR expression in human NASH. J Hepatol 2009;50:789‐796. [DOI] [PubMed] [Google Scholar]
- 96. Anuka E, Gal M, Stocco DM, Orly J. Expression and roles of steroidogenic acute regulatory (StAR) protein in ‘non‐classical’, extra‐adrenal and extra‐gonadal cells and tissues. Mol Cell Endocrinol 2013;371:47‐61. [DOI] [PubMed] [Google Scholar]
- 97. Serviddio G, Bellanti F, Tamborra R, Rollo T, Capitanio N, Romano AD, et al. Uncoupling protein‐2 (UCP2) induces mitochondrial proton leak and increases susceptibility of non‐alcoholic steatohepatitis (NASH) liver to ischaemia‐reperfusion injury. Gut 2008;57:957‐965. [DOI] [PubMed] [Google Scholar]
- 98. Hirano T, Kaplowitz N, Tsukamoto H, Kamimura S, Fernandez‐Checa JC. Hepatic mitochondrial glutathione depletion and progression of experimental alcoholic liver disease in rats. Hepatology 1992;16:1423‐1427. [DOI] [PubMed] [Google Scholar]
- 99. Garcia‐Ruiz C, Morales A, Ballesta A, Rodés J, Kaplowitz N, Fernández‐Checa JC. Effect of chronic ethanol feeding on glutathione and functional integrity of mitochondria in periportal and perivenous rat hepatocytes. J Clin Invest 1994;94:193‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Garcia‐Ruiz C, Morales A, Colell A, Ballesta A, Rodes J, Kaplowitz N, et al. Feeding S‐adenosyl‐L‐methionine attenuates both ethanol‐induced depletion of mitochondrial glutathione and mitochondrial dysfunction in peri‐portal and perivenous rat hepatocytes. Hepatology 1995;21:207‐214. [DOI] [PubMed] [Google Scholar]
- 101. Colell A, Coll O, García‐Ruiz C, París R, Tiribelli C, Kaplowitz N, et al. Tauroursodeoxycholic acid protects hepatocytes from ethanol‐fed rats against tumor necrosis factor‐induced cell death by replenishing mitochondrial glutathione. Hepatology 2001;34:964‐971. [DOI] [PubMed] [Google Scholar]
- 102. Zhao P, Kalhorn TF, Slattery JT. Selective mitochondrial glutathione depletion by ethanol enhances acetaminophen toxicity in rat liver. Hepatology 2002;36:326‐335. [DOI] [PubMed] [Google Scholar]
- 103. Zhao P, Slattery JT. Effects of ethanol dose and ethanol withdrawal on rat liver mitochondrial glutathione: implication of potentiated acetaminophen toxicity in alcoholics. Drug Metab Dispos 2002;30:1413‐1417. [DOI] [PubMed] [Google Scholar]
- 104. Varatharajalu R, Garige M, Leckey LC, Arellanes‐Robledo J, Reyes‐Gordillo K, Shah R, et al. Adverse signaling of scavenger receptor class B1 and PGC1s in alcoholic hepatosteatosis and steatohepatitis and protection by betaine in rat. Am J Pathol 2014;184:2035‐2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Guidot DM, Brown LAS. Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol‐fed rats. Alcohol Clin Exp Res 2000;24:1070‐1076. [PubMed] [Google Scholar]
- 106. Musso G, Anty R, Petta S. Antioxidant therapy and drugs interfering with lipid metabolism: could they be effective in NAFLD patients? Curr Pharm Des 2013;19:5297‐5313. [PubMed] [Google Scholar]
- 107. Sarkhy AA, Al‐Hussaini AA, Nobili V. Does vitamin E improve the outcomes of pediatric nonalcoholic fatty liver disease? A systematic review and meta‐analysis. Saudi. J Gastroenterol 2014;20:143‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lavine JE, Schwimmer JB, Van Natta ML, Molleston JP, Murray KF, Rosenthal P, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: the TONIC randomized controlled trial. JAMA 2011;305:1659‐1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Stewart S, Prince M, Bassendine M, Hudson M, James O, Jones D, et al. A randomized trial of antioxidant therapy alone or with corticosteroids in acute alcoholic hepatitis. J Hepatol 2007;47:277‐283. [DOI] [PubMed] [Google Scholar]
- 110. Nguyen‐Khac E, Thevenot T, Piquet MA, Benferhat S, Goria O, Chatelain D, et al. Glucocorticoids plus N‐acetylcysteine in severe alcoholic hepatitis. N Engl J Med 2011;365:1781‐1789. [DOI] [PubMed] [Google Scholar]
- 111. Tkachenko P, Maevskaya M, Pavlov A, Komkova I, Pavlov C, Ivashkin V. Prednisolone plus S‐adenosil‐l‐methionine in severe alcoholic hepatitis. Hepatol Int 2016;10:983‐987. [DOI] [PubMed] [Google Scholar]
- 112. Mato JM, Martínez‐Chantar ML, Lu SC. S‐adenosylmethionine metabolism and liver disease. Ann Hepatol 2013;12:183‐189. [PMC free article] [PubMed] [Google Scholar]
- 113. Mato JM, Camara J, Fernandez de Paz J, Caballería L, Coll S, Caballero A, et al. S‐adenosylmethionine in alcoholic liver cirrhosis: a randomized, placebo‐controlled, double‐blind, multicenter clinical trial. J Hepatol 1999;30:1081‐1089. [DOI] [PubMed] [Google Scholar]
- 114. Salvemini D, Riley DP, Cuzzocrea S. SOD mimetics are coming of age. Nat Rev Drug Discovery 2002;1:367‐374. [DOI] [PubMed] [Google Scholar]
- 115. Batinić‐Haberle I, Rebouças JS, Spasojević I. Superoxide dismutase mimics: chemistry, pharmacology, and therapeutic potential. Antioxid Redox Signal 2010;13:877‐918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Coudriet GM, Delmastro‐Greenwood MM, Previte DM, Marré ML, O’Connor EC, Novak EA, et al. Treatment with a Catalytic Superoxide Dismutase (SOD) mimetic improves liver steatosis, insulin sensitivity, and inflammation in obesity‐induced type 2 diabetes. Antioxidants 2017;4:85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Melov S, Schneider JA, Day BJ, Hinerfeld D, Coskun P, Mirra SS, et al. A novel neurological phenotype in mice lacking mitochondrial manganese superoxide dismutase. Nat Genetics 1998;18:159‐163. [DOI] [PubMed] [Google Scholar]
- 118. Mansouri A, Tarhuni A, Larosche I, Reyl‐Desmars F, Demeilliers C, Degoul F, et al. MnSOD overexpression prevents liver mitochondrial DNA depletion after an alcohol binge but worsens this effect after prolonged alcohol consumption in mice. Dig Dis 2010;28:756‐775. [DOI] [PubMed] [Google Scholar]
- 119. Larosche I, Lettéron P, Berson A, Fromenty B, Huang TT, Moreau R, et al. Hepatic mitochondrial DNA depletion after an alcohol binge in mice: probable role of peroxynitrite and modulation by manganese superoxide dismutase. J Pharmacol Exp Therap 2010;332:886‐897. [DOI] [PubMed] [Google Scholar]
- 120. Eguchi A, Yoshitomi T, Lazic M, Johnson CD, Vong LB, Wree A, et al. Redox nanoparticles as a novel treatment approach for inflammation and fibrosis associated with nonalcoholic steatohepatitis. Nanomedicine 2015;10:2697‐2708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M. Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 2007;6:280‐293. [DOI] [PubMed] [Google Scholar]