

ABSTRACT
Nanomaterials have gained a rapid increase in use in a variety of applications that pertain to many aspects of human life. The majority of these innovations are centered on medical applications and a range of industrial and environmental uses ranging from electronics to environmental remediation. Despite the advantages of NPs, the knowledge of their toxicological behavior and their interactions with the cellular machinery that determines cell fate is extremely limited. This review is an attempt to summarize and increase our understanding of the mechanistic basis of nanomaterial interactions with the cellular machinery that governs cell fate and activity. We review the mechanisms of NP-induced necrosis, apoptosis and autophagy and potential implications of these pathways in nanomaterial-induced outcomes.
Abbreviations: Ag, silver; CdTe, cadmium telluride; CNTs, carbon nanotubes; EC, endothelial cell; GFP, green fluorescent protein; GO, graphene oxide; GSH, glutathione; HUVECs, human umbilical vein endothelial cells; NP, nanoparticle; PEI, polyethylenimine; PVP, polyvinylpyrrolidone; QD, quantum dot; ROS, reactive oxygen species; SiO2, silicon dioxide; SPIONs, superparamagnetic iron oxide nanoparticles; SWCNT, single-walled carbon nanotubes; TiO2, titanium dioxide; USPION, ultra-small super paramagnetic iron oxide; ZnO, zinc oxide.
KEYWORDS: Apoptosis, autophagy, cell death, lysosome, nanoparticles; nanotoxicity, necrosis, stress
Introduction
The prefix ‘nano’ means dwarf in Greek, and the term ‘nanotechnology’ refers to advanced technologies on the nanometer scale that present potentials to revolutionize many aspects of human biology. Nanotechnology encompasses topics ranging from nanodevices, nanosensors and nanorobots to nanomedicines. Nanoparticles (NPs) are defined as particles with at least one dimension less than 100 nm. Nanomaterials demonstrate high surface-to-volume ratios, which renders unlimited surface modification and optical abilities [1–3]. Due to their unique physico-chemical characteristics, NPs have attracted huge attention in the field of nanomedicine. Advantages of NPs in this context include their ability to easily penetrate across cell barriers, preferential accumulation in specific organelles and cells, and theranostic (both therapy and diagnostic) properties, as well as their capacity for fine tuning [4,5]. Given these astonishing properties, NPs are under extensive investigation in order to utilize them as a carrier for genes or drugs, in imaging, for tissue engineering and even as single therapeutic agents to treat human diseases [6].
Despite these advantages, the interactions of NPs with living cells are complex and still far from fully understood [3]. NPs not only enter the cells at the site of deposition but can also reach distant organs through a variety of mechanisms [7]. NPs from many different compositions, such as metals, metal oxides, carbons, silica, and quantum dots show cytotoxic effects in different biological systems [3,8–11]. NP-induced cytotoxic effects correlate with NP composition, concentration, size, surface charge, surface area, functionalization, dispersion states and protein corona [11,12].
In this review, we discuss the interaction of NPs with cell fate pathways (necrosis/necroptosis, apoptosis, and autophagy) in order to provide a comprehensive state-of-the-art review on their eventual adverse and/or positive effects in human cells and on their therapeutic potentials in human diseases.
Cell death: why and how?
Since the advent of nanotechnology, the pathophysiological and possible cytotoxic effects of NPs are major concerns that have resulted in uncertainties about their benefits [13]. On the one hand, based on their doses and physico-chemical characteristics, NPs have the capability of producing reactive oxygen species (ROS) or otherwise initiating signaling pathways that in addition to regulating autophagy, can eventually modulate different cell fates, including necrosis, necroptosis, apoptosis, and mitotic catastrophe (Figure 1 and Tables 1–3) [4,6]. Thus, understanding nanotoxicity mechanisms is very important for the safe design of NP-based systems.
Figure 1.
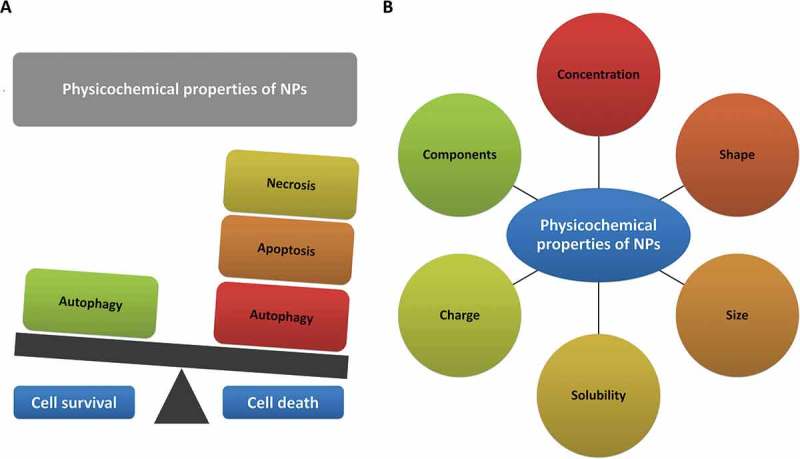
NPs, based on their doses and physico-chemical characteristics, can modulate different cell fates including necrosis, apoptosis, and autophagy. (a) Nanoparticles can induce cell death or foster cell survival. (b) Physicochemical properties of nanoparticles affect cell fate.
Table 1.
Physico-chemical characteristics of nanoparticles affect necrosis.
| Nanoparticles | Particle size | Shape | Charge | Dose | Exposure time | Cell lines/in vivo models | Major outcomes | Ref |
|---|---|---|---|---|---|---|---|---|
| Ag NPs | 10–200 nm | 1–50 µg/ml | 4 h, 24 h | L-929 fibroblast cells | Necrosis and apoptosis. | [110] | ||
| Ag NPs | 15 nm | EC50: 8.75 µg/ml | 48 h | Mouse spermatogonial stem C18–4 cells | Reduction in mitochondrial function and finally necrosis. | [111] | ||
| Ag NPs | 7–20 nm | 6.25–60 µg/ml | 24 h | Human fibrosarcoma HT-1080 and skin/carcinoma A431 cells | Reduced cell viability, oxidative stress, necrosis. | [77] | ||
| Ag NPs | 7–20 nm | 25–100 µg/ml | 24 h | Primary mouse fibroblasts and liver cells | Oxidative stress and necrosis at high doses (100 μg/mL in primary fibroblasts and 500 μg/mL in primary liver cells). | [112] | ||
| Ag NPs | 30–50 nm | Spherical, multi-facetted or slightly elongated shapes. | 2.5–15 µg/ml | 24 h | A549 human lung carcinoma epithelial-like cell line | Increase in necrosis/late apoptosis | [113] | |
| PVP-coated Ag NPs | 69 ± 3 nm | Approximately spherical, multi-facetted, or slightly elongated shapes. | 0 to 7.5 µg/ml | 0–24 h | Human leukemia THP-1 cells | ROS-induced apoptosis. | [76] | |
| Au NPs | 1.4 and 15 nm | 100 mM | 24 h, 48 h | Human cervical cancer HeLa cells | Oxidative stress, mitochondrial damage and triggered cell death by necrosis. | [82] | ||
| Au NPs | 10–50 nm | 10- and 20-nm GNPs show spherical shape whereas the 50-nm GNPs show a hexagonal shape. | 50 or 100 µl | 3 or 7 days | Male Wistar-Kyoto rats | Glutathione depletion, ROS generation, and necrosis. | [114] | |
| Au NPs | 0.8–15 nm | 30 to 56 µM | 24 h, 48 h | Connective tissue fibroblasts, epithelial cells, macrophages, and melanoma cells | Rapid cell death by necrosis. | [83] | ||
| Au NPs | 1.5 nm | Positively charged, neutral, and negatively charged. | 10 µg/ml and the neutral at 25 µg/ml. | - | Human epidermal keratinocyte HaCaT cells | Charged NPs induce cell death through apoptosis whereas neutral NPs lead to necrosis. | [84] | |
| Fullerene/C60 | 100 nm | Spindle-like shape. | 0.25 and 1 μg/ml | 24 h | Rat glioma cell line C6 and the human glioma cell line U251 | Reactive oxygen species-mediated necrotic cell damage. | [74] | |
| Fullerene/C60 | 96.3 nm | Nano-C60 (0.25 μg/ml) or C60(OH)n (250 μg/ml) | 24 h | L929 mouse fibroblast cells | Oxidative stress followed by rapid necrotic cell death. | [115] | ||
| Fullerene/C60 | 291 and 282 nm | Net zero charge on the fullerol in all solvents. | 0–50 μM | 24 h | Human lens epithelial cells (HLE B-3) | Necrosis. | [116] | |
| 1 nm | >25 μM | 72 h | Human embryonic lung fibroblasts (HELFs) | Necrosis is the dominant mechanism of death. | [117] | |||
| ZnO | 20 nm | 1, 5, 10, 40 or 80 m g/cm2 | 4 h, 24 h | Murine macrophage RAW 264.7. cells, Primary macrophages obtained from bone marrow of NCF1/p47 phox NADPH oxidase- and NFE2L2-deficient mice | Oxidative DNA damage followed with necrosis. | [107] | ||
| ZnS NPs | 80–120 nm | Symmetric, spherical. | Negative surface charge. | 25 and 50 μg/ml | 24 h | Human acute myeloid leukemia KG1a cells | Disruption of the mitochondrial membrane and ATP depletion followed by necrosis. | [118] |
| ZnO NPs | 40–100 nm | 10, 15, 20, 25, 30 μg/mL | 12 h, 24 h | SH-SY5Y human neuroblastoma | Necrosis by LOX-mediated ROS production elevation. | [119] | ||
| ZnO NPs | <150 nm | 15, 30, 100 μg/mL | 24, 48, 72, 96 h | Human epidermal keratinocyte HaCaT cells and human cervical cancer HeLa cells | Acute cytoskeletal collapse that triggers necrosis. | [109] | ||
| Realgar quantum dots (RQDs) | 5.48 nm | 10, 20, 40, 80, and 160 µg/ml | 48 h | Human endometrial cancer JEC cells | ER stress- or mitochondrial-dependent apoptosis and necrosis. | [120] | ||
| Ag(2)S QDs | 5.4 nm | Negative charge. | 6.25,12.5, 25, 50 and 100 µg/ml | 72 h | L929 mouse fibroblast cells | ROS-dependent apoptosis/necrosis. | [121] | |
| Cationic quantum dots | 16 and 9 nm | Cationic. | Up to 1.2 μM | 24 h | Human cervical cancer HeLa cells | Physical cell membrane rupture/necrosis that can lead to inflammation and subsequent cell death. | [122] | |
| Realgar quantum dots | 5.48 ± 1.09 nm | 20, 40 and 80 μg/ml | 6 h | Human hepatocellular HepG2 cells and human normal liver (L02) cells | Apoptosis and necrosis. | [123] | ||
| Si/SiO2 quantum dots | 6 and 8 nm | Spherical. | 25 and 200 μg/mL | 24–72 h | Lung fibroblast cells MRC-5 cell line | Apoptosis rate was much higher than the necrosis rate. | [124] | |
| CdTe quantum dots | 2–10 nm | Negative. | 0, 25, 50, 100, 200 nmol/L | 24 h | Human embryonic kidney cells (HEK293) and human cervical cancer HeLa cells | Mitochondrial permeability transition (MPT) and necrosis. | [108] |
Table 3.
Physico-chemical characteristics of nanoparticles affect autophagy.
| Physiochemical properties |
Dose and time of exposure |
Signaling pathways/mechanism |
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Nanomaterials | Size | Hydro-dynamic size | Shape | Charge/zeta potential | Functional groups | Dose | Time (h) |
Cell models | Upregulated | Downregulated | Autophagy flux or blockage | Ref |
| Ag NPs | 8–14 nm | Sphere | - | PVP | 8 µg/ml; dose dependent | 24 | Murine IL3-dependent pro-B cell line (Ba/F3 cells) | LC3-II, ROS | SQSTM1, p-MTOR | Flux | [314] | |
| 18 nm | 4–20 nm | - | - | - | 24 | Ovarian cancer cells | ROS, ATG5, ATG7 | ATG10, Vps30/Atg6, Atg3 | ? | [315] | ||
| 26.5 | 58.8 | Sphere | −12.9 ± 1.1 | PVP | 10 µg/ml | 24 | Cervical cancer cell line HeLa | LC3-II, PtdIns3K dependent | SQSTM1 | Flux | [316] | |
| 15 nm | 65.47 ± 1.0 | Sphere | −17.4 ± 0.92 | - | 100 µM | 24 | U-251 human glioblastoma cell line | LC3-II | SQSTM1 | Flux | [317] | |
| Au NPs | - | 43 ± 15 | Sphere | - | mPEG | 64.7 µg/ml | 24 | MCF-7 breast cancer cell line | ROS, LC3-II SQSTM1, alkalinization of lysosomes | - | Blockage | [318] |
| - | 31.57 | Sphere | −9.28 ± 1.32 | PEG/SV40 | 7.5 nM | 24 | Cervical cancer cell line HeLa | LC3-II | - | Eventually flux | [319] | |
| 5 | - | Sphere | −47.7 ± 10.6 | Citrate | 50 nM | 24 | Renal epithelial cell line HK-2 | LC3-II, BECN1, ROS | - | Eventually flux | [320] | |
| 20 | 22.1 | Sphere | −40.6 ± 2.1 | Citrate | 1 nM | 72 | Human fetal lung fibroblast (MRC-5) cells | LC3-II, ATG7, lipid hydroperoxides, ROS | - | Eventually flux | [283] | |
| Silica NPs | 5–15 nm | sphere | −32.6 to −35.1 mV | - | 50 and 100 µg/mL | 24 and 48 h | Human glioma LBC3 cell line | LC3-II, ATG5 | Eventually flux | [321] | ||
| 62 nm | 105 | sphere | 43 mV | - | 25, 50, 75 and 100 µg/mL | 24 h | Human hepatoma HepG2 Cells | LC3-II, ROS | Eventually flux | [322] | ||
| 62.1 | Sphere | - | 50 and 100 µg/ml | 24 | HUVECs | LC3-II, iNOS, CRP, IL1B, and IL6 | eNOS, NO, p-AKT, p-MTOR | Eventually flux | [323] | |||
| 57.66 | 107.94 | Sphere | −30.33 | - | 50 µg/ml | 24 | HUVECs | LC3-II, SQSTM1, NFE2L2, ROS, p-MAPK/JNK and MAPK/p38. | p-MAPK/ERK, p-PI3K, p-AKT, and p-MTOR | Blockage | [324] | |
| ZnO NPs | 20 | - | Sphere | - | - | 30 µg/ml | 12 | Human ovarian carcinoma cell line SKOV3 | LC3-II, ROS | - | Eventually flux | [242] |
| 50 | 278.8 | Sphere | −11.5 | - | 30 µg/ml | 24 | Lung carcinoma A549 cell line | LC3-II, SQSTM1 | - | Blockage | [325] | |
| 50 | 278.8 | Sphere | −11.5 | - | 2.5 μg/ml | 24 | Mouse macrophage | LC3-II, BECN1, ROS | NFE2L2, p-AKT, p-PI3K and p-MTOR | Flux | [270] | |
| 15.38 ± 1.47 nm (width) and 82.34 ± 14.23 | - | Rod shaped | - | - | 10 μg/ml | 24–72 | JB6 Cl 41-5a | BECN1, ATG5 and LC3-II, ROS | - | Eventually flux | [326] | |
| Graphene | Diameter: 56.6 ± 8.7 Height: 1.9 ± 0.8 |
- | Oval shaped | - | GQD | 200 μg/ml | 20 | Malignant glioblastoma cell line U251 | LC3-II, ROS | SQSTM1 | Flux | [280] |
| 1.0–1.2 | 350 | Sheet | - | GO | 100 μg/ml | 24 | RAW264.7 macrophage cell line | LC3-II, BECN1, IL2, IL10, IFNG and TNF | - | Eventually flux | [269] | |
| 1.5–5.5 | - | Multi-layer sheets | - | GQD | 100–200 μg/ml | 24 | Human monocytic cell line THP-1 | BECN1, LC3-II, BAX, BAD, CASP3, CASP9, p-MAPK and p-RELA, ROS | BCL2 | Eventually flux | [327] | |
| 65.5 ± 16.3 | - | Sheet | - | GO | 25 μg/mL | 12 | HUVECs | BECN1, LC3-II, | SQSTM1 | Flux | [328] | |
| Fullerene/C60 | - | - | Sphere | - | PEG, | 10 μM | 24 | Neuro-2A | LC3-II, p-AMPK | ROS, BCL2 | Eventually flux | [284] |
| - | - | Sphere | - | PTX | 10 μM | 24 | Neuro-2A | LC3-II | ROS, BCL2 | Eventually flux | [284] | |
| 7.1 | - | Sphere | C60(OH) | 100 μg/ml | 24 | HUVECs | LC3-II, LDH | cleavage of CASP3, PARP | Eventually flux | [330] | ||
| 20–100 | - | Sphere | - | - | 0.5 μg/ml | 24 | Cervical cancer cell line HeLa, primary mouse embryonic fibroblasts | LC3-II, acidic vesicles, ATG5 | - | Eventually flux | [274] | |
| Carbon nanotubes | 1–2 | - | Tube | - | carboxyl | 0.05 μg/ml | 24 | Primary glial cells | LC3-II | p-MTOR, p-ULK1, SQSTM1 | Flux | [331] |
| 1–2 | - | Tube | - | COOH | 1 mg/ml | 24 | Lung carcinoma A549 cell line | LC3-II | p-MTOR, p-AKT | Eventually flux | [227] | |
| Diameter: 10–20 nm Length: 2 μm |
- | Multi-wall | - | - | 20 mg/kg/day | 14 days | Rat hippocampal | LC3-II, BECN1 | GRIN2B/NR2B | Flux | ||
On the other hand, there is emerging evidence that cytotoxic potentials of NPs might be exploited in the treatment of multiple diseases and disorders, because dysregulation of cell death pathways is a common feature of cancer, neurodegenerative and neurological diseases [14,15] and thus cell death modulatory effects of NPs may have therapeutic value [6,16]. Apoptosis, necrosis, necroptosis and autophagy, are the focus of current research for new therapeutic pathway discovery in human diseases.
Regulated cell death pathways
Necrosis
Necrosis has long been considered to be the result of nonspecific cell injury resulting from trauma. Accordingly, it was viewed as being primarily accidental and an uncontrolled type of cell death that was not driven by specific signaling events. Various pathological conditions, such as trauma, exposure to toxins, ischemia, viral or bacterial infection and neurodegenerative disorders can induce necrotic cell death.
There is initially a sublethal phase in the pathogenesis of cell injury from which a cell can recover or, alternatively, cells may pass a ‘point of no return’ (permeabilization of the mitochondrial membranes) [17]. Morphologically, the cell displays a series of changes in the early (reversible) stage that includes ‘change of hydropic’, ‘degeneration of feathery’, ‘cloudy swelling’ or ‘vacuolar degeneration’ [9,14]. The point of no return is associated with a crucial separation between the outer and inner mitochondrial membranes followed by the irreversible loss of oxidative phosphorylation capacity [17]. Finally, in the late stages of necrosis, the cytoplasm loses contents and takes on a homogeneous eosinophilic appearance (as with ground glass), irregularities in the membrane of cytoplasmic organelles, mitochondrial swelling, increased matrix density, the formation of vacuoles, and the deposit of calcium phosphates. At the nuclear level, chromatin patterns are seen with pyknosis (chromatic condensation), karyorrhexis (nuclear fragmentation) and karyolysis (complete chromatin disruption) [18,19].
Necroptosis
Recent research has revealed the existence of caspase-independent cell death pathways that occur in response to a wide range of stimuli, and are initiated as a result of specific signaling events [20]. In this context, regulated necrosis (necroptosis) is a complex process that is not triggered by one signaling cascade but rather is caused by the interactions resulting from the activation of several signaling pathways; in turn, these various pathways may be provoked by a wide range of stimuli (Figure 2). The term necroptosis is one form of necrosis that involves death receptor activation and that can be inhibited by blocking the kinase activity of RIPK1 (receptor interacting serine/threonine kinase 1) [21,22]. In other words, necroptosis is a regulated process of cell death that involves ligand binding to TNF (tumor necrosis factor) death domain receptors (Figure 2). A common element of these receptor systems are proteins containing a RIPK homology interaction motif that recruits and activates RIPK3, leading to the activation of MLKL (mixed lineage kinase domain like pseudokinase). With regard to the TNF family members, the initiator is RIPK1; TICAM1/TRFI plays this role for TLR3 (toll like receptor 3)-TLR4, whereas for ZBP1/DAI (Z-DNA binding protein 1), there is a RIPK homology interaction motif domain within the cytosolic sensor [23]. Entry into necroptosis can occur as part of many human pathologies ranging from viral infections to neuronal excitotoxicity, which lead to neurological disorders such as Huntington, Parkinson, and Alzheimer diseases [24]. The same ligands that can activate apoptosis including TNF/TNFα, FASLG/FasL, and TNFSF10/TRAIL (TNF superfamily member 10), can induce necroptosis [25], typically through the action of RIPK1 and RIPK3 [26]. RIPK3 interacts with and modulates RIPK1 kinase activity; RIPK1 in the context of a RIPK1-RIPK3 complex (necrosome) activates RIPK3, which in turn activates MLKL to trigger necroptosis. A wide range of necrotic mediators are activated through RIP1 kinase activity. In other cases, the same mediators can be activated by the triggering of TLR3, TLR4 and NLRP33 or by DNA damage [27].
Figure 2.
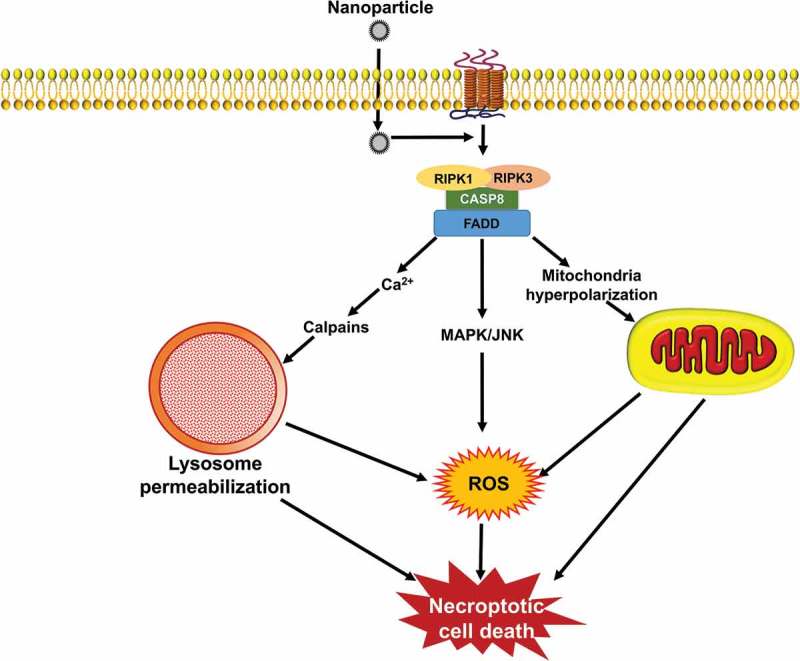
Signaling pathways that induce necroptosis. During necroptosis, different stimuli are sensed by the cell receptor apoptotic signaling pathway including, TNF/TNFα, FASLG/FasL, and TNFSF10/TRAIL that subsequently lead to the activation of RIPK1 and RIPK3 kinases. The process is followed through different signaling pathways, such as ROS generation, mitochondrial hyperpolarization, lysosomal impairment, and finally ends up with signs of necrotic cell death. NPs can induce necroptosis by stimulation of DNA damage and organelle dysfunctions as well as ROS production.
Apoptosis
The concept of apoptosis was first introduced to describe a morphologically distinct type of cell death that occurs in hepatocytes under physiological conditions [28,29]. Morphological signs of this type of cell death are described as membrane blebbing, cell shrinking, chromatin condensation and inter-nucleosomal DNA fragmentation, and formation of small vesicles named apoptotic bodies (Figure 3) [30,31]. In particular, a plasma membrane phosphatidylserine flip-flop from the inner layer to the outer side represents the signal that is detected by macrophages for engulfing apoptotic cells [29,32].
Figure 3.
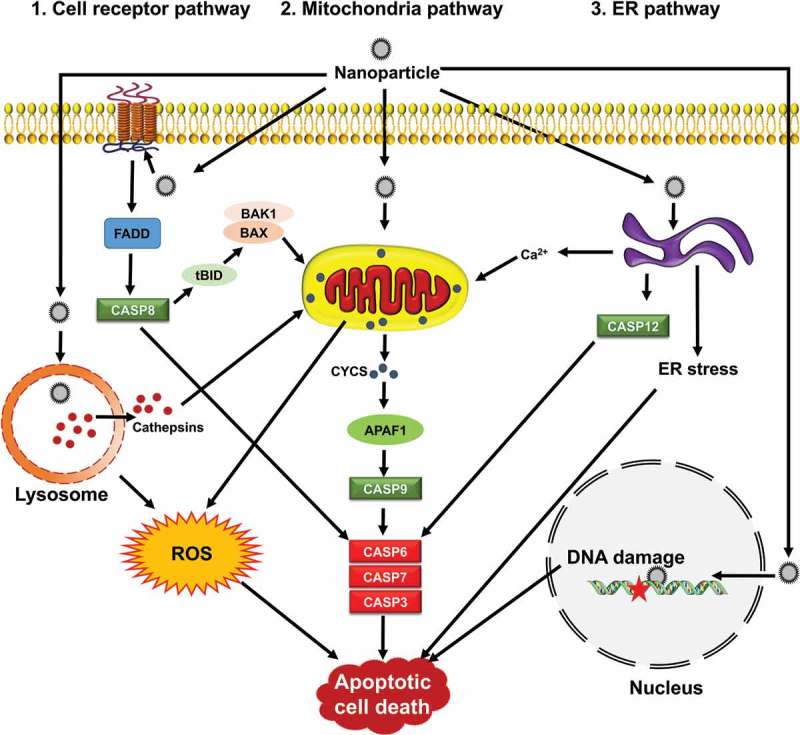
Signaling pathways by which NPs induce apoptosis. Apoptosis is mediated through 3 main apoptotic pathways including cell receptor, mitochondria and ER, that converge upon activating caspases. Mitochondria are central during apoptosis and can act as amplifiers of the cell receptor pathway through CASP8-mediated BID clevage (truncated BID, tBID) and BAX or BAK1 activation. NPs can induce apoptosis via organelle dysfunction, ROS generation, ER stress and DNA damage.
Mechanism(s) of apoptosis in mammalian cells
Apoptosis occurs by 3 distinct pathways, involving death receptors, mitochondria, or the endoplasmic reticulum (ER) (Figure 3) [29–34]. The activation of effector cysteinyl aspartate proteases (caspases), including CASP8 (caspase 8), CASP9, and CASP12 lead in turn to executor enzymes CASP3, CASP6 and CASP7 [35]. All morphological and biochemical changes related to apoptosis are mediated by these caspases.
In the death receptor or extrinsic apoptotic pathway, the superfamily of death receptor transmembrane proteins, including TNFRSF1A/TNFR1 (TNF receptor superfamily member 1A), FAS/CD95 (Fas cell surface death receptor), TNFRSF10A/TRAILR-1 (TNF receptor superfamily member 10a) and TNFRSF10B/TRAIL-R2 mediate the death signaling. Upon ligand binding, trimerization and clustering of receptors occurs to recruit adapter proteins such as FADD (Fas associated via death domain) and facilitates their binding to proCASP8 to form the death-inducing signaling complex/DISC [30–32,34]. Autocatalytic activation of CASP8 leads to cleavage and activation of CASP3, resulting in initiating the execution phase of apoptosis (Figure 3).
The intrinsic pathway of apoptosis is initiated by mitochondrial membrane permeabilization, various types of oxidative stress including hypoxia, DNA damage, and growth-factor deprivation [36]. Various members of the BCL2 superfamily of proteins can control mitochondrial integrity. These proteins are classified as pro-apoptotic and anti-apoptotic. BAX, BID, BAK1, BAD, PMAIP1/Noxa, and BBC3/Puma are members of the pro-apoptotic family, and BCL2, BCL2L1/Bcl-xl, MCL1, and BCL2A1 are members of the anti-apoptotic family proteins [37]. Pro-apoptotic BAX and BAK1 dimerize, and insert into the outer mitochondrial membrane, triggering the intrinsic pathway of apoptosis. Following mitochondrial permeabilization, CYCS (cytochrome c, somatic), is released into the cytosol, where it binds to APAF1 (apoptotic peptidase activating factor 1) that starts the formation of the apoptosome [38]. The apoptosome aids in the recruitment and activation of CASP9, which subsequently activates the downstream caspases CASP3, CASP6, and CASP7, and ultimately leads to apoptosis [30,31,34,39].
The third pathway of apoptosis is activated by various injuries from ER stressors. To cope with the ER stress, an adaptive response—the unfolded protein response (UPR)—is activated, and if the stress cannot be resolved the UPR leads to apoptosis [33,34,40]. It is thought that ER-induced apoptosis is mediated through activation of CASP12 in rodents (Figure 3). The human homolog of CASP12 was silenced during evolution and CASP4 may fulfill its function. ER-resident BCL2-family proteins regulate apoptosis through either direct or indirect (ER Ca2+ signaling) pathways [33,34].
Induction of apoptosis
It has become evident that there is complex and close crosstalk between different apoptotic pathways. For example, the extrinsic pathway can activate the mitochondrial pathway, and alternatively the cell death receptor pathway may active the intrinsic pathway of apoptosis. This amplification loop occurs by the CASP8-mediated cleavage of BID, a pro-apoptotic BCL2 family protein [41]. Also, recent evidence suggest that BBC3/Puma and PMAIP1/Noxa both contribute to ER-mediated apoptosis via crosstalk with the mitochondrial apoptotic pathway [32,40].
A wide variety of stimuli can induce apoptosis [42,43]. These include different physico-chemical stresses such as chemicals, DNA damaging agents, removal of nutrients, free radical-generating compounds (e.g., H2O2), oxygen, certain growth factors, and pro-inflammatory cytokines, in addition to normal physiological processes such as development and aging [43–49]. Some chemicals play a role in activating pro-apoptotic BCL2 members such as BAX or BAK1 [41,50–52]. ROS—including hydrogen peroxide (H2O2), free hydroxyl radicals (OH•), superoxide anions (O2•-), and nitric oxide (NO•), as well as new species produced by combinations of ROS (i.e., NO• and O2•-)—also have a major role in this process [53–57]. The particular response depends on the specific stimuli, which will in turn result in the production of different ROS species [58].
Autophagy
Autophagy definition and classification
Autophagy is primarily an adaptive process in cells, serving a cytoprotective function with the goal of survival under conditions of nutrient starvation and ATP deficiency. There are 2 fundamental types of autophagy, selective and nonselective; the selective process can be further categorized based on the cargo and mechanism of sequestration. Briefly, macroautophagy is the best characterized and perhaps the primary autophagic pathway (Figure 4). In this process, cytoplasmic cargos are sequestered through the action of a phagophore, which expands and matures to form a double-membrane vesicle (autophagosome). The autophagosome may fuse with an endosome and ultimately with a lysosome (or the vacuole in yeast or plants) to form an autolysosome. In microautophagy the cargo is directly engulfed by protrusion and/or septation of the lysosome/vacuole membrane. Macroautophagy in particular can be nonselective, but it can also be highly selective, whereas microautophagy has been characterized primarily as a selective process. The selectivity of macroautophagy depends on receptor proteins that typically bind ligands on the target, or are integral within the target membrane. A scaffold or adaptor then links the receptor with the autophagic machinery, usually via interaction with an Atg8-family protein, providing a physical connection with the phagophore. The type of sequestered cargo is used as the basis for naming the different types of selective autophagy, such as mitophagy and pexophagy for the selective autophagic degradation of mitochondria and peroxisomes, respectively. There are several models for mitophagy in mammalian cells involving different receptor proteins including BNIP3, BNIP3L/NIX, FUNDC1 and PRKN/PARK2/Parkin, depending on the stress condition and cell type [59]. There are also distinct types of selective autophagy that use protein machinery that does not overlap with that used for macroautophagy or microautophagy. One example of such a process is chaperone-mediated autophagy (CMA). During CMA, which is induced as a secondary starvation response after macroautophagy, cytosolic proteins with a particular recognition motif bind the HSPA8/HSC70 chaperone leading to unfolding (which is one significant distinction relative to macroautophagy). The unfolded substrates are translocated directly across the lysosome surface through a channel formed by LAMP2A (lysosomal associated membrane protein 2A) in a process that involves lumenal HSPA8; the substrate protein is subsequently degraded [60]. The remainder of this review focuses on macroautophagy, which we refer to as autophagy.
Figure 4.
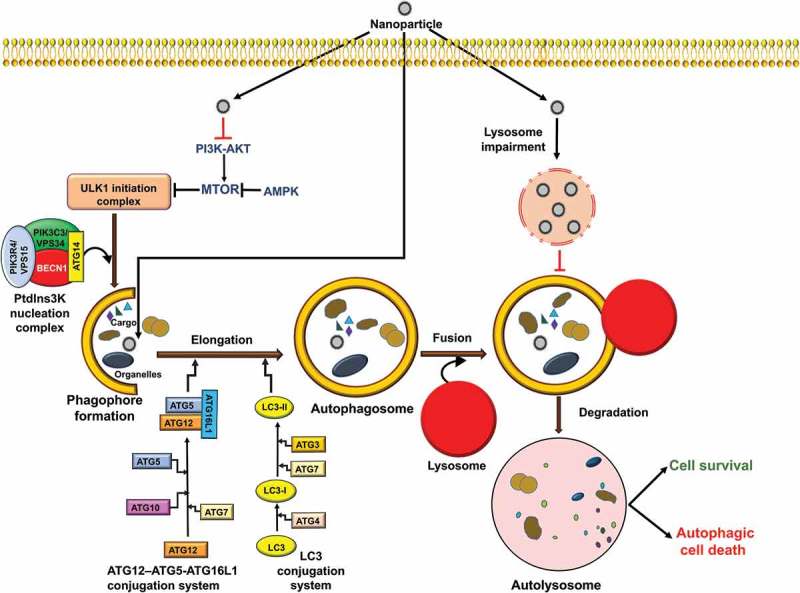
Autophagy signaling pathways induced by NPs. During autophagy, specific intracellular cargo is sequestered by phagophores. Autophagy is initiated by the activation of ULK1 and subsequent induction of a nucleation complex, including the class III PtdIns3K. Maturation and elongation require conjugation of ATG12 to ATG5, and of Atg8-family proteins including LC3 to phosphatidylethanolamine. The completed phagophore forms an autophagosome; subsequent fusion with the lysosome, releases its contents into the lumen where the cargo is degraded and the resulting macromolecules are released back into the cytoplasm. Autophagy induction may lead to cell survival or cell death, depending on the cell content or type of stimuli. NPs can induce autophagic cell death through modulation of the MTOR pathway or may trigger autophagy blockage via lysosomal impairment.
Molecular signaling pathways towards autophagy
There are various molecular signaling pathways that regulate the induction and magnitude of autophagy. Some of the principle players include MTOR (mechanistic target of rapamycin kinase), which is the primary negative regulator of autophagy, and 2 autophagy-promoting kinases, 5ʹ AMP-activated protein kinase (AMPK) and the class III phosphatidylinositol-3-kinase (PtdIns3K) (Figure 4). Based in large part on the data from yeast, a general model for autophagy involves a cascade starting with one or more phagophore assembly sites (i.e., nucleation sites for the phagophore membrane) that involve the recruitment of an initiation complex consisting of ULK1, ATG13, ATG101 and RB1CC1/FIP200 and the PtdIns3K complex (BECN1, PIK3C3/VPS34, PIK3R4/VPS15/p150, ATG14 and NRBF2) [61]. Following the nucleation stage, which is still not well understood, the cascade leads to phagophore expansion and autophagosome formation; the expansion process is relatively unique and provides tremendous flexibility with regard to cargo capacity. The expansion and maturation stage are traditionally thought to involve 2 ubiquitin-like conjugation systems comprised of ATG12–ATG5-ATG16L1, an Atg8 family protein (an LC3 or GABARAP isoform) and the processing, activating or conjugating enzymes ATG4, ATG7, ATG3 and ATG10 (Figure 4). Recent work suggests that in mammalian cells these components may be involved in a late stage of autophagy [62]. The completed autophagosome may fuse with an endosome to form an amphisome, and either compartment then fuses with a lysosome. The amphisome is a single-membrane compartment, whereas if a double-membrane autophagosome fuses directly with the lysosome the inner membrane is subsequently degraded [63–65]. The cargo is broken down by hydrolases and the products are released into the cytosol through permeases [66]. In mammalian cells transport of an autophagosome towards a lysosome occurs via microtubules and the motor protein dynein [67,68].
In other words, during starvation and other conditions that induce autophagy such as stress, infection, development and differentiation, the autophagy cascade will start in part by the activation of AMPK, which will phosphorylate components of the ULK1 kinase and PtdIns3K complexes, and the inhibition of MTOR (which otherwise phosphorylates these components at inhibitory sites); however, other mechanisms in addition to those involving AMPK and MTOR can play a role. One important link between autophagy and apoptosis is BCL2, an anti-apoptotic protein that inhibits the function of BECN1 in autophagy. Under short-term stress conditions MAPK8/JNK1 phosphorylation of BCL2 causes its dissociation from BECN1, allowing activation of the PtdIns3K and progression of autophagy; long-term phosphorylation may cause additional inactivation of BCL2 leading to apoptosis [69].
Cell fates triggered by NPs
Overall, structural and physicochemical properties of NPs (Tables 1, 2 and 3) can influence the modalities of cell death that are induced by them. Although the mechanisms of toxicity of NPs based on surface charge, crystallinity, ligand specificity, and surface chemistry, are complex, much research is underway to determine properties and functional groups that can have an influence on cellular outcomes and biological responses of NPs. In most cases, the cytotoxic potential of NPs can be dictated by their crystal structure [70], but also by shape, size, surface reactivity, composition of chemical, surface charge, presence of transition metals [71], nano-topography and surface roughness [72]. Therefore, in order to more safely design and manufacture NPs, it is necessary to ensure extensive characterization of their physico-chemical properties [73,74].
Table 2.
Physico-chemical characteristics of nanoparticles affect apoptosis.
| NPs | Size | Shape | Functional groups | Dose | Time of exposure | Cell model | Major outcomes | Ref |
|---|---|---|---|---|---|---|---|---|
| Ag NPs | 5.56 nm | 100, 200, and 400 ppm | 10 days | Rat Hippocampus | Upregulation of BAX. Downregulation of BCL2. | [173] | ||
| 18 nm | 11.0 μg/ml | 24 h | Baby hamster kidney BHK-21 cell line and human colorectal adenocarcinoma HT-29 cell line | Upregulation of BAX, BAD, BAK1, TP53, and MYC. Downregulation of BCL2 and BCL2L1. | [136] | |||
| 5 to 10 nm | 2, 4, 6, 8, 10 μg/ml | 1.0, 7.0, 9.5, 12.8 and 24.5 at 0, 6, 12, 24, and 48 h | Human Chang liver cells | Upregulation of BAX. Downregulation of BCL2. | [174] | |||
| 59 nm | 0.9–7.4 μg/mL | 24 h | Human bronchial epithelial BEAS-2B cell line | Upregulation of BAX. Downregulation of BCL2. | [175] | |||
| 20 to 100 nm | 50 to 200 μM | 24, 48 and 72 h | Human colon cancer HCT116 cell line | Upregulation of TP53 and CDKN1A/p21. | [176] | |||
| Ag-CS NCs (chitosan) | 24 μg/ml | Human colorectal adenocarcinoma HT-29 cell line | Downregulation of BCL2 | [177] | ||||
| 1–100 nm | 100 μg/ml | Up to 72 h | NIH3T3 fibroblast cells | ROS production and MAPK/JNK activation. | [178] | |||
| 1–100 nm | 50 μg/ml | Up to 72 h | Mouse embryonic fibroblast NIH3T3 cell line | Induced release of CYCS into the cytosol, translocation of BAX to mitochondria and decreased BCL2 expression. | [137] | |||
| Au NPs | 200 µg/ml | 24 h | Human breast epithelial MCF-7 cells | Upregulation of BAX and TP53. Downregulation of BCL2. | [179] | |||
| 20.52 nm | Plant extracts of Zataria multiflora | 0–400 μg/ml) | 48 h | Human cervical cancer HeLa cells and normal BMSC cells | [180] | |||
| AuNP20 was 18.6 nm and AuNP70 was 67.5 nm | 0 to 100 μg/ml | 24 h | Human polymorphonuclear leukocytes (PMNs) | [181] | ||||
| 13 nm | GNPs (10 μM), BSO (0.5 mM) or GNPs (10 μM) +BSO (0.5 mM) | 48 h | Lung carcinoma A549 cell line | Upregulation of BAX and TP53. Downregulation of BCL2. | [92] | |||
| 8 nm and 37 nm | 50 nM | 24, 48 or 72 h | Human liver HL7702 cells | Upregulation of BAX, CYCS. | [182] | |||
| 1–20 nm | Abutilon indicum leaf powder | 180, 360, 540 and 720 μg/ml | 24, 48 h | Human colon carcinoma HT-29 and HaCaT keratinocytes | Expression of LMNA and PARP. | [183] | ||
| size <20 nm | 100 ng–1 μg/ml | 24 h | C2C12 myoblasts | [184] | ||||
| Carbon Nanotubes | 400–1000 m2/g Length: 0.1–1 µm Width: 0.8–1.2 nm |
SWCNT | 0–8.34 µg/cm2 | 48 h | Non-tumorigenic human bronchial epithelial BEAS-2B | [185] | ||
| 450 nm | Polyethylene glycol (PEG) MWCNT-PEG |
5, 10, 50 µg/ml | 1 h followed by laser irradiation (3 min, 808 nm, 2W/cm2 | Human pancreatic adenocarcinoma cells (PANC-1) |
[186] | |||
| MWCNT: L: 24 ± 12 µm D: 20 ± 7 nm AT-MWCNT: L: 10 ± 6 µm D: 20 ± 9 nm |
MWCNT Acid-treated MWCNTs (AT-MWCNT) |
25–100 µg/ml | 48 h | RAW264.7 macrophage cell line | ROS production. | [187] | ||
| Acid-MWCNT L: 0.3–0.6 µm D: 10–20 nm Taurine- L: 0.3–0.6 µm D: 10–20 nm |
Acid-MWCNTs Taurine MWCNTs (tau-MWCNTs) |
0, 5, 20, 40, and 80 µg/ml for 12 or 24 h | 12, 48 h | RAW264.7 macrophage cell line | ROS production. | [188] | ||
| SWCNTs | 0.125–10 µg/ml | 24, 48 h | Normal rat kidney epithelial cells (NRK-52E) | Elevated protein levels for TP53 and CDKN1A. Arrest at G0/G1 phase, CDK2, CDK6, p-RB1. |
[189] | |||
| L: 401nm | MWCNT | 0.5–100 µg/ml | 6–72 h | Lung carcinoma A549 cell line | ROS production. Upregulation of TP53, CDKN1A, BAX. Downregulation of BCL2. |
[190] | ||
| MWCNT | 5 µg/ml | 12, 24 h | Rat lung epithelial cells | ATP production. Cytochrome-c oxidase and NFKB activation. |
[191] | |||
| L: 12 µm D: 15 nm |
MWCNTs-COOH | 40, 200, 400 μg/ml | 24, 48, 72, 96 h | Normal human dermal fibroblast cells | DNA damage. | [192] | ||
| MWCNTs | 0.5–10 μg/ml | Rat lung epithelial cells | ROS production. | [193] | ||||
| L: 0.5–2 µm D: 20–50 nm |
MWCNT | 0, 1, 10 or 100 µg | Rat (in vivo) | [194] | ||||
| L: 1–5 µm D: 20–40 nm |
MWCNT | 1 and 10 ng/cell | 24–12 h | T lymphocytes from healthy human | [195] | |||
| CuO | 22 nm | 10 µg/ml | 24 h | Human hepatoma HepG2 Cells | Upregulation of TP53. Downregulation of BCL2. | [140] | ||
| 20 nm | 2, 5, 10, and 25 µg/ml | 24 h | Chronic myeloid leukemia K562 cell line | Upregulation of BAX. Downregulation of BCL2. | [130] | |||
| 15 nm | Copper ferrite NP | 5, 25 and 100 μg/ml | 24 h | Human breast cancer MCF-7 cells | [197] | |||
| 35 nm | 50–100 μg/ml | 24 h | Human hepatoma HepG2 cells and lung carcinoma A549 cell line | [198] | ||||
| 60–80 nm | up to 4.5 μg/ml | 18 h | A375 human melanoma cell line | [199] | ||||
| Fullerene/C60 | 96 nm in PBS, 142 nm in DMEM |
Polyhydroxylated (fullerenols, C 60[OH]n) |
10–200 µM | 48, 72 h | Lung carcinoma A549 cell line | Not cytotoxic. Antioxidant through increase in NFE2L2. |
[200] | |
| C60-bis (N,N-dimethylpyrrolidinium iodide) | HL-60 promyelocytic leukemia cell line | [201] | ||||||
| hydrodynamic sizes (radius) were 4.7 and 40.1 nm | Hydroxylated fullerene (fullerol) | 1, 10 and 100 µg/ml | Caenorhabditis elegans | [202] | ||||
| 150 nm | C60 (C[COOH] 2)2 |
Cerebral microvessel endothelial cells | p-MAPK/JNK, activation of AP-1 Decrease in apoptosis. |
[203] | ||||
| Graphene | GO: 10 nm ~ 1 nm thickness |
nanosheets | [204] | |||||
| Thickness: GO: 0.6 nm rGO: 0.9 nm Size: GO: 219 nm rGO: 122.4 nm |
nanosheets | 0, 10, 20, 40, 60, 80, and 100 µg/ml | 6, 12, 24, 48 h | Phaeochromocytoma (PC12) cells | Cell cycle arrest. | [205] | ||
| 3.125–100 µg/ml | 24, 48 h | Osteosarcoma cell lines, HOS and KHOS | [206] | |||||
| GQDs 1.5–5.5 nm (mean: 3.03 nm) |
1–200 µg/ml | 24, 48 h | THP-1 human monocytic cell line | ROS production. Increase in BAX, BAD, BECN1, and LC3-I/II. Downregulation of BCL2. |
[207] | |||
| Quantum Dots | 2.2 ± 0.25 nm 3.5 ± 0.49 nm |
CdTe QDs | 80 μg/ml | 24 h | L929 mouse fibroblasts | ROS production. Upregulation of MAPK/p38, MAPK/JNK, MAPK/ERK. |
[208] | |
| 2.2 nm CdSe QDs 2.2 nm CdTe QDs 3.5 nm CdTe QDs |
5.85–150 μg/ml | 24 h | L929 mouse fibroblasts | Oxidative stress. Genotoxicity. |
[209] | |||
| Realgar QD: 5.48 ± 1.09 nm |
7.5–60 µg/ml | 6 h | Human hepatoma HepG2 Cells, L02 normal liver cell line | Upregulation of BAX. Downregulation of BCL2. | [210] | |||
| 7.3 ± 1.2 nm | CdTe-QDs | 0.001–10 µg/ml | 24 h | Human hepatoma HepG2 Cells | ROS production. Upregulation of MAPK/p38, MAPK/ERK, MAPK/JNK. |
[211] | ||
| 6.0 ± 0.5-nm | CuO QDs | 5–20 µg/ml | 8, 16, 24 h | C2C12 myoblasts | [212] | |||
| 2.0 nm to 2.5 nm | Spherical core/shell | CdSe QDs | 0.6–9.6 µg/ml | Mouse epidermal cells (JB6 cells) |
Upregulation of AIF. G1/S arrest. |
[213] | ||
| Realgar QDs: 5.48 ± 1.09 nm |
Realgar (As4S4) | 10–160 µg/ml | 48 h | Human endometrial cancer JEC cells | Upregulation of HSPA5/HSPA5 (gene/protein). ER stress. |
[120] | ||
| 3.5 nm | CdSe QD | 150 and 300 nM | IMR-32 human neuroblastoma cell line | Upregulation of BAX and p-MAPK/JNK. Downregulation of BCL2, p-MAPK/ERK, HSP90, RAF1, RAS. |
[215] | |||
| Iron Oxide | 25 nm | for 24 h | 25, 50 and 100 g/ml | A431 human epidermoid carcinoma and lung carcinoma A549 cell line | [216] | |||
| 21.5 nm | DSPE-PEG micelles | 2, 3, 7 and 14 days | 5 mg Fe/kg bw and 15 mg Fe/kg bw | Rat lung tissue | Upregulation of BAX. Downregulation of BCL2. | [217] | ||
| 41.5 nm | Magnetic iron oxide nanoparticles (M-FeNPs) | 24 h | 6.25, 12.5, 25, and 50 μg/ml | RAW264.7 macrophage cell line | Upregulation of BCL2, SQSTM1, LC3, ERN1, p-ERN1, DDIT3, and SOD2. Downregulation of BECN1 and BAX. |
[218] | ||
| Cold atmospheric plasma and iron nanoparticles | 24 h | 1.48–750 ppm | Human breast cancer cells (MCF-7) | Upregulation of BAX. Downregulation of BCL2. | [219] | |||
| 24, 48 h | 10, 30, 60, and 120 μg/ml | Human breast cancer cells (MCF-7) | [220] | |||||
| Fe3O4 | 24 h | 100 μg/ml | Human hepatoma HepG2 Cells and lung carcinoma A549 cell line | Upregulation of TP53. | [221] | |||
| SeNPs | Ch-SeNPs 40–60 nm | Spherical | 0.1–5 mg/l | 72h | Human hepatoma HepG2 Cells | Slow migration rate. No apoptosis induction. Cell cycle arrest at S-G2/M phase. Inhibition of CDK1. |
[222] | |
| Tf-SeNPs | 72h | MCF-7 breast cancer cells, Human hepatoma HepG2 cells, A375 human melanoma cell line, HUVECs |
ROS production. | [223] | ||||
| Spherical | 80 µg/ml | HK-2 normal human kidney cells | High mitochondrial activity. No cytotoxicity. |
[224] | ||||
| 10–20 (12.4) nm | 0–100 µM | 24h 48h |
Lung carcinoma A549 cell line, MCF-7 breast cancer cells, androgen-independent prostate cancer cell DU-145, androgen-dependent LNCaP, embryonic kidney HEK-239 |
Suppression of AR (androgen receptor) through phosphorylation of AKT. | [225] | |||
| 50 nm Aggregate: 442 nm |
Spherical | 4–20 µg/ml | 72 h | Human cervical cancer HeLa cells, HK-2 normal human kidney cells | [226] | |||
| Silica | 19, 43, 68 nm | 100 μg/ml | 24 h | Human hepatoma HepG2 Cells | Upregulation of MAPK/JNK and TP53. Downregulation of BCL2. |
[227] | ||
| 15, 30 nm | 10 and 15 μg/ml | 24 h | HaCaT keratinocytes | Upregulation of SERPINB5 | [228] | |||
| 21 nm | 200–600 μg/ml | 12 & 24 h | L02 normal liver cell line | Upregulation of BAX and TP53. Downregulation of BCL2. | [229] | |||
| 20 nm | 50–200 μg/ml | 24 h | HUVECs | Upregulation of MAPK/JNK and TP53. Downregulation of BCL2. |
[230] | |||
| 20 nm | 160 and 320 μg/ml | 24 h | Huma hepatoma HepG2 Cells | Upregulation of TP53. | [231] | |||
| 20 nm | 1 mg/ml | 24 h | Buffalo rat liver cells | Upregulation of BAX and TP53. Downregulation of BCL2. | [232] | |||
| 14 nm | 1–200 μg/ml | 24 h | Human hepatoma HepG2 Cells | Upregulation of BAX and TP53. Downregulation of BCL2. |
[132] | |||
| 15 nm | 25–200 μg/ml | 72 h | Epidermoid carcinoma A431 cells | [233] | ||||
| 20 nm | 62.5–250 μg/ml | 48 h | Human hepatic cell line (HL-7702) | Upregulation of BAX and TP53. Downregulation of BCL2. | [234] | |||
| 43 nm | 25–100 μg/ml | 24 h | Human hepatoma HepG2 Cells | Release of CYCS into the cytosol. Downregulation of BCL2. |
[235] | |||
| ZnO | 21, 34 nm | 15 μg/ml | 24 h | Human hepatoma HepG2 cells, lung carcinoma A549 cell line, human bronchial epithelial BEAS-2B cells | Upregulation of BAX and TP53. Downregulation of BCL2. | [236] | ||
| 22.5 nm | 20 and 25 μg/ml | 24 h | BJ human neonatal foreskin | TP53 (after TP53 knockdown, the cells become more resistant to NP-induced cell death, and they increase cell-cycle progression). | [237] | |||
| 23, 47 nm | 2.5, 10, 25, 50, and 100 μg/ml | 4, 24 h | Human dermal fibroblast | Upregulation of MAPK/p38 and TP53. | [238] | |||
| 30 nm | 20 μg/ml | 12 h | Human hepatoma HepG2 Cells | Upregulation of BAX and TP53. Downregulation of BCL2. | [239] | |||
| 17 nm | 5, 10, and 20 μg/ml | 24–48 h | Human skin melanoma (A375) | [240] | ||||
| 52 nm | 5, 10, 25, 50 and 100 μg/ml | 24–48 h | Lung carcinoma A549 cell line | Upregulation of BAX and TP53. Downregulation of BIRC5/survivin and BCL2. | [134] | |||
| 40 nm at 60 mg/L, and 60 nm at 120 mg/L | 60, 90, or 120 mg/L nano-ZnO | 96 hpf | Zebrafish embryos | [241] | ||||
| 20 nm | 30 μg/ml | 12 h | Human ovarian cancer cells (SKOV3) | Upregulation of BAX and TP53. Downregulation of BCL2. | [242] | |||
| 70 nm | 8, 15, 25 and 50 μg/ml | 12 h | Human aortic endothelial cells | Upregulation of BAX and FAS. Downregulation of BCL2. | [243] | |||
| 60 nm | 0, 2.5, 5.0 and 10.0 μg/ml | 2 h | Retinal ganglion RGC-5 cells | [244] | ||||
| 13 nm | 50 μg/ml | 24 h | Human hepatoma HepG2 cells | Upregulation of BAX and TP53. Downregulation of BCL2. | [245] |
The specific cell response to the presence of NPs is complex and determined by many diverse factors. For example low concentrations of silver (Ag) NPs induce necrosis and apoptosis, whereas necrosis alone is triggered at higher concentrations [75]. Also the exposure time of the Ag NPs dictate the mode of cell death (apoptosis or necrosis) in different experimental models, such as human skin, fibrosarcoma, and testicular carcinoma cells [76–78]. It should also be mentioned that the mode of cell death that is induced is cell type-specific [79,80].
In the present review a comprehensive literature survey on the cell fate induced by NPs and the molecular mechanisms involved will be reported.
Necrosis triggered by NPs
NPs and necrosis and/or necroptosis
There are conflicting examples in the literature about necrotic effects of NPs because on the one hand most reports only studied losing cell viability without focusing on the exact mode of cell death, and on the other hand in some conditions apoptosis can be followed by secondary necrosis, thus leading to an incorrect interpretation [70].
For instance, it was found that both crystal structure and size affect the mechanism of cell death induced by titanium dioxide (TiO2) NPs in mouse keratinocyte cells [81]. This study demonstrated that TiO2 NPs with 100% anatase crystal structure elicit necrosis in a size-independent manner, whereas TiO2 NPs with rutile crystal structure induce apoptosis. In a systematic study by Pan et al., gold NPs show size-dependent cytotoxicity; smaller-sized NPs (< 1.4 nm) are more cytotoxic and induce necrosis, whereas NPs larger than 1.4-nm particles often induce apoptosis [82,83]. Interestingly, the 15-nm gold NPs do not show any cytotoxic effects in different cell lines and are actually inert with regard to toxicity. Moreover, surface charge can also be a major factor in determination of the cell death modalities induced by NPs. Schaeublin et al. [84] showed that charged gold NPs induce apoptotic cell death, whereas neutral NPs trigger necrosis. Another study demonstrated that cationic carrier-induced cell necrosis is dependent on the nanocarrier surface cationic charge [85]. The toxicity is related to the shape of polyaniline (PANI). NPs with 4 various aspect ratios were assessed with regard to their effect on fibroblast cells of human lung and it was shown that low-aspect ratio PANI NPs induce necrosis more than the others [86].
Moreover, recent evidence shows that cytotoxic effects of germanium NPs (4 nm) are blocked by necrostatin-1, suggesting that NPs may also induce necroptosis [87]. A very recent paper reports that selenium (Se) NPs induce ROS-mediated necroptosis in PC-3 cells following cellular internalization [88].
Other parameters that may affect the triggering of necrosis include exposure time and concentration (Table 1). For example, nano-C60 fullerene at high doses induces ROS-mediated necrosis, whereas an ROS-independent autophagic cell death is observed with low-dose NPs in glioma cells [74].
Pathways of NP-triggered necrosis
From the presented information, it is clear that advances in knowledge concerning the causes that may influence NP-induced necrosis will need more accurate and precise studies [89–91]. Nevertheless, a pro-oxidant pathway is one of the main mechanisms involved in NP-induced necrosis. Indeed, various NPs exhibit toxicity dependent on oxidative stress. ROS produced by NP exposure can lead to lipid peroxidation, protein denaturation and oxidative DNA damage. Excess free radical generation causes the reduction of mitochondrial membrane potential and leads to mitochondrial membrane damage, causing necrotic cell death [71,87]. For example, water-soluble germanium NPs trigger necrosis through an increase of the cellular calcium level, which subsequently leads to an increase at the level of ROS. Also, gold NPs (1.4 nm) induce necrosis through mitochondrial damage and oxidative stress, resulting not only from ROS but also induce a decrease in the intracellular antioxidant defense system as seen in HeLa cells [82]. Similar data have been presented by Liu et al. who reported that necrosis is an essential mechanism of cell death induced by gold NPs in lung cancer cells with a low level of intracellular glutathione (GSH). Therefore, there is evidence that triggering necrosis induced by NPs can be a beneficial approach for treating cancer [92].
High concentrations of Ag NPs induce necrosis in the breast cancer MCF-7 cell line [75] and can have a toxic effect on the respiratory system through reduced GSH levels, and increased ROS generation [93]. Generally, Ag NP cytotoxicity depends on dose, time, temperature, surface coatings, size and cell type [94]. Ag-NP exposure causes reduction in antioxidant enzymes such as GSH, in elevated levels of intracellular ROS and the consequent elevated expression of ROS-responsive genes, and lipid peroxidation, leading to DNA damage, necrosis and apoptosis [95,96]. Both Ag NP degradation into ions, and ROS generation depend on size; and polyvinylpyrrolidone (PVP)-coated Ag NP size negatively correlates with ROS level, necrosis and apoptosis and decreased cell viability [97]. ROS generation and silver ion release are 2 crucial factors mediating cytotoxicity that lead to Ag NP-mediated necrosis and apoptosis. Although both Ag NPs and Ag ions result in increased ROS and oxidative stress throughout the cell, Ag NPs cause stronger oxidative damage in cellular membranes and organelles, such as mitochondria, lysosomes and the nucleus, which directly leads to apoptosis or necrosis [94]. In the presence of oxygen, Ag ions catalyze ROS generation and Ag NPs themselves can induce a process to release Ag ions, in addition to generating ROS and causing oxidative stress in vitro [98]. The studies show that Ag NPs (15 and 100 nm) induce a significant depletion of reduced GSH, and cause an increase in ROS levels and reduced mitochondrial membrane potential in BRL 3A rat liver cells and rat alveolar macrophages [99,100]. Furthermore, 25-nm Ag NPs cause a significant increase in ROS both in vitro and in vivo. Recently, ROS generation was detected in the MC3T3-E1 and PC12 cell lines in a manner dependent on particle size and concentration [79,101]. As already stated, NP size can influence the cell response. Citrated coated Ag NPs of 13 nm induce higher cell membrane damage and ROS production while having a stronger bacteriostatic potential than the same particles of 17 nm. Thus, these NPs may be considered in cancer therapy [102].
Zinc oxide (ZnO) NPs increase basic ROS levels of macrophages and induced oxidative DNA damage-mediated necrosis. Therefore, clinically, ZnO NPs through this mechanism can help the immune system in the clearance of inhaled particulates during inflammation [87]. A novel mechanism by which cationic nanocarriers such as polyethylenimine (PEI), cationic liposomes, and chitosan, lead to rapid necrosis is related to their positive surface charges. In fact, acute cationic nanocarrier-induced necrosis occurs via an interaction with the Na+/K+-ATPase and is associated with the exposure of molecular patterns dependent on mitochondrial damage that lead to inflammatory responses. Although the toxicity of nanocarriers with positive surface charges hinders their clinical applications, the understanding of cationic carrier-induced acute cell necrosis can assist in the better and safer design of nanocarriers in drug delivery systems and also the development of screening assays and rapid assessment of biomaterials [84].
Another mechanism involved in necrosis induced by NPs is induction of endothelial cell (EC) dysfunction through release of VWF (von Willebrand factor). EC dysfunction induced by NPs, can also be due to the formation of ROS, inflammatory cytokines including IL6 (interleukin 6) and IL8 and/or the activation of the system of coagulation that causes pulmonary and ischemic cardiovascular diseases [87]. Many studies revealed that silica NPs induce pulmonary inflammation through dysfunction of ECs with the clotting cascade activation that leads to increase of blood coagulability in vivo [103]. In addition, silica NPs participate in ROS production and inflammatory responses in vitro [104–107].
Another pathway involved in necrosis triggered by NPs is the rapid degradation of NPs in lysosomes and the subsequent destabilization of this organelle that will allow the toxic substance to enter into the cytosol, and finally lead to cell necrosis. For example, cadmium telluride (CdTe) quantum dots (QDs) induce mitochondrial permeability transition via the increase of ROS, which leads to swelling of mitochondria and collapse of the membrane potential. Because of their large surface:volume ratios, adding QDs into the culture medium causes serum proteins to attach to the QD surface. This absorption alters the properties of surface and size, and leads to the cellular uptake of the QDs through the endocytic pathway mediated by clathrin. After that, QDs translocate in cells via endocytic vesicles and then are rapidly degraded in lysosomes, leading to cell necrosis. Therefore, considering the cytotoxicity mechanism of CdTe QDs can provide valuable data for the safe use of QDs in the future [108].
Tubulin and actin are 2 factors involved in necrosis induced by certain NPs such as different nanostructures of ZnO, including ZnO commercial NPs and custom‐made nanowires of ZnO. Extra levels of cytoplasmic zinc have cytotoxic effects. Moreover, tubulin and actin, important cellular cytoskeleton proteins involved in migration, cell division and maintenance of cellular architecture, control zinc homeostasis in the cells. Following ZnO intracellular dissolution, actin microfilaments and microtubules undergo dramatic structural modifications and an acute cytoskeletal collapse that trigger a rapid necrotic process in most cells. There might be possible health risks for these NPs on skin and mucosa in children that cause significant cell damage and aneuploidies that eventually lead to cell transformation and cancer [109].
Apoptosis triggered by NPs
NPs induce intrinsic and extrinsic apoptotic pathways
ROS generation induced by NPs causes damage to DNA, proteins and organelles, including mitochondria. Damaged mitochondria lead to induction of intrinsic and then extrinsic apoptosis pathways [125]. The central role of ROS in various cellular functions, including cell cycle regulation, proliferation, self-renewal, differentiation and apoptosis is well known [126]. TP53/p53 can protect the genome during stressful conditions, such as ROS-mediated DNA damage. The primary function of this bodyguard of the genome is to induce cell cycle arrest in order to allow time for repairing damage. If the DNA damage cannot be repaired the cell responds by shifting to apoptosis [127]. Apoptosis is regulated by the BCL2-family proteins that are comprised of pro-apoptotic and anti-apoptotic members as discussed above (see Mechanism(s) of apoptosis in mammalian cells) [128]. The BAX:BCL2 ratio determines the threshold of cell death in response to an apoptotic stimulus. An increase in the BAX:BCL2 ratio decreases the cellular resistance to apoptotic stimuli, leading to caspase-mediated apoptosis [129,130].
NPs can induce oxidative stress in cells through different mechanisms: (i) direct generation of ROS by their physico-chemical properties, (ii) indirect generation of ROS and reactive nitrogen species (RNS) by stimulating inflammatory cells, (iii) indirectly through changes on mitochondrial integrity such as through effects on NADPH oxidase or cellular calcium homeostasis, and (iv) ROS generation by releasing ions or soluble compounds from certain types of NPs (e.g., metal oxides) [131].
Similar to necrosis, NP-induced apoptosisis is also a function of the NPs physico-chemical structure (Table 2). For example, metal NPs, such as copper oxide/CuO, cadmium oxide/CdO, and TiO2 NPs, exhibit different apoptotic potency [32].
NP-induced apoptosis is size, concentration and time dependent. Ahmad et al. [132], reported that 14-nm silica NPs induce a dose-dependent depletion of GSH and ROS-mediated apoptosis response in human liver HepG2 cells. In addition, the cell cycle regulatory gene TP53 and apoptotic genes (BAX and CASP3) are upregulated, whereas BCL2 is downregulated in cells exposed to silica NPs in a dose-dependent manner [132].
Ye et al. [229] reported that silicon dioxide (SiO2) NPs regulated the expression of the TP53 and BAX genes, in an NP dose-, size- and exposure time-dependent manner. SiO2 NPs show ROS-mediated oxidative stress and consequently apoptosis in L-02 cells [229].
ZnO NPs dose and time dependency was shown by Ahamed et al. who reported ZnO NP upregulation of BAX and downregulation of BIRC5/survivin and BCL2 in lung cancer cells (A549 cells) [134]. Ag NPs induce oxidative stress resulting in genotoxicity in a TP53/p53-dependent manner in a variety of experimental systems such as adult cells, stem cells and cancer cells [135–138].
Liu et al. [139] showed that high doses of superparamagnetic iron oxide nanoparticles (SPIONs)-induce mitochondrial apoptosis by increasing the BAX:BCL2 ratio, by the activation of CASP9 and CASP3 and by downregulating HSPA/HSP70 and HSP90 survival factors [139].
NPs induce mitochondrial apoptosis
The mitochondrial pathway of apoptosis is one of the important mechanisms that contributes to the cytotoxic effects of NPs. Several NPs such as TiO2, CuO, ZnO, SPIONs and silica NPs affect mitochondrial pathways [130,140–143]. Yan et al. [206] showed that graphene and single-walled carbon nanotubes (SWCNT) are more cytotoxic than graphene-SWCNT hybrids, and the 3D nanostructures induce a ROS-mediated mitochondrial apoptotic pathway in osteosarcoma cells. Low-dose exposure to silica NPs causes epigenetic toxicity associated with mitochondrial apoptosis in human bronchial epithelial BEAS-2B cells. Zou et al. [145] showed that the silica NPs over 30 passages significantly hypermethylate the promoters of the CREB3L1 and BCL2 genes.
Ag NPs trigger activation of the TP53 protein, which in turn increases the expression level of BAX, BAD and BAK1, causing mitochondrial membrane leakage and release of CYCS. In parallel, the apoptosis inducer MYC/c-MYC is upregulated, and anti-apoptotic genes, such as BCL2 and BCL2L1, are downregulated. These signaling pathways trigger cell blebbing [136]. In another study, Hsin et al. reported that Ag NPs induce mitochondrial apoptosis in NIH3T3 cells. The NPs generate ROS and trigger a JUN/c-Jun N-terminal kinase-dependent mechanism [115].
Permeabilization of the outer mitochondrial membrane occurs by exposing cells to graphene, resulting in a change in mitochondrial membrane potential [146]. Graphene, by increasing ROS generation, affects the MAPK and TGFB/TGF-β signaling pathways and activates CASP3. The carbon-based nanomaterial induces mitochondria-mediated apoptosis [147,148]. Similarly, low doses of reduced graphene oxide induce an early apoptosis via death-receptor and canonical mitochondrial pathways [149]. Graphene can act in different ways: GO leads to an ROS-dependent apoptotic pathway, and GO-COOH activates an ROS-independent apoptotic pathway. T lymphocytes undergo apoptosis after the membranes have been damaged by exposure to GO-PEI [150,151]. Gadolinium oxide induces mitochondrial apoptosis with increasing doses and exposure time in human neuronal (SH-SY5Y) cells. Ahmad et al. [152] demonstrated that the number of apoptotic cells is increased with an increase of the concentrations of nickel NPs in mouse epidermal JB6 cells.
NPs induce ER-mediated apoptosis
The ER has pivotal functions in cells for controlling protein folding and calcium homeostasis. The release of calcium into the cytosol from the ER occurs upon ER stress [153]. Transcription factor CREB phosphorylation, which induces the transcription of PPP2 (protein phosphatase 2), is induced by an increase in the calcium concentration. PPP2 regulates various critical cellular processes [154]; therefore, ER stress-inducible regulation of PPP2 contributes to the cytotoxicity of NPs [155].
High doses of NPs can generate ROS, damage membrane and disrupt calcium homeostasis to cause cell death [156,157]. Ag NPs induce transient changes in intracellular calcium in human lung fibroblasts [158]. Additionally, TiO2 NPs increase intracellular free calcium [159]. Cytotoxic NPs trigger ER stress, and the associated changes in calcium homeostasis may be an important aspect of the response that results in apoptosis [155].
ER stress causes accumulation of misfolded protein aggregates and triggers apoptosis through the blocking of nutrient transport to retinal cells [160], and occurs in response to ZnO NPs in human umbilical vein endothelial cells (HUVECs) [161], and also as a result of exposure to poly(lactic-co-glycolic acid) [162] and gold [163] NPs in human chronic myelogenous leukemia cells.
As suggested by the references above, ER stress can result in apoptosis. ATF4 induces DDIT3/CHOP (DNA damage inducible transcript 3) expression, and the expression of the apoptotic genes BCL2L11/BIM and BBC3/PUMA is induced by DDIT3 [155]. The ERN1/IRE1 pathway may induce apoptosis through the activation of MAP3K5/ASK1 (mitogen-activated protein kinase kinase kinase 5) and through interaction with TRAF2 (TNF receptor associated factor 2). Therefore, SiO2-NPs may exert hepatotoxic activity through ER stress. PMAIP1/Noxa [164], is another factor that can induce apoptosis—in this case through the USP9X-MCL1 pathway—in response to ER stress [165].
Multiple signaling pathways regulate inflammatory and immune responses. Among them are the NFKB and MAPK signaling pathways, involving MAPK1/ERK2-MAPK3/ERK1, which regulate inflammatory and immune responses [166]. The MAPK signaling pathway also plays an important role in cancer development and apoptosis [155].
TiO2 damages DNA and activates TP53 by being deposited in the membrane of the nucleus in HEK-293 cells [167]. TP53 is a key tumor suppressor that blocks the cell cycle in the G1/S phase. Through TP53, DNA damage causes the initiation of apoptosis [168]; SiO2 NPs significantly downregulate TP53 in Huh7 cells. Transcriptional downregulation of TP53 could contribute to the apoptotic or carcinogenic activity of SiO2 NPs [155].
In one study, Simard et al. reported that Ag NPs cause protein misfolding, leading to ER stress in MCF-7 and T47-D cells. The NPs activate several caspases leading to apoptosis through constant activation of the UPR pathway [169]. Chen et al. [161] reported significant cellular ER stress induced by ZnO nanorods at noncytotoxic concentration. ZnO nanorods change both the transcriptome and proteome of HUVECs. The NPs in higher dosage (240 μM) result in an ER stress response before apoptosis induction [161]. Yang et al. [171] showed that hepatotoxicity of orally delivered ZnO NPs is mediated through an ER stress-mediated apoptotic signaling pathway and increased translation of related proteins in mice. In another study Kuang et al. [172] reported that ER stress-mediated apoptosis triggered by ZnO NPs is size dependent, and smaller ZnO NPs are more toxic than 90-nm ZnO NPs in murine liver.
Two-fold role of autophagy and dysregulated autophagy as a cytoprotective mechanism and death signal triggered by NPs
How do you set up an autophagy investigation for nanomaterials and what markers do you use?
The size of NPs, their functionalization and surface charge influence autophagy pathways and must be carefully evaluated before in vitro and in vivo investigations. In some cases (eventually as a biodegradation mechanism) [246,247], cationic NPs more than anionic ones [248] and some functionalizations such as arginine-glycine-aspartic acid (RGD) induce autophagosomes at higher levels [249]. However, other stressful factors that trigger autophagy such as cell membrane damage, and ROS production must be evaluated to give more information on the mechanism. After the characterization of NPs and evaluation of some eventual risk factors, to track autophagy, autophagosomes as double-membrane vesicles might be monitored via electron microscopy [250].
To accurately evaluate autophagy activity, the analysis should be categorized into 2 separate elements: static and dynamic measurements. The importance of differentiating between dynamic and static measurements is easily seen when considering the most common types of assays that are used for monitoring autophagy—the presence of autophagosomes based on fluorescence and electron microscopy, and the level of LC3 based on western blot. In particular, the increase or accumulation of these autophagic markers can be an indication of autophagic induction; however, because both autophagosomes and LC3 are degraded in the terminal stages of autophagy, an increase of either component can also be the result of a block in turnover or flux, resulting from a defect in autophagosome or amphisome fusion with the lysosome or in degradation within the lysosome.
Nonetheless, static measurements can be useful if used appropriately. For example, the phosphorylation status of MTOR, BCL2, AMPK, and ULK1 at specific sites as assessed by western blot correlate with autophagy induction. In addition, the evaluation of gene expression via real-time PCR can provide another valid method for assessing induction when used in combination with other assays. Monitoring the complete process of autophagy requires an assessment of flux. This can be achieved by examining the turnover of LC3 and cargo proteins such as SQSTM1/p62 by western blot in the presence and absence of fusion, acidification or lysosomal protease inhibitors, or by following long-lived protein turnover [251]. Fluorescence techniques can be used that rely on tandem mRFP/mCherry-green fluorescent protein (GFP)-LC3 or the more recent GFP-LC3-RFP-LC3∆G [252]. Thus, similar approaches can be used to determine the effects of NPs on auto-phagic flux. In particular, cells exposed or not to NPs can also be treated with fusion inhibitors such as bafilomycin A1, alkalinizing agents such as chloroquine, or protease inhibitors including E64-d, leupeptin and pepstatin A. It is also important to note that some NPs are sequestered into the lysosome and may induce lysosomal dysfunction. Thus, when using NPs it may be worthwhile to monitor lysosomal pH and degradative capacity. Neutral red, acridine orange and LysoSensor dyes can be used as pH-dependent lysosomotropic indicators, whereas DQ-BSA and Magic Red™ can be used to follow degradative activity [253].
NPs and autophagy
Autophagy has a close crosstalk with other cell death and signaling pathways; thus, cellular outcomes induced by autophagic NPs are very complex [250]. Although NPs primarily induce macroautophagy, based on the increased formation of and detection of NPs within autophagosomes, it is difficult to classify autophagic effects of NPs as a cell death mechanism at first glance [4,250,254]. In most cases, cells respond to NPs as an endosomal pathogen; the NPs are ubiquitinated leading to sequestration by a phagophore. Interestingly, size and concentrations of the nanomaterials influence the autophagic response [4,254]. This fact may explain why it is typical to observe a particle present in autophagosomes on the nano rather than micro scale [255–257]. The consequence of internalization of NPs and their accumulation within autophagosomes can be the disturbance of autophagy flux via interference with vesicle trafficking and the cytoskeleton, and a decrease or inhibition of lysosomal stability and enzyme activity. All these events suppress fusion of autophagosomes with lysosomes and ultimately induce autophagy blockage. The cumulative result of NP-induced autophagy blockage can be an accumulation of damaged DNA, proteins and organelles that in turn can increase the risk of cancer [258] and neurodegenerative diseases [259,260].
Conversely, some NPs, such as manganese NPs [261], core-shell of Fe@Au NPs and TiO2 NPs [4,262], induce a functional autophagy response in cells that may terminate in cell death. This autophagy flux response is mostly reported for those NPs that can augment ROS levels in the cells and subsequently may stimulate mitochondria-dependent autophagic cell death, depending on their physico-chemical properties (Table 3) [4,254]. In this scenario, autophagic effects of NPs may have therapeutic value [4,263].
The role of different types of NPs in inducing the autophagy pathway
NPs are taken up into cells and treated almost the same as a bacterium or any other particulate pathogen with a goal of degrading them. These NPs are ubiquitinated allowing them to interact with receptor proteins such as SQSTM1/p62; the latter typically contain an LC3-interacting region, a short motif that allows them to bind LC3 or another Atg8-family protein, resulting in sequestration by a phagophore [264]. Based on the above discussion, the influence of NPs on autophagy may be categorized in 2 distinct parts: An increase of auto-phagosome formation and flux, or autophagic dysfunction. In both categories, the NPs typically increase the LC3-II protein; however, in the case of dysfunction there will also be an increase in SQSTM1/p62, which is no longer degraded via autophagy. For example, SWCNT [265], carboxylated multi-walled carbon nanotubes [266], graphene oxide, PAMAM dendrimer [248], gold [267] and iron oxide [255] NPs induce autophagosome accumulation and block autophagy flux. Other NPs that lead to the accumulation of autophagosomes and autophagy blockage as can be seen with fullerene and its derivatives, Au NPs [260], may even disrupt lysosomal trafficking [268]. Interestingly, autophagosome activation resulting from exposure to graphene oxide NPs involves the TLR pathway [269]. However, fullerene at nanocytotoxic concentration may induce autophagy and increase the killing potential of chemotherapeutics, and even may be influential in treatment of neurodegenrative disorders. In fact, the effects of NPs show two-fold behavior based on concentration—they may either disrupt autophagy or induce auto-phagy [250].
Cationic dendrimers, nano-scale and non-agglomerated quantum dots, alumina NPs, fullerenes, negatively charged zinc oxide NPs [270], Au NPs (22 nm), silica NPs, carbon nanotubes (CNTs), TiO2 NPs, ultra-small super paramagnetic iron oxide (USPION) NPs, palladium NPs and some others induce autophagy in part by inhibition of MTOR or by inducing the expression and/or phosphorylation of autophagy-related and BCL2-family proteins involved in autophagy [259].
Because one of the triggering mechanisms of autophagy activation is via ROS production, metallic NPs such as Ag [271], Au [272] and Cu may be involved. Au NPs induce autophagy but act through different mechanisms such as perturbation of the cytoskeleton, blocking fusion, and alkalinization of the lysosome, causing lysosomal dysfunction and subsequent autophagy blockage [255]. Iron core and gold shell NPs induce a transient loss of mitochondrial membrane-potential in normal cells and irreversible ones in cancer cells as a mechanism that triggers autophagy by ROS production and involvement of the MTOR signaling pathway [262]. Nano alumina results in an increase in the permeability of the blood-brain barrier due to decreased expression of tight-junction proteins that is related to the triggering of autophagy and cytotoxicity [273]. However, this NP is considered as a candidate in anti-tumor therapy due to its being scavenged as an antigen by T cells, so that less antigen is needed overall to provoke adequate T cell production [246]. Similarly, Ag nanowire, manganese NPs, neodymium(III) oxide (Nd2O3), samarium oxide, europium oxide, gadolinium oxide, cerium dioxide, lanthanum oxide and terbium oxide NPs also induce autophagy [250].
Fullerenes induce the production of ROS and damage to mitochondria [274] and the ER, resulting in autophagy induction. Autophagy activation by fullerenes and its derivatives, especially C60(Nd) [275], makes them ideal NPs for cancer therapy in combination with drugs such as doxorubicin; these NPs can induce cell mortality in the breast cancer MCF-7 cell line by the involvement of ATG5 and ROS production [274].
It should be mentioned that the autophagic effects of NPs are highly dependent on their physico-chemical characterization, including size, dispersity and surface charge and as well as their concentrations. One of the best examples is TiO2, a principle component of cosmetics, that induces both auto-phagy flux and blockage in a size-dependent manner [276]. NP functionalization also affects cancer cell viability. For example, Liu et al. showed that COOH-CNT induces autophagy and mortality in human lung adenocarcinoma through a decrease of phosphorylated (i.e., active) MTOR, whereas PABS-CNTs and PEG-CNT do not have this effect [227]. Besides, specific surface characteristics such as nanotopography (vertical aligned nanotubular surface in the range of approximately 100 nm) as compared to flat surface dictates a reversible and temporal MTOR-independent autophagy in osteoblast cells to modulate differentiation via cell membrane stretch [277].
Increase and decrease of autophagic flux by NPs
Increase of autophagic flux by NPs and their functionalization
Increase and decrease of autophagy flux is defined by the increase and decrease in autophagic degradation activity [95]. It seems that increase of acidification in impaired lysosomes leads to restoration and increase of autophagic flux. Based on this finding, polymeric NPs such as PLGA NPs are favorably taken up by lysosomes and their degradation produces an acidic pH [278]. Trudeau et al., prepared a photoactivable NP that in the presence of water and UV irradiation turns to an acidic NP. They demonstrated that an increase of acidic pH in impaired lysosomes leads to an increase of pH and restoration and increase of autophagic flux along with a decrease of SQSTM1 [279]. In addition, graphene QDs increase auto-phagy flux when irradiated by 470-nm blue light as assessed by LC3-II increase and SQSTM1 decrease [280]. Furthermore, QDs increase autophagy flux based on their chemical formulation; CdSe QDs induce a stronger autophagy flux than InGa QDs, while the size of the former was larger than that of the latter [281].
Another important point is that some NPs increase autophagy flux to compete with oxidative stress. As a result, for gold NPs there are different reports on its decrease [282] and increase [283] of autophagy flux. Fe@AuO NPs increase autophagy flux through the MTOR signaling pathway in response to ROS production. Of greater interest, however, is that Fe@AuO NPs behave selectively in normal and tumor cells and lead to tumor cell death [267]. Among ZnO, FeO and TiO2 NPs, only TiO2 NPs increase autophagy flux in response to oxidative stress in a size-dependent manner [276].
Functionalized fullerene (poly ethylene glycol [PEG] or pentoxifylline) may induce autophagy and increase autophagy flux that may lead to elimination of β-amyloid, resulting in inhibition of Alzheimer symptom development [284]. Finally, studies indicate that there are different behaviors of functionalized CNTs. Although, COOH-CNT induces autophagy flux through the AKT-MTOR pathway both PEG and poly-aminobenzene sulfonic acid CNTs do not induce autophagy [227].
Decrease of autophagy flux by NPs
Nanoparticles lead to autophagy dysfunction through disruption of microtubules and actin
The cascade of autophagy may be disrupted via interfering with delivery to the lysosome due to microtubule and or motor protein dynein destruction. It has been disclosed that disturbance in actin polymerization influences autophagosome trafficking. A variety of NPs such as TiO2, polystyrene, silicon, Au NPs [285], CNT [286] and USPION [287] possess binding affinity to actin, resulting in alterations in its polymerization potential. Studies indicate that fullerene [288], TiO2 [289] and Au NPs [290] inhibit microtubule polymerization by the formation of hydrogen bonds with tubulin heterodimers. SWCNT [291], fullerol [260] and silicon dioxide NPs [292], alter autophagy directly via bonding or indirectly via expression of actin modulatory proteins. However, in some studies, it was disclosed that Au NPs cause damage to the cytoskeleton but not via tubulin and actin [285], and Fe2O3 NPs decrease the number of actin filaments [293], whereas Fe3O4 NPs—via bonding to tubulin dimers—change microtubule polymerization [294], also resulting in a decrease of VCL (vinculin) spots and disorganized tubulin and actin structures, along with autophagy dysfunction in blood outgrowth endothelial cells, HUVECs, and neural progenitor cells as a possible reason for toxicity [287,295].
Lysosomal dysfunction by NPs
As indicated above, NPs tend to become trapped within autophagosomes and lysosomes due to their size, ubiquitination and similarity in some aspects to particulate pathogens. Thus, they can directly and indirectly induce dysfunctionality in autophagy that may lead to cell death and many pathological conditions [259].
Lysosomal dysfunction by NPs may be derived from lysosome membrane permeabilization. Oxidative stress by lysosomal-iron mediation and release of lysosomal hydrolases and cathepsins can lead to permeabilization of the mitochondrial outer membrane [253] that in turn, via ROS production, induces apoptosis or necrosis. In addition, lysosomal dysfunction by NPs may be due to a lack of vacuolar-type H+-ATPase activity, increase of pH, bio-persistence and inhibition of functional lysosomal enzymes, which can lead to lysosomal storage disorders, such as sphingolipidoses [296] as reported for fullerenes [297] and dendrimers [298].
Based on the literature, those NPs that cause lysosomal dysfunction include CNT, negative-charge Au NPs (5 nm), negative-charge Ag NPs (25 nm), negative and neutral TiO2 NPs (5–500 nm), fullerenes (150 nm), PEGyated NPs, ZnO NPs, positive-charge dendrimers (5 nm) and positive-charged polystyrene (110 nm) [259]. Cationic NPs, such as cationic dendrimers (G5) [299], act, at least in part, as ‘proton sponges’, resulting in the accumulation of protons in the lysosome and subsequent organelle osmotic swelling followed by lysosomal rupture [80]. Other NPs destabilize the lysosome and induce dysfunctionality in different ways [272,300]. For instance, ZnO NPs (release of Zn2+) [300], TiO2 [301] and polystyrene NPs [302], induce intracellular ROS production, fullerol blocks lysosomal trafficking by affecting actin [260], and Au NPs alkalinize the lysosome (acting as a proton pump inhibitor) and block fusion with autophagosomes [255].
In any form, dysfunctionality of lysosomes may influence autophagy by inhibiting fusion between lysosomes and auto-phagosomes, leading to overloading and accumulation of autophagosome in cells [11,303]. Along these lines, autophagic dysfunction may be derived from excessive induction of autophagy and/or blockade in its flux, which can lead to cell death and some pathologies such as Parkinson [305], Alzheimer [306], and Crohn diseases, as well as amyotrophic lateral sclerosis [253]. Accordingly, dysfunctionality of auto-phagy as a type of cell death may result in apoptosis and in some cases necrosis. However, in normal conditions there are some shared factors that maintain homeostasis in cells. For example the activation of MAPK8/JNK1 as a kinase to phosphorylate BCL2 induces autophagy (via BECN1 release) in the short term, but can ultimately induce BAX and BAK1 leading to apoptosis [307]. Blocking autophagy through inhibition of BECN1 function may result in cancer as seen in breast, ovarian and prostate cancers, eventually due to accumulation of ROS, inflammation and stressful conditions in the affected cells [258]. It is worth noting that although autophagic dysfunction increases the chance of cancer, administration of an autophagy blocker along with chemotherapeutic anti-cancer drugs synergistically improves anti-tumor efficacy in part by decreasing the tolerance of cancer cells to starvation and stress [308].
In conclusion, NPs are taken up into cells as a particulate external material often resulting in a stress response that induces autophagy. However, the physiological condition of the cell may be changed due to the existence of the NP, which causes a block in autophagy. In turn, this block can lead to the accumulation of damaged, depolarized and dysfunctional mitochondria and an increase in ROS resulting in further cellular damage and finally NLRP3 inflammasome activation [269,309]. However, it must be considered that, in a therapeutic context, lysosomal dysfunction is not always a disadvantage; for example, as seen with drug delivery to a target site in cells by nano carriers via an endosomal escape pathway similar to gene delivery by cationic dendrimers or chitosan NPs [310], delivery of alumina NPs with a special antigen on the surface as a targeted vaccine to dendritic cells to enhance anti-tumor effects via lysosomal dysfunction [227], administration of Au NPs and SPION for cancer treatment, and successful co-administration of fullerene and doxorubicin in killing breast cancer cells [311].
Size-dependent effects of NPs on autophagy
Because physico-chemical properties of NPs such as their functionalization, and the chemical structure of NPs have been discussed in the above sections, in the present section the influence of particle size on autophagy will be discussed separately. It is worth mentioning that when the effect of particle size on autophagy is investigated, it is critical to consider cellular uptake and to compare different NPs in normalized conditions. For example Kenzaoui et al., investigated the effect on autophagy of FeO (8–9 nm), silica NPs (25 and 50 nm), TiO2 (21 nm) and PLGA NPs (150 nm). The results showed that just uncoated FeO and TiO2 NPs induce autophagy, whereas these NPs induced oxidative stress in human brain-derived endothelial cells [276]. The important point is that only silica, FeO and TiO2 had been internalized so it is not appropriate to suggest that larger PLGA NPs of 150 nm do not induce autophagy; PLGA NPs of 150 nm could not induce autophagy because they were not internalized into cells. The authors emphasized that those NPs that induce aggregation and oxidative stress (uncoated FeO and TiO2) in cell culture medium, are more potent to induce autophagy and their autophagic potentials were not in good agreement with DNA damage [276]. Stern et al., reported that larger QDs of CdSe (5.1 nm) induce significantly higher autophagy activity (increase of LC3-II) as compared to InGa QD (3.7 nm) at their IC50 concentration. However, it is not clear as to whether their cellular uptake profiles are similar or not; nonetheless they postulated that this effect is related to the severity of induction of oxidative stress in cells [281]. Li et al., compared cytotoxic and autophagic potential of 3 silica NPs of 40, 60 and 200 nm. The results showed that the smallest NPs induce significantly higher cell mortality than others with the highest cellular uptake, whereas 60-nm NPs give the second highest score. However, this trend continues in ROS production and dynamic autophagic index of LC3-I to LC3-II conversion as well, and they are completely size dependent [312]. The important point is that it is not clear whether the increase of higher autophagy potential by 40-nm particle size is related to its autophagic potential or to higher cellular uptake that results in a higher load of NPs within cells.
In contrast, others demonstrated that smaller QDs (QD525) are more efficient in autophagy induction than larger QDs (QD605) as a cytoprotective mechanism and may not be useful for prolonged cell tracking relative to QD605 [256]. An interesting point is related to the independence of vacuolization and autophagy in which larger-sized particles of rare earth oxides of ~800 nm can induce both autophagy and vacuolization, whereas smaller ones of ~200 nm only induce autophagy [313]. Thus, consideration of just vacuolization by TEM without a dynamic (i.e., flux) investigation may led to an incorrect interpretation.
Based on the above-mentioned findings, it might be said that the decision on the influence of particle size on autophagy is strongly dependent on other techniques that are performed in parallel as part of the investigation, and the analysis should be not only by static investigation (i.e., TEM) but also should include dynamic autophagic assays, analysis of cellular uptake, and consideration of IC50.
Conclusions and remarks
Nanoparticles as building blocks of nanotechnology have led to rapidly increasing applications in different fields. NPs can affect important cellular outcomes, including cell cycle, proliferation, differentiation and cell death. Although understanding the mechanisms of these pathways is important as we discussed in the Introduction, it should be noted that cytotoxic effects of NPs are highly dependent on their physico-chemical properties including, dispersity state, size, shape and charge. Also, some NPs may induce different forms of cell death pathways, depending on their physico-chemical properties. For example, TiO2 NPs can induce all 3 pathways of apoptosis in a dose-, size- and shape-dependent manner. Another important issue that should be taken into account is that the effects of NPs can change in biological systems due to effects of proteins and other compounds. This so-called ‘protein corona’ effect is an important topic in nontoxicity. The protein corona can have an effect on size and act as a protective shield against toxic effects of NPs such as inflammation and ROS-induced toxicity in the cell. All these things suggest that more research and optimized systems are need to define the exact mode of toxicity of a certain type of NP, and many cautions should be considered in deciphering the effects on NPs, particularly when we are going to use them as therapeutic tools.
Funding Statement
This work was supported by the HHS | NIH | National Institute of General Medical Sciences (NIGMS) [GM053396].
Acknowledgments
Reza Mohammadinejad is thankful for the financial support of Kerman University of Medical Sciences. Amin Moosavi acknowledges National Institute for Medical Research Development (NIMAD) grant 940943 and financial supports of National Institute of Genetic Engineering and Biotechnology and Iranian National Science Foundation. Daniel J. Klionsky is supported by NIH grant GM053396.
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Buzea C, Pacheco II, Robbie K.. Nanomaterials and nanoparticles: sources and toxicity. Biointerphases. 2007;2(4):MR17–MR71. [DOI] [PubMed] [Google Scholar]
- [2].Carlson C, Hussein SM, Schrand AM, et al. Unique cellular interaction of silver nanoparticles: size-dependent generation of reactive oxygen species. J Phys Chem B. 2008;112(43):13608–13619. [DOI] [PubMed] [Google Scholar]
- [3].Ray PC, Yu H, Fu PP. Toxicity and environmental risks of nanomaterials: challenges and future needs. J Environ Sci Health C Environ Carcinogenesis Ecotoxicology Rev. 2009;27(1):1–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Moosavi MA, Sharifi M, Ghafary SM, et al. Photodynamic N-TiO2 nanoparticle treatment induces controlled Ros-mediated autophagy and terminal differentiation of leukemia cells. Sci Rep. 2016;6:34413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kamaly N, Xiao Z, Valencia PM, et al. Targeted polymeric therapeutic nanoparticles: design, development and clinical translation. Chem Soc Rev. 2012;41(7):2971–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Davis ME, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7(9):771. [DOI] [PubMed] [Google Scholar]
- [7].Connor EE, Mwamuka J, Gole A, et al. Gold nanoparticles are taken up by human cells but do not cause acute cytotoxicity. Small. 2005;1(3):325–327. [DOI] [PubMed] [Google Scholar]
- [8].Özel RE, Alkasir RSJ, Ray K, et al. Comparative evaluation of intestinal nitric oxide in embryonic zebrafish exposed to metal oxide nanoparticles. Small. 2013;9(24):4250–4261. [DOI] [PubMed] [Google Scholar]
- [9].Goodman CM, McCusker CD, Yilmaz T, et al. Toxicity of gold nanoparticles functionalized with cationic and anionic side chains. Bioconjug Chem. 2004;15(4):897–900. [DOI] [PubMed] [Google Scholar]
- [10].Griffitt RJ, Luo J, Gao J, et al. Effects of particle composition and species on toxicity of metallic nanomaterials in aquatic organisms. Environ Toxicol Chem. 2008;27(9):1972–1978. [DOI] [PubMed] [Google Scholar]
- [11].Sohaebuddin SK, Thevenot PT, Baker D, et al. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part Fibre Toxicol. 2010;7 DOI: 10.1186/1743-8977-7-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wang S, Lu W, Tovmachenko O, et al. Challenge in understanding size and shape dependent toxicity of gold nanomaterials in human skin keratinocytes. Chem Phys Lett. 2008;463(1–3):145–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gwinn MR, Vallyathan V. Nanoparticles: health effects—pros and cons. Environ Health Perspect. 2006;114(12):1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tavakol S, Mousavi SMM, Tavakol B, et al. Mechano-transduction signals derived from self-assembling peptide nanofibers containing long motif of laminin influence neurogenesis in in-vitro and in-vivo. Mol Neurobiol. 2017;54(4):2483–2496. [DOI] [PubMed] [Google Scholar]
- [15].Tavakol S, Shakibapour S, Bidgoli SA. The level of testosterone, vitamin D, and irregular menstruation more important than omega-3 in non-symptomatic women will define the fate of multiple scleroses in future. Mol Neurobiol. 2016;55(1):1–8. [DOI] [PubMed] [Google Scholar]
- [16].Tavakol S, Musavi SMM, Tavakol B, et al. Noggin along with a self-assembling peptide nanofiber containing long motif of laminin induces tyrosine hydroxylase gene expression. Mol Neurobiol. 2017;54(6):4609–4616. [DOI] [PubMed] [Google Scholar]
- [17].Galluzzi L, Blomgren K, Kroemer G. Mitochondrial membrane permeabilization in neuronal injury. Nat Rev Neurosci. 2009;10(7):481–494. [DOI] [PubMed] [Google Scholar]
- [18].Alvarez A, Lacalle J, Cañavate M, et al. Cell death. A comprehensive approximation. Necrosis. Microsc Science, Technology, Appl Educ. 2010;2:1017–1024. [Google Scholar]
- [19].Chaabane W, User SD, El-Gazzah M, et al. Autophagy, apoptosis, mitoptosis and necrosis: interdependence between those pathways and effects on cancer. Arch Immunol Ther Exp (Warsz). 2013;61(1):43–58. [DOI] [PubMed] [Google Scholar]
- [20].Duprez L, Wirawan E, Berghe TV, et al. Major cell death pathways at a glance. Microbes Iinfection. 2009;11(13):1050–1062. [DOI] [PubMed] [Google Scholar]
- [21].Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4(5):313–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135(7):1161–1163. [DOI] [PubMed] [Google Scholar]
- [23].Grootjans S, Berghe TV, Vandenabeele P. Initiation and execution mechanisms of necroptosis: an overview. Cell Death Differ. 2017;24(7):1184–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Vandenabeele P, Galluzzi L, Berghe TV, et al. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11(10):700. [DOI] [PubMed] [Google Scholar]
- [25].Conrad M, Angeli JPF, Vandenabeele P, et al. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Festjens N, Berghe TV, Cornelis S, et al. RIP1, a kinase on the crossroads of a cell’s decision to live or die. Cell Death Differ. 2007;14(3):400–410. [DOI] [PubMed] [Google Scholar]
- [27].Vanlangenakker N, Berghe TV, Krysko DV, et al. Molecular mechanisms and pathophysiology of necrotic cell death. Curr Mol Med. 2008;8(3):207–220. [DOI] [PubMed] [Google Scholar]
- [28].Kerr J. A histochemical study of hypertrophy and ischaemic injury of rat liver with special reference to changes in lysosomes. J Pathol Bacteriol. 1965;90(2):419–435. [DOI] [PubMed] [Google Scholar]
- [29].Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nat Rev Mol Biol. 2001;2(7):545–550. [DOI] [PubMed] [Google Scholar]
- [30].Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35(4):495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Jayakiran M. Apoptosis-biochemistry: a mini review. J Clin Exp Pathol. 2015;5(1):1–4. [Google Scholar]
- [32].Fink SL, Cookson BT. Apoptosis, pyroptosis, and necrosis: mechanistic description of dead and dying eukaryotic cells. Infect Immun. 2005;73(4):1907–1916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ghavami S, Hashemi M, Ande SR, et al. Apoptosis and cancer: mutations within caspase genes. J Med Genet. 2009;46(8):497–510. [DOI] [PubMed] [Google Scholar]
- [34].Samali A, Zhivotovsky B, Jones D, et al. Apoptosis: cell death defined by caspase activation. Cell Death Differ. 1999;6(6):495. [DOI] [PubMed] [Google Scholar]
- [35].Coleman ML, Sahai EA, Yeo M, et al. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001;3(4):339–345. [DOI] [PubMed] [Google Scholar]
- [36].Birnbaum M, Clem R, Miller L. An apoptosis-inhibiting gene from a nuclear polyhedrosis virus encoding a polypeptide with Cys/His sequence motifs. J Virol. 1994;68(4):2521–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Black RA, Kronheim SR, Sleath PR. Activation of interleukin-1β by a co-induced protease. FEBS Lett. 1989;247(2):386–390. [DOI] [PubMed] [Google Scholar]
- [38].Black S, Kadyrov M, Kaufmann P, et al. Syncytial fusion of human trophoblast depends on caspase 8. Cell Death Differ. 2004;11(1):90. [DOI] [PubMed] [Google Scholar]
- [39].Takahashi R, Deveraux Q, Tamm I, et al. A single BIR domain of XIAP sufficient for inhibiting caspases. J Biol Chem. 1998;273(14):7787–7790. [DOI] [PubMed] [Google Scholar]
- [40].White E. Death-defying acts: a meeting review on apoptosis. Genes Dev. 1993;7(12):2277–2284. [DOI] [PubMed] [Google Scholar]
- [41].Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. 2009;122(4):437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Toivola D, Strnad P, Habtezion A, et al. Intermediate filaments take the heat as stress proteins. Trends Cell Biol. 2010;20(2):79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wyllie AH. “Where, O death, is thy sting?” A brief review of apoptosis biology. Mol Neurobiol. 2010;42(1):4–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Arboleda G, Morales LC, Benítez B, et al. Regulation of ceramide-induced neuronal death: cell metabolism meets neurodegeneration. Brain Res Rev. 2009;59(2):333–346. [DOI] [PubMed] [Google Scholar]
- [45].Assefa Z, Van Laethem A, Garmyn M, et al. Ultraviolet radiation-induced apoptosis in keratinocytes: on the role of cytosolic factors. Biochim Biophys Acta. 2005;1755(2):90–106. [DOI] [PubMed] [Google Scholar]
- [46].Dmitrieva NI, Burg MB. Hypertonic stress response. Mutat Res. 2005;569(1):65–74. [DOI] [PubMed] [Google Scholar]
- [47].Greijer A, Van der Wall E.. The role of hypoxia inducible factor 1 (HIF-1) in hypoxia induced apoptosis. J Clin Pathol. 2004;57(10):1009–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Raj GV, Barki-Harrington L, Kue PF, et al. Guanosine phosphate binding protein coupled receptors in prostate cancer: a review. J Urol. 2002;167(3):1458–1463. [PubMed] [Google Scholar]
- [49].Zheng M, Zhu W, Han Q, et al. Emerging concepts and therapeutic implications of β-adrenergic receptor subtype signaling. Pharmacol Ther. 2005;108(3):257–268. [DOI] [PubMed] [Google Scholar]
- [50].Chipuk JE, Green DR. How do BCL-2 proteins induce mitochondrial outer membrane permeabilization? Trends Cell Biol. 2008;18(4):157–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Khoury CM, Greenwood MT. The pleiotropic effects of heterologous Bax expression in yeast. Biochim Biophys Acta. 2008;1783(7):1449–1465. [DOI] [PubMed] [Google Scholar]
- [52].Lalier L, Cartron P-F, Juin P, et al. Bax activation and mitochondrial insertion during apoptosis. Apoptosis. 2007;12(5):887–896. [DOI] [PubMed] [Google Scholar]
- [53].Orrenius S, Gogvadze V, Zhivotovsky B. Mitochondrial oxidative stress: implications for cell death. Annu Rev Pharmacol Toxicol. 2007;47:143–183. [DOI] [PubMed] [Google Scholar]
- [54].Pourova J, Kottova M, Voprsalova M, et al. Reactive oxygen and nitrogen species in normal physiological processes. Acta Physiologica. 2010;198(1):15–35. [DOI] [PubMed] [Google Scholar]
- [55].Fulda S, Gorman AM, Hori O, et al. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010(214074):1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Gutteridge J, Halliwell B. Free radicals and antioxidants in the year 2000: a historical look to the future. Ann N Y Acad Sci. 2000;899(1):136–147. [DOI] [PubMed] [Google Scholar]
- [57].Winterbourn CC, Hampton MB. Thiol chemistry and specificity in redox signaling. Free Radic Biol Med. 2008;45(5):549–561. [DOI] [PubMed] [Google Scholar]
- [58].Thorpe GW, Fong CS, Alic N, et al. Cells have distinct mechanisms to maintain protection against different reactive oxygen species: oxidative-stress-response genes. Proc Natl Acad Sci U S A. 2004;101(17):6564–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Dengjel J, Abeliovich H. Roles of mitophagy in cellular physiology and development. Cell Tissue Res. 2017;367(1):95–109. [DOI] [PubMed] [Google Scholar]
- [60].Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. 2010;221(1):3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Cohignac V, Landry MJ, Boczkowski J, et al. Autophagy as a possible underlying mechanism of nanomaterial toxicity. Nanomaterials. 2014;4(3):548–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Tsuboyama K, Koyama-Honda I, Sakamaki Y, et al. The ATG conjugation systems are important for degradation of the inner autophagosomal membrane. Science (80-). 2016;354(6315):1036–1041. [DOI] [PubMed] [Google Scholar]
- [63].Berg TO, Fengsrud M, Strømhaug PE, et al. Isolation and characterization of rat liver amphisomes evidence for fusion of autophagosomes with both early and late endosoMES. J Biol Chem. 1998;273(34):21883–21892. [DOI] [PubMed] [Google Scholar]
- [64].Gutierrez MG, Vázquez CL, Munafó DB, et al. Autophagy induction favours the generation and maturation of the Coxiella‐replicative vacuoles. Cell Microbiol. 2005;7(7):981–993. [DOI] [PubMed] [Google Scholar]
- [65].Renna M, Schaffner C, Winslow AR, et al. Autophagic substrate clearance requires activity of the syntaxin-5 SNARE complex. J Cell Sci. 2011;124(3):469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tanida I, Minematsu-Ikeguchi N, Ueno T, et al. Lysosomal turnover, but not a cellular level, of endogenous LC3 is a marker for autophagy. Autophagy. 2005;1(2):84–91. [DOI] [PubMed] [Google Scholar]
- [67].Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16(8):461–472. [DOI] [PubMed] [Google Scholar]
- [68].Webb JL, Ravikumar B, Rubinsztein DC. Microtubule disruption inhibits autophagosome-lysosome fusion: implications for studying the roles of aggresomes in polyglutamine diseases. Int J Biochem Cell Biol. 2004;36(12):2541–2550. [DOI] [PubMed] [Google Scholar]
- [69].Wei Y, Pattingre S, Sinha S, et al. JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell. 2008;30(6):678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].De Stefano D, Carnuccio R, Maiuri MC. Nanomaterials toxicity and cell death modalities. J Drug Deliv. 2012;2012(167896):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Manke A, Wang L, Rojanasakul Y. Mechanisms of nanoparticle-induced oxidative stress and toxicity. Biomed Res Int. 2013;2013(942916):1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Tavakol S, Nikpour MR, Hoveizi E, et al. Investigating the effects of particle size and chemical structure on cytotoxicity and bacteriostatic potential of nano hydroxyapatite/chitosan/silica and nano hydroxyapatite/chitosan/silver; as antibacterial bone substitutes. J Nanopart Res. 2014;16(10):2622. [Google Scholar]
- [73].Andón FT, Fadeel B. Programmed cell death: molecular mechanisms and implications for safety assessment of nanomaterials. Acc Chem Res. 2012;46(3):733–742. [DOI] [PubMed] [Google Scholar]
- [74].Harhaji L, Isakovic A, Raicevic N, et al. Multiple mechanisms underlying the anticancer action of nanocrystalline fullerene. Eur J Pharmacol. 2007;568(1):89–98. [DOI] [PubMed] [Google Scholar]
- [75].Ciftci H, Türk M, Tamer U, et al. Silver nanoparticles: cytotoxic, apoptotic, and necrotic effects on MCF-7 cells. Turkish J Biol. 2013;37(5):573–581. [Google Scholar]
- [76].Foldbjerg R, Olesen P, Hougaard M, et al. PVP-coated silver nanoparticles and silver ions induce reactive oxygen species, apoptosis and necrosis in THP-1 monocytes. Toxicol Lett. 2009;190(2):156–162. [DOI] [PubMed] [Google Scholar]
- [77].Arora S, Jain J, Rajwade J, et al. Cellular responses induced by silver nanoparticles: in vitro studies. Toxicol Lett. 2008;179(2):93–100. [DOI] [PubMed] [Google Scholar]
- [78].Asare N, Instanes C, Sandberg WJ, et al. Cytotoxic and genotoxic effects of silver nanoparticles in testicular cells. Toxicology. 2012;291(1):65–72. [DOI] [PubMed] [Google Scholar]
- [79].Kim TH, Kim M, Park HS, et al. Size‐dependent cellular toxicity of silver nanoparticles. J Biomed Mater Res. 2012;100(4):1033–1043. [DOI] [PubMed] [Google Scholar]
- [80].Xia T, Kovochich M, Liong M, et al. Cationic polystyrene nanosphere toxicity depends on cell-specific endocytic and mitochondrial injury pathways. ACS Nano. 2007;2(1):85–96. [DOI] [PubMed] [Google Scholar]
- [81].Braydich-Stolle LK, Schaeublin NM, Murdock RC, et al. Crystal structure mediates mode of cell death in TiO2 nanotoxicity. J Nanoparticle Res. 2009;11(6):1361–1374. [Google Scholar]
- [82].Pan Y, Leifert A, Ruau D, et al. Gold nanoparticles of diameter 1.4 nm trigger necrosis by oxidative stress and mitochondrial damage. Small. 2009;5(18):2067–2076. [DOI] [PubMed] [Google Scholar]
- [83].Pan Y, Neuss S, Leifert A, et al. Size‐dependent cytotoxicity of gold nanoparticles. Small. 2007;3(11):1941–1949. [DOI] [PubMed] [Google Scholar]
- [84].Schaeublin NM, Braydich-Stolle LK, Schrand AM, et al. Surface charge of gold nanoparticles mediates mechanism of toxicity. Nanoscale. 2011;3(2):410–420. [DOI] [PubMed] [Google Scholar]
- [85].Wei X, Shao B, He Z, et al. Cationic nanocarriers induce cell necrosis through impairment of Na+/K+-ATPase and cause subsequent inflammatory response. Cell Res. 2015;25(2):237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Oh W-K, Kim S, Kwon O, et al. Shape-dependent cytotoxicity of polyaniline nanomaterials in human fibroblast cells. J Nanosci Nanotechnol. 2011;11(5):4254–4260. [DOI] [PubMed] [Google Scholar]
- [87].Bauer AT, Strozyk EA, Gorzelanny C, et al. Cytotoxicity of silica nanoparticles through exocytosis of von Willebrand factor and necrotic cell death in primary human endothelial cells. Biomaterials. 2011;32(33):8385–8393. [DOI] [PubMed] [Google Scholar]
- [88].Sonkusre P, Cameotra SS. Biogenic selenium nanoparticles induce ROS-mediated necroptosis in PC-3 cancer cells through TNF activation. J Nanobiotechnology. 2017;15(1):43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Panzarini E, Mariano S, Dini L, editors. Glycans coated silver nanoparticles induces autophagy and necrosis in HeLa cells. AIP Conference Proceedings; 2015. AIP Publishing. [Google Scholar]
- [90].Panzarini E, Mariano S, Dini L, editors. Investigations of the toxic effects of glycans-based silver nanoparticles on different types of human cells. AIP Conference Proceedings; 2017. AIP Publishing. [Google Scholar]
- [91].Vergallo C, Panzarini E, Carata E, et al, editors. Cytotoxicity of β-D-glucose/sucrose-coated silver nanoparticles depends on cell type, nanoparticles concentration and time of incubation. AIP Conference Proceedings; 2016. AIP Publishing. [Google Scholar]
- [92].Liu M, Gu X, Zhang K, et al. Gold nanoparticles trigger apoptosis and necrosis in lung cancer cells with low intracellular glutathione. J Nanopart Res. 2013;15(8):1745. [Google Scholar]
- [93].Xia T, Kovochich M, Brant J, et al. Comparison of the abilities of ambient and manufactured nanoparticles to induce cellular toxicity according to an oxidative stress paradigm. Nano Lett. 2006;6(8):1794–1807. [DOI] [PubMed] [Google Scholar]
- [94].Zhang T, Wang L, Chen Q, et al. Cytotoxic potential of silver nanoparticles. Yonsei Med J. 2014;55(2):283–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Asharani P, Hande MP, Valiyaveettil S. Anti-proliferative activity of silver nanoparticles. BMC Cell Biol. 2009;10(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Tran QH, Le A-T. Silver nanoparticles: synthesis, properties, toxicology, applications and perspectives. Adv Nat Sci: Nanosci Nanotech. 2013;4(3):033001. [Google Scholar]
- [97].Li L, Sun J, Li X, et al. Controllable synthesis of monodispersed silver nanoparticles as standards for quantitative assessment of their cytotoxicity. Biomaterials. 2012;33(6):1714–1721. [DOI] [PubMed] [Google Scholar]
- [98].Kawata K, Osawa M, Okabe S. In vitro toxicity of silver nanoparticles at noncytotoxic doses to HepG2 human hepatoma cells. Environ Sci Technol. 2009;43(15):6046–6051. [DOI] [PubMed] [Google Scholar]
- [99].Carlson C, Hussain SM, Schrand AM, et al. Unique cellular interaction of silver nanoparticles: size-dependent generation of reactive oxygen species. J Phys Chem B. 2008;112(43):13608–13619. [DOI] [PubMed] [Google Scholar]
- [100].Hussain S, Hess K, Gearhart J, et al. In vitro toxicity of nanoparticles in BRL 3A rat liver cells. Toxicol Vitro. 2005;19(7):975–983. [DOI] [PubMed] [Google Scholar]
- [101].Rahman M, Wang J, Patterson T, et al. Expression of genes related to oxidative stress in the mouse brain after exposure to silver-25 nanoparticles. Toxicol Lett. 2009;187(1):15–21. [DOI] [PubMed] [Google Scholar]
- [102].Tavakol S, Hoveizi E, Kharrazi S, et al. Organelles and chromatin fragmentation of human umbilical vein endothelial cell influence by the effects of zeta potential and size of silver nanoparticles in different manners. Artificial Cells, Nanomedicine, Biotechnology. 2017;45(4):817–823. [DOI] [PubMed] [Google Scholar]
- [103].Nemmar A, Nemery B, Hoet PH, et al. Silica particles enhance peripheral thrombosis: key role of lung macrophage–neutrophil cross-talk. Am J Respir Crit Care Med. 2005;171(8):872–879. [DOI] [PubMed] [Google Scholar]
- [104].Hamilton RF, Thakur SA, Holian A. Silica binding and toxicity in alveolar macrophages. Free Radic Biol Med. 2008;44(7):1246–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Lin W, Huang Y-W, Zhou X-D, et al. In vitro toxicity of silica nanoparticles in human lung cancer cells. Toxicol Appl Pharmacol. 2006;217(3):252–259. [DOI] [PubMed] [Google Scholar]
- [106].Park E-J, Park K. Oxidative stress and pro-inflammatory responses induced by silica nanoparticles in vivo and in vitro. Toxicol Lett. 2009;184(1):18–25. [DOI] [PubMed] [Google Scholar]
- [107].Wilhelmi V, Fischer U, Weighardt H, et al. Zinc oxide nanoparticles induce necrosis and apoptosis in macrophages in a p47phox-and Nrf2-independent manner. PLoS One. 2013;8(6):e65704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lai L, Jin J-C, Xu Z-Q, et al. Necrotic cell death induced by the protein-mediated intercellular uptake of CdTe quantum dots. Chemosphere. 2015;135:240–249. [DOI] [PubMed] [Google Scholar]
- [109].García-Hevia L, Valiente R, Martín-Rodríguez R, et al. Nano-ZnO leads to tubulin macrotube assembly and actin bundling, triggering cytoskeletal catastrophe and cell necrosis. Nanoscale. 2016;8(21):10963–10973. [DOI] [PubMed] [Google Scholar]
- [110].Kumar G, Degheidy H, Casey BJ, et al. Flow cytometry evaluation of in vitro cellular necrosis and apoptosis induced by silver nanoparticles. Food Chem Toxicol. 2015;85:45–51. [DOI] [PubMed] [Google Scholar]
- [111].Braydich-Stolle L, Hussain S, Schlager JJ, et al. In vitro cytotoxicity of nanoparticles in mammalian germline stem cells. Toxicol Sci. 2005;88(2):412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Arora S, Jain J, Rajwade J, et al. Interactions of silver nanoparticles with primary mouse fibroblasts and liver cells. Toxicol Appl Pharmacol. 2009;236(3):310–318. [DOI] [PubMed] [Google Scholar]
- [113].Foldbjerg R, Dang DA, Autrup H. Cytotoxicity and genotoxicity of silver nanoparticles in the human lung cancer cell line, A549. Arch Toxicol. 2011;85(7):743–750. [DOI] [PubMed] [Google Scholar]
- [114].Abdelhalim MAK, Jarrar BM. Gold nanoparticles induced cloudy swelling to hydropic degeneration, cytoplasmic hyaline vacuolation, polymorphism, binucleation, karyopyknosis, karyolysis, karyorrhexis and necrosis in the liver. Lipids Health Dis. 2011;10(1):166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Isakovic A, Markovic Z, Todorovic-Markovic B, et al. Distinct cytotoxic mechanisms of pristine versus hydroxylated fullerene. Toxicological Sci. 2006;91(1):173–183. [DOI] [PubMed] [Google Scholar]
- [116].Wielgus AR, Zhao B, Chignell CF, et al. Phototoxicity and cytotoxicity of fullerol in human retinal pigment epithelial cells. Toxicol Appl Pharmacol. 2010;242(1):79–90. [DOI] [PubMed] [Google Scholar]
- [117].Ershova E, Sergeeva V, Chausheva A, et al. Toxic and DNA damaging effects of a functionalized fullerene in human embryonic lung fibroblasts. Mutat Res Genet Toxicol Environ Mutagen. 2016;805:46–57. [DOI] [PubMed] [Google Scholar]
- [118].Dash SK, Ghosh T, Roy S, et al. Zinc sulfide nanoparticles selectively induce cytotoxic and genotoxic effects on leukemic cells: involvement of reactive oxygen species and tumor necrosis factor alpha. J Appl Toxicol. 2014;34(11):1130–1144. [DOI] [PubMed] [Google Scholar]
- [119].Kim J-H, Jeong MS, Kim D-Y, et al. Zinc oxide nanoparticles induce lipoxygenase-mediated apoptosis and necrosis in human neuroblastoma SH-SY5Y cells. Neurochem Int. 2015;90:204–214. [DOI] [PubMed] [Google Scholar]
- [120].Wang H, Liu Z, Gou Y, et al. Apoptosis and necrosis induced by novel realgar quantum dots in human endometrial cancer cells via endoplasmic reticulum stress signaling pathway. Int J Nanomedicine. 2015;10:5505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Zhang Y, Hong G, Zhang Y, et al. Ag2S quantum dot: a bright and biocompatible fluorescent nanoprobe in the second near-infrared window. ACS Nano. 2012;6(5):3695–3702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Yeh Y-C, Saha K, Yan B, et al. The role of ligand coordination on the cytotoxicity of cationic quantum dots in HeLa cells. Nanoscale. 2013;5(24):12140–12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Qin Y, Wang H, Liu ZY, et al. Realgar quantum dots induce apoptosis and necrosis in HepG2 cells through endoplasmic reticulum stress. Biomed Rep. 2015;3(5):657–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Stan MS, Memet I, Sima C, et al. Si/SiO 2 quantum dots cause cytotoxicity in lung cells through redox homeostasis imbalance. Chem Biol Interact. 2014;220:102–115. [DOI] [PubMed] [Google Scholar]
- [125].Ou L, Song B, Liang H, et al. Toxicity of graphene-family nanoparticles: a general review of the origins and mechanisms [journal article]. Part Fibre Toxicol. 2016. October 31;13(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Kim S, Ryu DY. Silver nanoparticle-induced oxidative stress, genotoxicity and apoptosis in cultured cells and animal tissues [Article]. J Appl Toxicol. 2013;33(2):78–89. [DOI] [PubMed] [Google Scholar]
- [127].Li T, Kon N, Jiang L, et al. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012;149(6):1269–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Antonsson B, Martinou JC. The Bcl-2 protein family. Exp Cell Res. 2000;256(1):50–57. [DOI] [PubMed] [Google Scholar]
- [129].Green DR, Reed JC. Mitochondria and apoptosis. Science (80- ). 1998;281(5381):1309–1312. [DOI] [PubMed] [Google Scholar]
- [130].Shafagh M, Rahmani F, Delirezh N. CuO nanoparticles induce cytotoxicity and apoptosis in human K562 cancer cell line via mitochondrial pathway, through reactive oxygen species and P53 [Article]. Iran J Basic Med Sci. 2015;18(10):993–1000. [PMC free article] [PubMed] [Google Scholar]
- [131].Kermanizadeh A, Chauché C, Brown DM, et al. The role of intracellular redox imbalance in nanomaterial induced cellular damage and genotoxicity: A review [Review]. Environ Mol Mutagen. 2015;56(2):111–124. [DOI] [PubMed] [Google Scholar]
- [132].Ahmad J, Ahamed M, Akhtar MJ, et al. Apoptosis induction by silica nanoparticles mediated through reactive oxygen species in human liver cell line HepG2. Toxicol Appl Pharmacol. 2012;259(2):160–168. [DOI] [PubMed] [Google Scholar]
- [133].Ye Y, Liu J, Xu J, et al. Nano-SiO2 induces apoptosis via activation of p53 and Bax mediated by oxidative stress in human hepatic cell line. Toxicol in Vitro. 2010;24:3–758. [DOI] [PubMed] [Google Scholar]
- [134].Ahamed M, Akhtar MJ, Raja M, et al. ZnO nanorod-induced apoptosis in human alveolar adenocarcinoma cells via p53, survivin and bax/bcl-2 pathways: role of oxidative stress. Nanomedicine: Nanotechnology, Biol Med. 2011;7(6):904–913. [DOI] [PubMed] [Google Scholar]
- [135].Ahamed M, Karns M, Goodson M, et al. DNA damage response to different surface chemistry of silver nanoparticles in mammalian cells. Toxicol Appl Pharmacol. 2008;233(3):404–410. [DOI] [PubMed] [Google Scholar]
- [136].Gopinath P, Gogoi SK, Sanpui P, et al. Signaling gene cascade in silver nanoparticle induced apoptosis. Colloids Surf B Biointerfaces. 2010;77(2):240–245. [DOI] [PubMed] [Google Scholar]
- [137].Hsin YH, Chen CF, Huang S, et al. The apoptotic effect of nanosilver is mediated by a ROS- and JNK-dependent mechanism involving the mitochondrial pathway in NIH3T3 cells [Article]. Toxicol Lett. 2008;179(3):130–139. [DOI] [PubMed] [Google Scholar]
- [138].Panzarini E, Mariano S, Vergallo C, et al. Glucose capped silver nanoparticles induce cell cycle arrest in HeLa cells. Toxicol Vitro. 2017;41:64–74. [DOI] [PubMed] [Google Scholar]
- [139].Liu Y, Li X, Bao S, et al. Plastic protein microarray to investigate the molecular pathways of magnetic nanoparticle-induced nanotoxicity [Article]. Nanotechnology. 2013;24:17. [DOI] [PubMed] [Google Scholar]
- [140].Siddiqui MA, Alhadlaq HA, Ahmad J, et al. Copper oxide nanoparticles induced mitochondria mediated apoptosis in human hepatocarcinoma cells. PLoS One. 2013;8(8):e69534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Hsieh H-C, Chen C-M, Hsieh W-Y, et al. ROS-induced toxicity: exposure of 3T3, RAW264. 7, and MCF7 cells to superparamagnetic iron oxide nanoparticles results in cell death by mitochondria-dependent apoptosis. J Nanoparticle Res. 2015;17(2):71. [Google Scholar]
- [142].Zhao J, Bowman L, Zhang X, et al. Titanium dioxide (TiO2) nanoparticles induce JB6 cell apoptosis through activation of the caspase-8/Bid and mitochondrial pathways. J Toxicol Environ Health Part A. 2009;72(19):1141–1149. [DOI] [PubMed] [Google Scholar]
- [143].Ryu W-I, Park Y-H, Bae HC, et al. ZnO nanoparticle induces apoptosis by ROS triggered mitochondrial pathway in human keratinocytes. Mol Cell Toxicol. 2014;10(4):387–391. [Google Scholar]
- [144].Yan X, Yang W, Shao Z, et al. Triggering of apoptosis in osteosarcoma cells by graphene/single‐walled carbon nanotube hybrids via the ROS‐mediated mitochondrial pathway. J Biomed Mater Res Part A. 2017;105(2):443–453. [DOI] [PubMed] [Google Scholar]
- [145].Zou Y, Li Q, Jiang L, et al. DNA hypermethylation of CREB3L1 and Bcl-2 associated with the mitochondrial-mediated apoptosis via PI3K/Akt pathway in human BEAS-2B cells exposure to silica nanoparticles. PLoS One. 2016;11(6):e0158475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Hu W, Peng C, Luo W, et al. Graphene-based antibacterial paper. ACS Nano. 2010;4 DOI: 10.1021/nn101097v [DOI] [PubMed] [Google Scholar]
- [147].Li Y, Liu Y, Fu Y, et al. The triggering of apoptosis in macrophages by pristine graphene through the MAPK and TGF-beta signaling pathways. Biomaterials. 2012;33 DOI: 10.1016/j.biomaterials.2011.09.091 [DOI] [PubMed] [Google Scholar]
- [148].Duch MC, Budinger GR, Liang YT, et al. Minimizing oxidation and stable nanoscale dispersion improves the biocompatibility of graphene in the lung. Nano Lett. 2011;11 DOI: 10.1021/nl202515a [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Chatterjee N, Eom HJ, Choi J. A systems toxicology approach to the surface functionality control of graphene-cell interactions. Biomaterials. 2014;35 DOI: 10.1016/j.biomaterials.2013.09.108 [DOI] [PubMed] [Google Scholar]
- [150].Ding Z, Zhang Z, Ma H, et al. In vitro hemocompatibility and toxic mechanism of graphene oxide on human peripheral blood T lymphocytes and serum albumin. ACS Appl Mater Interf. 2014;6 DOI: 10.1021/am505084s [DOI] [PubMed] [Google Scholar]
- [151].Yao Y, Costa M. Genetic and epigenetic effects of nanoparticles. J Mol Genet Med. 2013;7(4):1–6. [Google Scholar]
- [152].Ahmad J, Alhadlaq HA, Siddiqui MA, et al. Concentration‐dependent induction of reactive oxygen species, cell cycle arrest and apoptosis in human liver cells after nickel nanoparticles exposure. Environ Toxicol. 2015;30(2):137–148. [DOI] [PubMed] [Google Scholar]
- [153].Christen V, Treves S, Duong FHT, et al. Activation of endoplasmic reticulum stress response by hepatitis viruses up-regulates protein phosphatase 2A. Hepatology. 2007;46(2):558–565. [DOI] [PubMed] [Google Scholar]
- [154].Janssens V, Goris J. Protein phosphatase 2A: A highly regulated family of serine/threonine phosphatases implicated in cell growth and signalling. Biochem J. 2001;353(3):417–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Christen V, Camenzind M, Fent K. Silica nanoparticles induce endoplasmic reticulum stress response, oxidative stress and activate the mitogen-activated protein kinase (MAPK) signaling pathway. Toxicol Rep. 2014. 2014 January 01;1:1143–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ariano P, Zamburlin P, Gilardino A, et al. Interaction of spherical silica nanoparticles with neuronal cells: size-dependent toxicity and perturbation of calcium homeostasis. Small. 2011;7(6):766–774. [DOI] [PubMed] [Google Scholar]
- [157].Napierska D, Thomassen LCJ, Lison D, et al. The nanosilica hazard: another variable entity. Part Fibre Toxicol. 2010;7 DOI: 10.1186/1743-8977-7-39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].AshaRani PV, Hande MP, Valiyaveettil S. Anti-proliferative activity of silver nanoparticles. BMC Cell Biol. 2009;10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Koeneman BA, Zhang Y, Westerhoff P, et al. Toxicity and cellular responses of intestinal cells exposed to titanium dioxide. Cell Biol Toxicol. 2010;26(3):225–238. [DOI] [PubMed] [Google Scholar]
- [160].Qian Sun WZ, Zhang W, Li Q, et al. Zinc deficiency mediates alcohol-induced apoptotic cell death in the liver of rats through activating ER and mitochondrial cell death pathways. Am J Physiol Gastrointest Liver Physiol. 2015. May 1;308(9):G757–G766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Chen R, Huo L, Shi X, et al. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. ACS Nano. 2014;8(3):2562–2574. [DOI] [PubMed] [Google Scholar]
- [162].Hou CC, Tsai TL, Su WP, et al. Pronounced induction of endoplasmic reticulum stress and tumor suppression by surfactant-free poly(lactic-co-glycolic acid) nanoparticles via modulation of the PI3K signaling pathway. Int J Nanomedicine. 2013;8:2689–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Tsai YY, Huang YH, Chao YL, et al. Identification of the nanogold particle-induced endoplasmic reticulum stress by omic techniques and systems biology analysis. ACS Nano. 2011;5(12):9354–9369. [DOI] [PubMed] [Google Scholar]
- [164].Rosebeck S, Sudini K, Chen T, et al. Involvement of Noxa in mediating cellular ER stress responses to lytic virus infection. Virology. 2011;417(2):293–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Yan J, Zhong N, Liu G, et al. Usp9x- and Noxa-mediated Mcl-1 downregulation contributes to pemetrexed-induced apoptosis in human non-small-cell lung cancer cells. Cell Death Dis. 2014;5:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Didonato JA, Mercurio F, Karin M. NF-κB and the link between inflammation and cancer. Immunol Rev. 2012;246(1):379–400. [DOI] [PubMed] [Google Scholar]
- [167].Meena R, Rani M, Pal R, et al. Nano-TiO2-induced apoptosis by oxidative stress-mediated DNA damage and activation of p53 in human embryonic kidney cells. Appl Biochem Biotechnol. 2012;167(4):791–808. [DOI] [PubMed] [Google Scholar]
- [168].Amaral JD, Castro RE, Steer CJ, et al. p53 and the regulation of hepatocyte apoptosis: implications for disease pathogenesis. Trends Mol Med. 2009;15(11):531–541. [DOI] [PubMed] [Google Scholar]
- [169].Simard J-C, Durocher I, Girard D. Silver nanoparticles induce irremediable endoplasmic reticulum stress leading to unfolded protein response dependent apoptosis in breast cancer cells [journal article]. Apoptosis. 2016. November 01;21(11):1279–1290. [DOI] [PubMed] [Google Scholar]
- [170].Chen R, Huo L, Shi X, et al. Endoplasmic reticulum stress induced by zinc oxide nanoparticles is an earlier biomarker for nanotoxicological evaluation. Acs Nano. 2014;8:2574. [DOI] [PubMed] [Google Scholar]
- [171].Yang X, Shao H, Liu W, et al. Endoplasmic reticulum stress and oxidative stress are involved in ZnO nanoparticle-induced hepatotoxicity. Toxicol Lett. 2015;234(1):40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kuang H, Yang P, Yang L, et al. Size dependent effect of ZnO nanoparticles on endoplasmic reticulum stress signaling pathway in murine liver. J Hazard Mater. 2016;317:119–126. [DOI] [PubMed] [Google Scholar]
- [173].Ghooshchian M, Khodarahmi P, Tafvizi F. Apoptosis-mediated neurotoxicity and altered gene expression induced by silver nanoparticles. Toxicol Ind Health. 2017;33(10):757–764. [DOI] [PubMed] [Google Scholar]
- [174].Piao MJ, Kang KA, Lee IK, et al. Silver nanoparticles induce oxidative cell damage in human liver cells through inhibition of reduced glutathione and induction of mitochondria-involved apoptosis. Toxicol Lett. 2011;201(1):92–100. [DOI] [PubMed] [Google Scholar]
- [175].Kim HR, Da YS, Park YJ, et al. Silver nanoparticles induce p53-mediated apoptosis in human bronchial epithelial (BEAS-2B) cells. J Toxicol Sci. 2014;39(3):401–412. [DOI] [PubMed] [Google Scholar]
- [176].Satapathy SR, Mohapatra P, Preet R, et al. Silver-based nanoparticles induce apoptosis in human colon cancer cells mediated through p53. Nanomedicine. 2013;8(8):1307–1322. [DOI] [PubMed] [Google Scholar]
- [177].Sanpui P, Chattopadhyay A, Ghosh SS. Induction of apoptosis in cancer cells at low silver nanoparticle concentrations using chitosan nanocarrier. ACS Appl Mater Interfaces. 2011;3(2):218–228. [DOI] [PubMed] [Google Scholar]
- [178].Hsin Y-H, Chen C-F, Huang S, et al. The apoptotic effect of nanosilver is mediated by a ROS-and JNK-dependent mechanism involving the mitochondrial pathway in NIH3T3 cells. Toxicol Lett. 2008;179(3):130–139. [DOI] [PubMed] [Google Scholar]
- [179].Selim ME, Hendi AA. Gold nanoparticles induce apoptosis in MCF-7 human breast cancer cells. Asian Pac J Cancer Prev. 2012;13(4):1617–1620. [DOI] [PubMed] [Google Scholar]
- [180].Baharara J, Ramezani T, Divsalar A, et al. Induction of apoptosis by green synthesized gold nanoparticles through activation of caspase-3 and 9 in human cervical cancer cells. Avicenna J Med Biotechnol. 2016;8(2):75. [PMC free article] [PubMed] [Google Scholar]
- [181].Noël C, Simard J-C, Girard D. Gold nanoparticles induce apoptosis, endoplasmic reticulum stress events and cleavage of cytoskeletal proteins in human neutrophils. Toxicol Vitro. 2016;31:12–22. [DOI] [PubMed] [Google Scholar]
- [182].Gao W, Xu K, Ji L, et al. Effect of gold nanoparticles on glutathione depletion-induced hydrogen peroxide generation and apoptosis in HL7702 cells. Toxicol Lett. 2011;205(1):86–95. [DOI] [PubMed] [Google Scholar]
- [183].Mata R, Nakkala JR, Sadras SR. Polyphenol stabilized colloidal gold nanoparticles from Abutilon indicum leaf extract induce apoptosis in HT-29 colon cancer cells. Colloids Surf B Biointerfaces. 2016;143:499–510. [DOI] [PubMed] [Google Scholar]
- [184].Wahab R, Dwivedi S, Khan F, et al. Statistical analysis of gold nanoparticle-induced oxidative stress and apoptosis in myoblast (C2C12) cells. Colloids Surf B Biointerfaces. 2014;123:664–672. [DOI] [PubMed] [Google Scholar]
- [185].Pongrakhananon V, Luanpitpong S, Stueckle TA, et al. Carbon nanotubes induce apoptosis resistance of human lung epithelial cells through FLICE-inhibitory protein. Toxicol Sci. 2015. February;143(2):499–511. PubMed PMID: 25412619; PubMed Central PMCID: PMCPMC4306727. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Mocan T, Matea CT, Cojocaru I, et al. Photothermal treatment of human pancreatic cancer using PEGylated multi-walled carbon nanotubes induces apoptosis by triggering mitochondrial membrane depolarization mechanism. J Cancer. 2014;5(8):679–688. PubMed PMID: 25258649; PubMed Central PMCID: PMCPMC4174512. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [187].Ye S, Jiang Y, Zhang H, et al. Multi-walled carbon nanotubes induce apoptosis in RAW 264.7 cell-derived osteoclasts through mitochondria-mediated death pathway. J Nanosci Nanotechnol. 2012. March;12(3):2101–2112. PubMed PMID: 22755027; eng. [DOI] [PubMed] [Google Scholar]
- [188].Wang X, Guo J, Chen T, et al. Multi-walled carbon nanotubes induce apoptosis via mitochondrial pathway and scavenger receptor. Toxicol In Vitro. 2012. September;26(6):799–806. PubMed PMID: 22664788; eng. [DOI] [PubMed] [Google Scholar]
- [189].Nam CW, Kang SJ, Kang YK, et al. Cell growth inhibition and apoptosis by SDS-solubilized single-walled carbon nanotubes in normal rat kidney epithelial cells. Arch Pharm Res. 2011. April;34(4):661–669. PubMed PMID: 21544732; eng. [DOI] [PubMed] [Google Scholar]
- [190].Srivastava RK, Pant AB, Kashyap MP, et al. Multi-walled carbon nanotubes induce oxidative stress and apoptosis in human lung cancer cell line-A549. Nanotoxicology. 2011. June;5(2):195–207. PubMed PMID: 20804439; eng. [DOI] [PubMed] [Google Scholar]
- [191].Ravichandran P, Baluchamy S, Sadanandan B, et al. Multiwalled carbon nanotubes activate NF-kappaB and AP-1 signaling pathways to induce apoptosis in rat lung epithelial cells. Apoptosis. 2010. December;15(12):1507–1516. PubMed PMID: 20694747; eng. [DOI] [PubMed] [Google Scholar]
- [192].Patlolla A, Knighten B, Tchounwou P. Multi-walled carbon nanotubes induce cytotoxicity, genotoxicity and apoptosis in normal human dermal fibroblast cells. Ethn Dis. 2010. Winter;20(Suppl 1):S1–65–72 PubMed PMID: 20521388; PubMed Central PMCID: PMCPMC2902968. eng. [PMC free article] [PubMed] [Google Scholar]
- [193].Ravichandran P, Periyakaruppan A, Sadanandan B, et al. Induction of apoptosis in rat lung epithelial cells by multiwalled carbon nanotubes. J Biochem Mol Toxicol. 2009. Sep-Oct;23(5):333–344. PubMed PMID: 19827037; eng. [DOI] [PubMed] [Google Scholar]
- [194].Elgrabli D, Abella-Gallart S, Robidel F, et al. Induction of apoptosis and absence of inflammation in rat lung after intratracheal instillation of multiwalled carbon nanotubes. Toxicology. 2008. November 20;253(1–3):131–136. PubMed PMID: 18834917; eng. [DOI] [PubMed] [Google Scholar]
- [195].Bottini M, Bruckner S, Nika K, et al. Multi-walled carbon nanotubes induce T lymphocyte apoptosis. Toxicol Lett. 2006. January 5;160(2):121–126. PubMed PMID: 16125885; eng. [DOI] [PubMed] [Google Scholar]
- [196].Shafagh M, Rahmani F, Delirezh N.. CuO nanoparticles induce cytotoxicity and apoptosis in human K562 cancer cell line via mitochondrial pathway, through reactive oxygen species and P53. Iranian J Basic Med Sci. 2015;18(10):993. [PMC free article] [PubMed] [Google Scholar]
- [197].Ahamed M, Akhtar MJ, Alhadlaq HA, et al. Copper ferrite nanoparticle-induced cytotoxicity and oxidative stress in human breast cancer MCF-7 cells. Colloids Surf B Biointerfaces. 2016;142:46–54. [DOI] [PubMed] [Google Scholar]
- [198].Ahmad J, Alhadlaq HA, Alshamsan A, et al. Differential cytotoxicity of copper ferrite nanoparticles in different human cells. J Appl Toxicol. 2016;36(10):1284–1293. [DOI] [PubMed] [Google Scholar]
- [199].Chakraborty R, Basu T. Metallic copper nanoparticles induce apoptosis in a human skin melanoma A-375 cell line. Nanotechnology. 2017;28(10):105101. [DOI] [PubMed] [Google Scholar]
- [200].Ye S, Chen M, Jiang Y, et al. Polyhydroxylated fullerene attenuates oxidative stress-induced apoptosis via a fortifying Nrf2-regulated cellular antioxidant defence system. Int J Nanomedicine. 2014;9:2073–2087. PubMed PMID: 24812508; PubMed Central PMCID: PMCPMC4010637. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Nishizawa C, Hashimoto N, Yokoo S, et al. Pyrrolidinium-type fullerene derivative-induced apoptosis by the generation of reactive oxygen species in HL-60 cells. Free Radic Res. 2009. December;43(12):1240–1247. 10.3109/13814780903260764. PubMed PMID: 19905986; eng. [DOI] [PubMed] [Google Scholar]
- [202].Cha YJ, Lee J, Choi SS. Apoptosis-mediated in vivo toxicity of hydroxylated fullerene nanoparticles in soil nematode Caenorhabditis elegans. Chemosphere. 2012. March;87(1):49–54. PubMed PMID: 22182706; eng. [DOI] [PubMed] [Google Scholar]
- [203].Lao F, Chen L, Li W, et al. Fullerene nanoparticles selectively enter oxidation-damaged cerebral microvessel endothelial cells and inhibit JNK-related apoptosis. ACS Nano. 2009. November 24;3(11):3358–3368. PubMed PMID: 19839607; eng. [DOI] [PubMed] [Google Scholar]
- [204].Ayoubi M, Naserzadeh P, Hashemi MT, et al. Biochemical mechanisms of dose-dependent cytotoxicity and ROS-mediated apoptosis induced by lead sulfide/graphene oxide quantum dots for potential bioimaging applications. Sci Rep. 2017. October 10;7(1):12896 PubMed PMID: 29018231; PubMed Central PMCID: PMCPMC5635035. eng DOI: 10.1038/s41598-017-13396-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Kang Y, Liu J, Wu J, et al. Graphene oxide and reduced graphene oxide induced neural pheochromocytoma-derived PC12 cell lines apoptosis and cell cycle alterations via the ERK signaling pathways. Int J Nanomedicine. 2017;12:5501–5510. PubMed PMID: 28814866; PubMed Central PMCID: PMCPMC5546784. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Yan X, Yang W, Shao Z, et al. Triggering of apoptosis in osteosarcoma cells by graphene/single-walled carbon nanotube hybrids via the ROS-mediated mitochondrial pathway. J Biomed Mater Res A. 2017. February;105(2):443–453. PubMed PMID: 27684494; eng. [DOI] [PubMed] [Google Scholar]
- [207].Qin Y, Zhou ZW, Pan ST, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-kappaB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015. January 2;327:62–76. PubMed PMID: 25446327; eng. [DOI] [PubMed] [Google Scholar]
- [208].Wu T, Zhan Q, Zhang T, et al. The protective effects of resveratrol, H2S and thermotherapy on the cell apoptosis induced by CdTe quantum dots. Toxicol In Vitro. 2017. June;41:106–113. PubMed PMID: 28219723; eng. [DOI] [PubMed] [Google Scholar]
- [209].Zhang T, Wang Y, Kong L, et al. Threshold dose of three types of Quantum Dots (QDs) induces oxidative stress triggers DNA damage and apoptosis in mouse fibroblast L929 cells. Int J Environ Res Public Health. 2015. October 26;12(10):13435–13454. PubMed PMID: 26516873; PubMed Central PMCID: PMCPMC4627041. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Qin YU, Wang H, Liu ZY, et al. Realgar quantum dots induce apoptosis and necrosis in HepG2 cells through endoplasmic reticulum stress. Biomed Rep. 2015. September;3(5):657–662. PubMed PMID: 26405541; PubMed Central PMCID: PMCPMC4534832. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Nguyen KC, Willmore WG, Tayabali AF. Cadmium telluride quantum dots cause oxidative stress leading to extrinsic and intrinsic apoptosis in hepatocellular carcinoma HepG2 cells. Toxicology. 2013. April 5;306:114–123. [DOI] [PubMed] [Google Scholar]
- [212].Amna T, Van Ba H, Vaseem M, et al. Apoptosis induced by copper oxide quantum dots in cultured C2C12 cells via caspase 3 and caspase 7: a study on cytotoxicity assessment. Appl Microbiol Biotechnol. 2013. June;97(12):5545–5553. PubMed PMID: 23467821; eng. [DOI] [PubMed] [Google Scholar]
- [213].Kong L, Zhang T, Tang M, et al. Apoptosis induced by cadmium selenide quantum dots in JB6 cells. J Nanosci Nanotechnol. 2012. November;12(11):8258–8265. PubMed PMID: 23421204; eng. [DOI] [PubMed] [Google Scholar]
- [214].Wang H, Liu Z, Gou Y, et al. Apoptosis and necrosis induced by novel realgar quantum dots in human endometrial cancer cells via endoplasmic reticulum stress signaling pathway. Int J Nanomedicine. 2015;10:5505–5512. DOI: 10.2147/ijn.s83838 PubMed PMID: 26357474; PubMed Central PMCID: PMCPMC4560518. eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Chan WH, Shiao NH, Lu PZ. CdSe quantum dots induce apoptosis in human neuroblastoma cells via mitochondrial-dependent pathways and inhibition of survival signals. Toxicol Lett. 2006. December 15;167(3):191–200. PubMed PMID: 17049762; eng. [DOI] [PubMed] [Google Scholar]
- [216].Ahamed M, Alhadlaq AH, Alam J, et al. Iron oxide nanoparticle-induced oxidative stress and genotoxicity in human skin epithelial and lung epithelial cell lines. Curr Pharm Des. 2013;19(37):6681–6690. [DOI] [PubMed] [Google Scholar]
- [217].Radu M, Din IM, Hermenean A, et al. Exposure to iron oxide nanoparticles coated with phospholipid-based polymeric micelles induces biochemical and histopathological pulmonary changes in mice. Int J Mol Sci. 2015;16(12):29417–29435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Park E-J, Choi D-H, Kim Y, et al. Magnetic iron oxide nanoparticles induce autophagy preceding apoptosis through mitochondrial damage and ER stress in RAW264. 7 cells. Toxicol Vitro. 2014;28(8):1402–1412. [DOI] [PubMed] [Google Scholar]
- [219].Jalili A, Irani S, Mirfakhraie R. Combination of cold atmospheric plasma and iron nanoparticles in breast cancer: gene expression and apoptosis study. Onco Targets Ther. 2016;9:5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Alarifi S, Ali D, Alkahtani S, et al. Iron oxide nanoparticles induce oxidative stress, DNA damage, and caspase activation in the human breast cancer cell line. Biol Trace Elem Res. 2014;159(1–3):416–424. [DOI] [PubMed] [Google Scholar]
- [221].Ahamed M, Alhadlaq HA, Khan MM, et al. Selective killing of cancer cells by iron oxide nanoparticles mediated through reactive oxygen species via p53 pathway. J Nanopart Res. 2013;15(1):1225. [Google Scholar]
- [222].Estevez H, Garcia-Lidon JC, Luque-Garcia JL, et al. Effects of chitosan-stabilized selenium nanoparticles on cell proliferation, apoptosis and cell cycle pattern in HepG2 cells: comparison with other selenospecies. Colloids Surf B Biointerfaces. 2014. October 1;122:184–193. PubMed PMID: 25038448; eng. [DOI] [PubMed] [Google Scholar]
- [223].Huang Y, He L, Liu W, et al. Selective cellular uptake and induction of apoptosis of cancer-targeted selenium nanoparticles. Biomaterials. 2013. September;34(29):7106–7116.PubMed PMID: 23800743; eng. [DOI] [PubMed] [Google Scholar]
- [224].Li Y, Li X, Wong YS, et al. The reversal of cisplatin-induced nephrotoxicity by selenium nanoparticles functionalized with 11-mercapto-1-undecanol by inhibition of ROS-mediated apoptosis. Biomaterials. 2011. December;32(34):9068–9076. PubMed PMID: 21864903; eng. [DOI] [PubMed] [Google Scholar]
- [225].Kong L, Yuan Q, Zhu H, et al. The suppression of prostate LNCaP cancer cells growth by selenium nanoparticles through Akt/Mdm2/AR controlled apoptosis. Biomaterials. 2011. September;32(27):6515–6522. PubMed PMID: 21640377; eng. [DOI] [PubMed] [Google Scholar]
- [226].Zheng JS, Zheng SY, Zhang YB, et al. Sialic acid surface decoration enhances cellular uptake and apoptosis-inducing activity of selenium nanoparticles. Colloids Surf B Biointerfaces. 2011. March;83(1):183–187. PubMed PMID: 21145219; eng. [DOI] [PubMed] [Google Scholar]
- [227].Liu H, Zhang Y, Yang N, et al. A functionalized single-walled carbon nanotube-induced autophagic cell death in human lung cells through Akt–TSC2-mTOR signaling. Cell Death Dis. 2011;2(5):e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Yang X, Liu J, He H, et al. SiO 2 nanoparticles induce cytotoxicity and protein expression alteration in HaCaT cells. Part Fibre Toxicol. 2010;7(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Ye Y, Liu J, Xu J, et al. Nano-SiO 2 induces apoptosis via activation of p53 and Bax mediated by oxidative stress in human hepatic cell line. Toxicol Vitro. 2010;24(3):751–758. [DOI] [PubMed] [Google Scholar]
- [230].Liu X, Sun J. Endothelial cells dysfunction induced by silica nanoparticles through oxidative stress via JNK/P53 and NF-κB pathways. Biomaterials. 2010;31(32):8198–8209. [DOI] [PubMed] [Google Scholar]
- [231].Lu X, Qian J, Zhou H, et al. In vitro cytotoxicity and induction of apoptosis by silica nanoparticles in human HepG2 hepatoma cells. Int J Nanomedicine. 2011;6:1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Xue Y, Chen Q, Ding T, et al. SiO2 nanoparticle-induced impairment of mitochondrial energy metabolism in hepatocytes directly and through a Kupffer cell-mediated pathway in vitro. Int J Nanomedicine. 2014;9:2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Ahamed M. Silica nanoparticles-induced cytotoxicity, oxidative stress and apoptosis in cultured A431 and A549 cells. Hum Exp Toxicol. 2013;32(2):186–195. [DOI] [PubMed] [Google Scholar]
- [234].Zuo D, Duan Z, Jia Y, et al. Amphipathic silica nanoparticles induce cytotoxicity through oxidative stress mediated and p53 dependent apoptosis pathway in human liver cell line HL-7702 and rat liver cell line BRL-3A. Colloids Surf B Biointerfaces. 2016;145:232–240. [DOI] [PubMed] [Google Scholar]
- [235].Sun L, Li Y, Liu X, et al. Cytotoxicity and mitochondrial damage caused by silica nanoparticles. Toxicol Vitro. 2011;25(8):1619–1629. [DOI] [PubMed] [Google Scholar]
- [236].Akhtar MJ, Ahamed M, Kumar S, et al. Zinc oxide nanoparticles selectively induce apoptosis in human cancer cells through reactive oxygen species. Int J Nanomedicine. 2012;7:845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Ng KW, Khoo SP, Heng BC, et al. The role of the tumor suppressor p53 pathway in the cellular DNA damage response to zinc oxide nanoparticles. Biomaterials. 2011;32(32):8218–8225. [DOI] [PubMed] [Google Scholar]
- [238].Meyer K, Rajanahalli P, Ahamed M, et al. ZnO nanoparticles induce apoptosis in human dermal fibroblasts via p53 and p38 pathways. Toxicol Vitro. 2011;25(8):1721–1726. [DOI] [PubMed] [Google Scholar]
- [239].Sharma V, Anderson D, Dhawan A. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria mediated apoptosis in human liver cells (HepG2). Apoptosis. 2012;17(8):852–870. [DOI] [PubMed] [Google Scholar]
- [240].Alarifi S, Ali D, Alkahtani S, et al. Induction of oxidative stress, DNA damage, and apoptosis in a malignant human skin melanoma cell line after exposure to zinc oxide nanoparticles. Int J Nanomedicine. 2013;8:983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Zhao X, Ren X, Zhu R, et al. Zinc oxide nanoparticles induce oxidative DNA damage and ROS-triggered mitochondria-mediated apoptosis in zebrafish embryos. Aquatic Toxicol. 2016;180:56–70. [DOI] [PubMed] [Google Scholar]
- [242].Bai D-P, Zhang X-F, Zhang G-L, et al. Zinc oxide nanoparticles induce apoptosis and autophagy in human ovarian cancer cells. Int J Nanomedicine. 2017;12:6521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Liang S, Sun K, Wang Y, et al. Role of Cyt-C/caspases-9, 3, Bax/Bcl-2 and the FAS death receptor pathway in apoptosis induced by zinc oxide nanoparticles in human aortic endothelial cells and the protective effect by alpha-lipoic acid. Chem Biol Interact. 2016;258:40–51. [DOI] [PubMed] [Google Scholar]
- [244].Guo D, Bi H, Liu B, et al. Reactive oxygen species-induced cytotoxic effects of zinc oxide nanoparticles in rat retinal ganglion cells. Toxicol Vitro. 2013;27(2):731–738. [DOI] [PubMed] [Google Scholar]
- [245].Wahab R, Siddiqui MA, Saquib Q, et al. ZnO nanoparticles induced oxidative stress and apoptosis in HepG2 and MCF-7 cancer cells and their antibacterial activity. Colloids Surf B Biointerfaces. 2014;117:267–276. [DOI] [PubMed] [Google Scholar]
- [246].Wang Z, Li J, Zhao J, et al. Toxicity and internalization of CuO nanoparticles to prokaryotic alga microcystis aeruginosa as affected by dissolved organic matter. Environ Sci Technol. 2011;45(14):6032–6040. [DOI] [PubMed] [Google Scholar]
- [247].Calzolai L, Franchini F, Gilliland D, et al. Protein− nanoparticle interaction: identification of the ubiquitin− gold nanoparticle interaction site. Nano Lett. 2010;10(8):3101–3105. [DOI] [PubMed] [Google Scholar]
- [248].Li C, Liu H, Sun Y, et al. PAMAM nanoparticles promote acute lung injury by inducing autophagic cell death through the Akt-TSC2-mTOR signaling pathway. J Mol Cell Biol. 2009;1(1):37–45. [DOI] [PubMed] [Google Scholar]
- [249].Zhang Y, Zheng F, Yang T, et al. Tuning the autophagy-inducing activity of lanthanide-based nanocrystals through specific surface-coating peptides. Nat Mater. 2012;11(9):817–826. [DOI] [PubMed] [Google Scholar]
- [250].Zhong W, Lü M, Liu L, et al. Autophagy as new emerging cellular effect of nanomaterials. Chin Sci Bull. 2013;58(33):4031–4038. [Google Scholar]
- [251].Klionsky DJ, Abdelmohsen K, Abe A, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2016;12(1):1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Kaizuka T, Morishita H, Hama Y, et al. An autophagic flux probe that releases an internal control. Mol Cell. 2016;64(4):835–849. [DOI] [PubMed] [Google Scholar]
- [253].Kroemer G, Jäättelä M. Lysosomes and autophagy in cell death control. Nat Rev Cancer. 2005;5(11):886–897. [DOI] [PubMed] [Google Scholar]
- [254].Lopes VR, Loitto V, Audinot J-N, et al. Dose-dependent autophagic effect of titanium dioxide nanoparticles in human HaCaT cells at non-cytotoxic levels. J Nanobiotechnology. 2016;14(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [255].Yokoyama T, Tam J, Kuroda S, et al. EGFR-targeted hybrid plasmonic magnetic nanoparticles synergistically induce autophagy and apoptosis in non-small cell lung cancer cells. PLoS One. 2011;6(11):e25507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Seleverstov O, Zabirnyk O, Zscharnack M, et al. Quantum dots for human mesenchymal stem cells labeling. A size-dependent autophagy activation. Nano Lett. 2006;6(12):2826–2832. [DOI] [PubMed] [Google Scholar]
- [257].Chen Y, Yang L, Feng C, et al. Nano neodymium oxide induces massive vacuolization and autophagic cell death in non-small cell lung cancer NCI-H460 cells. Biochem Biophys Res Commun. 2005;337(1):52–60. [DOI] [PubMed] [Google Scholar]
- [258].Tavakol S. Acidic pH derived from cancer cells may induce failed reprogramming of normal differentiated cells adjacent tumor cells and turn them into cancer cells. Med Hypotheses. 2014;83(6):668–672. [DOI] [PubMed] [Google Scholar]
- [259].Stern ST, Adiseshaiah PP, Crist RM. Autophagy and lysosomal dysfunction as emerging mechanisms of nanomaterial toxicity. Part Fibre Toxicol. 2012;9(1):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Johnson-Lyles DN, Peifley K, Lockett S, et al. Fullerenol cytotoxicity in kidney cells is associated with cytoskeleton disruption, autophagic vacuole accumulation, and mitochondrial dysfunction. Toxicol Appl Pharmacol. 2010;248(3):249–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [261].Ngwa HA, Kanthasamy A, Gu Y, et al. Manganese nanoparticle activates mitochondrial dependent apoptotic signaling and autophagy in dopaminergic neuronal cells. Toxicol Appl Pharmacol. 2011;256(3):227–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [262].Wu Y-N, Yang L-X, Shi X-Y, et al. The selective growth inhibition of oral cancer by iron core-gold shell nanoparticles through mitochondria-mediated autophagy. Biomaterials. 2011;32(20):4565–4573. [DOI] [PubMed] [Google Scholar]
- [263].Panzarini E, Inguscio V, Tenuzzo BA, et al. Nanomaterials and autophagy: new insights in cancer treatment. Cancers. 2013;5(1):296–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [264].Zheng YT, Shahnazari S, Brech A, et al. The adaptor protein p62/SQSTM1 targets invading bacteria to the autophagy pathway. J Immunol. 2009;183(9):5909–5916. [DOI] [PubMed] [Google Scholar]
- [265].Wan B, Wang Z-X, Lv Q-Y, et al. Single-walled carbon nanotubes and graphene oxides induce autophagosome accumulation and lysosome impairment in primarily cultured murine peritoneal macrophages. Toxicol Lett. 2013;221(2):118–127. [DOI] [PubMed] [Google Scholar]
- [266].Orecna M, De Paoli SH, Janouskova O, et al. Toxicity of carboxylated carbon nanotubes in endothelial cells is attenuated by stimulation of the autophagic flux with the release of nanomaterial in autophagic vesicles. Nanomedicine: Nanotechnology, Biol Med. 2014;10(5):e939–e948. [DOI] [PubMed] [Google Scholar]
- [267].Khan MI, Mohammad A, Patil G, et al. Induction of ROS, mitochondrial damage and autophagy in lung epithelial cancer cells by iron oxide nanoparticles. Biomaterials. 2012;33(5):1477–1488. [DOI] [PubMed] [Google Scholar]
- [268].Chen HH, Yu C, Ueng TH, et al. Acute and subacute toxicity study of water-soluble polyalkylsulfonated C60 in rats. Toxicol Pathol. 1998;26(1):143–151. [DOI] [PubMed] [Google Scholar]
- [269].Chen G-Y, Yang H-J, Lu C-H, et al. Simultaneous induction of autophagy and toll-like receptor signaling pathways by graphene oxide. Biomaterials. 2012;33(27):6559–6569. [DOI] [PubMed] [Google Scholar]
- [270].Roy R, Singh SK, Chauhan L, et al. Zinc oxide nanoparticles induce apoptosis by enhancement of autophagy via PI3K/Akt/mTOR inhibition. Toxicol Lett. 2014;227(1):29–40. [DOI] [PubMed] [Google Scholar]
- [271].AshaRani P, Low Kah Mun G, Mp H, et al. Cytotoxicity and genotoxicity of silver nanoparticles in human cells. ACS Nano. 2008;3(2):279–290. [DOI] [PubMed] [Google Scholar]
- [272].Hussain S, Thomassen LC, Ferecatu I, et al. Carbon black and titanium dioxide nanoparticles elicit distinct apoptotic pathways in bronchial epithelial cells. Part Fibre Toxicol. 2010;7(1):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Zhang Y, Li X, Huang Z, et al. Enhancement of cell permeabilization apoptosis-inducing activity of selenium nanoparticles by ATP surface decoration. Nanomedicine. 2013. January;9(1):74–84. PubMed PMID: 22542821; eng. [DOI] [PubMed] [Google Scholar]
- [274].Zhang Q, Yang W, Man N, et al. Autophagy-mediated chemosensitization in cancer cells by fullerene C60 nanocrystal. Autophagy. 2009;5(8):1107–1117. [DOI] [PubMed] [Google Scholar]
- [275].Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Kenzaoui BH, Bernasconi CC, Guney-Ayra S, et al. Induction of oxidative stress, lysosome activation and autophagy by nanoparticles in human brain-derived endothelial cells. Biochem J. 2012;441(3):813–821. [DOI] [PubMed] [Google Scholar]
- [277].Song W, Shi M, Dong M, et al. Inducing temporal and reversible autophagy by nanotopography for potential control of cell differentiation. ACS Appl Mater Interfaces. 2016;8(49):33475–33483. [DOI] [PubMed] [Google Scholar]
- [278].Baltazar GC, Guha S, Lu W, et al. Acidic nanoparticles are trafficked to lysosomes and restore an acidic lysosomal pH and degradative function to compromised ARPE-19 cells. PLoS One. 2012;7(12):e49635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Trudeau KM, Colby AH, Zeng J, et al. Lysosome acidification by photoactivated nanoparticles restores autophagy under lipotoxicity. J Cell Biol. 2016;214(1):25–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Markovic ZM, Ristic BZ, Arsikin KM, et al. Graphene quantum dots as autophagy-inducing photodynamic agents. Biomaterials. 2012;33(29):7084–7092. [DOI] [PubMed] [Google Scholar]
- [281].Stern ST, Zolnik BS, McLeland CB, et al. Induction of autophagy in porcine kidney cells by quantum dots: a common cellular response to nanomaterials? Toxicological Sci. 2008;106(1):140–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Ma X, Wu Y, Jin S, et al. Gold nanoparticles induce autophagosome accumulation through size-dependent nanoparticle uptake and lysosome impairment. ACS Nano. 2011;5(11):8629–8639. [DOI] [PubMed] [Google Scholar]
- [283].Li JJ, Hartono D, Ong C-N, et al. Autophagy and oxidative stress associated with gold nanoparticles. Biomaterials. 2010;31(23):5996–6003. [DOI] [PubMed] [Google Scholar]
- [284].Lee C-M, Huang S-T, Huang S-H, et al. C60 fullerene-pentoxifylline dyad nanoparticles enhance autophagy to avoid cytotoxic effects caused by the β-amyloid peptide. Nanomedicine: Nanotechnology, Biol Med. 2011;7(1):107–114. [DOI] [PubMed] [Google Scholar]
- [285].Mironava T, Hadjiargyrou M, Simon M, et al. Gold nanoparticles cellular toxicity and recovery: effect of size, concentration and exposure time. Nanotoxicology. 2010;4(1):120–137. [DOI] [PubMed] [Google Scholar]
- [286].Walker VG, Li Z, Hulderman T, et al. Potential in vitro effects of carbon nanotubes on human aortic endothelial cells. Toxicol Appl Pharmacol. 2009;236(3):319–328. [DOI] [PubMed] [Google Scholar]
- [287].Wu X, Tan Y, Mao H, et al. Toxic effects of iron oxide nanoparticles on human umbilical vein endothelial cells. Int J Nanomedicine. 2010;5:385–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Ratnikova TA, Nedumpully Govindan P, Salonen E, et al. In vitro polymerization of microtubules with a fullerene derivative. ACS Nano. 2011;5(8):6306–6314. [DOI] [PubMed] [Google Scholar]
- [289].Gheshlaghi ZN, Riazi GH, Ahmadian S, et al. Toxicity and interaction of titanium dioxide nanoparticles with microtubule protein. Acta Biochim Biophys Sin (Shanghai). 2008;40(9):777–782. [PubMed] [Google Scholar]
- [290].Choudhury D, Xavier PL, Chaudhari K, et al. Unprecedented inhibition of tubulin polymerization directed by gold nanoparticles inducing cell cycle arrest and apoptosis. Nanoscale. 2013;5(10):4476–4489. [DOI] [PubMed] [Google Scholar]
- [291].Shams H, Holt BD, Mahboobi SH, et al. Actin reorganization through dynamic interactions with single-wall carbon nanotubes. ACS Nano. 2013;8(1):188–197. [DOI] [PubMed] [Google Scholar]
- [292].Okoturo-Evans O, Dybowska A, Valsami-Jones E, et al. Elucidation of toxicity pathways in lung epithelial cells induced by silicon dioxide nanoparticles. PLoS One. 2013;8(9):e72363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [293].Pisanic TR, Blackwell JD, Shubayev VI, et al. Nanotoxicity of iron oxide nanoparticle internalization in growing neurons. Biomaterials. 2007;28(16):2572–2581. [DOI] [PubMed] [Google Scholar]
- [294].Dadras A, Riazi GH, Afrasiabi A, et al. In vitro study on the alterations of brain tubulin structure and assembly affected by magnetite nanoparticles. JBIC J Biol Inorg Chem. 2013;18(3):357–369. [DOI] [PubMed] [Google Scholar]
- [295].Soenen SJ, Nuytten N, De Meyer SF, et al. High Intracellular iron oxide nanoparticle concentrations affect cellular cytoskeleton and focal adhesion kinase‐mediated signaling. Small. 2010;6(7):832–842. [DOI] [PubMed] [Google Scholar]
- [296].Bellettato CM, Scarpa M. Pathophysiology of neuropathic lysosomal storage disorders. J Inherit Metab Dis. 2010;33(4):347–362. [DOI] [PubMed] [Google Scholar]
- [297].Ueng T-H, Kang -J-J, Wang H-W, et al. Suppression of microsomal cytochrome P450-dependent monooxygenases and mitochondrial oxidative phosphorylation by fullerenol, a polyhydroxylated fullerene C 60. Toxicol Lett. 1997;93(1):29–37. [DOI] [PubMed] [Google Scholar]
- [298].Shcharbin D, Jokiel M, Klajnert B, et al. Effect of dendrimers on pure acetylcholinesterase activity and structure. Bioelectrochemistry. 2006;68(1):56–59. [DOI] [PubMed] [Google Scholar]
- [299].Thomas TP, Majoros I, Kotlyar A, et al. Cationic poly (amidoamine) dendrimer induces lysosomal apoptotic pathway at therapeutically relevant concentrations. Biomacromolecules. 2009;10(12):3207–3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [300].Cho W-S, Duffin R, Howie SE, et al. Progressive severe lung injury by zinc oxide nanoparticles; the role of Zn 2+ dissolution inside lysosomes. Part Fibre Toxicol. 2011;8(1):27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Hamilton RF, Wu N, Porter D, et al. Particle length-dependent titanium dioxide nanomaterials toxicity and bioactivity. Part Fibre Toxicol. 2009;6(1):35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Lunov O, Syrovets T, Loos C, et al. Amino-functionalized polystyrene nanoparticles activate the NLRP3 inflammasome in human macrophages. ACS Nano. 2011;5(12):9648–9657. [DOI] [PubMed] [Google Scholar]
- [303].Settembre C, Fraldi A, Jahreiss L, et al. A block of autophagy in lysosomal storage disorders. Hum Mol Genet. 2008;17(1):119–129. [DOI] [PubMed] [Google Scholar]
- [304].SK Sohaebuddin, PT Thevenot, Baker D, et al. Nanomaterial cytotoxicity is composition, size, and cell type dependent. Part Fibre Toxicol. 2010;7(1):22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [305].Geisler S, Holmström KM, Treis A, et al. The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy. 2010;6(7):871–878. [DOI] [PubMed] [Google Scholar]
- [306].Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early alzheimer disease and regulates amyloid β accumulation in mice. J Clin Invest. 2008;118(6):2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [307].Wei Y, Sinha SC, Levine B. Dual role of JNK1-mediated phosphorylation of Bcl-2 in autophagy and apoptosis regulation. Autophagy. 2008;4(7):949–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Livesey KM, Tang D, Zeh HJ, et al. Autophagy inhibition in combination cancer treatment. Curr Opin Investig Drugs. 2009;10(12):1269–1279. [PubMed] [Google Scholar]
- [309].Zhou R, Yazdi AS, Menu P, et al. A role for mitochondria in NLRP3 inflammasome activation. Nature. 2011;469(7329):221–225. [DOI] [PubMed] [Google Scholar]
- [310].Pietersz GA, Tang C-K, Apostolopoulos V. Structure and design of polycationic carriers for gene delivery. Mini Rev Med Chem. 2006;6(12):1285–1298. [DOI] [PubMed] [Google Scholar]
- [311].Wei P, Zhang L, Lu Y, et al. C60 (Nd) nanoparticles enhance chemotherapeutic susceptibility of cancer cells bymodulation of autophagy. Nanotechnology. 2010;21(49):495101. [DOI] [PubMed] [Google Scholar]
- [312].Li Q, Hu H, Jiang L, et al. Cytotoxicity and autophagy dysfunction induced by different sizes of silica particles in human bronchial epithelial BEAS-2B cells. Toxicol Res (Camb). 2016;5(4):1216–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Zhang Y, Yu C, Huang G, et al. Nano rare-earth oxides induced size-dependent vacuolization: an independent pathway from autophagy. Int J Nanomedicine. 2010;5:601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Zhu L, Guo D, Sun L, et al. Activation of autophagy by elevated reactive oxygen species rather than released silver ions promotes cytotoxicity of polyvinylpyrrolidone-coated silver nanoparticles in hematopoietic cells. Nanoscale. 2017;9(17):5489–5498. [DOI] [PubMed] [Google Scholar]
- [315].Zhang X-F, Gurunathan S. Combination of salinomycin and silver nanoparticles enhances apoptosis and autophagy in human ovarian cancer cells: an effective anticancer therapy. Int J Nanomedicine. 2016;11:3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Lin J, Huang Z, Wu H, et al. Inhibition of autophagy enhances the anticancer activity of silver nanoparticles. Autophagy. 2014;10(11):2006–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Wu H, Lin J, Liu P, et al. Is the autophagy a friend or foe in the silver nanoparticles associated radiotherapy for glioma? Biomaterials. 2015;62:47–57. [DOI] [PubMed] [Google Scholar]
- [318].Lin Y-X, Gao Y-J, Wang Y, et al. pH-sensitive polymeric nanoparticles with gold (I) compound payloads synergistically induce cancer cell death through modulation of autophagy. Mol Pharm. 2015;12(8):2869–2878. [DOI] [PubMed] [Google Scholar]
- [319].Tsai T-L, Hou -C-C, Wang H-C, et al. Nucleocytoplasmic transport blockage by SV40 peptide-modified gold nanoparticles induces cellular autophagy. Int J Nanomedicine. 2012;7:5215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Ding F, Li Y, Liu J, et al. Overendocytosis of gold nanoparticles increases autophagy and apoptosis in hypoxic human renal proximal tubular cells. Int J Nanomedicine. 2014;9:4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Krętowski R, Kusaczuk M, Naumowicz M, et al. The effects of silica nanoparticles on apoptosis and autophagy of glioblastoma cell lines. Nanomaterials. 2017;7(8):230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Yu Y, Duan J, Yu Y, et al. Silica nanoparticles induce autophagy and autophagic cell death in HepG2 cells triggered by reactive oxygen species. J Hazard Mater. 2014;270:176–186. [DOI] [PubMed] [Google Scholar]
- [323].Duan J, Yu Y, Yu Y, et al. Silica nanoparticles induce autophagy and endothelial dysfunction via the PI3K/Akt/mTOR signaling pathway. Int J Nanomedicine. 2014;9:5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [324].Guo C, Yang M, Jing L, et al. Amorphous silica nanoparticles trigger vascular endothelial cell injury through apoptosis and autophagy via reactive oxygen species-mediated MAPK/Bcl-2 and PI3K/Akt/mTOR signaling. Int J Nanomedicine. 2016;11:5257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Zhang J, Qin X, Wang B, et al. Zinc oxide nanoparticles harness autophagy to induce cell death in lung epithelial cells. Cell Death Dis. 2017;8:cddis2017337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Yu K-N, Yoon T-J, Minai-Tehrani A, et al. Zinc oxide nanoparticle induced autophagic cell death and mitochondrial damage via reactive oxygen species generation. Toxicol Vitro. 2013;27(4):1187–1195. [DOI] [PubMed] [Google Scholar]
- [327].Qin Y, Zhou Z-W, Pan S-T, et al. Graphene quantum dots induce apoptosis, autophagy, and inflammatory response via p38 mitogen-activated protein kinase and nuclear factor-κB mediated signaling pathways in activated THP-1 macrophages. Toxicology. 2015;327:62–76. [DOI] [PubMed] [Google Scholar]
- [328].Lim M-H, Jeung IC, Jeong J, et al. Graphene oxide induces apoptotic cell death in endothelial cells by activating autophagy via calcium-dependent phosphorylation of c-Jun N-terminal kinases. Acta biomaterialia. 2016;46:191–203. [DOI] [PubMed] [Google Scholar]
- [329].C-M Lee, S-T Huang, S-H Huang, et al. C 60 fullerene-pentoxifylline dyad nanoparticles enhance autophagy to avoid cytotoxic effects caused by the β-amyloid peptide. Nanomed. 2011;7(1):107–114. [DOI] [PubMed] [Google Scholar]
- [330].Yamawaki H, Iwai N. Cytotoxicity of water-soluble fullerene in vascular endothelial cells. Am J Physiology-Cell Physiol. 2006. [DOI] [PubMed] [Google Scholar]
- [331].Xue X, Wang L-R, Sato Y, et al. Single-walled carbon nanotubes alleviate autophagic/lysosomal defects in primary glia from a mouse model of alzheimer’s disease. Nano Lett. 2014;14(9):5110–5117. [DOI] [PMC free article] [PubMed] [Google Scholar]