

Abstract
Nonalcoholic fatty liver disease (NAFLD) is a complex disease dictated by both genetic and environmental factors. While insulin resistance (IR) is a key pathogenic driver, two common genetic variants in patatin‐like phospholipase domain containing 3 (PNPLA3) and transmembrane 6 superfamily member 2 (TM6SF2) also impart significant risk for disease progression. Traditional approaches to NAFLD risk stratification rely on biomarkers of fibrosis, an end result of disease progression. We hypothesized that by combining genetics and a novel measurement for IR we could predict disease progression by the NAFLD activity score (NAS) and histologic presence of significant fibrosis. A total of 177 patients with biopsy‐proven NAFLD were enrolled in this cross‐sectional study. PNPLA3 I148M and TM6SF2 E167K genotypes were determined by TaqMan assays. The enhanced lipoprotein IR index (eLP‐IR) was calculated from serum biomarkers using nuclear magnetic resonance (NMR) spectroscopy. Multivariate regression models were used to study the relationships between genetics, IR, and histologic features of NAFLD. In the multivariate analysis, the eLP‐IR was strongly associated with histologic features of NAFLD activity and hepatic fibrosis (P < 0.001 to 0.02) after adjustment for potential confounders. PNPLA3 148M and TM6SF2 E167K genotypes were significantly associated with steatosis (P = 0.003 and P = 0.02, respectively). A combination of the eLP‐IR and genetic score was able to predict the presence of NAS ≥3 with an area under the receiver operating characteristic curve (AUROC) of 0.74. Adding age to this model predicted stages 3‐4 liver fibrosis with an AUROC of 0.82. Conclusion: This proof‐of‐concept study supports the hypothesis that genetics and IR are major determinants of NAFLD severity and demonstrates the feasibility of a new risk stratification paradigm using exclusively pathogenic factors.
Abbreviations
- ALT
alanine aminotransferase
- APRI
AST‐to‐platelet ratio index
- AST
aspartate aminotransferase
- AUROC
area under the receiver operator characteristic curve
- BCAA
branched‐chain amino acid
- BMI
body mass index
- CI
confidence interval
- eLP‐IR
enhanced lipoprotein insulin resistance index
- FIB‐4
fibrosis‐4
- GlycA
glycoprotein acetylation
- HDL
high‐density lipoprotein
- IR
insulin resistance
- MBOAT7
membrane bound O‐acyltransferase domain containing 7
- NAFLD
nonalcoholic fatty liver disease
- NAS
nonalcoholic fatty liver disease activity score
- NASH
nonalcoholic steatohepatitis
- NFS
nonalcoholic fatty liver disease fibrosis score
- NMR
nuclear magnetic resonance
- PNPLA3
patatin‐like phospholipase domain containing 3
- SNP
single‐nucleotide polymorphism
- TM6SF2
transmembrane 6 superfamily 2
NAFLD is the most common cause of chronic liver disease worldwide, affecting up to 25% of the global population and a third of the U.S. population.1, 2, 3 Together with this growing epidemic, morbidity from NAFLD is on the rise with a 170% increase between 2004 and 2013 in the number of cases of nonalcoholic steatohepatitis (NASH)‐related cirrhosis in patients on the liver transplant wait list. This makes NASH cirrhosis the second leading transplant indication in the United States.4
A major challenge in managing NAFLD is to identify patients that will progress to cirrhosis because a significant portion of patients are at low risk of disease progression. NASH, characterized by ballooning degeneration and lobular inflammation, is generally thought of as the progressive form of the disease with a higher risk of fibrosis progression and cirrhosis.5 IR is central to the development of NAFLD and its progression through multiple mechanisms, including increased hepatocellular de novo lipogenesis, adipose tissue lipolysis, and low‐grade systemic inflammation.6, 7 Recent studies suggest that genetics also plays a pivotal role in NAFLD pathogenesis. Several genetic polymorphisms have been identified that influence the development and progression of NAFLD, including an I148M variant in PNPLA3, an E167K variant in TM6SF2, and an rs641738 C>T variant in membrane bound O‐acyltransferase domain containing 7 (MBOAT7).8, 9, 10
Currently, the primary strategy for NAFLD risk stratification centers on the diagnosis of NASH and the staging of liver fibrosis. Although the diagnosis of NASH still requires a liver biopsy, a handful of options are available to assess fibrosis, especially advanced fibrosis (metavir stages 3‐4), including noninvasive biomarkers and special imaging modalities, such as transient elastography (TE) and magnetic resonance elastography (MRE).11 However, these tests have not been fully adopted by primary care physicians who see the majority of patients with NAFLD. They are also variably limited by cost, accessibility, and suboptimal performance, especially in capturing patients at early stages of disease. An alternative strategy for NAFLD risk stratification is to identify pathogenic risk factors that contribute to disease progression. In addition to genetics, IR measured by the homeostasis model assessment of IR (HOMA‐IR) has been shown to predict hepatic steatosis.12 The eLP‐IR is another validated means of assessing IR that takes into account circulating levels of eight lipoprotein parameters, a marker of systemic inflammation called glycoprotein acetylation (GlycA), and small‐molecule metabolites related to IR.13 Demographic factors, the NAFLD fibrosis score (NFS), and cytokeratin 18 (CK‐18) measurement of apoptosis were also included in the original model.
We hypothesized that our progressive understanding of NAFLD biology may allow us to predict NAFLD severity using non–liver‐centric factors related to pathogenesis, namely genetics and IR. In this proof‐of‐concept study, we test the hypothesis that the combination of a genetic score and IR can be predictive of NAFLD activity and that these two pathologic factors, together with the duration of disease, can predict the stage of liver fibrosis (Fig. 1).
Figure 1.
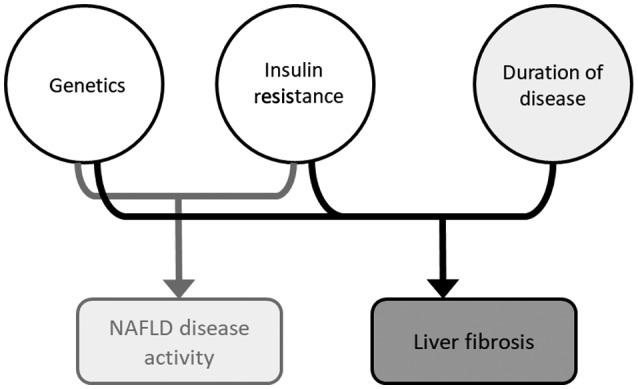
A model of pathophysiology‐based risk stratification. Based on our current understanding of NAFLD pathogenesis, a combination of IR and genetic predisposition (eLP‐IR and genetic score in our model) leads to hepatic steatosis and inflammation (i.e., NAFLD activity). The duration of disease activity (approximated by age in our model) leads to hepatic fibrosis.
Patients and Methods
Patient Population
Patients were derived from a prospective NAFLD registry at Beth Israel Deaconess Medical Center (BIDMC) that was started in 2009. Patients were enrolled after obtaining informed consent, and the diagnosis of NAFLD was confirmed by liver biopsy on all the patients in the registry. Patients with other forms of chronic liver diseases, alternative causes of fatty liver, or the consumption of alcohol greater than 20 g daily were excluded. Laboratory tests, blood collection, and medical history were performed at enrollment, and liver biopsy was performed within 3 months of the index visit. All patients (n = 177) who had undergone genetic testing and eLP‐IR were enrolled in the study. Among those, 129 had fasting blood samples that were used in the NMR measurements. The study was approved by the BIDMC institutional review board. The entire study was conducted in compliance with the ethical principles described in the 2013 revision of the Declaration of Helsinki.
Liver Biopsy
Nontargeted liver biopsy was performed under ultrasound guidance. Biopsy results were interpreted by staff pathologists specializing in hepatopathology and blinded to the genetic and eLP‐IR data. All liver biopsies were assessed and reported in a standardized fashion, including fibrosis stages (1‐4) and NAS (0‐8) calculated based on the degrees of hepatic steatosis, lobular inflammation, and ballooning degeneration.14
Calculation of eLP‐IR Scores
NMR spectra were collected using proton NMR on a 400‐MHz Vantera Clinical Analyzer as described.15 The NMR MetaboProfile analysis, which reports concentrations of lipoprotein particles and sizes as well as several metabolites (e.g., glucose, alanine, total branched‐chain amino acids [BCAAs], valine, leucine, and isoleucine), was performed using the recently developed LP4 deconvolution algorithm. The eLP‐IR was developed by taking into account changes that occur in eight NMR‐measured lipoprotein parameters, GlycA (a marker of systemic inflammation), and BCAAs, all of which are altered in patients who are insulin resistant. Lipoprotein particles, including the low‐density lipoprotein (LDL) and high‐density lipoprotein (HDL) subclasses generated from the LP4 algorithm, have been calibrated to be closer to the absolute concentrations of LDL and HDL particles than those reported by the previous LP2 and LP3 algorithms. The NMR MetaboProfile (LP4) algorithm was used to reanalyze stored NMR spectra and calculate values for LP‐IR (the predecessor to eLP‐IR), medium triglyceride‐rich lipoprotein particles, small HDL particles ranging from 7.4 to 8.7 nm, GlycA, and BCAA, using described methods.13, 16 The eLP‐IR correlates with HOMA‐IR in the Multi‐Ethnic Study of Atherosclerosis.13
DNA Extraction and Single‐Nucleotide Polymorphism Genotyping
Genomic DNA was extracted from frozen, human, whole‐blood samples with phenol chloroform and was resuspended in double‐distilled water. Single‐nucleotide polymorphism (SNP) rs738409 in the PNPLA3 gene, rs58542926 in TM6SF2, and rs641738 in MBOAT7 were genotyped by TaqMan allelic discrimination using predesigned TaqMan SNP genotyping assays. Among the three genotypes, rs738409 in PNPLA3 and rs641738 in MBOAT7 were in Hardy‐Weinberg equilibrium, while rs58542926 in TM6SF2 was not. Assuming a baseline prevalence of 0.2 in fibrosis or NASH, this study has 80% power to detect a 1.9‐fold, 2.0‐fold, and 2.0‐fold increase in the odds ratio in association with rs738409, rs58542926, and rs641738 variants, respectively, using dominant models.
NAFLD Genetic Score
Based on the associations between PNPLA3, TM6SF2, and MBOAT7 genotypes and histology in regression analysis, we created a genetic score by assigning 1 point for either heterozygotes or homozygotes of PNPLA3 and TM6SF2 minor alleles. Those individuals with neither allele were assigned a score of 0, and those with at least one allele of both PNPLA3 and TM6SF2 minor alleles were assigned a score of 2. The MBOAT7 genotype was not included in the genetic score because its association was limited only to lobular inflammation and not to other histologic features of NAFLD in our cohort.
Serum CK‐18 Measurement
CK‐18 levels were measured using the PEVIVA in vitro immunoassay M30 Apoptosense enzyme‐linked immunosorbent assay (ELISA) kit (DiaPharma, West Chester, OH) as described.17 Serum CK‐18 concentration was expressed in U/L (1 U/L = 1.24 pM recombinant protein standard).
Noninvasive Fibrosis Indices
The NFS was calculated using the following formula: −1.675 + 0.037 × age (years) + 0.094 × body mass index (BMI; kg/m2) + 1.13 × diabetes (yes = 1, no = 0) + 0.99 × aspartate aminotransferase (AST)/alanine aminotransferase (ALT) ratio – 0.013 × platelet count (×109/L) – 0.66 × albumin (g/dL), as described by Angulo et al.18 The AST‐to‐platelet ratio index (APRI) was calculated using the formula ([AST {IU/L} / 40] / platelet count [×109/L]) × 100, as described by Lin et al.19 Fibrosis‐4 (FIB‐4) was calculated using the formula (age [years] × AST [IU/L]) / (platelet count [×109/L] × square root[ALT {IU/L}]), as described by Sterling et al.20
Statistical Analysis
Univariate linear regression was performed using histologic scores of hepatic steatosis, lobular inflammation, ballooning degeneration, NAS, and fibrosis stage as continuous dependent variables. Sex, hypertension, hypercholesterolemia, diabetes, and PNPLA3 I148M, TM6SF2 E167K, and MBOAT7 rs641738 heterozygous and homozygous genotypes were treated as categorical variables; age, BMI, ALT, and eLP‐IR were treated as continuous variables. Although the analysis was adjusted for population stratification due to the lack of genome‐wide coverage of genotypes, we did not find an association of ethnicity with either fibrosis or NAS by using chi‐square tests. Multivariable linear regression was first performed to assess the association of eLP‐IR, PNPLA3 L148M, and TM6SF2 E167K with histologic outcome variables of steatosis, inflammation, ballooning degeneration scores, NAS, and fibrosis stage while controlling for age, sex, hypertension, diabetes, ALT, and BMI. The PNPLA3 I148M genotype was analyzed under an additive genetic model, and TM6SF2 E167K was analyzed using a dominant genetic model due to the low number of E167K homozygotes. A pathophysiologic model consisting of eLP‐IR and genetic score was then assessed by linear regression for an association with NAS. Only fasting samples were used for analyses that included eLP‐IR.
Logistic regression was used to assess the ability of the pathophysiologic model to predict the histologic outcomes of high‐grade hepatic steatosis (score 3), lobular inflammation (score ≥2), ballooning degeneration (score 2), moderate disease activity (NAS ≥3), and high‐grade disease activity (NAS ≥5) using C‐statistics of the receiver operator curve (ROC). These were compared to logistic regression models using CK‐18. A conventional model consisting of BMI, diabetes, and ALT was also created, and logistic regression was used to compare a conventional approach to a pathophysiologic approach in the prediction of NAS ≥5. Age, a proxy for the duration of disease, was added to the pathophysiologic model to predict fibrosis stage, which was compared to models using FIB‐4, APRI, NFS, and CK‐18. The comparison of the AUROC was performed using the 95% confidence interval (CI) calculated by bootstrapping. Internal validation was performed using bootstrap resampling with 1,000 iterations. This method has been shown to produce stable and low‐biased estimates of predictive accuracy with better efficiency than split‐sample modeling. We also used the cohort of patients with nonfasting samples (n = 48) as an independent validation. We recognize that there is potential misclassification bias in this validation cohort as the prandial state can influence the eLP‐IR because the very low density lipoprotein particle profile is one of the components required to calculate the eLP‐IR. All analyses were performed in Stata 11.2 (StataCorp, College Station, TX).
Results
Patient Characteristics
The clinical characteristics of 177 patients with NAFLD are summarized in Table 1 The mean age of the patient population was 55.4, 40.1% were female patients, and 13.6% were Hispanic. There was a high prevalence of metabolic syndrome, with 45.7%, 41.8%, and 28.8% carrying a diagnosis of hypertension, dyslipidemia, and diabetes, respectively. The average BMI was 34.1. The prevalence of PNPLA3 I148M genotypes was 37.3% CC, 41.2% GC, and 21.4% GG. The prevalence of TM6SF2 E167K genotypes was 79.1% CC, 19.2% CT, and 1.1% TT. The majority of patients had at least one minor allele of either PNPLA3 or TM6SF2 (60.5%) corresponding to a genetic score of 1, and 11.3% of patients had both minor alleles (genetic score of 2). The majority of patients had NASH (74.6%) with a mean NAS of 4.6. Roughly a third of patients (31.6%) had no fibrosis on biopsy, and 7.9% had cirrhosis.
Table 1.
Background Characteristics of the Study Population (n = 177)
| Characteristic | Mean ± SD or n (%) |
|---|---|
| Age (years) | 55.4 ± 12.5 |
| Female | 71 (40.1) |
| Hispanic | 24 (13.6) |
| Hypertension | 81 (45.7) |
| Hypercholesterolemia | 74 (41.8) |
| Diabetes | 51 (28.8) |
| BMI (kg/m2) | 34.1 ± 6.5 |
| ALT (IU/L) | 74.8 ± 50.6 |
| Fibrosis stage | |
| 0 | 56 (31.6) |
| 1 | 39 (22) |
| 2 | 50 (28.2) |
| 3 | 18 (10.2) |
| 4 | 14 (7.9) |
| NASH | |
| Simple steatosis | 45 (25.4) |
| NASH | 132 (74.6) |
| NAS | 4.6 ± 1.5 |
| PNPLA3 | |
| CC | 66 (37.3) |
| GC | 73 (41.2) |
| GG | 38 (21.4) |
| TM6SF2 | |
| CC | 140 (79.1) |
| CT | 34 (19.2) |
| TT | 2 (1.1) |
| Genetic score* | |
| 0 | 50 (28.2) |
| 1 | 107 (60.5) |
| 2 | 20 (11.3) |
| eLP‐IR † | 67.0 ± 22.5 |
Genetic score of 0 assigned for those carrying neither PNPLA3 nor TM6SF2 minor alleles, 1 assigned for carrying either minor allele (regardless of heterozygosity or homozygosity), and 2 assigned for carriers of both PNPLA3 and TM6SF2 minor alleles.
Calculated only using fasting samples (n = 129).
Association of Genetics and IR with Histologic Disease Activity of NAFLD
We first performed univariate analyses of the NAFLD‐related variables for associations with the histologic measurements of NAFLD activity, i.e., steatosis, lobular inflammation, ballooning degeneration, and NAS. ALT, diabetes, eLP‐IR, and PNPLA3 I148M genotype were significantly associated with NAS in univariate analyses (Table 2). eLP‐IR and ALT were significantly associated with all three histologic features of NAFLD (Supporting Table S2). While both PNPLA3 I148M and TM6SF2 E167K genotypes were significantly associated with higher scores of steatosis, only PNPLA3 I148M was significantly associated with lobular inflammation. In a multivariate analysis, only eLP‐IR remained statistically associated with NAS as well as all three histologic features. Homozygosity for PNPLA3 I148M remained a significant factor in association with NAS (Table 2). MBOAT7 T allele carriers in rs641738 were noted to have a positive association with a higher score of lobular inflammation (β coefficient, 0.23; 95% CI, 0.001‐0.46; P = 0.049) but not with steatosis or ballooning degeneration. Interestingly, age was negatively associated with steatosis in both univariate and multivariate analysis but not lobular inflammation or ballooning (Supporting Table S2). In keeping with prior studies that have not shown an association between PNPLA3 and TM6SF2 minor alleles and IR,8, 9 there was no correlation between eLP‐IR and genotype on linear regression in our cohort.
Table 2.
Multivariate Analysis of the Association of eLP‐IR and Genetics With Increased NAS
| Univariate Analysis | Multivariate Analysis | |||||
|---|---|---|---|---|---|---|
| β | 95% CI | P Value | β* | 95% CI | P Value | |
| Conventional | ||||||
| Age | −0.01 | −0.03 to 0.01 | 0.16 | −0.01 | −0.02 to 0.01 | 0.4 |
| Sex (% female) | 0.57 | 0.13 to 1.0 | 0.01 | 0.4 | 0.04 to 0.8 | 0.03 |
| BMI | 0.04 | 0.002 to 0.07 | 0.03 | 0.02 | −0.01 to 0.06 | 0.12 |
| Hypertension | 0.24 | −0.19 to 0.68 | 0.27 | 0.18 | −0.25 to 0.62 | 0.41 |
| Diabetes | 0.62 | 0.16 to 1.08 | 0.01 | 0.19 | −0.27 to 0.65 | 0.41 |
| ALT (IU/L) | 0.01 | 0.005 to 0.01 | <0.001 | 0.01 | 0.002 to 0.01 | 0.004 |
| Pathogenesis based | ||||||
| eLP‐IR | 0.01 | 0.02 to 0.03 | <0.001 | 0.02 | 0.02 to 0.03 | <0.001 |
| PNPLA3 CC | REF | REF | ||||
| CG | 0.78 | 0.3 to 1.25 | 0.002 | 0.36 | −0.07 to 0.79 | 0.1 |
| GG | 0.88 | 0.3 to 1.45 | 0.003 | 0.75 | 0.24 to 1.25 | 0.004 |
| TM6SF2 CC | REF | REF | ||||
| CT or TT | 0.29 | −0.21 to 0.79 | 0.26 | 0.37 | −0.05 to 0.79 | 0.09 |
Abbreviation: REF, reference.
Adjusted β coefficient calculated from multivariate models with all variables in this table.
Association of Genetics, IR, and Age with Hepatic Fibrosis
eLP‐IR was also strongly associated with higher stages of liver fibrosis in both univariate and multivariate analyses (Table 3). Consistent with previous studies, age, ALT, and features of the metabolic syndrome (BMI and diabetes) were also found to have a positive association with hepatic fibrosis. In comparison, PNPLA3, TM6SF2, and MBOAT7 minor alleles in separation were not significantly associated with hepatic fibrosis (Table 3).
Table 3.
Multivariate Analysis of the Association Between eLP‐IR and Genetics and Fibrosis Stage in NAFLD
| Univariate Analysis | Multivariate Analysis | |||||
|---|---|---|---|---|---|---|
| β | 95% CI | P Value | β* | 95% CI | P Value | |
| Conventional | ||||||
| Age | 0.02 | 0.004 to 0.03 | 0.01 | 0.02 | 0.001 to 0.03 | 0.04 |
| Sex (% female) | −0.04 | −0.42 to 0.33 | 0.82 | −0.36 | −0.7 to ‐0.03 | 0.03 |
| BMI | 0.04 | 0.01 to 0.07 | 0.003 | 0.04 | 0.01 to 0.06 | 0.01 |
| Hypertension | 0.82 | 0.47 to 1.16 | <0.001 | 0.45 | 0.07 to 0.83 | 0.02 |
| Diabetes | 1.04 | 0.67 to 1.41 | <0.001 | 0.65 | 0.24 to 1.05 | 0.002 |
| ALT (IU/L) | 0.005 | 0.00 to 0.01 | 0.01 | 0.01 | 0.001 to 0.01 | 0.01 |
| Pathogenesis based | ||||||
| eLP‐IR | 0.01 | 0.004 to 0.02 | 0.004 | 0.01 | 0.001 to 0.02 | 0.03 |
| PNPLA3 CC | REF | REF | ||||
| CG | 0.37 | −0.05 to 0.78 | 0.09 | 0.18 | −0.2 to 0.56 | 0.36 |
| GG | 0.34 | −0.16 to 0.84 | 0.18 | 0.37 | −0.07 to 0.82 | 0.1 |
| TM6SF2 CC | REF | REF | ||||
| CT or TT | 0.07 | −0.36 to 0.49 | 0.75 | 0.03 | −0.34 to 0.4 | 0.87 |
Adjusted β coefficient calculated from multivariate models with all variables in this table.
Abbreviation: REF, reference.
A Pathogenesis‐Based Model to Predict NAFLD Activity and Hepatic Fibrosis
We proposed a pathogenesis‐based model to predict NAFLD activity and hepatic fibrosis based on the hypothesis that genetics and IR synergistically predict NAFLD activity and that these two factors together with the duration of disease can predict liver fibrosis (Fig. 1).
To simplify the working model, a numeric genetic score was generated to capture the cumulative effect of having both PNPLA3 I148M and TM6SF2 E167K genotypes, where 1 point was assigned for the presence of either minor allele and 2 points for the presence of both. We observed only a modest dose effect of the homozygous I148M genotype compared to the heterozygous PNPLA3 genotype in predicting NAS and no effect in predicting fibrosis; as a result, those with either PNPLA3 or TM6SF2 minor alleles were assigned a score of 1 irrespective of homozygosity. There was, however, significant synergism between the PNPLA3 and TM6SF2 genotypes on both NAS and hepatic fibrosis (Table 4), with an estimated increase of 1.6 points in NAS in the presence of both PNPLA3 and TM6SF2 minor alleles compared to 0.7 NAS points in the presence of either allele. Despite the lack of associations with PNPLA3 and TM6SF2 genotypes individually with liver fibrosis, the presence of both minor alleles irrespective of homozygosity was associated with an estimated 0.9 increase in fibrosis stage. In a sensitivity analysis, we compared patients with homozygous PNPLA3 I148M to those carrying TM6SF2 E167K in addition to one allele of PNPLA3 I148M or homozygous PNPLA3 I148M; the latter two groups of patients had higher coefficients for NAS (0.9 versus 1.3 versus 2.2, respectively) (Supporting Table S3). In comparison, the genetic association with liver fibrosis was primarily driven by a synergistic effect between PNPLA3 I148M and TM6SF2 E167K, whereas PNPLA3 I148M homozygosity had no additive impact (Supporting Table S3).
Table 4.
Pathophysiology‐Based Models Predicting NAS and Fibrosis Stage
| NAS | |||
|---|---|---|---|
| β* | 95% CI | P Value | |
| eLP‐IR | 0.02 | 0.01 to 0.03 | <0.001 |
| Genetic score | |||
| 1 | 0.72 | 0.2 to 1.23 | 0.01 |
| 2 | 1.57 | 0.77 to 2.37 | <0.001 |
| Fibrosis stage | |||
| β* | 95% CI | P Value | |
| Age | 0.03 | 0.01 to 0.04 | 0.001 |
| eLP‐IR | 0.02 | 0.01 to 0.02 | <0.001 |
| Genetic score | |||
| 1 | 0.09 | −0.35 to 0.54 | 0.7 |
| 2 | 0.87 | 0.18 to 1.57 | 0.01 |
Adjusted β coefficient calculated using eLP‐IR and genetic score for NAS and using age, eLP‐IR, and genetic score for fibrosis stage.
We then investigated whether the pathophysiology‐based model can predict NAFLD disease activity and, if so, how such a model would perform. Using logistic regression of eLP‐IR and genetic score to predict NAFLD disease activity, the pathophysiologic model predicted NAS ≥3 and NAS ≥5 with an AUROC of 0.74 (bias‐corrected, 0.74) and 0.71 (bias‐corrected, 0.70), respectively (Fig. 2A,B). To assess the predictive value of this model, we selected three cutoff points for prediction of NAS ≥3: a high cutoff point (>1.717) representing patients with high IR (eLP‐IR ≥70) and at least one genetic risk allele (genetic risk score 1); and two low cutoff points representing patients with low IR (eLP‐IR ≤30) with at least one genetic risk allele (genetic risk score 1; cutoff, <0.597) and high IR with no genetic risk allele (genetic risk score 0; cutoff, <0.714). Our cohort had a prevalence of 79.1% of NAS ≥3. Using the high cutoff, our model had a positive predictive value (PPV) of 92.5% for NAS ≥3 (Supporting Table S4). Using either low cutoff points returned similar results with a negative predictive value (NPV) of 82.1% and 84.0% for low IR and low genetic risk cutoffs, respectively (Supporting Table S4). We then performed several sensitivity and validation tests. We first examined whether the genetic score and eLP‐IR had additive value in predicting NAFLD disease activity by comparing the two‐component pathophysiology‐based model to the genetic score and eLP‐IR in separation to predict NAS ≥3, NAS ≥5, and NASH. As shown in Supporting Fig. S1, the combination of the two biomarkers was superior to either alone. Next, we examined how the pathophysiology‐based model performs in comparison to conventional methods. The point estimate of AUROC was higher than that of CK‐18, a proposed marker for NASH, although this difference was not statistically significant by bootstrap estimates of 95% CI. In a comparative analysis, a model using conventional parameters consisting of BMI, presence of diabetes, and ALT in the prediction of NAS ≥5 had an AUROC of 0.70. However, when the pathophysiology‐based model parameters were included with the conventional parameters in the same logistic regression, the conventional parameters were no longer an independent predictor. We also examined whether the predictive value is reproducible and found that the pathophysiology model performed similarly with an AUROC of 0.76 (95% CI, 0.59‐0.93) in prediction of NAS ≥3 and 0.65 (95% CI, 0.44‐087) for NAS ≥5 in the cohort with nonfasting samples (n = 48).
Figure 2.
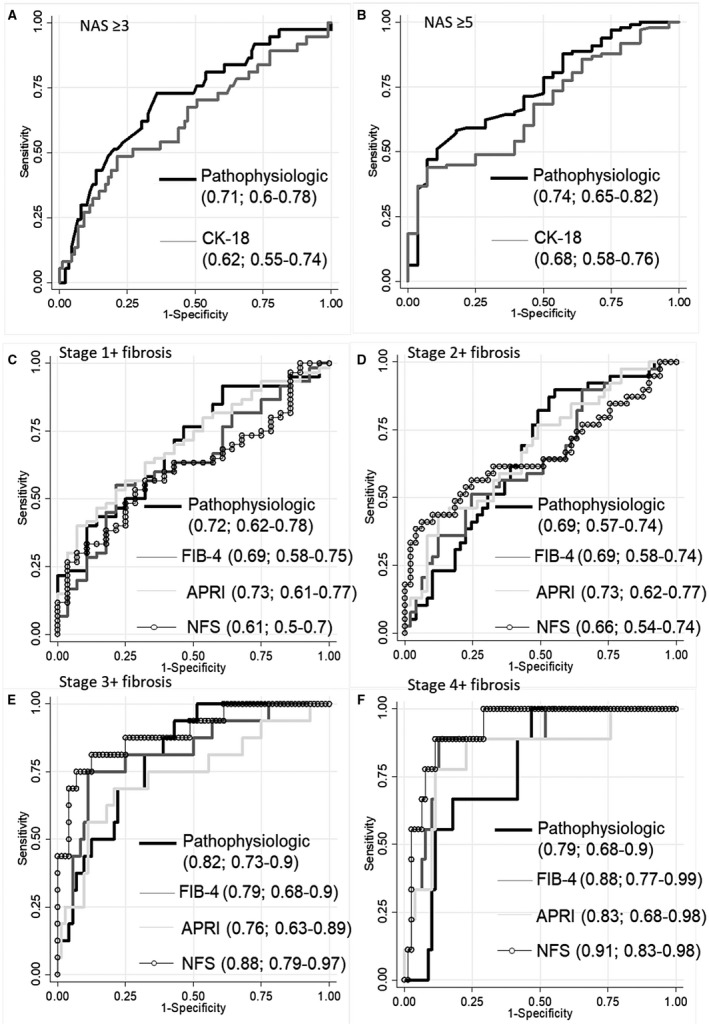
Comparison of a pathophysiology‐based model to current predictive biomarkers for NAFLD activity and fibrosis stage. ROC comparing a pathophysiologic model (eLP‐IR and genetic score) and CK‐18 in the prediction of (A) NAS ≥3 and (B) NAS ≥5, respectively. (C‐F) ROC comparing the pathophysiologic model (eLP‐IR, genetic score, and age) and conventional fibrosis indices FIB‐4, APRI, and NFS in predicting stages 1+, 2+, 3+, and 4 fibrosis. AUROC and CIs for pathophysiologic and conventional models are shown in each panel.
As we hypothesized, a model combining age, genetic score, and eLP‐IR can predict liver fibrosis, with AUROCs of 0.73 (bias‐corrected, 0.73), 0.69 (bias‐corrected, 0.68), 0.82 (bias‐corrected, 0.8), and 0.79 (bias‐corrected, 0.77) in predicting stages 1+, 2+, 3+, and 4 fibrosis, respectively (Fig. 2C‐F). In our cohort, a high cutoff point (>0.837) representing patients with high IR (eLP‐IR ≥70) and at least one genetic risk allele (genetic risk score 1) had a PPV of 79.5% for stage 1+ fibrosis (Supporting Table S5). Two low cutoff points representing patients with low IR (eLP‐IR ≤30) with at least one genetic risk allele (genetic risk score 1; cutoff, <–0.363) or high IR with no genetic risk allele (genetic risk score 0; cutoff, <0.475) had an NPV of 70.5% and 77.9%, respectively (Supporting Table S5).
This pathophysiology‐based model performed similarly to common noninvasive fibrosis scores (FIB‐4, APRI, and NFS) in the prediction of stages 1+, 2+, and 3+ fibrosis with AUROCs of 0.61‐0.73, 0.65‐0.73, and 0.76‐0.88, respectively, but statistically inferiorly to NFS in the prediction of stage 4 fibrosis (AUROC, 0.91) (Fig. 2F). We also evaluated the predictive ability of genetic score and eLP‐IR alone in the prediction of fibrosis stage (Supporting Fig. S2). The synergistic effect of these factors is especially apparent in an advanced stage (3‐4) where individually either one is a relatively poor predictor with an AUROC of 0.65 and 0.6 for eLP‐IR and genetic score, respectively. Together they are able to predict stage 3‐4 fibrosis with an AUROC of 0.82. In an external validation, this model predicted stage 1+ fibrosis with an AUROC of 0.63 (95% CI, 0.44‐0.71) in the nonfasting cohort.
Discussion
Growing knowledge of NAFLD suggests this is a complex disease most commonly driven by a combination of genetic and acquired factors related to IR. Based on this framework, it is reasonable to say that a combination of genetics and IR can effectively predict NAFLD activity and, when adding the duration of disease, can predict liver fibrosis (Fig. 1).
In risk stratifying patients with chronic diseases, two types of strategy are commonly used. One strategy is to monitor organ failure and identify patients by biomarkers or functional measurements. Examples of this strategy include the pulmonary function test for chronic obstructive pulmonary disease or the glomerular filtration rate for chronic kidney disease. The second strategy is to identify risk factors or pathogenic causes for the disease outcome. An example of this strategy includes the Framingham risk score in cardiac risk stratification. A pathogenesis‐based approach may have the ability to capture patients earlier in the disease process and be more easily implemented in a primary care setting when organ‐specific testing modalities are only used by specialists. The recent American Association for the Study of Liver Diseases guidance statement recommends against screening for NAFLD even among high‐risk populations, in part due to a lack of cost‐effective tools suitable in a primary care setting.21 At the same time, the guideline recommends clinicians maintain a high index of suspicion for advanced fibrosis. The current strategies for NAFLD risk stratification are liver centric (based on identifying the hepatic consequences of disease, i.e., NASH and liver fibrosis), mostly occur in a gastroenterology or hepatology subspecialty clinic, and often require a liver biopsy. Noninvasive assessments, such as biomarkers, TE, and MRE, similarly focus on the consequences of liver disease for risk stratification. Even prior scoring systems that incorporate genetics have relied on liver‐centric consequences, such as platelet counts, to refine their predictive ability.22, 23 In this proof‐of‐concept study, we demonstrate the feasibility of NAFLD risk stratification using risk factors rather than measurements of disease state.
A pathogenesis‐based approach is not liver centric in that it does not require liver‐specific imaging or biochemical markers of liver inflammation, such as ALT. The success of a pathogenesis‐based approach depends on the understanding of disease pathophysiology. Only three components are relevant in this model: duration of disease, genetic predisposition, and IR, a term well versed by primary care physicians in the management of diabetes and cardiovascular diseases (Fig. 1). Our data suggest that PNPLA3 I148M and TM6SF2 E167K variants are highly prevalent among patients with NAFLD. Both the genetic score and IR are strongly associated with all three histologic features of NAFLD activity and can effectively predict NAS. NAFLD activity impacts the rate of disease progression but is largely independent from the duration of disease. In our cohort, age is not associated with NAS in the multivariate model. In fact, age has a small but significant negative association with steatosis. The same genetics and IR‐based model predict hepatic fibrosis when adding age, a proxy for the duration of disease. We observed a synergy of PNPLA3 and TM6SF2 minor alleles in predicting NAFLD activity and, to a greater extent, liver fibrosis. The pathophysiology‐based model performs similarly to common fibrosis indices in the prediction of advanced (stages 1‐3) fibrosis, although this model lags behind in stage 4. This is not surprising given the reliance of these fibrosis scores on markers of portal hypertension, such as platelet counts.
The current pathogenesis‐based model predicts steatosis better than lobular inflammation and ballooning degeneration. This highlights a potential gap in our understanding of the pathophysiology of steatohepatitis. Both PNPLA3 and TM6SF2 variants and IR are associated with increased hepatic steatosis, but the precise relation with inflammation and cellular injury is not as well defined. The functions of PNPLA3 and TM6SF2 are yet to be fully elucidated, although both I148M and E167K variants may impact intrahepatic lipid handling and lipoprotein assembly.24, 25 Genetic factors that influence inflammatory response to hepatic fat may better capture inflammation and cellular injury, although a recent Mendelian randomization study by Dongiovanni et al.26 demonstrated that steatosis is the major determinant of fibrosis independent of inflammation. MBOAT7 has demonstrated a statistically significant association with hepatic steatosis, necroinflammation, and fibrosis in the Dallas Heart Study and Liver Biopsy Cross‐Sectional Cohort using a single‐SNP trait‐association study design.10 We only observed its association with lobular inflammation, which is in keeping with the lack of association to ballooning, and modest associations to steatosis and fibrosis in a previous study.10 The addition of other candidate genes associated with inflammation and fibrosis in future studies may improve the prediction of inflammation and cellular injury.
A pathophysiology‐based model for risk stratification in NAFLD has several advantages over the traditional liver‐centric method. The foremost is its simplicity. Such a model is likely to predict both NAFLD activity and liver fibrosis. No histologic variables other than NASH have been proven to predict fibrosis progression.27, 28, 29, 30 A predictor that captures not only fibrosis but also disease activity, i.e., the rate of fibrosis progression, is desperately needed to identify the “rapid progressors” who may develop cirrhosis rapidly in the absence of fibrosis or identifiable histologic features on initial biopsy.28, 29, 30 Secondly, NAFLD is a multiorgan disease with implications for diabetes and cardiovascular risk. A pathophysiology‐based model can be integrated in the risk stratification of cardiovascular disease and other complications associated with NAFLD and the metabolic syndrome. For example, PNPLA3 and TM6SF2 polymorphisms have been associated with lower cardiovascular risk in NAFLD,31, 32, 33 and eLP‐IR is predictive of incident diabetes as is its predecessor LP‐IR.34, 35, 36 The model can be implemented in a primary care setting akin to the use of lipid profile and blood pressure to predict the occurrence of diabetes and to assess cardiovascular risk, using the atherosclerotic cardiovascular disease risk calculator. It is also flexible and modifiable as our understanding of NAFLD pathophysiology expands.
Previous attempts to predict histology using genetic and liver‐centric factors have been successful at predicting either NASH or fibrosis but not both.22, 23, 37, 38 Those that have relied on aminotransferases have been more successful at predicting NASH with an AUROC of 0.73 to 0.76,22, 37, 38 similar to our model, which predicts NAS ≥5 with an AUROC of 0.74. The addition of platelets and the interferon‐λ4 genotype is successful at predicting advanced (stages 3‐4) fibrosis (AUROC, 0.81),23 which is also similar to ours (AUROC, 0.82); however, the advantage of our pathogenesis‐based model is that it predicts both. Combining our model with liver‐centric measurements (i.e., NFS) improves the performance of both, with an AUROC of 0.73, 0.74, 0.93, and 0.92 for stages ≥1‐4 fibrosis, respectively (Supporting Fig. S3). However, our model does not serve as a prediction tool yet but rather a demonstration of the feasibility and validity of our current understanding of the pathogenesis of NAFLD and NASH fibrosis.
Pathogenesis‐based risk stratification has its limitations. The performance of a pathogenesis‐based approach is determined by the fidelity of the pathogenic model. It has the advantage of capturing the majority of patients fitting in the pathogenic model, but it also risks missing patients with rarer NAFLD pathogenesis, such as abetalipoproteinemia, hypobetalipoproteinemia, glycogen storage disease, and lipodystrophy.39 The model proposed here is hypothesis generating and not a final product. The use of a biopsy‐required registry potentially has selection bias toward more advanced disease because both patients and providers are more likely to pursue liver biopsy at higher clinical concerns of disease severity. The lack of genome‐wide genotyping coverage limits the adjustment for population stratification. A pathophysiology‐based model needs to be calibrated in larger cohorts and independently validated for generalizability. Finally, a pathogenesis‐based model should be refined in the measurements of all three components (genetics, IR, and duration of disease), using longitudinal cohorts to improve its prediction of fibrosis progression and liver‐related outcomes.
In conclusion, NAFLD is a complex disease dictated in large part by both genetic and acquired factors. Our proof‐of‐concept study demonstrates the feasibility of using a strategy based solely on these pathophysiologic underpinnings to predict histologic features of NAFLD severity. A pathogenesis‐based approach shows promise as a new strategy to risk stratify patients with NAFLD in a general practice setting.
Potential conflict of interest
Dr. Connelly, Dr. Shalaurova, and Dr. Otvos are employed by LabCorp. Dr. Connelly owns stock in LabCorp. Dr. Afdhal consults and advises for Gilead and Ligand and consults for Merek, Shionogi, and Echosens; he is employed by and owns stock in Spring Bank and Trio. The other authors have nothing to report.
Supporting information
Supported in part by the National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health (grants K23DK083439 to M.L. and R01DK100425 to M.A.H.), American College of Gastroenterology Clinical Research Award (to Z.G.J.), and American Association for the Study of Liver Diseases Alan Hofmann Clinical and Translational Research Award (to Z.G.J.).
Contributor Information
Z. Gordon Jiang, Email: zgjiang@bidmc.harvard.edu.
Michelle Lai, Email: mlai@bidmc.harvard.edu.
References
Author names in bold designate shared co‐first authorship.
- 1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease‐meta‐analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016;64:73‐84. [DOI] [PubMed] [Google Scholar]
- 2. Browning JD, Szczepaniak LS, Dobbins R, Nuremberg P, Horton JD, Cohen JC, et al. Prevalence of hepatic steatosis in an urban population in the United States: impact of ethnicity. Hepatology 2004;40:1387‐1395. [DOI] [PubMed] [Google Scholar]
- 3. Lazo M, Hernaez R, Eberhardt MS, Bonekamp S, Kamel I, Guallar E, et al. Prevalence of nonalcoholic fatty liver disease in the United States: the Third National Health and Nutrition Examination Survey, 1988–1994. Am J Epidemiol 2013;178:38‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wong RJ, Aguilar M, Cheung R, Perumpail RB, Harrison SA, Younossi ZM, et al. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015;148:547‐555. [DOI] [PubMed] [Google Scholar]
- 5. Dam‐Larsen S, Franzmann M, Andersen IB, Christoffersen P, Jensen LB, Sørensen TI, et al. Long term prognosis of fatty liver: risk of chronic liver disease and death. Gut 2004;53:750‐755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Musso G, Cassader M, De Michieli F, Rosina F, Orlandi F, Gambino R. Nonalcoholic steatohepatitis versus steatosis: adipose tissue insulin resistance and dysfunctional response to fat ingestion predict liver injury and altered glucose and lipoprotein metabolism. Hepatology 2012;56:933‐942. [DOI] [PubMed] [Google Scholar]
- 7. Berk PD, Verna EC. Nonalcoholic fatty liver disease: lipids and insulin resistance. Clin Liver Dis 2016;20:245‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Romeo S, Kozlitina J, Xing C, Pertsemlidis A, Cox D, Pennacchio LA, et al. Genetic variation in PNPLA3 confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2008;40:1461‐1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kozlitina J, Smagris E, Stender S, Nordestgaard BG, Zhou HH, Tybjærg‐Hansen A, et al. Exome‐wide association study identifies a TM6SF2 variant that confers susceptibility to nonalcoholic fatty liver disease. Nat Genet 2014;46:352‐356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mancina RM, Dongiovanni P, Petta S, Pingitore P, Meroni M, Rametta R, et al. The MBOAT7‐TMC4 variant rs641738 increases risk of nonalcoholic fatty liver disease in individuals of European descent. Gastroenterology 2016;150:1219‐1230.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alkhouri N, Feldstein AE. Noninvasive diagnosis of nonalcoholic fatty liver disease: are we there yet? Metabolism 2016;65:1087‐1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Isokuortti E, Zhou Y, Peltonen M, Bugianesi E, Clement K, Bonnefont‐Rousselot D, et al. Use of HOMA‐IR to diagnose non‐alcoholic fatty liver disease: a population‐based and inter‐laboratory study. Diabetologia 2017;60:1873‐1882. [DOI] [PubMed] [Google Scholar]
- 13. Shalaurova I, Connelly MA, Garvey WT, Otvos JD. Lipoprotein insulin resistance index: a lipoprotein particle‐derived measure of insulin resistance. Metab Syndr Relat Disord 2014;12:422‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, et al. Nonalcoholic Steatohepatitis Clinical Research Network. Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313‐1321. [DOI] [PubMed] [Google Scholar]
- 15. Jeyarajah E, Cromwell W, Otvos JD. Lipoprotein particle analysis by nuclear magnetic resonance spectroscopy. Clin Lab Med 2006;26:847‐870. [DOI] [PubMed] [Google Scholar]
- 16. Otvos JD, Shalaurova I, Wolak‐Dinsmore J, Connelly MA, MacKey RH, Stein JH, et al. GlycA: a composite nuclear magnetic resonance biomarker of systemic inflammation. Clin Chem 2015;61:714‐723. [DOI] [PubMed] [Google Scholar]
- 17. Feldstein AE, Wieckowska A, Lopez AR, Liu YC, Zein NN, Mccullough AJ. Cytokeratin‐18 fragment levels as noninvasive biomarkers for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 2009;50:1072‐1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Angulo P, Hui JM, Marchesini G, Bugianesi E, George J, Farrell GC, et al. The NAFLD fibrosis score: a noninvasive system that identifies liver fibrosis in patients with NAFLD. Hepatology 2007;45:846‐854. [DOI] [PubMed] [Google Scholar]
- 19. Lin ZH, Xin YN, Dong QJ, Wang Q, Jiang XJ, Zhan SH, et al. Performance of the aspartate aminotransferase‐to‐platelet ratio index for the staging of hepatitis C‐related fibrosis: an updated meta‐analysis. Hepatology 2011;53:726‐736. [DOI] [PubMed] [Google Scholar]
- 20. Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al.; APRICOT Clinical Investigators . Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006;43:1317‐1325. [DOI] [PubMed] [Google Scholar]
- 21. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, et al. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018;67:328‐357. [DOI] [PubMed] [Google Scholar]
- 22. Hyysalo J, Männistö VT, Zhou Y, Arola J, Kärjä V, Leivonen M, et al. A population‐based study on the prevalence of NASH using scores validated against liver histology. J Hepatol 2014;60:839‐846. [DOI] [PubMed] [Google Scholar]
- 23. Eslam M, Hashem AM, Romero‐Gomez M, Berg T, Dore GJ, Mangia A, et al. International Liver Disease Genetics Consortium (ILDGC). FibroGENE: a gene‐based model for staging liver fibrosis. J Hepatol 2016;64:390‐398. [DOI] [PubMed] [Google Scholar]
- 24. Pirazzi C, Adiels M, Burza MA, Mancina RM, Levin M, Ståhlman M, et al. Patatin‐like phospholipase domain‐containing 3 (PNPLA3) I148M (rs738409) affects hepatic VLDL secretion in humans and in vitro. J Hepatol 2012;57:1276‐1282. [DOI] [PubMed] [Google Scholar]
- 25. Dongiovanni P, Petta S, Maglio C, Fracanzani AL, Pipitone R, Mozzi E, et al. Transmembrane 6 superfamily member 2 gene variant disentangles nonalcoholic steatohepatitis from cardiovascular disease. Hepatology 2015;61:506‐514. [DOI] [PubMed] [Google Scholar]
- 26. Dongiovanni P, Stender S, Pietrelli A, Mancina RM, Cespiati A, Petta S, et al. Causal relationship of hepatic fat with liver damage and insulin resistance in nonalcoholic fatty liver. J Intern Med 2018;283:356‐370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wong VW, Wong GL, Choi PC, Chan AW, Li MK, Chan HY, et al. Disease progression of non‐alcoholic fatty liver disease: a prospective study with paired liver biopsies at 3 years. Gut 2010;59:969‐974. [DOI] [PubMed] [Google Scholar]
- 28. Adams LA, Sanderson S, Lindor KD, Angulo P. The histological course of nonalcoholic fatty liver disease: a longitudinal study of 103 patients with sequential liver biopsies. J Hepatol 2005;42:132‐138. [DOI] [PubMed] [Google Scholar]
- 29. Pais R, Charlotte F, Fedchuk L, Bedossa P, Lebray P, Poynard T, et al. LIDO Study Group. A systematic review of follow‐up biopsies reveals disease progression in patients with non‐alcoholic fatty liver. J Hepatol 2013;59:550‐556. [DOI] [PubMed] [Google Scholar]
- 30. Singh S, Allen AM, Wang Z, Prokop LJ, Murad MH, Loomba R. Fibrosis progression in nonalcoholic fatty liver vs nonalcoholic steatohepatitis: a systematic review and meta‐analysis of paired‐biopsy studies. Clin Gastroenterol Hepatol 2015;13:643‐654.e1‐e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Simons N, Isaacs A, Koek GH, Kuč S, Schaper NC, Brouwers MCGJ. PNPLA3, TM6SF2, and MBOAT7 genotypes and coronary artery disease. Gastroenterology 2017;152:912‐913. [DOI] [PubMed] [Google Scholar]
- 32. Jiang ZG, Tapper EB, Kim M, Connelly MA, Krawczyk SA, Yee EU, et al. Genetic determinants of circulating lipoproteins in nonalcoholic fatty liver disease. J Clin Gastroenterol 2018;52:444‐451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kahali B, Liu Y‐L, Daly AK, Day CP, Anstee QM, Speliotes EK. TM6SF2: catch‐22 in the fight against nonalcoholic fatty liver disease and cardiovascular disease? Gastroenterology 2015;148:679‐684. [DOI] [PubMed] [Google Scholar]
- 34. MacKey RH, Mora S, Bertoni AG, Wassel CL, Carnethon MR, Sibley CT, et al. Lipoprotein particles and incident type 2 diabetes in the multi‐ethnic study of atherosclerosis. Diabetes Care 2015;38:628‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Harada PHN, Demler OV, Dugani SB, Akinkuolie AO, Moorthy MV, Ridker PM, et al. Lipoprotein insulin resistance score and risk of incident diabetes during extended follow‐up of 20 years: The Women’s Health Study. J Clin Lipidol 2017;11:1257‐1267.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dugani SB, Akinkuolie AO, Paynter N, Glynn RJ, Ridker PM, Mora S. Association of lipoproteins, insulin resistance, and rosuvastatin with incident type 2 diabetes mellitus: secondary analysis of a randomized clinical trial. JAMA Cardiol 2016;1:136‐145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nobili V, Donati B, Panera N, Vongsakulyanon A, Alisi A, Dallapiccola B, et al. A 4‐polymorphism risk score predicts steatohepatitis in children with nonalcoholic fatty liver disease. J Pediatr Gastroenterol Nutr 2014;58:632‐636. [DOI] [PubMed] [Google Scholar]
- 38. Leon‐Mimila P, Vega‐Badillo J, Gutierrez‐Vidal R, Villamil‐Ramirez H, Villareal‐Molina T, Larrieta‐Carrasco E, et al. A genetic risk score is associated with hepatic triglyceride content and non‐alcoholic steatohepatitis in Mexicans with morbid obesity. Exp Mol Pathol 2015;98:178‐183. [DOI] [PubMed] [Google Scholar]
- 39. Kneeman JM, Misdraji J, Corey KE. Secondary causes of nonalcoholic fatty liver disease. Therap Adv Gastroenterol 2012;5:199‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials