

ABSTRACT
Determining prostate cancer (PCa) aggressiveness and reclassification are critical events during the treatment of localized disease and for patients undergoing active surveillance (AS). Since T cells play major roles in cancer surveillance and elimination, we aimed to identify genetic biomarkers related to T cell cancer immune response which are predictive of aggressiveness and reclassification risks in localized PCa. The genotypes of 3,586 single nucleotide polymorphisms (SNPs) from T cell cancer immune response pathways were analyzed in 1762 patients with localized disease and 393 who elected AS. The aggressiveness of PCa was defined according to pathological Gleason score (GS) and D’Amico criteria. PCa reclassification was defined according to changes in GS or tumor characteristics during subsequent surveillance biopsies. Functional characterization and analysis of immune phenotypes were also performed. In the localized PCa cohort, seven SNPs were significantly associated with the risk of aggressive disease. In the AS cohort, another eight SNPs were identified as predictors for aggressiveness and reclassification. Rs1687016 of PSMB8 was the most significant predictor of reclassification. Cumulative analysis showed that a genetic score based on the identified SNPs could significantly predict risk of D’Amico high risk disease (P-trend = 2.4E-09), GS4 + 3 disease (P-trend = 1.3E-04), biochemical recurrence (P-trend = 0.01) and reclassification (P-trend = 0.01). In addition, the rs34309 variant was associated with functional somatic mutations in the PI3K/PTEN/AKT/MTOR pathway and tumor lymphocyte infiltration. Our study provides plausible evidence that genetic variations in T cell cancer immune response can influence risks of aggressiveness and reclassification in localized PCa, which may lead to additional biological insight into these outcomes.
Abbreviations: PCa, prostate cancer; AS, active surveillance; GS, Gleason score; PSA, prostate specific antigen; TCGA, The Cancer Genome Atlas; SNP, single nucleotide polymorphisms; UFG, unfavorable genotype.
Keywords: Aggressiveness, active surveillance, biochemical recurrence, single nucleotide polymorphisms, T cell cancer immune response, prostate cancer, PI3K signaling pathway
Introduction
Treatment decisions for localized prostate cancer (PCa) are guided by risk stratification based on disease aggressiveness.1 Clinical factors such as pathological Gleason score (GS) and serum prostate specific antigen (PSA) values are known to predict outcomes such as recurrence and metastasis;2 however, risk determination remains imperfect. For example, approximately 20–30% of men who elected active surveillance for low risk PCa experienced reclassification or disease upgrading during 5 years of follow-up.3
Genomic-level changes can partially explain the variation seen in PCa, as several chromosomal regions have been implicated in disease aggressiveness.4 Despite this, no specific genetic mutation has been conclusively validated as a predictor of risk in localized PCa. Multi-stage genome wide scans have identified single nucleotide polymorphisms (SNPs) that are associated with PCa diagnosis, although few are associated with PCa aggressiveness.5 Hypothesis-driven work has also identified SNPs associated with PCa aggressiveness in intermediate-risk disease, reclassification on active surveillance (AS), and post-treatment outcomes.6–9 Despite these findings, the contribution of germline variants to PCa aggressiveness and progression of low risk disease remains largely unexplored.
Immune cells are the major force of cancer surveillance and elimination.10 Multiple immune-related treatments such as sipuleucel-T and checkpoint inhibitors have shown potential promise in the treatment of metastatic castration-resistant PCa.11,12 Although recent clinical trials of a CTLA4 inhibitor (ipilimumab) have indicated minimal impact on overall survival,13,14 the treatment involving autologous cellular immunotherapy has already been approved to treat advanced PCa, while more definitive studies are being planned to determine efficacy of other checkpoint blockade treatment. Furthermore, androgen ablation may increase prostate T cell infiltration in both normal and malignant prostate tissue15 and PD-L1 expression may serve as an independent indicator of biochemical recurrence in primary PCa,16 lending evidence to the role that the innate adaptive immune system may play in PCa development or progression.
T cell cancer immune response is dependent on the full anti-tumor life cycle of T cells, from priming to immune-related function,10 much of which is under genetic control. Intra-tumor T cells, rather than B cells, are reported to be associated with clinical outcome in PCa.17 Also, there is evidence that T cells, but less so for other immune cells, play a key role in the anti-cancer immunity.10,18 In light of the importance of T cell cancer immune response in PCa prognosis, we hypothesized that germline differences in T cell cancer immune response pathways may be associated with PCa aggression and reclassification. In this multi-phase study, we aimed to investigate the association of cancer immune pathway genetic variation with localized PCa aggressiveness and reclassification risk. We first compiled a comprehensive panel of germline genetic variants of 312 T cell cancer immune response genes and investigated the association between variant alleles and risk of localized PCa diagnosis in a MD Anderson Cancer Center PCa (MDACC-PCa) patient cohort (phases I and II). We then evaluated associations of these SNPs with reclassification risk in patients undergoing AS from an ongoing prospective trial (phase III). We calculated genotypic risk scores and then examined their association with PCa-related outcomes. Based on these findings, we performed functional characterization of identified SNPs and implicated possible mechanisms. To our knowledge, this is the first integrated, large-scale investigation of the association of T cell cancer immune response-related genetic variations with PCa aggressiveness, outcomes, and immune phenotypes.
Results
Patient characteristics
Clinicopathologic characteristics for the two patient cohorts are listed in Table 1. There were 1,762 and 393 patients in phases I/II and phase III, respectively. In the phase I cohort, 598(33.9%) patients had low risk PCa and 330(18.7%) had high risk according to D’Amico risk classification. When grouped based on GS, 657(37.3%) patients had GS6 or lower and 218(12.4%) patients had GS8 or higher PCa. Among the 887 patients in the cohort with GS7 disease, 647(36.7%) patients had GS3 + 4 on biopsy and 240(13.6%) had GS4 + 3. Of 1180 patients who received local therapy, 96 developed biochemical recurrence (Table S1). Among the 393 patients in the AS cohort used for phase III of the study, most had low-risk characteristics, including 325(82.7%) with GS6 diseases, 349(88.8%) patients with T1 stage and 379(96.4%) patients with PSA <10 ng/ml. After a median follow up time of 47.4 months, the PCa in 127(32.4%) of these patients was reclassified.
Table 1.
Clinical characteristics of localized PCa study cohorts.
| MDACC-PCa cohort |
AS cohort |
|
|---|---|---|
| Characteristics | N (%) | N (%) |
| Total | 1762 | 393 |
| Age at diagnosis | ||
| Mean(SD) | 61.6 (7.9) | 64.2 (8.3) |
| Tumor Stage (T), N (%) | ||
| T1 | 1,109 (62.94) | 349 (88.8) |
| T2 | 575 (32.63) | 44 (11.2) |
| T3-T4 | 69 (3.92) | 0 |
| Unknown | 9 (0.51) | 0 |
| PSA level at diagnosis, ng/mla,b | ||
| <4 or <2.5 | 442 (25.11) | 89 (22.7) |
| 4–9.9 or 2.5–3.9 | 1,108 (62.95) | 108 (27.5) |
| 10–19.9 or 4–9.9 | 145 (8.24) | 182 (46.3) |
| ≥20 or ≥10 | 65 (3.69) | 14 (3.6) |
| Biopsy-proven GS | ||
| ≤6 | 657 (37.29) | 325 (82.7) |
| 3 + 4 | 647 (36.72) | 60 (15.3) |
| 4 + 3 | 240 (13.62) | 8 (2.0) |
| ≥8 | 218 (12.37) | 0 |
| D’Amico risk group | ||
| Low | 598 (33.94) | 315 (80.2) |
| Intermediate | 829 (47.05) | 73 (18.6) |
| High | 330 (18.73) | 5 (1.3) |
| Not grouped | 5 (0.28) | 0 |
| Disease reclassificationb | ||
| Yes | - | 127 (32.4) |
| No | - | 265 (67.6) |
| Biochemical recurrence | ||
| Yes | 96 (8.14) | - |
| No | 1,084 (91.86) | - |
| Treatment | ||
| Radical prostatectomy | 918 (52.10) | - |
| Radiotherapy | 378 (21.45) | - |
| Surveillance or unknownc | 429 (24.35) | - |
| Otherd | 37 (2.10) | - |
| Follow-up time | ||
| Median(Range) | 47.4 (2.9–125.8) | - |
aDifferent criteria were used to categorize patients by PSA levels between study populations. Retrospective case series: <4 versus 4–9.9 versus 10–19.9 versus ≥20 ng/ml; AS cohort: <2.5 versus 2.5–3.9 versus 4–9.9 versus ≥10 ng/ml.
bPSA information of two patients in MDACC-PCa cohort were missing, while disease reclassification information of one patients in AS cohort was not available.
cPatients undergoing active surveillance/watchful waiting or whose initial treatment information was unavailable.
dCryoablation, high intensity focused ultrasound, transurethral resection of prostate or androgen deprivation therapy.
Association of SNPs with PCa aggressiveness and reclassification
The study scheme is displayed in Figure 1. In the phase I analysis, 73 SNPs were found to be significantly associated with both GS≥8 risk and high D’Amico risk (Table S2). In phase II, SNPs associated with GS4 + 3 = 7 were evaluated in intermediate risk patients only. When combined with results from phase I, seven SNPs in six loci were found to be associated with aggressive cancer and risk of GS4 + 3 (Table 2). In phase III of the study, another eight candidate SNPs (unique from those identified in phase II) were found to be both associated with disease reclassification in the AS cohort and disease aggressiveness as determined in phase I of the study (Table 3). Among these SNPs, rs16871026 demonstrated the strongest association with PCa aggressiveness (GS≥8 risk: odds ratio [OR] 3.52, 95% confidence interval [CI]: 1.40–8.84, P = 0.007) and reclassification (hazard ratio [HR] 2.68, 95%CI: 1.28–5.59, P = 0.009). Also, rs1687106 and rs34309 were significantly associated with reclassification time. Specifically, for rs16871026, time to reclassification for the AA+AG genotype was 41.0 months versus >60 months for GG genotypes (P = 0.03). For rs34309, time to reclassification for the AA genotype was 49.9 months versus >60 months for the AG+GG genotype (P = 0.03) (Figure 2A, B).
Figure 1.
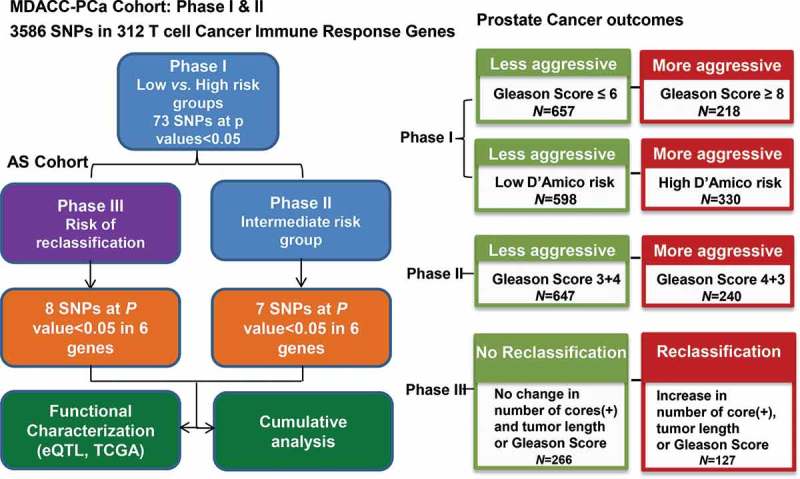
Schematic of study design. Two cohorts of patients with localized PCa, who were followed up prospectively, were included in this study. In phase I and II, we aimed to elucidate SNPs that were associated with PCa aggressiveness using the MDACC-PCa cohort of patients who underwent PCa treatment. Two criteria (GS≥8 vs.GS≤6 and D’Amico high risk vs. low risk) were used to define aggressive disease in phase I, while the risk of GS4 + 3 in GS7 patients was evaluated in phase II; In phase III, we investigated SNPs associated with disease reclassification in the AS cohort.
Table 2.
Selected SNPs significantly associated with PCa aggressiveness (GS ≥8 versus GS≤6 or D’Amico high versus low) and GS 4 + 3 = 7 risk.
| GS≥8 vs. GS≤6 |
GS 4 + 3 vs. GS 3 + 4 |
||||||
|---|---|---|---|---|---|---|---|
| Gene | SNP | Location | Model | OR (95% CI)c | P value | OR (95% CI)c | P value |
| NFATC1 | rs25656 | Coding, 3ʹUTRa | REC | 0.40 (0.19–0.85) | 0.017 | 0.31 (0.15–0.62) | 0.001 |
| NFATC1 | rs160189b | Intron | REC | 0.44 (0.19–0.99) | 0.048 | 0.30 (0.15–0.61) | 0.001 |
| MAP3K1 | rs187899492 | Intron | DOM | 3.76 (1.44–9.81) | 0.007 | 2.89 (1.42–5.88) | 0.003 |
| TRAF2 | rs6560652 | Intron | DOM | 0.58 (0.36–0.91) | 0.018 | 0.63 (0.45–0.88) | 0.007 |
|
CD48 |
rs10489639 |
Intron |
ADD |
0.72 (0.53–0.97) |
0.033 |
0.74 (0.59–0.94) |
0.012 |
| D’Amico high vs. low |
GS 4 + 3 vs. GS 3 + 4 |
||||||
| Gene |
SNP |
Location |
Model |
OR (95% CI)d |
P value |
OR (95% CI)c |
P value |
| NFATC1 | rs25656 | Coding, 3ʹUTRa | REC | 0.51 (0.29–0.90) | 0.021 | 0.31 (0.15–0.62) | 0.001 |
| NFATC1 | rs160189a | Intron | REC | 0.51 (0.28–0.91) | 0.024 | 0.30 (0.15–0.61) | 0.001 |
| RHOA | rs11706370 | Intron | REC | 0.47 (0.25–0.89) | 0.021 | 0.55 (0.30–0.99) | 0.048 |
| CASP8 | rs36039030 | Intron | DOM | 0.30 (0.09–0.98) | 0.047 | 0.29 (0.09–0.99) | 0.048 |
aLocation varied depending on transcript isoforms. Most contain synonymous SNP involving Pro662/Pro649.
Minor transcript contains either nonsynonymous SNP His>Arg at codon 634 or 3ʹUTR SNP.
bSNP in high linkage disequilibrium (r2 > 0.8) with rs25656.
cModels adjusted for age, clinical T stage and PSA level at diagnosis.
dModels adjusted for age and PSA level at diagnosis.
Table 3.
Selected SNPs significantly associated with aggressiveness risk in MDACC-PCa patient cohort and reclassification risk in AS cohort.
| MDACC-PCa patient cohort (N = 1762) |
AS Cohort (N = 393) |
||||||
|---|---|---|---|---|---|---|---|
| GS≥8 vs. GS≤6 |
Risk of reclassification |
||||||
| Gene name | SNP | location | Model | OR (95% CI)b | P value | HR (95% CI)d | P value |
| PSMB8 | rs16871026 | Intron | DOM | 3.52 (1.40–8.84) | 0.007 | 2.68 (1.28–5.59) | 0.009 |
| NFATC2 | rs75164249 | Intron | DOM | 1.74 (1.06–2.85) | 0.029 | 1.60 (1.02–2.52) | 0.041 |
| NFATC2 | rs55990504a | Intron | DOM | 1.68 (1.00–2.84) | 0.050 | 1.74 (1.09–2.78) | 0.020 |
|
PSMD11 |
rs4132610 |
Intron |
ADD |
1.34 (1.00–1.81) |
0.050 |
1.32 (1.00–1.74) |
0.047 |
| D’Amico high vs. low |
Risk of reclassification |
||||||
| Gene name |
SNP |
location |
Model |
OR (95% CI)c |
P value |
HR (95% CI)d |
P value |
| ITGAL | rs3087438 | Intron | DOM | 0.68 (0.49–0.95) | 0.025 | 0.65 (0.45–0.95) | 0.027 |
| PIK3R1 | rs34309 | Intron | REC | 1.63 (1.05–2.52) | 0.030 | 1.84 (1.13–3.00) | 0.014 |
| PSMD3 | rs4065321 | Intron | DOM | 0.69 (0.49–0.98) | 0.037 | 0.66 (0.44–0.99) | 0.045 |
| PSMD3 | rs12453334 | Intron | REC | 1.59 (1.00–2.51) | 0.046 | 1.74 (1.04–2.89) | 0.034 |
aSNP in linkage disequilibrium (r2 = 0.81) with rs75164249.
bThe model was adjusted with age, clinical T stage and PSA levels at diagnosis.
cThe Models adjusted for age and PSA level at diagnosis.
dThe model was adjusted for age, GS score, clinical T stage and PSA levels at diagnosis.
Figure 2.
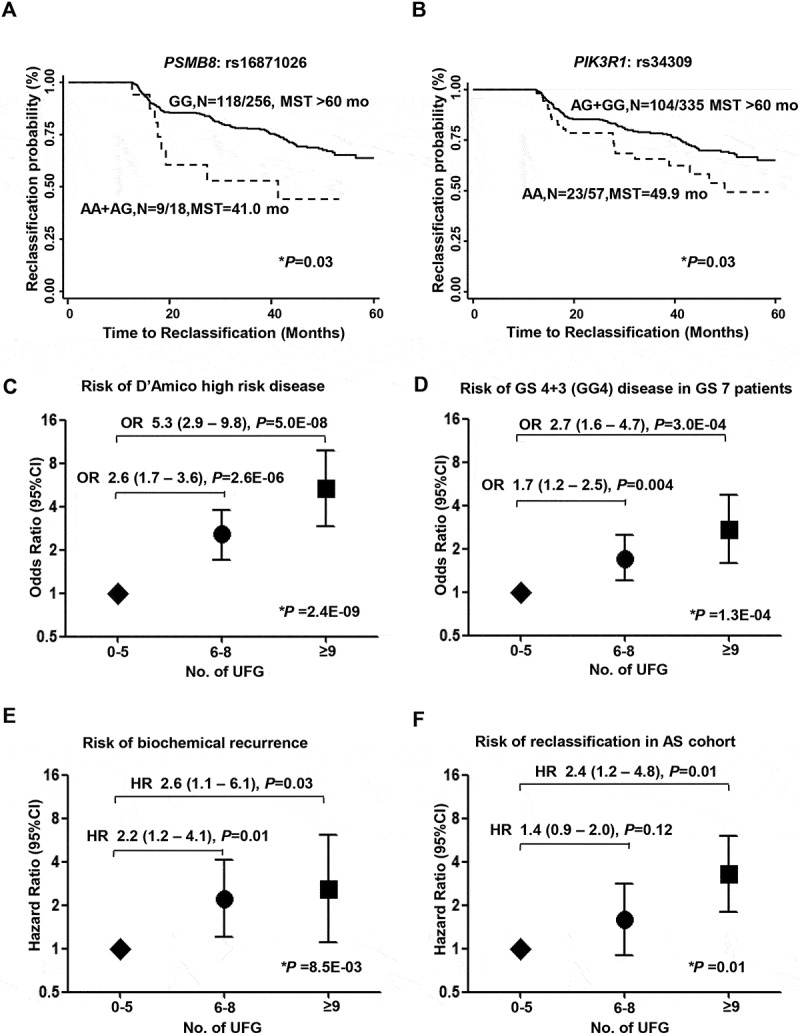
Genetic variants in T cell cancer immune response genes associated with risk of aggressiveness, biochemical recurrence and AS in PCa patients. A. Patients with rs16871026 (AA+AG) (dash line) showed significantly decreased reclassification-free survival compare to rs16871026 (GG) (solid line) patients (HR = 2.68, 95%CI = 1.28–5.59, median survival time: 41.0 months vs. >60 months, P log-rank = 0.03); B. Patients with rs34309 AA genotype (dash line) showed significantly reduced reclassification-free survival compared to rs34309 (AG+GG) (solid line) patients (HR = 1.84, 95%CI = 1.13–3.00, median survival time: 49.9 months versus >60 months, P log-rank = 0.03); C–F. Cumulative analysis was conducted to assess the effects of polygenetic UFGs on PCa outcomes. Patients with 0–5 UFGs, 6–8 UFGs and ≥9 UFGs were defined as low (diamond), intermediate (circle) and high genotypic risk (square). C. Polygenetic UFG was associated with high D’Amico risk disease (P for trend = 2.5E-09); D. GS4 + 3 disease in GS 7 patients (P for trend = 1.3E-04); E. biochemical recurrence (P for trend = 0.01); and F. Disease reclassification in patients on AS of PCa (P for trend = 0.01). SNPs were included based on results of all significant variants found in phases I-III of both cohorts, which included: rs25656, rs187899492, rs6560652, rs10489639, rs11706370, rs36039030, rs16871026, rs75164249, rs4132610, rs3087438, rs34309, rs4065321, rs12453334. * indicates P < 0.05.
Cumulative analysis of the effects of unfavorable genotypes on PCa outcomes
Unfavorable genotype (UFG) analyses were conducted based on the identified 13 candidate SNPs from phases I-III in both study populations, which included NFATC1:rs25656, MAP3K1:rs187899492, TRAF2:rs6560652, CD48:rs10489639, RHOA:rs11706370, CASP8:rs36039030, PSMB8:rs16871026, NFATC2:rs75164249, PSMD11:rs4132610, ITGAL:rs3087438, PIK3R1:rs34309, PSMD3:rs4065321, and PSMD3:rs12453334. Patients were divided into tertiles based on the total number of UFGs, with higher number of UFGs corresponding to increased risk. In the MDACC-PCa cohort, compared to the low genotypic risk group, the intermediate risk and high risk groups demonstrated 2.6 fold (95%CI 1.7–3.8) and 5.3 fold (95%CI 2.9–9.8) increased risk of D’Amico high risk disease (P-trend = 2.4E-09), 1.7 fold (95%CI 1.2–2.5) and 2.7 fold (95%CI 1.6–4.7) increased risk of GS4 + 3 in the GS7 patients (P-trend = 1.30E-04) and 2.2 fold (95%CI 1.2–4.1) and 2.6 fold (95%CI 1.2–6.1) increased risk of biochemical recurrence (P-trend = 0.01), respectively. In the AS cohort, patients from the intermediate genotypic risk group and high genotypic risk group also demonstrated 1.4 fold (95%CI:0.9–2.0) and 2.4 fold (95%CI:1.2–4.8) greater reclassification risk, respectively, compared to low risk patients (P-trend = 0.01). All results of the cumulative analysis are shown in Figure 2C-F and Table S3.
Rs34309 is associated with functional mutations in the PI3K/PTEN/AKT/MTOR pathway
To further investigate potential mechanisms through which identified SNPs may increase the risk of PCa aggressiveness, we performed functional characterization studies in silico. We assessed somatic mRNA expression for genes related to SNPs present in the UFG analysis, leveraging The Cancer Genome Atlas (TCGA) data involving 499 prostate tumors and 52 normal tissue samples. The analysis revealed the expression of MAP3K1, PIK3R1, NFATC2 and RHOA were significantly down-regulated in tumor samples, whereas that of CASP8, PSMD11, TRAF2 and PSMD3 were significantly up-regulated in tumors (P < 0.01 in all cases; Figure S1). No differences were found for PSMB8, CD48, NFATC1 and ITGAL genes (Figure S1). Additionally, no significant expression quantitative trait loci (eQTL) associations were observed in prostate tissues from the Genotype-Tissue Expression (GTEx) database.
A SNP found in the PIK3R1 gene, rs34309, was associated with both PCa aggressiveness and disease reclassification in this study. PIK3R1 is a frequently mutated gene in advanced PCa19 and plays an important role in T cell function.20 We further assessed the association between genotypes of rs34309 and somatic mutations in the PI3K/PTEN/AKT/MTOR pathway using genotype and mutation data from TCGA. Distribution of functional mutations differed significantly between UFG (rs34309-AA) and non-UFG carriers (rs34309-AG+GG)(chi-square test, P = 0.03). A higher fraction of functional mutations (60% [6/10] vs. 27% [27/99]) were observed in UFG carriers compared to non-carriers (Figure 3, Figure S2, and Table S4).
Figure 3.
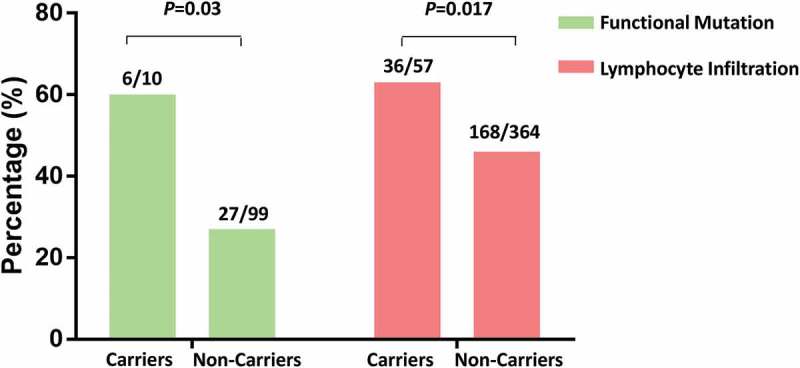
Rs34309 variant carrier status and presence of functional somatic mutations in the PI3K/PTEN/AKT/MTOR signaling pathway (green bars) and tumor lymphocyte infiltration (red bars) in the TCGA prostate cancer patient cohort. In UFG (rs34309-AA) carriers, 60% of tumor mutations (6/10) contained functional mutations, while only 27% of tumor mutations (27/99) had functional mutations in non-carriers (rs34309-AG+GG)(chi-square test, P = 0.03). Similarly, the proportion of tumors with lymphocyte infiltration was significantly higher in UFG-carriers (63.2%, 36/57) than non-carriers (46.2%, 168/364) (chi-square test, P = 0.017). Detailed distributions of mutations and lymphocyte infiltration were depicted in Table S4 and Table S5, respectively.
Additionally, proportion of tumors with lymphocytic infiltrate was significantly elevated in UFG carriers compared to non-carriers among all available TCGA cases (N = 421, 63.2% vs. 46.2%, P = 0.017) (Figure 3, Table S5).
Discussion
In this study, we demonstrated that germline genetic variants related to T cell cancer immune response may be associated with disease aggressiveness and reclassification among patients with localized PCa. Furthermore, genotype-derived risk grouping successfully stratified patients who were at increased risk of aggressiveness and reclassification following enrollment onto AS.
While a number of studies have identified SNPs associated with PCa diagnosis,21 few SNPs have been shown to be associated with PCa aggressiveness and patient reclassification after AS enrollment. Our group previously demonstrated that rs2735839, a SNP related to the KLK gene, was associated with risk of GS4 + 3 disease compared to GS3 + 4.7 A study using data on 33 PCa-risk associated SNPs also indicated that the addition of genotypic data to clinical factors may improve the identification of aggressive PCa at diagnosis.22 In terms of outcomes after PCa diagnosis, a group in Chicago demonstrated that the risk allele of one SNP (rs11568818) might be associated with PCa upgrading at time of surgery and after AS enrollment.6 Additionally, three SNPs were recently shown to be associated with post-prostatectomy reclassification in patients on AS.23 However, to our knowledge, no prior study has systematically examined the association of genetic variants in T cell cancer immune response pathways with PCa aggressiveness and reclassification.
Anti-tumor T cell response is poorly characterized in PCa patients. Studies have demonstrated associations between genetic variants in the inflammation pathway and PCa aggressiveness.24 In the present study, seven variants from six genes were associated with PCa aggressiveness, whereas another eight SNPs in seven genes were associated with reclassification risk. All 13 of these genes are involved in antigen presentation and T cell co-stimulation pathways, suggesting underlying biological mechanisms.
Among the identified genetic variants, rs25656 exhibited the strongest association with PCa aggressiveness. This SNP is located in NFATC1, a gene that encodes a nuclear factor involved in immune response through T cell receptor signaling and NFAT activation.25 An interesting feature of this variant is its varied functional annotation depending on unique NFATC1 isoforms. Rs25656 is located in the coding regions for most transcripts, though is also present in the 3ʹ untranslated region for one minor transcript. In terms of protein translation, this SNP is synonymous at proline codon 662 or 649 depending on transcript length. However, in one truncated transcript, the SNP is nonsynonymous, with the variant allele causing conversion of histidine to arginine at codon 634, though Polyphen prediction indicates that this substitution is unlikely to have harmful effects on protein function. Interestingly, rs25656 is located in a CpG island potentially affecting methylation status,26 although no eQTL effects have been observed for this variant on NFATC1 expression in cells or tissues. Nevertheless, alterations in NFAT family of genes may affect tumor cell proliferation, invasion, metastasis, and drug resistance, which would impact cancer development and progression.27 Further studies are needed to identify the causal variant and the functional mechanism for this SNP’s association with PCa aggressiveness.
The PSMB8 SNP, rs16871026, displayed the most significant association with aggressiveness and reclassification risk. PSMB8 is a key immunoproteasome component that plays a critical role in the process of antigen presentation for T cell mediated anti-tumor immunity.28 Inhibition of PSMB8 may deplete T cell mediated anti-tumor immunity in vitro,28 and PSMB8 is also reported as a predictor of radiation sensitivity in rectal cancer.29 It is therefore biologically plausible that rs16871026 may affect PCa aggressiveness and reclassification through altered function of PSMB8, though further study is still warranted.
The PIK3R1 SNP, rs34309, was also found to be associated with PCa aggressiveness and reclassification risk in our study. PIK3R1 encodes a regulatory subunit in the PI3K signaling pathway, which plays a critical role in T and B cell development.30 Although ubiquitously expressed, inactivating mutations of this gene could result in immune deficiency by means of impaired lymphocyte function.20 This finding supports the gene’s essential but unique role in primary immunity. However, we cannot rule out the possibility that the association of the PIK3R1 variant might be due to the gene’s functional impact on tumor cells. Further studies are needed to identify the underlying biological mechanism for the observed association. Furthermore, PIK3R1 is a known tumor suppressor gene that is frequently mutated in PCa,19 potentially causing aberrant PI3K/PTEN/AKT/MTOR signaling activation.31 In our study, rs34309 genotype was not associated with PIK3R1 expression in normal prostate tissues in eQTL analysis, although this analysis was potentially limited by sample size. However, we observed a considerably higher percentage of patients with functional somatic mutations in PI3K/PTEN/AKT/MTOR pathway genes and patients with lymphocyte infiltration among UFG carriers than among non-carriers. The latter finding is intriguing and counter-intuitive to the general observation that increased number of tumor infiltrating lymphocytes (TILs) correlating with improved prognosis in different cancer types.32 However, investigators have seen conflicting results in PCa cases, with low TIL density as an independent predictor of shortened progression-free survival in one study33 but strong expression of intra-tumoral T cells correlating with shortened biochemical recurrence-free survival in other studies.17,34,35 Differences in methodologies (e.g. H&E [hemotoxylin and eosin] staining versus immunohistochemistry), measured endpoint (e.g. progression versus biochemical recurrence versus survival), location of infiltrate, and immune cell subsets analyzed (e.g. T versus B cells, cytotoxic versus regulatory) may have affected these results. The lack of lymphocyte classification in the TCGA data precluded us from performing detailed subgroup analyses. However, one study of TILs in PCa tumor samples indicates that the composite immune cell population may be skewed toward a suppressive regulatory Treg phenoptype, potentially explaining the association between increased TIL and poor outcome.36
Although our study has distinct advantages in terms of its use of prospective cohorts, relatively large sample size, and moderate-term follow-up time, we acknowledge several limitations. First, there were few reclassification events in the AS cohort, limiting our ability to detect genotypic differences associated with reclassification. Second, tumor and prostate size information were not available, which may have introduced biopsy-related errors into tissue sample grading. Third, the lack of prostate magnetic resonance imaging (MRI) evaluation for some patients prior to baseline and repeat biopsies during AS may have led to inaccuracies in measurements of reclassification and/or upgrading. Additionally, only non-Hispanic whites were included in this study as other groups had few available patients. Future studies are needed to explore associations in other racial/ethnic groups, such as African Americans who have higher risk of aggressive PCa. Due to the exploratory nature of our study, we included associations that were non-significant after adjustment for multiple testing; therefore, it is possible that some findings may contain false positive results. Nevertheless, the identification of candidate genes and SNPs across multiple phases and functional characterization by bioinformatic analyses add validity to the study. Finally, our results were based solely on patients treated at a single institution; further validation in large independent cohorts is required.
In summary, we identified a panel of SNPs related to T cell cancer immune response pathways that were associated with disease aggressiveness and reclassification in patients with localized PCa. A combined genomic risk score using the sum of patient risk alleles enabled stratification into groups that were independently associated with risk of cancer aggressiveness, biochemical recurrence, and disease reclassification. Moreover, a genetic variant in PIK3R1 correlated with functional somatic mutations in the PI3K/PTEN/AKT/MTOR pathway and tumor lymphocyte infiltration, suggesting potential genotype-phenotype interactions. While future validation is warranted, these findings may bring additional insights into biological mechanisms of PCa aggressiveness and biochemical recurrence, which may enable improved risk stratification and eventual personalization of localized PCa treatment.
Materials and methods
Study population and data collection
This study used data from two PCa cohorts enrolled at The University of Texas MD Anderson Cancer Center (MDACC), both of which have been previously described.7,9 In brief, both cohorts involved non-Hispanic white men with previously untreated PCa. The first cohort (MDACC-PCa cohort) recruited patients who underwent treatment for localized PCa from 2003 to 2013, while the second cohort (AS cohort) included only those patients who were enrolled in an ongoing, institutional AS trial that began in February 2006 (registered with clinical.trail.gov: NCT00490763). Clinical data, including diagnosis date, PSA level at diagnosis, biopsy-proven GS, clinical tumor stage, treatment details, tissue pathology, and follow up information were collected from medical records. Age at diagnosis was defined as age at date of first positive biopsy. Biochemical recurrence (BCR) after local therapy was defined as a single measure of PSA ≥ 0.2 ng/ml after radical prostatectomy,37 and a PSA rise of 2 ng/mL or more above the nadir PSA in patients who received radiotherapy.38 Patients in the MDACC-PCa cohort who received radical prostatectomy or radiotherapy were include in the subgroup used for the BCR analysis. All pathologic slides from outside institutions were reviewed by a single genitourinary pathologist at MDACC. In all cases, the GS assessed at MDACC was used. The study was approved by the MDACC Institutional Review Board. Each subject consented to having their clinical data obtained and providing blood samples for DNA extraction for research purposes.
Study design
Phases I and II: exploratory analysis to determine genotypes associated with pca aggressiveness
We first aimed to elucidate SNPs that were associated with PCa aggressiveness using the MDACC PCa cohort of patients who underwent PCa treatment. In phase I, we applied two criteria in order to define aggressiveness based on initial patient biopsy GS, clinical stage, and PSA at diagnosis: GS≥8 or high D’Amico risk was defined as more aggressive, while GS≤6 (grade group 1) or low D’Amico risk was defined as less aggressive, respectively. D’Amico risk stratification39 is defined as follows: low risk (T1-T2a and GS≤6 and PSA < 10 ng/ml), intermediate risk, (T2b or GS = 7 [grade group 2 and 3], or PSA ≥10–20 ng/ml) and high risk (≥T2c or GS 8–10 [grade group 4 and 5], or PSA > 20 ng/ml). In phase II, we separately evaluated SNPs associated with PCa aggressiveness within the subgroup of intermediate risk patients (GS7) by comparing patients with GS3 + 4 disease (grade group 2) to GS4 + 3 (grade group 3).
Phase III: investigation of snps associated with disease reclassification on active surveillance
The cohort of patients used in this portion of the study was recruited from the aforementioned ongoing clinical trial at MDACC. Patients were eligible for this phase of the study who were enrolled in AS, had GS ≤ 7 (grade group 3 or lower), and at least 1 repeat biopsy during follow-up. The AS trial protocol has been previously described.40 In short, all patients were evaluated at baseline and every 6 months by clinical examination including digital rectal examination (DRE) and laboratory studies (serum PSA). Prostate biopsies were repeated every 1–2 years; if the biopsy was negative, then the following year’s biopsy was omitted (unless requested by the patient). Due to the low number of patients progressed from GS6 to GS7 disease, the primary outcome of interest was disease reclassification. This was defined as an increase in number of positive cores or tumor length outside of the study entry criteria, or increase in GS on repeat biopsy. Patients without reclassification were censored on March 31, 2015 when the dataset was prepared for analysis. The time to event was defined as the time from the date of diagnosis to the date of reclassification, last follow-up, or censor.
Genes of interest in t cell cancer immune response pathways
Based on the stepwise cancer immune cycle of T cells,10 we generated T cell cancer immune response pathways and genes by extensively searching the keywords of each step (antigen presentation, T cell priming/activation, T cell trafficking, T cell infiltration, T cell recognition and T cell cytotoxicity) in the KEGG (http://www.genome.jp/kegg/), Biocarta (https://cgap.nci.nih.gov/Pathways/BioCarta_Pathways) and Reactome (http://www.reactome.org/) databases. Furthermore, previously published T cell cancer immune response genes10,41 and gene lists used on commercially customized gene panels (Nanostring: nCounter® PanCancer Immune Profiling Panel, NanoString Technologies; HTG: HTG EdgeSeq Immuno-Oncology Assay, HTG Molecular Diagnostics) were included. All identified pathways and genes were pooled, and only genes mentioned by two or more sources were used in the final gene list. A total of 312 genes from 25 pathways were selected (Table S6). The chromosome positions of gene start and end areas were obtained from USCS Genome Browser (http://genome.ucsc.edu/, build version: GRCh37/hg19).
Genotyping and quality control
All DNA samples were extracted from peripheral whole blood using the QIAamp DNA extraction Kit (QIAGEN). Custom Infinium OncoArray-500K Beadchip was used to genotype both populations. Assays were run on the iScan system (illumina). Genotyping data were analyzed and exported using the Genome Studio software (illumina). All subjects had a call rate >95%. The following exclusion criteria were applied to all samples: gender disparity (as identified by checking X chromosome), non-localized disease, ethnicity other than non-Hispanic white, histology other than adenocarcinoma, and samples from identical subjects. The mean concordance rate of replicated samples was 99.2%. SNPs with minor allele frequency (MAF) <0.01 (n = 83,738) and call rate <0.90 (n = 2,945) were excluded. A total of 412,487 SNPs in the OncoArray dataset remained after strict quality control and were included. Finally, genotyping data of 3586 SNPs found within 10kb of upstream and downstream flanking regions for each gene of interest were extracted from the OncoArray dataset and included in the study.
Functional characterization and immune phenotype association
Bio-informative analysis was applied using the TCGA and GETx databases. eQTL analysis was conducted using the GTEx portal (https://www.gtexportal.org/home/).42 Gene level expression based on RNA-Seq data (normalized, RSEM level 3), somatic mutation (level 3), SNP (level 2) and clinical data for each sample were downloaded directly from the TCGA data portal and analyzed using Firebrowse API (http://firebrowse.org/). SNP function was analyzed with SNPnexus (http://snp-nexus.org/), which includes in silico analysis of CpG islands and predicted effect of non-synonymous coding SNP on protein function using SIFT and Polyphen.43,44 We defined functional mutations as DNA deletions that cause codon frame shift or missing codons which remain in frame, splice site mutations, non-sense mutations, and missense mutations that were predicted in http://mutationassessor.org/r3/with medium or high functional impact score (FI score). In contrast, non-functional mutations included mutations located in intron or other non-coding regions of RNA transcripts, and silent mutations. Lymphocyte infiltration information was obtained from TCGA. Patients with lymphocyte infiltration (>0%) were considered as positive, while the remaining patients were considered as negative. Chi-square test was used in the association analysis.
Statistical analysis
In phase I, all SNPs were separately evaluated for association with aggressive disease (D’Amico high risk vs. low risk and GS≥8 vs. GS≤6) using unconditional multivariable logistic regression adjusted by age at diagnosis (continuous), PSA levels at diagnosis (categorical) and clinical T stage (T1 to T2a vs. T2b+). Only age and PSA levels were adjusted in models that used D’Amico high-risk versus low-risk groups as the endpoint. Phase II involved similar analyses applied to patients that had GS7 disease, specifically examining genotype differences between patients with GS 4 + 3 disease vs. 3 + 4 using same statistical approach as done in phase I. In phase III, all SNPs of interest were examined for association with time to AS reclassification using multivariate cox proportional hazard models adjusted for age, GS, T stage and baseline PSA level. Kaplan–Meier analyses and log-rank tests were used to calculate reclassification-free survival differences between individual genotypes. The proportional hazard assumption was verified by plotting and testing the Schoenfeld residuals, and through inclusion of time varying covariates in the models (interaction terms for GS, T stage and natural logarithms of time).
The cumulative effects of UFGs were assessed for association with aggressiveness, reclassification, and post-treatment biochemical recurrence by combining genotype data from individual SNPs identified in phases I-III that were significantly associated with aggressiveness and reclassification of PCa. Specifically, SNPs that were significantly associated with either GS≥8 or D’Amico high risk disease (phase I) and GS4 + 3 disease (phase II) were included. Additionally, SNPs that were associated with time to reclassification (phase III) and either of the phase I aggressiveness analyses were included. For highly linked SNPs (r2 > 0.8), the SNP with smaller P value was selected. UFGs were selected for each included SNP dependent on the best model (dominant, recessive, or additive). The UFG sum was calculated for each patient and patients were stratified into tertiles based on the total number UFGs present. Patients in each risk group were analyzed for association with disease aggressiveness, GS 7 disease stratification, reclassification while on active surveillance, and biochemical recurrence after PCa treatment following methods described previously.
All data were analyzed and visualized with Excel (Microsoft office 2013), R software (v3.4.1), PLINK (v1.07), and STATA (v13, STATA Corp). All P values were two-sided, with values less than 0.05 considered statistically significant.
Funding Statement
This work was supported by National Cancer Institute (P50 CA140388), MD Anderson Cancer Center Prostate Cancer SPORE and Center for Translational and Public Health Genomics (CTPHG), Duncan Family Institute. Oncoarray for AS cohort was supported by PRACTICAL and the GAME-On OncoArray funding; Duncan Family Institute; HHS | NIH | National Cancer Institute (NCI) [P50 CA140388].
Acknowledgments
We like to thank Donald Norwood of the Department of Scientific Publications at MD Anderson Cancer Center for his helpful assistance in language editing of the manuscript.
Disclosure of interest
The authors have no conflicts of interest to disclose.
Supplemental Material
Supplemental data for this article can be accessed here.
References
- 1.Carroll PR, Parsons JK, Andriole G, Bahnson RR, Castle EP, Catalona WJ, Dahl DM, Davis JW, Epstein JI, Etzioni RB, et al. 2016. NCCN guidelines insights: prostate cancer early detection, version 2.2016. J Natl Compr Canc Netw 14(5):509–519. doi: 10.6004/jnccn.2016.0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.D’Amico AV, Whittington R, Malkowicz SB, Cote K, Loffredo M, Schultz D, Chen MH, Tomaszewski JE, Renshaw AA, Wein A, et al. 2002. Biochemical outcome after radical prostatectomy or external beam radiation therapy for patients with clinically localized prostate carcinoma in the prostate specific antigen era. Cancer 95(2):281–286. doi: 10.1002/cncr.10657. [DOI] [PubMed] [Google Scholar]
- 3.Thomsen FB, Brasso K, Klotz LH, Roder MA, Berg KD, Iversen P.. 2014. Active surveillance for clinically localized prostate cancer–a systematic review. J Surg Oncol 109(8):830–835. doi: 10.1002/jso.23584. [DOI] [PubMed] [Google Scholar]
- 4.Barbieri CE, Bangma CH, Bjartell A, Catto JW, Culig Z, Gronberg H, Luo J, Visakorpi T, Rubin MA. 2013. The mutational landscape of prostate cancer. Eur Urol 64(4):567–576. doi: 10.1016/j.eururo.2013.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eeles R, Goh C, Castro E, Bancroft E, Guy M, Al Olama AA, Easton D, Kote-Jarai Z. 2014. The genetic epidemiology of prostate cancer and its clinical implications. Nat Rev Urol 11(1):18–31. doi: 10.1038/nrurol.2013.266. [DOI] [PubMed] [Google Scholar]
- 6.Kearns JT, Lapin B, Wang E, Roehl KA, Cooper P, Catalona WJ, Helfand BT. 2016. Associations between icogs single nucleotide polymorphisms and upgrading in both surgical and active surveillance cohorts of men with prostate cancer. Eur Urol 69(2):223–228. doi: 10.1016/j.eururo.2015.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.He Y, Gu J, Strom S, Logothetis CJ, Kim J, Wu X. 2014. The prostate cancer susceptibility variant rs2735839 near KLK3 gene is associated with aggressive prostate cancer and can stratify gleason score 7 patients. Clin Cancer Res 20(19):5133–5139. doi: 10.1158/1078-0432.CCR-14-0661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Parikh H, Wang Z, Pettigrew KA, Jia J, Daugherty S, Yeager M, Jacobs KB, Hutchinson A, Burdett L, Cullen M, et al. 2011. Fine mapping the KLK3 locus on chromosome 19q13.33 associated with prostate cancer susceptibility and psa levels. Hum Genet 129(6):675–685. doi: 10.1007/s00439-011-0953-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shu X, Ye Y, Gu J, He Y, Davis JW, Thompson TC, Logothetis CJ, Kim J, Wu X. 2016. Genetic variants of the wnt signaling pathway as predictors of aggressive disease and reclassification in men with early stage prostate cancer on active surveillance. Carcinogenesis 37(10):965–971. doi: 10.1093/carcin/bgw082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen DS, Mellman I. 2013. Oncology meets immunology: the cancer-immunity cycle. Immunity 39(1):1–10. doi: 10.1016/j.immuni.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 11.Schweizer MT, Drake CG. 2014. Immunotherapy for prostate cancer: recent developments and future challenges. Cancer Metastasis Rev 33(2–3):641–655. doi: 10.1007/s10555-013-9479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. 2010. Sipuleucel-T immunotherapy for castration-resistant prostate cancer. N Engl J Med 363(5):411–422. doi: 10.1056/NEJMoa1001294. [DOI] [PubMed] [Google Scholar]
- 13.Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, Van Den Eertwegh AJ, Krainer M, Houede N, Santos R, Mahammedi H, et al. 2014. Ipilimumab versus placebo after radiotherapy in patients with metastatic castration-resistant prostate cancer that had progressed after docetaxel chemotherapy (CA184-043): a multicentre, randomised, double-blind, phase 3 trial. Lancet Oncol 15(7):700–712. doi: 10.1016/S1470-2045(14)70189-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, et al. 2017. Randomized, double-blind, phase iii trial of ipilimumab versus placebo in asymptomatic or minimally symptomatic patients with metastatic chemotherapy-naive castration-resistant prostate cancer. J Clin Oncol 35(1):40–47. doi: 10.1200/JCO.2016.69.1584. [DOI] [PubMed] [Google Scholar]
- 15.Mercader M, Bodner BK, Moser MT, Kwon PS, Park ES, Manecke RG, Ellis TM, Wojcik EM, Yang D, Flanigan RC, et al. 2001. T cell infiltration of the prostate induced by androgen withdrawal in patients with prostate cancer. Proc Natl Acad Sci USA 98(25):14565–14570. doi: 10.1073/pnas.251140998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gevensleben H, Dietrich D, Golletz C, Steiner S, Jung M, Thiesler T, Majores M, Stein J, Uhl B, Muller S, et al. 2016. The immune checkpoint regulator PD-L1 is highly expressed in aggressive primary prostate cancer. Clin Cancer Res 22(8):1969–1977. doi: 10.1158/1078-0432.CCR-15-2042. [DOI] [PubMed] [Google Scholar]
- 17.Flammiger A, Bayer F, Cirugeda-Kuhnert A, Huland H, Tennstedt P, Simon R, Minner S, Bokemeyer C, Sauter G, Schlomm T, et al. 2012. Intratumoral T but not B lymphocytes are related to clinical outcome in prostate cancer. APMIS 120(11):901–908. doi: 10.1111/j.1600-0463.2012.02924.x. [DOI] [PubMed] [Google Scholar]
- 18.Palucka AK, Coussens LM. 2016. The basis of oncoimmunology. Cell 164(6):1233–1247. doi: 10.1016/j.cell.2016.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. 2014. Organoid cultures derived from patients with advanced prostate cancer. Cell 159(1):176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Deau MC, Heurtier L, Frange P, Suarez F, Bole-Feysot C, Nitschke P, Cavazzana M, Picard C, Durandy A, Fischer A, et al. 2014. A human immunodeficiency caused by mutations in the PIK3R1 gene. J Clin Invest 124(9):3923–3928. doi: 10.1172/JCI75746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ishak MB, Giri VN. 2011. A systematic review of replication studies of prostate cancer susceptibility genetic variants in high-risk men originally identified from genome-wide association studies. Cancer Epidemiol Biomarkers Prev 20(8):1599–1610. doi: 10.1158/1055-9965.EPI-11-0312. [DOI] [PubMed] [Google Scholar]
- 22.Kader AK, Sun J, Reck BH, Newcombe PJ, Kim ST, Hsu FC, D’Agostino RB Jr., Tao S, Zhang Z, Turner AR, et al. 2012. Potential impact of adding genetic markers to clinical parameters in predicting prostate biopsy outcomes in men following an initial negative biopsy: findings from the reduce trial. Eur Urol 62(6):953–961. doi: 10.1016/j.eururo.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McGuire BB, Helfand BT, Kundu S, Hu Q, Banks JA, Cooper P, Catalona WJ. 2012. Association of prostate cancer risk alleles with unfavourable pathological characteristics in potential candidates for active surveillance. BJU Int 110(3):338–343. doi: 10.1111/j.1464-410X.2011.10750.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kwon EM, Salinas CA, Kolb S, Fu R, Feng Z, Stanford JL, Ostrander EA. 2011. Genetic polymorphisms in inflammation pathway genes and prostate cancer risk. Cancer Epidemiol Biomarkers Prev 20(5):923–933. doi: 10.1158/1055-9965.EPI-10-0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Northrop JP, Ho SN, Chen L, Thomas DJ, Timmerman LA, Nolan GP, Admon A, Crabtree GR. 1994. Nf-at components define a family of transcription factors targeted in T-cell activation. Nature 369(6480):497–502. doi: 10.1038/369497a0. [DOI] [PubMed] [Google Scholar]
- 26.Kerkel K, Spadola A, Yuan E, Kosek J, Jiang L, Hod E, Li K, Murty VV, Schupf N, Vilain E, et al. 2008. Genomic surveys by methylation-sensitive SNP analysis identify sequence-dependent allele-specific DNA methylation. Nature Genet 40(7):904–908. doi: 10.1038/ng.174. [DOI] [PubMed] [Google Scholar]
- 27.Shou J, Jing J, Xie J, You L, Jing Z, Yao J, Han W, Pan H. 2015. Nuclear factor of activated t cells in cancer development and treatment. Cancer Lett 361(2):174–184. doi: 10.1016/j.canlet.2015.03.005. [DOI] [PubMed] [Google Scholar]
- 28.Mayes K, Alkhatib SG, Peterson K, Alhazmi A, Song C, Chan V, Blevins T, Roberts M, Dumur CI, Wang XY, et al. 2016. BPTF depletion enhances T-cell-mediated antitumor immunity. Cancer Res 76(21):6183–6192. doi: 10.1158/0008-5472.CAN-15-3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ha YJ, Tak KH, Kim CW, Roh SA, Choi EK, Cho DH, Kim JH, Kim SK, Kim SY, Kim YS, et al. 2017. PSMB8 as a candidate marker of responsiveness to preoperative radiation therapy in rectal cancer patients. Intl J Radiat Oncol Biol Phys 98(5):1164–1173. doi: 10.1016/j.ijrobp.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 30.Lucas CL, Chandra A, Nejentsev S, Condliffe AM, Okkenhaug K. 2016. PI3Kdelta and primary immunodeficiencies. Nature Rev Immunol 16(11):702–714. doi: 10.1038/nri.2016.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheung LW, Hennessy BT, Li J, Yu S, Myers AP, Djordjevic B, Lu Y, Stemke-Hale K, Dyer MD, Zhang F, et al. 2011. High frequency of PIK3R1 and PIK3R2 mutations in endometrial cancer elucidates a novel mechanism for regulation of pten protein stability. Cancer Discov 1(2):170–185. doi: 10.1158/2159-8290.CD-11-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jochems C, Schlom J. 2011. Tumor-infiltrating immune cells and prognosis: the potential link between conventional cancer therapy and immunity. Exp Biol Med (Maywood) 236(5):567–579. doi: 10.1258/ebm.2011.011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vesalainen S, Lipponen M, Talja M, Syrjänen K. 1994. Histological grade, perineural infiltration, tumour-infiltrating lymphocytes and apoptosis as determinants of long-term prognosis in prostatic adenocarcinoma. Eur J Cancer 30(12):1797–1803. doi: 10.1016/0959-8049(94)E0159-2. [DOI] [PubMed] [Google Scholar]
- 34.Karja V, Aaltomaa S, Lipponen P, Isotalo T, Talja M, Mokka R. 2005. Tumour-infiltrating lymphocytes: A prognostic factor of PSA-free survival in patients with local prostate carcinoma treated by radical prostatectomy. Anticancer Res 25:4435–4438. [PubMed] [Google Scholar]
- 35.Ness N, Andersen S, Valkov A, Nordby Y, Donnem T, Al-Saad S, Busund LT, Bremnes RM, Richardsen E. 2014. October Infiltration of CD8+ lymphocytes is an independent prognostic factor of biochemical failure-free survival in prostate cancer. Prostate 74(14):1452–1461. doi: 10.1002/pros.22862. [DOI] [PubMed] [Google Scholar]
- 36.Sfanos KS, Bruno TC, Maris CH, Xu L, Thoburn CJ, DeMarzo AM, Meeker AK, Isaacs WB, Drake CG. 2008. Phenotypic analysis of prostate-infiltrating lymphocytes reveals TH17 and Treg skewing. Clin Cancer Res 14(11):3254–3261. doi: 10.1158/1078-0432.CCR-07-5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pound CR, Partin AW, Eisenberger MA, Chan DW, Pearson JD, Walsh PC. 1999. Natural history of progression after psa elevation following radical prostatectomy. JAMA 281(17):1591–1597. doi: 10.1001/jama.281.17.1591. [DOI] [PubMed] [Google Scholar]
- 38.Bruce JY, Lang JM, McNeel DG, Liu G. 2012. Current controversies in the management of biochemical failure in prostate cancer. Clin Adv Hematol Oncol 10(11):716–722. [PubMed] [Google Scholar]
- 39.D’Amico AV, Whittington R, Malkowicz SB, Schultz D, Blank K, Broderick GA, Tomaszewski JE, Renshaw AA, Kaplan I, Beard CJ, et al. 1998. Biochemical outcome after radical prostatectomy, external beam radiation therapy, or interstitial radiation therapy for clinically localized prostate cancer. JAMA 280(11):969–974. doi: 10.1001/jama.280.11.969. [DOI] [PubMed] [Google Scholar]
- 40.Davis JW, Ward JF 3rd, Pettaway CA, Wang X, Kuban D, Frank SJ, Lee AK, Pisters LL, Matin SF, Shah JB, et al. 2016. Disease reclassification risk with stringent criteria and frequent monitoring in men with favourable-risk prostate cancer undergoing active surveillance. BJU Int 118(1):68–76. doi: 10.1111/bju.13193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singer M, Wang C, Cong L, Marjanovic ND, Kowalczyk MS, Zhang H, Nyman J, Sakuishi K, Kurtulus S, Gennert D, et al. 2016. A distinct gene module for dysfunction uncoupled from activation in tumor-infiltrating t cells. Cell 166(6):1500–1511 e1509. doi: 10.1016/j.cell.2016.08.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Consortium GT. 2013. The genotype-tissue expression (GTEX) project. Nature Genet 45(6):580–585. doi: 10.1038/ng.2653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kumar P, Henikoff S, Ng PC. 2009. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4(7):1073–1081. doi: 10.1038/nprot.2009.86. [DOI] [PubMed] [Google Scholar]
- 44.Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. 2010. A method and server for predicting damaging missense mutations. Nat Methods 7(4):248–249. doi: 10.1038/nmeth0410-248. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.