

Abstract
Background:
Mitochondrial reactive oxygen species (ROS) contribute to inflammation and vascular remodeling during atherosclerotic plaque formation. C57BL/6N (6N) and C57BL/6J (6J) mice display distinct mitochondrial redox balance due to the absence of nicotinamide nucleotide transhydrogenase (NNT) in 6J mice. We hypothesize that differential NNT expression between these animals alters plaque development.
Methods:
6N and 6J mice were treated with AAV8-PCSK9 (adeno-associated virus serotype 8/proprotein convertase subtilisin/kexin type 9) virus leading to hypercholesterolemia, increased low-density lipoprotein, and atherosclerosis in mice fed a high-fat diet (HFD). Mice were co-treated with the mitochondria-targeted superoxide dismutase mimetic MitoTEMPO to assess the contribution of mitochondrial ROS to atherosclerosis.
Results:
Baseline and HFD-induced vascular superoxide is increased in 6J compared to 6N mice. MitoTEMPO diminished superoxide in both groups demonstrating differential production of mitochondrial ROS among these strains. PCSK9 treatment and HFD led to similar increases in plasma lipids in both 6N and 6J mice. However, 6J animals displayed significantly higher levels of plaque formation. MitoTEMPO reduced plasma lipids but did not affect plaque formation in 6N mice. In contrast, MitoTEMPO surprisingly increased plaque formation in 6J mice.
Conclusion:
These data indicate that loss of NNT increases vascular ROS production and exacerbates atherosclerotic plaque development.
Keywords: Atherosclerosis, Nicotinamide nucleotide, transhydrogenase, Mitochondria, Reactive oxygen species, Proprotein convertase subtilisin/kexin type 9 virus
Introduction
Atherosclerosis, a progressive chronic inflammatory disease of the vessel wall, is regulated by oxidative stress throughout the course of disease development. While reactive oxygen species (ROS) can be generated at multiple sites in the cell, increasing evidence implicates mitochondrial ROS as a major factor in multiple steps during atherosclerotic plaque formation, including endothelial activation, lipoprotein modification, macrophage function, and smooth-muscle recruitment [1–3]. Mitochondrial ROS generation occurs during oxidative phosphorylation primarily at complexes I and III, leading to increased formation of superoxide (O2·−) and hydrogen peroxide (H2O2) [4]. Mitochondria possess multiple antioxidant systems that function to maintain the balance between ROS production and consumption. In the mitochondrial matrix, O2·− is efficiently converted to H2O2 by the activity of manganese superoxide dismutase (MnSOD) [5–7]. H2O2 is further detoxified to water by multiple thiol-dependent reductive pathways in the mitochondria that include the thioredoxin, peroxiredoxin, and glutathione peroxidase systems. Alterations in MnSOD expression in atherosclerosis-prone ApoE−/− mice and the regulation of plaque formation by downstream antioxidant enzymes (e.g., glutathione peroxidase-1, thioredoxin-2, peroxiredoxin-3, glutaredoxin-2) suggest mitochondrial ROS play a critical role in atherosclerosis [8–10]. Interestingly, all of the antioxidant systems downstream of MnSOD in the mitochondria are either directly or indirectly dependent on NADPH to provide the reducing equivalents to maintain mitochondrial redox balance.
The mitochondria possess several enzymatic sources of NADPH including nicotinamide nucleotide transhydrogenase (NNT), NADP-dependent isocitrate dehydrogenase, NADPH-dependent malic enzyme, and glutamate dehydrogenase [11–16]. NNT is a ubiquitous mitochondrial inner membrane protein that couples proton translocation across the mitochondrial inner membrane to a redox reaction that reduces mitochondrial NADP+ while oxidizing NADH [17–19]. The commonly used C57BL/6J (6J) mouse strain possesses a mutation in NNT leading to a loss of NNT activity and reduced mitochondrial NADPH levels [20–22], whereas the related C57BL/6N (6N) mice show normal NNT activity. The loss of NNT in 6J mitochondria results in a pro-oxidative phenotype, characterized by increased hydrogen peroxide production and a reduced GSH/GSSG ratio compared to the 6N animals [23]. We previously demonstrated that 6J mice show enhanced susceptibility to angiotensin II-induced hypertension compared to 6N mice, and blunting mitochondrial oxidant stress with the MnSOD mimetic MitoTEMPO alleviates this differential response [24]. While 6J is commonly used for atherosclerosis studies, the functional consequence of altered NNT activity between these 2 C57BL/6 substrains has not been explored in the context of atherosclerosis.
To assess whether these substrain differences affect atherosclerotic plaque formation, we used the recombinant adeno-associated virus serotype 8 (rAAV) encoding a gain-of-function mutant (D377Y) of proprotein convertase subtilisin/kexin type 9 (PCSK9), a regulator of hepatic low-density lipoprotein (LDL) receptor trafficking and expression [25]. Overexpression of PCSK9 results in hypercholesterolemia and susceptibility to diet-induced atherosclerosis in multiple animal models [25, 26]. In the present study, we demonstrate that 6N and 6J mice show both differential susceptibility to diet-induced atherosclerosis and differential responsiveness to the MnSOD mimetic MitoTEMPO.
Materials and Methods
AAV8-PCSK9 Viral Vector Preparation
DNA for pAAV/D377Y-mPCSK9 (Addgene plasmid No. 58376), a gift from Jacob Bentzon [25], was packaged into adeno-associated virus serotype 8 (AAV8) using helper and capsid plasmids from the University of Pennsylvania [27, 28]. Viral stocks were sterilized via Millipore Millex-GV syringe filter (Billerica, MA, USA), titered by dot blot assay, aliquoted, and stored frozen until use. The final product will be referred to as PCSK9.
Animal Procedures and Tissue Collection
All experimental procedures involving animals were performed according to the criteria outlined by the National Institutes of Health and approved by the Institutional Animal Care and Use Committee of the LSU Health Sciences Center – Shreveport. Male 6N and 6J mice (The Jackson Laboratory) were fed a standard chow diet. Between 8 and 9 weeks of age, mice were given retro-orbital injections of 3 × 1010 vector genomes of AAV8-PCSK9. Two weeks following the AAV8-PCSK9 injection the mice were switched to a high-fat, Western diet (TD 88137; Harlan-Teklad, Madison, WI, USA) that contained 21% fat by weight (0.15% cholesterol and 19.5% casein without sodium cholate) for 8 weeks. Weight was monitored weekly. After 4 weeks of feeding a high-fat diet (HFD), a subset of the 6N and 6J mice received implants of Alzet (Cuperino, CA) pumps (Micro-Osmotic Pump, Model 1004) that contained 0.8 mg/kg/day Mito-TEMPO (Enzo Life Sciences) under isoflurane anesthesia (5% on induction; 2% for maintenance during surgery), and the HFD feeding was resumed for an additional 4 weeks.
After 8 weeks on HFD, all mice received an intraperitoneal injection of dihydroethidium (Sigma) 1 h prior to sacrifice (details in “Superoxide Measurements”). The mice were then euthanized by exsanguination and pneumothorax under isoflurane anesthesia. Blood was collected by vena cava puncture into heparinized blood collection tubes, centrifuged, and plasma was isolated and frozen at −80 °C until analysis. The left carotid sinus was immediately collected and flash frozen for tissue superoxide measurements by HPLC. Mice were perfused with phosphate-buffered saline to remove residual blood from the circulation. The aortic root and aorta were excised, placed in 4% phosphate-buffered formaldehyde, and analyzed for plaque size and composition. The liver was removed and placed in 4% phosphate-buffered formaldehyde until pathological analysis.
Blood Plasma Analysis
Blood plasma was analyzed for total cholesterol (Wako), high-density lipoprotein cholesterol (Wako), and triglycerides (Pointe Scientific) using commercially available kits. LDL cholesterol was calculated using the Friedewald equation.
Liver Pathology Analysis
Livers were fixed in phosphate-buffered 4% formaldehyde, embedded in paraffin, and cut into 5-μm sections. Hemotoxylin-eosin and Masson trichrome staining was performed to assess lipid content, inflammation, and fibrosis. Slides were provided to a pathologist for blinded scoring. Livers were scored on the NASH Clinical Research Network Scoring System [29].
Histology and Image Quantification
Aortic roots were fixed in phosphate-buffered 4% formaldehyde, embedded in paraffin, and cut into 5-μm sections. Russell-Movat Pentachrome staining was performed to determine plaque area. Images were collected using an Olympus BX40 microscope, and quantification of the lesion area inside the internal elastic lamina was determined using Nikon Elements imaging software.
Immunofluorescence Staining
Aortic roots were fixed in phosphate-buffered 4% formaldehyde, embedded in paraffin, and cut into 5-μm sections. All sections within each regimen were taken from the same site at equal distance from an anatomical landmark (initiation of valve leaflets). Tissue sections were rehydrated, and antigen retrieval was performed in 10 mm citrate buffer (0.1 m citric acid and 0.1 m sodium citrate) using a microwave oven. Sections were blocked in blocking buffer (1% bovine serum albumin in phosphate-buffered saline) for at least 1 h at room temperature. Sections were rinsed with Tris-buffered saline with 0.1% Tween-20 three times. Primary antibodies were added and incubated overnight at 4 °C. Sections were then rinsed with 3 washes of Tris-buffered saline with 0.1% Tween-20. Secondary antibodies (all diluted 1:200) were added and allowed to incubate for 2 h at room temperature. Sections were washed in phosphate-buffered saline, then incubated with 3,3′-diaminobenzidine (Molecular Probes, D-3571) at a dilution of 1:50,000 for 10 min at room temperature. Slides were imaged using a Nikon Eclipse Ti inverted epifiuorescence microscope equipped with a Photometries CollSNAP120 ES2 camera, and images were prepared using Nikon Elements software. Primary antibodies used include: 1:10,000 Mac2 (Accurate Chem., CL8942AP), 1:200 smooth-muscle actin (Sigma Aldrich, C6198), and 1:250 von Willebrand factor (Dako, A0082). Secondary antibodies used included: Alexa Fluor® 647 donkey anti-rabbit IgG (A31573) and Alexa Fluor® 488 donkey anti-rat IgG (A21208) purchased from Life Technologies.
Oil Red O Staining
Aortas were excised, cleaned of adventitia, and stained for atherosclerotic lesions using 0.5% oil red O stain prepared in 60% isopropanol. The images were photographed using a DS-Fil camera (Nikon) on a multizoom AZ100 microscope (Nikon), and the plaque burden was analyzed using Nikon Elements software and expressed as a percentage of total aortic area.
Superoxide Measurements
Tissue superoxide production was measured using the dihydroethidium HPLC method as previously reported [30]. One hour prior to sacrifice, all mice were injected intraperitoneally with 300 μL of dihydroethidium (10 mg/kg). Left carotid sinuses were obtained at the time of sacrifice and were flash frozen. For HPLC tissues were homogenized in a 50-mm phosphate buffer (pH 7.4) with 5 mm potassium cyanide, divided into 2 fractions, and one half was precipitated using acidified methanol; 2-OH-E+ was enriched from the supernatant of the second fraction after precipitating protein, using a microcolumn preparation of Dowex 50WX-8 cation exchange resin and eluted with 10 n HCl. The 2-OH-E+ product was then measured using fluorescence detection (ex: 490; em: 567) with a Shimadzu UFLC HPLC system. 2-OH-E+ superoxide production was normalized to total protein and reported as picomoles per milligram of protein.
Statistical Analysis
Data are given as means ± SEM; n indicates the number of mice. Statistical comparisons between groups were performed using GraphPad Prism software. Superoxide levels, plasma lipids, plaque measurements, and immunofluorescence data were analyzed by 1-way ANOVA with a Bonferroni multiple comparison test. A 1-way ANOVA was performed with Dunn’s multiple comparison test to evaluate liver pathology.
Results
Vascular Superoxide Production Is Increased in 6J Mice on HFD
Elevated mitochondrial ROS contribute to vascular inflammation and atherosclerosis [31, 32]; however, it is unclear how distinct mitochondrial functional properties may impact the development of atherosclerosis. To test this, we utilized 6N and 6J mice that have documented differences in mitochondrial functional properties [23, 24, 33, 34]. The 6J mice contain a naturally occurring in-frame 5-exon deletion within the Nnt gene that results in the loss of NNT protein production [35]. NNT is a mitochondrial inner membrane protein that catalyzes the transhydrogenation between NADH and NADP+. We have previously shown that this mutation leads to unique mitochondrial bioenergetic profiles in primary vascular endothelial cells isolated from these animals [24], and these variations may contribute to differences in atherosclerotic plaque progression. AAV8-PCSK9 injection was used to disrupt hepatic lipid handling and make the animals susceptible to diet-induced atherogenesis. Two weeks after AAV8-PCSK9 injection, the 6N and 6J animals were placed on an HFD for 8 weeks with a subset of each group receiving the mitochondrial SOD mimetic MitoTEMPO (0.8 mg/kg/day) for the final 4 weeks of the experiment. To determine how superoxide levels were impacted by HFD and co-treatment with HFD and MitoTEMPO, we quantified the oxidation of dihydroethidium to the superoxide-specific oxidation product (2-OH-HE+) in the left carotid sinus by HPLC (Fig. 1). Interestingly, even in untreated animals, vascular superoxide production was significantly higher in the 6J animals when compared to the 6N confirming mitochondrial functional variation as a regulator of vascular superoxide production. Furthermore, AAV8-PCSK9 treatment followed by 8 weeks of HFD significantly increased superoxide production in both animal groups; however, this effect was exacerbated in the 6J animals. Co-treatment with MitoTEMPO in the final 4 weeks of the experimental period was able to completely block HFD-induced increases in vascular superoxide production in both groups implicating the mitochondria as the major source of the ROS.
Fig. 1.
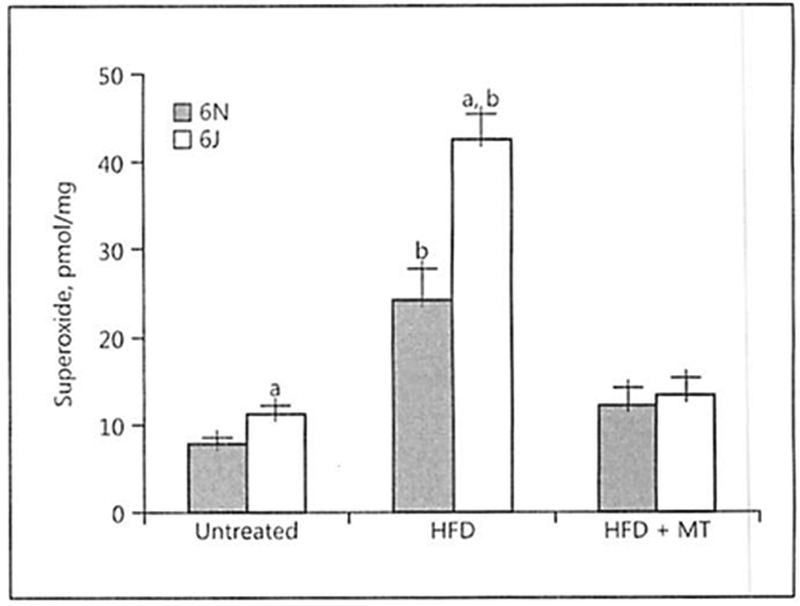
High-fat diet (HFD) exacerbates the increased vascular superoxide production observed in C57BL/6J mice. Vascular superoxide production was determined in untreated mice, those with AAV8-PCSK9 virus (3 × 1010 vector genomes) for 2 weeks and then subjected to HFD for 8 weeks, or HFD for 4 weeks followed by co-treatment with HFD and MitoTEMPO (MT; 0.8 mg/kg/day) for the final 4 weeks of the experiment. Superoxide was quantified in the left carotid sinus of the mice by exposing the animals to dihydroethidium at 10 mg/kg i.p. for 1 h and measuring the superoxide-specific oxidation product 2-OH-HE+ by HPLC. Values are means ± SEM of ≥6–8 animals. a p < 0.05 versus 6N; b p < 0.05 versus untreated.
AAV8-PCSK9 Treatment Leads to Disruption of Cholesterol Handling in Both 6N and 6J Mice
Two weeks of AAV8-PCSK9 treatment led to increased plasma cholesterol in both groups; however, there were no significant differences in plasma cholesterol between the 6N and 6J animals before or after AAV8-PCSK9 treatment (Fig. 2a). We observed no significant differences in weight gain among any of the treatment groups (Fig. 2b) and no marked differences in plasma lipids were detected in animals treated with FIFD alone except that 6J animals had significantly fewer triglycerides compared to 6N (Fig. 2c). Interestingly, MitoTEMPO treatment resulted in a significant decrease in total cholesterol, triglycerides, and LDL in the 6N animals that was not observed in 6J mice (Fig. 2c). This unique response to MitoTEMPO in the 6N mice led us to consider whether the livers of these mice were differentially impacted by AAV8-PCSK9 treatment with HFD in the presence or absence of MitoTEMPO. Histological analysis of the livers indicated that there was no difference in steatosis (Fig. 3a) or ballooning (Fig. 3b), suggesting hepatic lipid handling is similar among all groups. Additionally, levels of liver inflammation (Fig. 3c) and fibrosis (Fig. 3d) were consistent in both 6N and 6J mice across all treatments. These data indicate that differences in mitochondrial function and the production of mitochondrial superoxide may contribute to a unique regulation of plasma lipids in the context of atherogenic development that is independent of liver dysfunction.
Fig. 2.
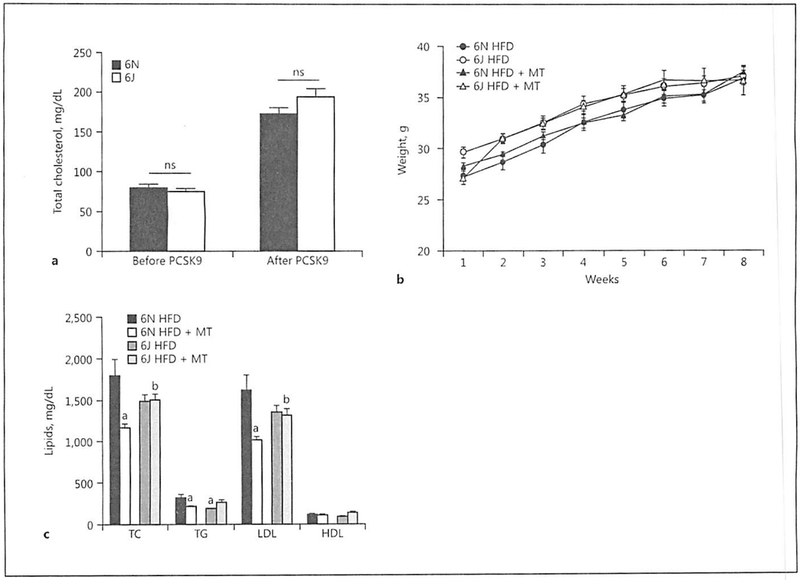
AAV8-PCSK9 treatment leads to disruption of cholesterol handling in both C57BL/6N and C57BL/6J mice, a Male C57BL/6N and C57BL/6J mice were treated with AAV8-PCSK9 virus (3 × 1010 vector genomes) for 2 weeks, and plasma cholesterol was determined by ELISA. b Mouse weight was monitored in AAV8-PCSK9 virus (3 × 1010 vector genomes)-infected mice fed a high-fat diet (HFD) for 8 weeks, or HFD for 4 weeks followed by co-treatment with HFD and MitoTEMPO (MT; 0.8 mg/kg/day) for the Final 4 weeks, c Plasma lipid analysis in C57BL/6N and C57BL/6J mice treated with AAV8-PCSK9 followed by HFD and HFD + MT. TC, total cholesterol; TG, triglycerides; LDL, low-density lipoprotein; HDL, high-density lipoprotein. Values are means ± SEM of 8 animals. a p < 0.05 versus 6N HFD; b p < 0.05 versus 6N HFD + MT.
Fig. 3.
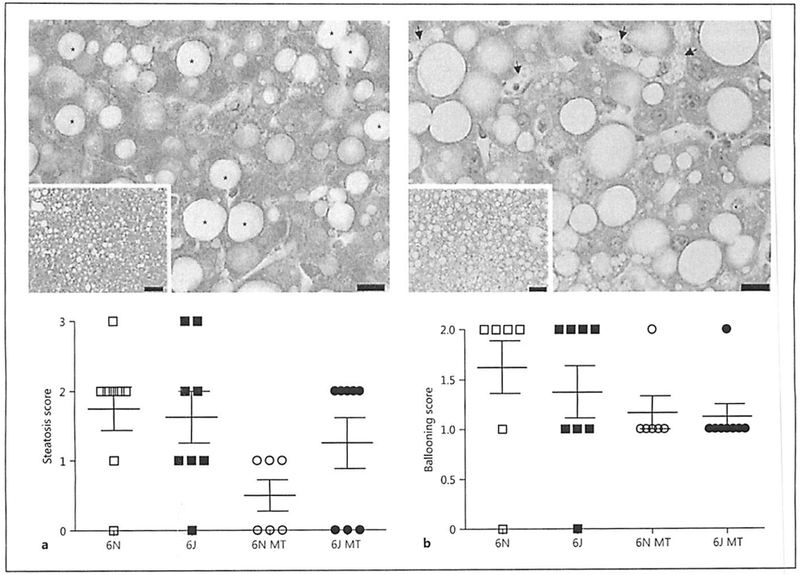
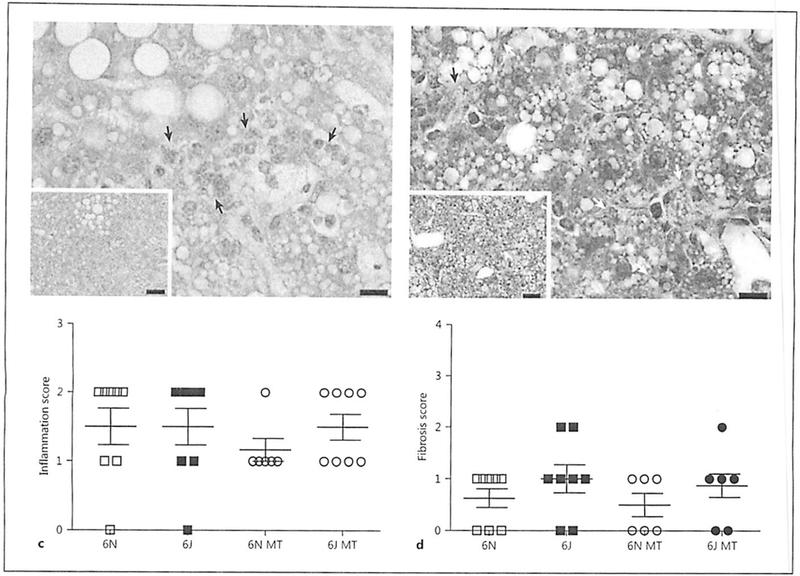
AAV8-PCSK9 and Mito-TEMPO treatment does not alter liver pathology. Male C57BL/6N and C57BL/6J mice were treated with AAV8-PCSK9 (3 × 1010 vector genomes) for 2 weeks and then subjected to a high-fat diet (HFD) for 8 weeks. A subset of animals was co-treated with MitoTEMPO (MT; 0.8 mg/kg/day) for the final 4 weeks of the experiment. All tissues were scored by a pathologist using the NASH Clinical Research Network Scoring System. Quantification of graded livers for steatosis (a), ballooning (b), and inflammation (c) with representative hematoxylin-eosin images. d Quantification of graded livers for fibrosis with representative Masson trichrome image. Scale bars are 10 and 50 μm for the 60× and 20× images, respectively. Values are means ± SEM of 6–8 animals.
Absence of NNT Increases Atherosclerotic Plaque Burden in 6J Mice on HFD
To determine whether the 6N and 6J mice show differential susceptibility to diet-induced atherosclerotic plaque formation, the aorta and aortic root were collected after 8 weeks of HFD, and atherosclerosis was assessed. En face staining of aortas with oil red O to visualize the atherosclerotic plaque area demonstrated that 6J mice show a significantly higher atherosclerotic burden in the aortic arch and in the whole aorta when compared to 6N mice (Fig. 4a, b). While MitoTEMPO treatment did not affect the atherosclerotic burden in whole aorta in either the 6N or 6J animals, MitoTEMPO treatment surprisingly exacerbated atherosclerotic plaque formation in the aortic root in the 6J mice despite normalizing vascular superoxide levels (Fig. 4a, c). In contrast, 6N mice showed a trend for reduced atherosclerosis following MiloTEMPO treatment in both the aortic arch and the whole aorta, although the differences did not reach statistical significance.
Fig. 4.
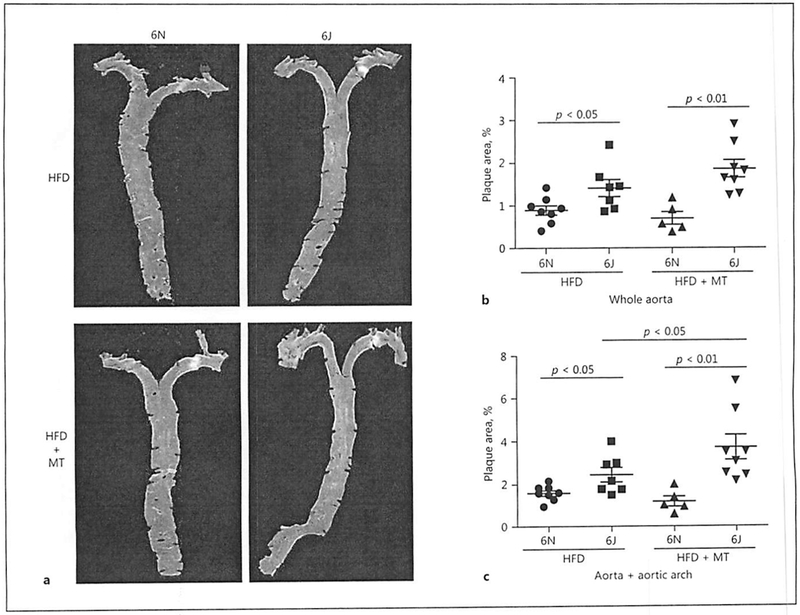
Absence of NNT increases atherosclerotic plaque formation in C57BL/6J mice on high-fat diet (HFD). Male C57BL/6N and C57BL/6J mice were treated with AAV8-PCSK9 virus (3 × 1010 vector genomes) for 2 weeks and then subjected to HFD for 8 weeks. A subset of animals was co-treated with MitoTEMPO (MT; 0.8 mg/kg/day) for the final 4 weeks of the experiment. a Representative images of oil red O-stained aortas from each group. Relative quantitation of plaque size in the whole aorta (b) and the aortic arch (c). Values are means ± SEM of 6–8 animals.
To confirm the changes in plaque formation observed with en face staining of aortas, cross-sections of the aortic root were analyzed for changes in plaque size between 6J and 6N mice. Consistent with the aorta, 6J animals placed on HFD show significantly larger plaques in the aortic root compared to the 6N animals (Fig. 5a, b). Similarly, MitoTEMPO treatment exacerbated plaque development in the 6J animals while having no appreciable impact on plaque development in the 6N mice (Fig. 5b). To determine whether changes in plaque composition accompanied differences in plaque size, cross-sections of the aortic roots were stained with markers for macrophages (Mac2), smooth-muscle cells (smooth-muscle actin), and endothelial cells (von Willebrand factor) and visualized by immunofluorescence (Fig. 6a). Quantification of fluorescence intensity indicates that plaque composition was not significantly different in any of the treatment groups (Fig. 6b–d) although we did observe a trend for increased smooth-muscle cell content in plaques from both the 6N and 6J mice treated with HFD and MitoTEMPO. These data demonstrate that distinct mitochondrial functional properties and the production of mitochondrial ROS influence the severity of HFD-induced atherosclerosis in 6N and 6J substrains.
Fig. 5.
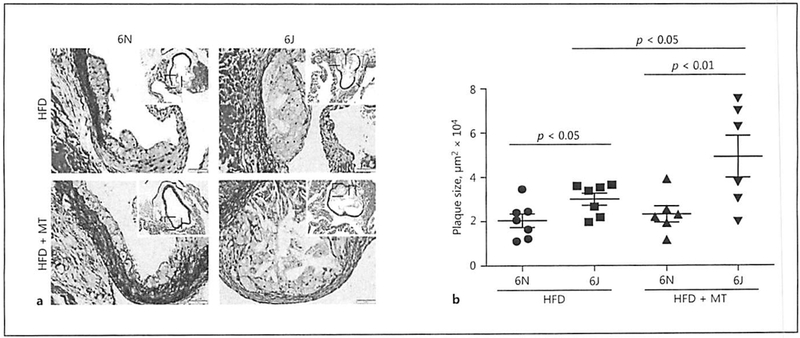
Plaque size is increased in C57BL/6J mice on high-fat diet (HFD). Male C57BL/6N and C57BL/6J were treated with AAV8-PCSK9 virus (3 × 1010 vector genomes) for 2 weeks and then subjected to HFD for 8 weeks. A subset of animals was co-treated with MitoTEMPO (MT; 0.8 mg/kg/day) for the final 4 weeks of the experiment. a Representative images of Movat Pentachrome-stained aortic sinus from each group. b Quantitation of plaque size. Values are means ± SEM of 6–8 animals.
Fig. 6.
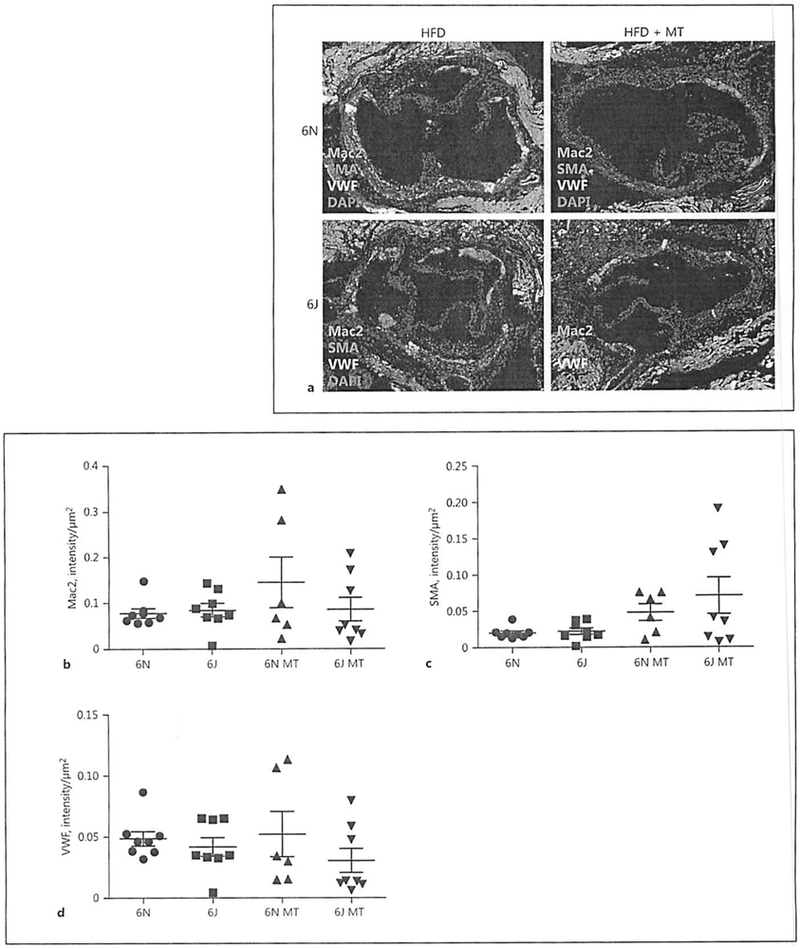
Cellular composition of aortic root plaques is similar among treatment groups. Male C57BL/6N and C57BL/6J mice were treated with AAV8-PCSK9 (3 × 1010 vector genomes) for 2 weeks and then subjected to a high-fat diet (HFD) for 8 weeks. A subset of animals was co-treated with MitoTEMPO (MT; 0.8 mg/kg/day) for the final 4 weeks of the experiment. a Representative immunofluorescence images visualizing macrophage area (Mac2 positive, green), smooth muscle (smooth-muscle actin, SMA positive, red), and endothelial (von Willebrand factor positive, VWF, white) localization within aortic root plaques. Quantification of macrophage (b), smooth muscle (c), and endothelial area (d) is provided. The scale bar is 200 μm at the ×20 magnification. Values are means ± SEM of 6–8 animals.
Discussion
Numerous studies have implicated mitochondria-derived ROS as a critical regulator of atherosclerotic development [32]. As mitochondrial superoxide generation is a byproduct of normal energy metabolism, the rale and/or levels of superoxide generation will be dictated by the functional properties of the mitochondria. However, it is unclear how variation in mitochondrial function may impact the production of superoxide and further alter the development of atherosclerosis in vivo. Previous work from our lab and others has demonstrated that the presence or absence of NNT in 6N and 6J mice results in unique mitochondrial functional properties [23, 24, 33, 34]. Additionally, we have shown that increased mitochondrial superoxide generation observed in the 6J mice is able to exacerbate angiotensin II-induced hypertension in these mice [24]. We decided to investigate whether differences in NNT expression and subsequent pro-oxidative mitochondrial phenotype influences atherosclerosis development using the rAAV encoding a gain-of-function mutant form (D377Y) of PCSK9. Hypercholesterolemia associated with this model led to increased vascular superoxide production, with an even greater increase observed in 6J mice lacking NNT. Furthermore, the loss of NNT augmented atherosclerosis independent of changes in atherosclerotic serum lipid profiles. Interestingly, even though MitoTEMPO treatment reduced hypercholesterolemia-elicited vascular superoxide production, it did not reduce atherosclerosis. In fact, MitoTEMPO exacerbated atherosclerosis severity in 6J mice. The ability of MitoTEMPO to reduce cholesterol in the 6N mice, but not the 6J, may have contributed to this observation. Histological analysis of livers from all groups indicates that this effect is independent of dysfunctional hepatic lipid handling associated with the PCSK9 and/or MitoTEMPO treatments in these mice. Reduced circulating cholesterol levels incited by scavenging mitochondrial ROS is an unexpected observation, and the link between mitochondrial ROS and the regulation of blood lipids in atherosclerosis remains completely undefined. Supporting our observations, Kim et al. [36] have also demonstrated that systemic scavenging of ROS with TEMPOL can also decrease plasma lipid levels in ApoE−/− mice treated with HFD. These data suggest another mechanism by which mitochondrial ROS may contribute to HFD-induced atherosclerosis that merits further exploration. It is possible that the increased plaque size observed in the HFD-treated 6J mice that received MitoTEMPO could be an adaptive response aimed at stabilization of the plaque to prevent plaque rupture. Previous work has demonstrated that treatment with TEMPOL increases plaque collagen content that is consistent with a more stable plaque [36]. However, immunocytofluorescence of the plaques from this study indicated that there was no difference in smooth muscle cell content in the plaques among any of the groups (Fig. 6) suggesting that MitoTEMPO in the 6J mice did not alter plaque stability. It is important to note that the 6J animal is not an NNT-specific knockout but rather a unique substrain of the C57BL/6 mouse, and as a result we cannot exclude other strain-specific differences as contributors to the pro-atherogenic phenotype we observe in the 6J mice [37]. However, the vast majority of studies that have investigated the development of metabolic disease in these mice have implicated NNT, changes in mitochondrial function, or alterations of ROS metabolism as a critical regulator of the differences observed amongst these mice [13, 38]. Our data are consistent with these studies and suggest that the loss of NNT impacts not only the development of atherosclerosis but also the effect that antioxidant treatment has on atherosclerotic development.
The contribution of ROS to the development of atherosclerosis has primarily been focused on NADPH oxidases as the source of ROS production [31]. Far less is known regarding the ability of mitochondrial ROS to impact the development of atherosclerosis in vivo. Previous studies have demonstrated that treatment with mitochondria-targeted antioxidants can reduce the vascular pathologies associated with atherosclerosis when given systemically [39]. However, it is unclear whether variation in mitochondrial functional properties will have any impact on the initiation or progression of atherosclerosis. The 6J animals display a pro-oxidative mitochondrial phenotype characterized by increased superoxide and hydrogen peroxide production and decreased glutathione levels and glutathione peroxidase activity [23, 24]. Consistent with a pro-oxidative mitochondrial phenotype we observed increased vascular superoxide production in untreated 6J animals compared to 6N ones (Fig. 1). Vascular superoxide was elevated in both groups in response to the HFD, but the production of superoxide was significantly higher in the 6J animals. As expected, co-treatment of the mice with MitoTEMPO was able to reduce vascular superoxide production to near baseline levels in both groups. These data demonstrate that mitochondrial ROS production differs among the 6N and 6J mice implicating the absence of NNT in the disruption of normal mitochondrial redox balance in the 6J mice. The HFD increased mitochondrial superoxide production in both groups, and again the effect was more severe in the 6J animals suggesting that the increased ROS production plays a key role in the observed differences in plaque development in 6J mice.
MitoTEMPO functions as a mitochondria-targeted SOD mimetic that will effectively scavenge mitochondrial superoxide with hydrogen peroxide as a putative product. In most circumstances, this hydrogen peroxide would then be quickly broken down by the mitochondrial antioxidant systems that include glutathione peroxidase and peroxiredoxins. However, we have previously shown that the absence of NNT in the 6J animals leads to a reduction in glutathione peroxidase activity [24]. Previous studies have demonstrated that a reduction in glutathione peroxidase activity is associated with an accelerated progression of atherosclerosis [9]. This observation agrees with the concept that the absence of NNT activity in the 6J cells will have an impact on antioxidant activities that are dependent upon NADPH for their reducing equivalents. In this case, glutathione peroxidase relies upon NADPH to maintain the reduced glutathione pools utilized for its catalytic cycle. The data presented here suggest that, in the presence of MitoTEMPO, superoxide production in the mitochondria of 6J cells will be quickly scavenged and may lead to increases in hydrogen peroxide, but due to impaired glutathione peroxidase activity, the hydrogen peroxide will persist. A similar study that compared the metabolic responses of the 6N and 6J mice to an HFD/high-sucrose diet observed increased hydrogen peroxide emission rates under all substrate conditions in muscle fibers from 6J compared to 6N mice [40]. While no significant differences were observed across multiple phenotypic indices of metabolic disease, this study supports the concept that the absence of NNT will increase the steady-state levels of mitochondrial hydrogen peroxide. In light of this study, our data illustrating the impact of NNT’s absence on plaque development suggests a more prominent role for NNT in regulating the contribution of mitochondrial ROS at sites of inflammation within the regions of the plaque itself. As hydrogen peroxide can easily diffuse across mitochondrial membranes, reduced NNT activity could lead to a longer half-life and an increased opportunity to contribute to cellular dysfunction. Supporting this concept, mice that overexpress both SOD1 and catalase have a greater reduction in atherosclerosis than the expression of just SOD1, highlighting the ability of hydrogen peroxide to contribute to atherosclerotic development and suggesting that the removal of both superoxide and hydrogen peroxide provides the greatest atheroprotective effect [41]. The data presented here demonstrate that variation in mitochondrial function and a pro-oxidative mitochondrial phenotype in 6J animals leads to more severe atherosclerotic disease and implicates hydrogen peroxide as an important mediator in atherosclerotic plaque development.
The significant contribution of ROS to plaque development suggests that antioxidant therapy should provide a therapeutic benefit with respect to atherosclerotic progression. However, the use of antioxidant treatments to reduce atherosclerosis has been met with mixed results. The most compelling evidence for a beneficial effect of antioxidants to reduce oxidative stress in the pathogenesis of atherosclerosis originates from animal models of disease. Studies have shown that decreasing ROS generation by transgenic expression of antioxidant enzymes such as SOD1, catalase, and glutathione reductase in general reduce atherosclerotic severity [41, 42]. Similarly, increased ROS associated with a loss of SOD2 or glutathione peroxidase activity enhances atherosclerotic development [8, 9, 43]. While these studies proposed that the mechanism of enhanced atherosclerosis in the SOD2+/− mice is likely due to changes in mitochondrial function rather than enhanced oxidative stress, the authors did not address to what extent mitochondrial function and ROS production may be linked. In humans, several antioxidant therapies including vitamin E, vitamin C, vitamin A and β-carotene have failed to provide an overall benefit, and meta-analysis suggests that there may be a potential increase in mortality associated with these supplements [44]. Interestingly, we demonstrate here that MitoTEMPO treatment failed to reduce atherosclerosis in 6N mice, and further exacerbated atherosclerosis in 6J mice. This is in contrast to earlier work demonstrating that 12-week MitoTEMPO therapy in aged ApoE−/− mice on HFD modestly reduced plaque burden in aortas [45]. Differences in the impact of MitoTEMPO treatment on atherosclerotic plaque development may be related to several factors. The mice used in our study were young compared to the aged ApoE−/− mice, and it is unclear how mitochondrial function and the propensity for mitochondrial ROS production may have changed over that time. Additionally, the models of diet-induced atherosclerosis used in these studies are unique. While both models disrupt normal lipid handling allowing the mice to be susceptible to diet-induced atherosclerosis, it is accomplished in very different ways. The AAV8-PSCK9 model shows remarkable consistency with other mouse models of atherosclerosis; however, some differences have been observed, and further studies are required to determine exactly how to assimilate these new data with what has previously been shown with established models.
We have shown that the absence of NNT and a pro-oxidative mitochondrial phenotype leads to increased atherosclerotic burden in response to HFD in the 6J mice. Interestingly, co-treatment with the mitochondria-targeted SOD mimetic MitoTEMPO lowered vascular superoxide production but exacerbated atherosclerosis in the 6J animals suggesting a pro-atherogenic role for hydrogen peroxide under these conditions. These data are consistent with our previous studies implicating NNT as a critical regulator of mitochondrial redox balance that, when lost, contributes to enhanced cardiovascular pathologies. As a central regulator of mitochondrial ROS levels through modulation of the mitochondrial antioxidant systems, our data suggest that NNT could be a novel effector protein that is uniquely capable of having a significant impact on ROS-driven endothelial dysfunction and plaque development associated with the development of atherosclerosis.
Footnotes
Disclosure Statement
The authors have no conflicts of interest to disclose.
References
- 1.Chen K, Keaney JFJ: Evolving concepts of oxidative stress and reactive oxygen species in cardiovascular disease. Curr Atheroscler Rep 2012;14:476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mabile L, Meilhac O, Escargueil-Blanc I, Troly M, Pieraggi MT, Salvayre R, Nègre-Salvayre A: Mitochondrial function is involved in LDL oxidation mediated by human cultured endothelial cells. Arterioscler Thromb Vasc Biol 1997;17:1575–1582. [DOI] [PubMed] [Google Scholar]
- 3.Madamanchi NR, Vendrov A, Runge MS: Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol 2005;25:29–38. [DOI] [PubMed] [Google Scholar]
- 4.Divakaruni AS, Brand MD: The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2010;26:192–205. [DOI] [PubMed] [Google Scholar]
- 5.Fukai T, Ushio-Fukai M: Superoxide dismutases: role in redox signaling, vascular function, and diseases. Antioxid Redox Signal 2011;15:1583–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Halliwell B, Gutteridge JMC: Free Radicals in Biology and Medicine, ed 4 New York, Oxford University Press, 2007. [Google Scholar]
- 7.Mavelli I, Rigo A, Federico R, Ciriolo MR, Rotilio G: Superoxide dismutase, glutathione peroxidase and catalase in developing rat brain. Biochem J 1982;204:535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis P, Stefanovic N, Pete J, Calkin AC, Giunti S, Thallas-Bonke V, Jandeleit-Dahm KA, Allen TJ, Kola I, Cooper ME, de Haan JB: Lack of the antioxidant enzyme glutathione peroxidase-1 accelerates atherosclerosis in diabetic apolipoprotein E-deficient mice. Circulation 2007;115:2178–2187. [DOI] [PubMed] [Google Scholar]
- 9.Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, Rossmann H, Tsimikas S, Reifenberg K, Cheng F, Lehr HA, Blankenberg S, Förstermann U. Münzel T, Lackner KJ: Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2007;27:850–857. [DOI] [PubMed] [Google Scholar]
- 10.Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, Sessa WC, Min W: Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. Am I Pathol 2007;170:1108–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Engel PC: Glutamate dehydrogenases: the why and how of coenzyme specificity. Neurochem Res 2014;39:426–432. [DOI] [PubMed] [Google Scholar]
- 12.Jo SH, Son MK, Koh HJ, Lee SM, Song IH, Kim YO, Lee YS, Jeong KS, Kim WB, Park JW, Song BJ, Huh TL: Control of mitochondrial redox balance and cellular defense against oxidative damage by mitochondrial NADP+-dependent isocitrate dehydrogenase. J Biol Chem 2001;276:16168–16176. [DOI] [PubMed] [Google Scholar]
- 13.Ronchi JA, Francisco A, Passos LA, Figueira TR, Castilho RF: The contribution of nicotinamide nucleotide transhydrogenase to peroxide detoxification is dependent on the respiratory state and counterbalanced by other sources of NADPH in liver mitochondria. J Biol Chem 2016;291:21073–20187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rydström J: Mitochondrial NADPH, transhydrogenase and disease. Biochim Biophys Acta 2006;1757:721–726. [DOI] [PubMed] [Google Scholar]
- 15.Teller JK, Fahien LA, Davis JW: Kinetics and regulation of hepatoma mitochondrial NAD(P) malic enzyme. J Biol Chem 1992;267:10423–10432. [PubMed] [Google Scholar]
- 16.Yin F, Sancheti H, Cadenas E: Silencing of nicotinamide nucleotide transhydrogenase impairs cellular redox homeostasis and energy metabolism in PC12 cells. Biochim Biophys Acta 2012;1817:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Earle SR, Fisher RR: A direct demonstration of proton translocation coupled to transhydrogenation in reconstituted vesicles. J Biol Chem 1980;255:827–830. [PubMed] [Google Scholar]
- 18.Olausson T, Fjellström O, Meuller J, Rydström J: Molecular biology of nicotinamide nucleotide transhydrogenase – a unique proton pump. Biochim Biophys Acta 1995;1231:1–19. [DOI] [PubMed] [Google Scholar]
- 19.Rydström J, Teixeira da Cruz A, Ernster L: Factors governing the steady state of the mitochondrial nicotinamide nucleotide transhydrogenase system. Biochem J 1970;116:12P–13P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aston-Mourney K, Wong N, Kebede M, Zraika S, Balmer L, McMahon JM, Fam BC, Favaloro J, Proietto J, Morahan G, Andrikopoulos S: Increased nicotinamide nucleotide transhydrogenase levels predispose to insulin hypersecretion in a mouse strain susceptible to diabetes. Diabetologia 2007;50:2676–2485. [DOI] [PubMed] [Google Scholar]
- 21.Freeman H, Shimomura K, Horner E, Cox RD. Ashcroft FM: Nicotinamide nucleotide transhydrogenase: a key role in insulin secretion. Cell Metabolism 2006;3:35–45. [DOI] [PubMed] [Google Scholar]
- 22.Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A, Mijat V, Goldsworthy M, Moir L, Haynes A, Quarterman J, Freeman HC, Ashcroft FM, Cox RD: A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia 2005;48:675–686. [DOI] [PubMed] [Google Scholar]
- 23.Ronchi JA, Figueira TR, Ravagnani FG, Oliveira HC, Vercesi AE, Castilho RF: A spontaneous mutation in the nicotinamide nucleotide transhydrogenase gene of C57BL/6J mice results in mitochondrial redox abnormalities. Free Radic Biol Med 2013;63:446–456. [DOI] [PubMed] [Google Scholar]
- 24.Leskov I, Neville A, Shen X, Pardue S, Kevil CG, Granger DN, Krzywanski DM: Nicotinamide nucleotide transhydrogenase activity impacts mitochondrial redox balance and the development of hypertension in mice. J Am Soc Hypertens 2016;S1933-1711:30601–30605. [DOI] [PubMed] [Google Scholar]
- 25.Bjørklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG, Bentzon JF: Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res 2014;114:1684–1689. [DOI] [PubMed] [Google Scholar]
- 26.Al-Mashhadi RH, Bjørklund MM, Mortensen MB, Christoffersen C, Larsen T, Falk E, Bentzon JF: Diabetes with poor glycaemic control does not promote atherosclerosis in genetically modified hypercholesterolaemic minipigs. Diabetologia 2015;58:1926–1936. [DOI] [PubMed] [Google Scholar]
- 27.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM: Clades of adeno-associated viruses are widely disseminated in human tissues. J Virol 2004;78:6381–6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Klein RL, Dayton RD, Tatom JB, Henderson KM, Henning PP: AAV8, 9, Rh10, Rh43 vector gene transfer in the rat brain: effects of serotype, promoter and purification method. Mol Ther 2008;16:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kleiner DE, Brunt EM, Van Natta M, Behling C, Contos MJ, Cummings OW, Ferrell LD, Liu YC, Torbenson MS, Unalp-Arida A, Yeh M, McCullough AJ, Sanyal AJ; Nonalcoholic Steatohepatitis Clinical Research Network: Design and validation of a histological scoring system for nonalcoholic fatty liver disease. Hepatology 2005;41:1313–1321. [DOI] [PubMed] [Google Scholar]
- 30.Zielonka J, Vasquez-Vivar J, Kalyanaraman B: Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat Protoc 2008:3:8–21. [DOI] [PubMed] [Google Scholar]
- 31.Li H, Horke S, Förstermann U: Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 2014;237:208–219. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Tabas I: Emerging roles of mitochondria ROS in atherosclerotic lesions: causation or association? J Atheroscler Thromb 2014;21:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Arkblad EL, Tuck S, Pestov NB, Dmitriev RI, Kostina MB, Stenvall J, Tranberg M, Rydström J: A Caenorhabditis elegans mutant lacking functional nicotinamide nucleotide transhydrogenase displays increased sensitivity to oxidative stress. Free Radic Biol Med 2005;38:1518–1525. [DOI] [PubMed] [Google Scholar]
- 34.Nickel AG, von Hardenberg A, Hohl M, Löffler JR, Kohlhaas M, Becker J, Reil JC, Kazakov A, Bonnekoh J, Stadelmaier M, Puhl SL, Wagner M, Bogeski I, Cortassa S, Kappl R, Pasieka B, Lafontaine M, Lancaster CR, Blacker TS, Hall AR, Duchen MR, Kästner L, Lipp P, Zeller T, Müller C, Knopp A, Laufs U, Böhm M, Hoth M, Maack C: Reversal of mitochondrial transhydrogenase causes oxidative stress in heart failure. Cell Metab 2015;22:472–484. [DOI] [PubMed] [Google Scholar]
- 35.Huang TT, Naeemuddin M, Elchuri S, Yamaguchi M, Kozy HM, Carlson EJ, Epstein CJ: Genetic modifiers of the phenotype of mice deficient in mitochondrial superoxide dismutase. Hum Mol Genet 2006;15:1187–1194. [DOI] [PubMed] [Google Scholar]
- 36.Kim CH, Mitchell JB, Bursill CA, Sowers AL, Thetford A, Cook JA, van Reyk DM, Davies MJ: The nitroxide radical TEMPOL prevents obesity, hyperlipidaemia, elevation of inflammatory cytokines, and modulates atherosclerotic plaque composition in apoE−/− mice. Atherosclerosis 2015;240:234–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Simon MM, Greenaway S, White JK, Fuchs H, Gailus-Durner V, Wells S, Sorg T, Wong K, Bedu E, Cartwright EJ, Dacquin R, Djebali S, Estabel J, Graw J, Ingham NJ, Jackson IJ, Lengeling A, Mandillo S, Marvel J, Meziane H, Preitner F, Puk O, Roux M, Adams DJ, Atkins S, Ayadi A, Becker L, Blake A, Brooker D, Cater H, Champy MF, Combe R, Danecek P, di Fenza A, Gates H, Gerdin AK, Golini E, Hancock JM, Hans W, et al. : A comparative phenotypic and genomic analysis of C57BL/6J and C57BL/6N mouse strains. Genome Biol 2010;14:R82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nicholson A, Reifsnyder PC, Malcolm RD, Lucas CA, MacGregor GR, Zhang W, Leiter EH: Diet-induced obesity in two C57BL/6 substrains with intact or mutant nicotinamide nucleotide transhydrogenase (Nnt) gene. Obesity (Silver Spring) 2010;18:1902–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dikalova AE, Bikineyeva AT, Budzyn K, Nazarewicz RR, McCann L, Lewis W, Harrison DG, Dikalov SI: Therapeutic targeting of mitochondrial superoxide in hypertension. Circ Res 2010;107:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher-Wellman KH, Ryan TE, Smith CD, Gilliam LA, Lin CT, Reese LR, Torres MJ, Neufer PD: A direct comparison of metabolic responses to high-fat diet in C57BL/6J and C57BL/6NJ mice. Diabetes 2016;65:3249–3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yang H, Roberts LJ, Shi MJ, Zhou LC, Ballard BR, Richardson A, Guo ZM: Retardation of atherosclerosis by overexpression of catalase or both Cu/Zn-superoxide dismutase and catalase in mice lacking apolipoprotein E. Circ Res 2004;95:1075–1081. [DOI] [PubMed] [Google Scholar]
- 42.Guo Z, Ran Q, Roberts LJ, Zhou L, Richardson A, Sharan C, Wu D, Yang H: Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic Biol Med 2008;44:343–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS: Mitochondrial integrity and function in atherogenesis. Circulation 2002;106:544–549. [DOI] [PubMed] [Google Scholar]
- 44.Vivekananthan DP, Penn MS, Sapp SK, Hsu A, Topol EJ: Use of antioxidant vitamins for the prevention of cardiovascular disease: meta-analysis of randomised trials. Lancet 2003;361:2017–2023. [DOI] [PubMed] [Google Scholar]
- 45.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR: NOX4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal 2015;23:1389–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]