

Abstract
Background:
In recent years, familial pheochromocytoma (PHEO) with germline mutations in the MAX (MYC Associated Factor X) gene has been reported in a few cases. Here we investigated a 25 years old patient with multiple PHEOs associated with a non-sense germline MAX mutation. Preoperative 18F-FDOPA PET/CT revealed bilateral adrenal involvement with multiple tumors. In addition both adrenal glands were found to have diffuse or nodular adrenal medullary hyperplasia (AMH), a histopathological feature previously described as a precursor of MEN2 and SDHB-related PHEOs but not MAX.
Materials and Methods:
After bilateral adrenalectomy, different paraffin-embedded and frozen samples were analyzed for allelic imbalances of the MAX gene using allelic quantification by pyrosequencing. The expression of the protein MAX was studied by immunohistochemistry.
Results:
All PHEOs but also nodular AMH exhibited a loss of the normal allele. By contrast, the diffuse AMH did not show Loss-of-Heterozygosity. Nevertheless, immunohistochemistry demonstrated loss of protein MAX expression in all samples including diffuse hyperplasia, suggesting a causative role of MAX mutation for both PHEOs and AMH.
Conclusion:
The present case shows that both nodular and diffuse AMH belongs to the spectrum of MAX-related disease. These data support the possible continuum between nodular AMH and PHEO, expanding the qualification of micro-PHEO to nodular AMH.
Keywords: Pheochromocytoma, MYC Associated Factor X, MAX, adrenal medullary hyperplasia
Introduction
Pheochromocytomas (PHEOs) are neural crest-derived catecholamine-secreting tumors arising from the adrenal medulla. They belong to the family of pheochromocytoma/paraganglioma (PPGL). Although about 14% of apparently sporadic PHEOs are caused by germline mutations [1]. Patients with bilateral PHEOs and/or family history, carry germline mutations in most cases. Hereditary PHEO primarily develops in the context of several familial tumor syndromes: von Hippel-Lindau disease (VHL), Multiple Endocrine Neoplasia type 2 (MEN2), Neurofibromatosis 1 (NF1), respectively due to mutation in the VHL, RET or NF1 genes, or familial PPGL associated with mutations in one of the genes encoding for the succinate dehydrogenase complex (SDHA-D, collectively named SDHx). Diffuse and nodular adrenal medullary hyperplasia (AMH) has been described as a precursor of PHEO in MEN2 as well as familial PPGL due to mutation in SDHB [2,3]. In the recent years, several additional genes have been added to the list of genes related to the PPGL susceptibility. Among them, MAX (MYC Associated Factor X) was identified as a PHEO susceptibility gene by whole-exome sequencing of three unrelated patients with PHEO and a family history of the disease [4]. In these cases there is a loss of MAX protein in the tumors most often caused by Loss-of-Heterozygosity (LOH), deleting the wild-type allele (uniparental disomy, partial deletions). This data indicates that MAX acts as a tumor-suppressor gene in PPGL tumorigenesis. This brief report first described a patient with MAX germline mutation causing both bilateral PHEOs and nodular or diffuse AMH with an extensive histopathological and molecular analysis.
Report
A 25-year old asymptomatic adopted Caucasian man with no relevant background medical history had been screened in 2013 for a hereditary mutation of the MAX gene. The screening was prompted after receiving news that his biological father with a MAX-related PHEO was deceased from complications of chronic alcoholism. The patient received genetic counselling and has signed the informed consent for genetic analysis. The genetic test was positive for a non-sense heterozygous mutation in MAX (c.97C>T, p.Arg33* ; NM_002382.3 ; primers available upon request) previously reported in one study [4]. The patient was lost to follow up without being informed of his results. Several months later, a cardiologist saw the patient after an episode of chest pain, for which an ischemic pathology was ruled out. He was eventually treated with Labetalol for hypertension with irregular follow-up. The patient finally underwent investigational workup for PHEO in 2015 after complaining of intense malaise, palpitations, and sweating while driving. At this point, he was informed of his genetic test results. Twenty-four-hour urinary (7.29 μmol/24h, Upper Reference Limit, URL<2.5) and plasma (2.45 nmol/L, URL <1.29) normetanephrine were elevated. Urinary metanephrine was slightly elevated (1.79 μmol/24h, URL<1.5) while plasma metanephrine was normal (0.37 nmol/L, URL<0.92). Plasma chromogranin A was also elevated (283 µg/L, URL<100). Abdominal computed tomography (CT) revealed bilateral adrenal masses, measuring 1.6 cm in the right, and 1.8 cm in the left. 18F-FDOPA PET/CT revealed 2 uptake foci in both adrenal glands. A laparoscopic bilateral adrenalectomy was performed. Histopathological analysis revealed bilateral multiple adrenal PHEOs (11 mm on the left side and 18 and 10 mm on the right side), bilateral diffuse AMH and left nodular AMH (Figure 1, 2A). Since diffuse or nodular AMH have been described in RET and SDHB-related PHEOs, we decided to evaluate whether MAX-related tumorigenesis may follow the same process.
Figure 1. Gross pathology showing bilateral PHEO with diffuse and nodular hyperplasia.
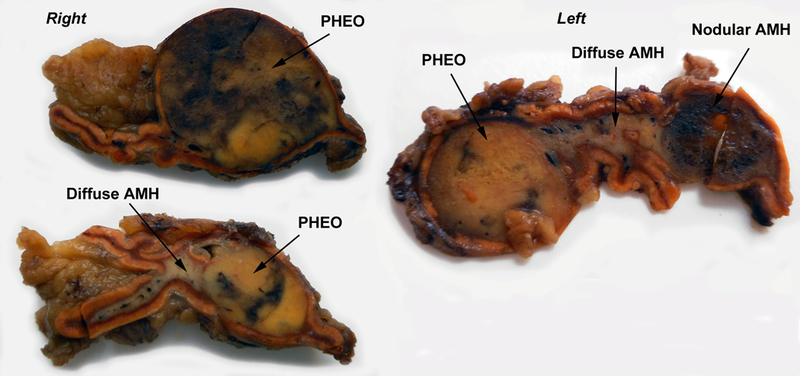
The right adrenal shows extension of gray medullary tissue into one of the alae of the gland. In the left adrenal, the medullary tissue between nodules appears diffusely expanded to more than double the thickness of the cortex.
Figure 2. Pathological findings.
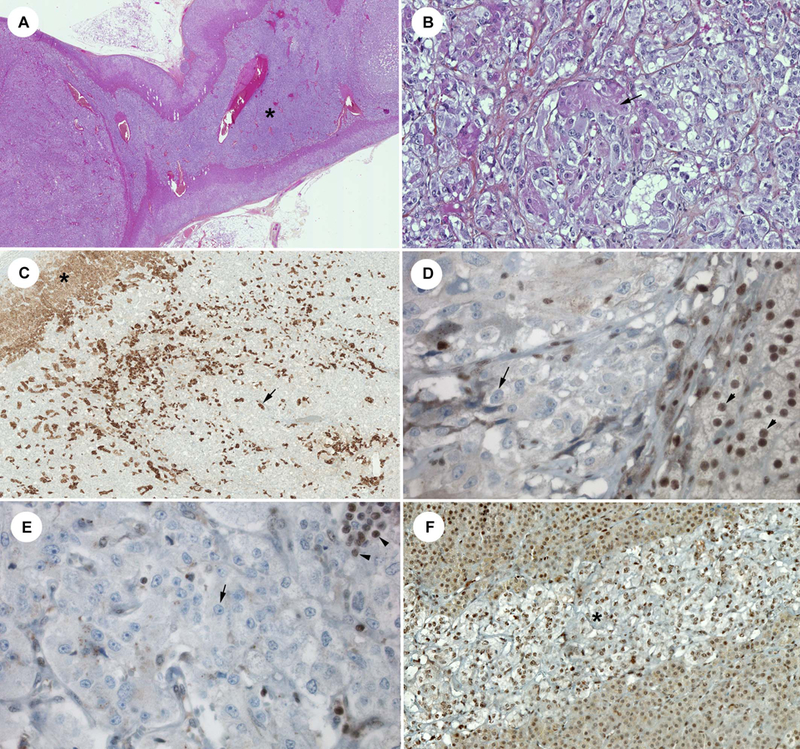
A: Hematoxylin-eosin-saffron staining (HES) showing centered over the left hyperplasia area (*), B: Left PHEO with cortical cells embedded in the tumor (arrow), C: LDL-R staining showing these cortical cells (arrow) (similar pattern than adrenal cortex : *). This finding may explained the increased % of WT alleles in this nodule, compared to other ones, D: protein MAX staining negative in the nodular AMH (arrow) (see the positive control in the cortex: arrowhead), E: protein MAX staining negative in the diffuse AMH area (arrow) (positive lymphocytes: arrowhead), F: protein MAX staining positive in an area of residual normal adrenal medulla (*).
Materials and methods:
Allelic quantification (AQ) by pyrosequencing:
Ten tissue specimens of both adrenal glands of the patient underwent MAX mutation analysis. Seven specimens were obtained from formalin-fixed paraffin-embedded (FFPE) tissues and corresponded to normal adrenal tissue (1 sample), diffuse AMH (2 samples), nodular AMH (1 sample) or PHEOs (3 samples). Additionally, three fresh frozen (FF) samples from the left or right normal adrenal gland and the nodular AMH were studied. After DNA extraction, all samples were originally analyzed for somatic allelic imbalances of the MAX gene using AQ by pyrosequencing (PyroMark MD96, Qiagen, Courtaboeuf, France).[5] Pyrosequencing is a real-time sequence-based technology useful for detection and quantification of sequence variants. After a PCR amplification step using a primer coupled with one biotinylated primer, pyrosequencing of the biotinylated DNA strand is performed on the instrument using a unique sequencing primer (PCR primers: forward 5’-TTCTAGGCTGACAAACGGGC-3’, reverse 5’-biot-CCTTCCCAATAGGTGAGTGCT-3’; sequencing primer: 5’-CGGGCTCATCATAATGCACTG-3’; design Primer3 software). Incorporation of the nucleotides by the DNApolymerase through elongation results in the release of light. A CCD camera captured and converted this signal in a pyrogram using the PyroMark™ MD software (Figure 4). For quantification of the sequence variants, the software determines an AQ in percentage, based on the respective light output level for each allelic variant. All AQ percentages given here refer to the percentage of the normal allele in the tissues.
Figure 4. Pyrogram and AQ results.
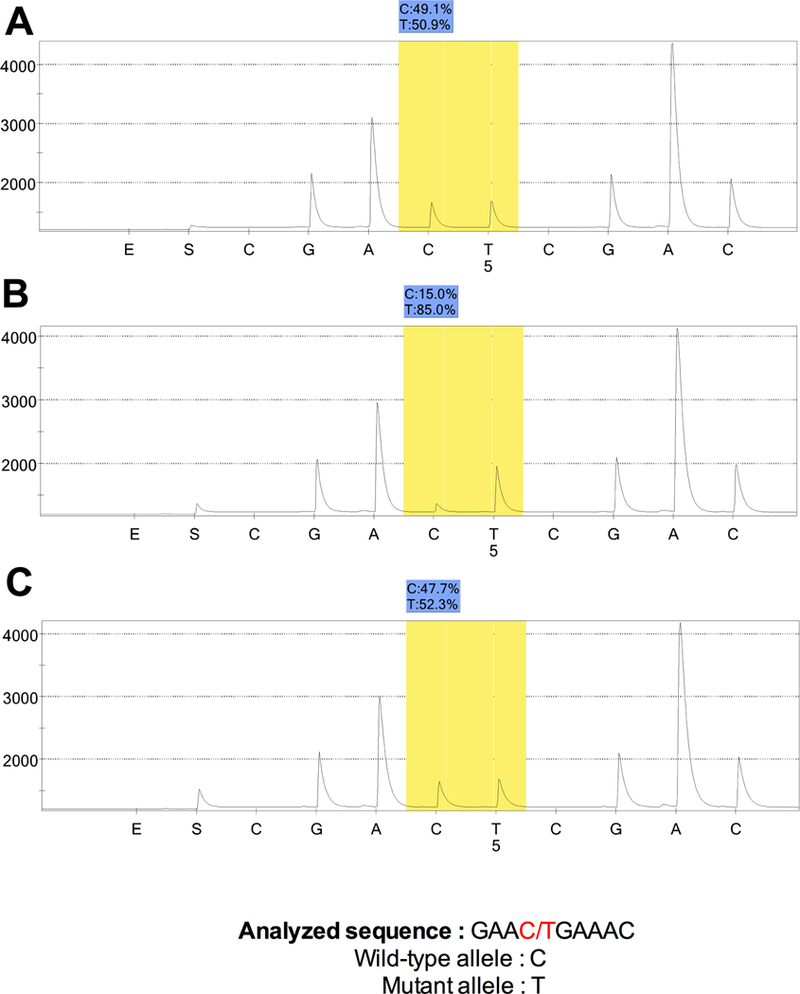
A: left normal adrenal gland, B: right PHEO, C: left diffuse AMH
Immunohistochemistry:
An immunostaining against the MAX protein was performed in the different regions of the adrenal glands with MAX polyclonal antibodies C-17 (Santa Cruz Biotechnology, Dallas, USA).
Statistical analysis:
The statistical analysis was performed with the Prism 6 Software (GraphPad, La Jolla, USA) using a Mann-Withney test.
Results:
The MAX immunostaining was negative in the PHEOs but also in the diffuse or nodular hyperplasia (Figure 2 D–E). Residual normal adrenal medulla identified in the same adrenals (Figure 2F) exhibited the same expression of the protein MAX than in 5 control adrenals.
Representative AQ are summarized in Figure 3. A heterozygous mutation profile was found in normal tissue samples (AQ of the FFPE-normal tissue 48.7% ± 3.6 (mean ± SD), AQ of the FF-normal tissue 51.7% ± 1.9). The comparison between the AQ values of normal DNA from FFPE and FF-samples confirmed the lack of impact of the FFPE-treatment on molecular allelic imbalance. As expected, all PHEOs and nodular AMH exhibited a loss of the normal allele (AQ: 20.1 to 35 %, p<0.05). The larger left PHEO was found to have a higher percentage of normal alleles compared to the both right PHEOs (respectively 35% vs. 20.7% ± 4.1) a finding that may be correlated to the presence of adrenocortical cells within PHEO (Figure 2B–C). Surprisingly, the AQ of diffuse AMH (AQ: 47.4% ± 3.1) was similar to the normal DNA from FFPE and FF samples, reflecting the absence of LOH of the MAX gene in this tissue.
Figure 3. Results of the AQ by pyrosequencing for the ten specimens.
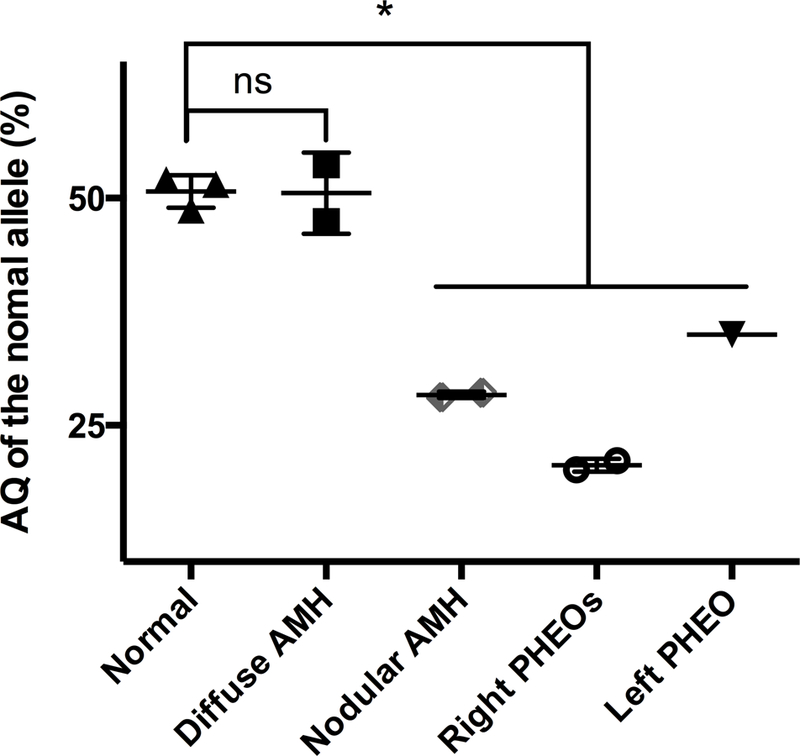
All normal specimens have an AQ for the wild-type allele consistent with the germline heterozygous MAX mutation (AQ: 50%). All PHEOs and nodular AMH are presented with a decreased AQ for the wild-type allele consistent with LOH (*p<0.05, Mann-Whitney test). The diffuse AMH profile is similar to the normal adrenal gland.
Discussion
The MAX gene has been described as a new susceptibility PPGL in adult [4] and pediatric patients [6]. Since the protein MAX antagonizes MYC activity, ablation of MAX’s transcriptional repression of MYC could lead to MYC-dependent cell transformation [7]. MYC may also regulate the antitumor immune response [8]. The non-sense heterozygous MAX mutation found in our patient leads to a premature stop codon with subsequent degradation of the mRNA transcript or a truncated protein with complete disruption of the MAX/MYC interaction (Supplemental Figure 1). The largest screening study performed on 1,694 PPGL patients without mutations in major susceptibility genes has reported pathogenic MAX mutations in 1.3% of patients with a mean age of occurrence of 32 years. All patients had developed PHEOs (13/19 either bilateral or multiple PHEOs in the same gland) with additional thoracic and abdominal PGLs in about 20% of cases. Thirty-seven percent had a family history of the disease and 10% had metastases [9]. In another study done on 153 patients with PPGLs under the age of 40 and/or with multiple tumors, a MAX mutation was detected in only one case with bilateral PHEOs [10].
Our patient had a classical clinical/biochemical phenotype with bilateral multiple PHEOs associated with predominant norepinephrine secretion. Both adrenals contained PHEOs of different sizes with diffusely hyperplasic areas. Pathological features of MAX-associated PPGLs have rarely been reported in the literature. There are typically multiple tumors in each adrenal gland with dumbbell appearance [11]. Immunohistochemistry for the protein MAX is negative in tumor cells with positive staining in adrenocortical cells. To our best knowledge, AMH has not been reported in these cases. In a recent study, the authors have shown that MAX-related PHEO can contain distinct morphological tumor regions with different gene-expression patterns [12].
In the normal adrenal gland, medullary tissue is typically located predominantly in the head of the gland. AMH is defined by prominent anatomic expansion of the medulla in transverse sections, extending into the adrenal alae and tail [13]. Involvement of the alae can be asymmetrical, as in the present case [14]. Often this expansion is both diffuse and nodular. Diffuse hyperplasia can be difficult to diagnose in subtle cases and requires morphometric analysis of the entire gland to demonstrate an overall cortex: medulla area greater than ~ 10:1. A subjective impression of diffuse hyperplasia from a single section can be misleading because the ratio typically varies from ~ 3:1 in the head of the gland to >25:1 approaching the tail [13].
However, the diagnosis of adrenal medullary nodules in the context of hyperplasia is controversial and is historically based on the diameter of the lesion. A nodule with a maximum diameter of < 1 cm is defined as nodular AMH, whereas a larger lesion is considered to a PHEO. In order to better characterize these lesions, MEN2-related AMH and PHEO were screened in a recent study for the presence of different chromosomal aberrations [2]. The authors found similar molecular aberrations in diffuse or nodular AMH and PHEOs, at similar frequencies, suggesting a molecular relationship between these entities. Consequently, they proposed to replace the term AMH with micro-PHEO to indicate adrenal medullary proliferations of less than 1 cm. This terminology is currently not formally endorsed. However, the continuum model of tumorigenesis is mostly admitted for MEN2 and has also been reported in a case of bilateral SDHB-related PHEO [3].
Apart from chromosomal losses causing LOH and second coding mutations, other potential mechanisms can account for the loss of expression of a given gene, such as promoter methylation or deep intronic variants. In the present case, the lack of LOH in the diffuse AMH suggests different molecular abnormalities than in PHEO or in nodular AMH. It has been reported that tumors carrying truncating MAX mutations can be identified on the basis of a negative immunohistochemical staining for MAX. In the present case, the absence of MAX immunostaining in diffuse and nodular AMH, regardless of the “second hit” occurring, clearly points to the involvement of the germline MAX mutation in AMH.
The most recent algorithms of genetic testing in PPGLs are based on clinical presentation, secretory phenotype and immunohistochemical characterization to guide the sequential genetic testing (in case of applying Sanger sequencing) [15]. Considering the costs of Sanger-based genetic diagnosis in PPGL (with more than 13 major susceptibility genes identified so far), it seems clear that IHC can be useful to guide the squencing of one the SDH genes or MAX (negative staining). Nowadays, in the era of panels for genetic testing, IHC has been proposed to guide the biological validation of germline or somatic variants [1,15]. This is really more important in the case of PHEO, taking into account the number of variants of unknown significance that can be found when all susceptibility genes are studied in one experiment.
In conclusion, the loss of the protein MAX expression would be sufficient to suggest the causative role of the MAX germline mutation for both diffuse or nodular AMH and a possible continuum between nodular AMH and PHEO. Moreover, these data support the distinction between nodular and diffuse AMH and the qualification of micro-PHEO for nodular AMH as previously observed in RET or SDHB-related PHEO.
Supplementary Material
Acknowledgement:
AST receicves gran support from The Pheo Para Alliance.
Source of support: the French Ministry of Health supports this work.
Footnotes
Compliance with Ethical Standards
Informed consent: Informed consent was obtained from all individual participants included in the study.
Conflict of interest declaration: The authors declare that they have no conflict of interest.
References
- 1.Curras-Freixes M, Inglada-Perez L, Mancikova V et al. Recommendations for somatic and germline genetic testing of single pheochromocytoma and paraganglioma based on findings from a series of 329 patients. J Med Genet 52: 647–656, 2015. [DOI] [PubMed] [Google Scholar]
- 2.Korpershoek E, Petri BJ, Post E et al. Adrenal medullary hyperplasia is a precursor lesion for pheochromocytoma in MEN2 syndrome. Neoplasia 16: 868–873, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Grogan RH, Pacak K, Pasche L, Huynh TT, Greco RS Bilateral adrenal medullary hyperplasia associated with an SDHB mutation. J Clin Oncol 29: e200–202, 2011. [DOI] [PubMed] [Google Scholar]
- 4.Comino-Mendez I, Gracia-Aznarez FJ, Schiavi F et al. Exome sequencing identifies MAX mutations as a cause of hereditary pheochromocytoma. Nat Genet 43: 663–667, 2011. [DOI] [PubMed] [Google Scholar]
- 5.Kwok CT, Hitchins MP Allele Quantification Pyrosequencing(R) at Designated SNP Sites to Detect Allelic Expression Imbalance and Loss-of-Heterozygosity. Methods Mol Biol 1315: 153–171, 2015. [DOI] [PubMed] [Google Scholar]
- 6.Cascon A, Inglada-Perez L, Comino-Mendez I et al. Genetics of pheochromocytoma and paraganglioma in Spanish pediatric patients. Endocr Relat Cancer 20: L1–L6, 2013. [DOI] [PubMed] [Google Scholar]
- 7.Cascon A, Robledo M MAX and MYC: a heritable breakup. Cancer Res 72: 3119–3124, 2012. [DOI] [PubMed] [Google Scholar]
- 8.Casey SC, Tong L, Li Y et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burnichon N, Cascon A, Schiavi F et al. MAX mutations cause hereditary and sporadic pheochromocytoma and paraganglioma. Clinical cancer research: an official journal of the American Association for Cancer Research 18: 2828–2837, 2012. [DOI] [PubMed] [Google Scholar]
- 10.Peczkowska M, Kowalska A, Sygut J et al. Testing new susceptibility genes in the cohort of apparently sporadic phaeochromocytoma/paraganglioma patients with clinical characteristics of hereditary syndromes. Clinical Endocrinology 79: 817–823, 2013. [DOI] [PubMed] [Google Scholar]
- 11.Kimura N A pathologist’s view: molecular profiles for diagnosing pheochromocytomas and paragangliomas. Int J Endocrine Onc: 193–200, 2015. [Google Scholar]
- 12.Flynn A, Dwight T, Harris J et al. Pheo-Type: A Diagnostic Gene-expression Assay for the Classification of Pheochromocytoma and Paraganglioma. J Clin Endocrinol Metab 101: 1034–1043, 2016. [DOI] [PubMed] [Google Scholar]
- 13.Lack EE Tumors of the Adrenal Gland and Extraadrenal Paraganglia Washington, DC: American Registry Of Pathology: 231–240, 2007. [Google Scholar]
- 14.DeLellis RA, Wolfe HJ, Gagel RF et al. Adrenal medullary hyperplasia. A morphometric analysis in patients with familial medullary thyroid carcinoma. Am J Pathol 83: 177–196, 1976. [PMC free article] [PubMed] [Google Scholar]
- 15.Favier J, Amar L, Gimenez-Roqueplo AP Paraganglioma and phaeochromocytoma: from genetics to personalized medicine. Nat Rev Endocrinol 11: 101–111, 2015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.