

ABSTRACT
Multiple immunotherapeutics have been approved for cancer patients, however advanced solid tumors are frequently refractory to treatment. We evaluated the safety and immunogenicity of a vaccination approach with multimodal oncolytic potential in non-human primates (NHP) (Macaca fascicularis). Primates received a replication-deficient adenoviral prime, boosted by the oncolytic Maraba MG1 rhabdovirus. Both vectors expressed the human MAGE-A3. No severe adverse events were observed. Boosting with MG1-MAGEA3 induced an expansion of hMAGE-A3-specific CD4+ and CD8+ T-cells with the latter peaking at remarkable levels and persisting for several months. T-cells reacting against epitopes fully conserved between simian and human MAGE-A3 were identified. Humoral immunity was demonstrated by the detection of circulating MAGE-A3 antibodies. These preclinical data establish the capacity for the Ad:MG1 vaccination to engage multiple effector immune cell populations without causing significant toxicity in outbred NHPs. Clinical investigations utilizing this program for the treatment of MAGE-A3-positive solid malignancies are underway (NCT02285816, NCT02879760).
Keywords: Oncolytic vaccination, MG1 Maraba, MAGE-A3, Cancer immunotherapy, endogenous immunity
Introduction
Immunotherapy is revolutionizing the care of cancer patients by (re-)educating the host’s immune system to selectively destroy malignant cells and establish long-lasting memory. Less than a decade ago, dacarbazine was considered standard-of-care treatment for metastatic melanoma with 5-year survivals of 5–10%.1,2 Since then, checkpoint inhibitors have supplanted dacarbazine and quadrupled long-term survival.3–6 Although checkpoint inhibitors have garnered much attention,1,2,7 other biotherapetics have been approved including immunostimulatory cytokines,8 monoclonal antibodies,9 cancer vaccines,10 oncolytic virotherapy 11,12 and chimeric antigen receptor (CAR) T-cell therapy.13 All these approaches invoke immunity through non-redundant mechanisms of action. In spite of this remarkable progress, many patients will continue to be diagnosed with incurable disease, thus dictating the need for next-generation medicines capable of engaging multiple effector mechanisms.
Ourselves and others have reported on innovative strategies that combine cancer vaccination with oncolytic virotherapy.14–22 Our approach relies on a heterologous prime:boost immunization involving two viruses expressing the same tumor-associated antigen (TAA).14,17,20 A replication-deficient type-5 human adenoviral (Ad) vaccine is administered intramuscularly to prime against a TAA. Subsequently, an oncolytic viral vaccine derived from the MG1 strain of the rhabdovirus Maraba, and expressing the same TAA, is administered intravenously. The therapeutic activity of oncolytic Maraba MG1 results from multimodal mechanisms: i) systemic administration delivers MG1 to the tumor bed resulting in selective direct oncolysis;14,20,23–27 ii) infection of tumors by MG1 reprograms the tumor microenvironment (TME) rendering it susceptible to the recruitment and activation of innate and adaptive mediators of antitumor immunity 14,28–30 and iii) circulating MG1 accesses secondary lymphoid organs and rapidly engages the central memory compartment thereby boosting Ad-primed adaptive antitumor immunity.31 Maraba MG1 has a well-established safety profile within rodents and demonstrates broad oncolytic activity against multiple human and murine cancer cell lines,14,20,23–25,32,33 primary tumor biopsies,14,23,28 as well as xenograft and syngeneic tumor models in mice.23–25,28,29 When administered as an oncolytic vaccine, MG1 exerts remarkable efficacy with complete regressions achieved in aggressive immunocompetent models of melanoma and HPV-positive cancer.14,20 The present work enabled appraisal of the safety and immunogenicity of oncolytic vaccination in an outbred population of cynomolgus macaques (Macaca fascicularis), with increased biologic relevance to humans, enabling the translation of this treatment into a first-in-man clinical trial.
Cancer-testis (CT) antigens have multiple properties making them a favorable target for immunotherapy. Melanoma-associated antigen A3 (MAGE-A3) is amongst the most intensely studied CT antigens in oncology.34 Vaccination against MAGE-A3 is theoretically auspicious as MAGE-A3 i) is not expressed in healthy adult tissues (with the exception of MHC-negative testicular germ cells and placental trophoblasts), ii) is aberrantly expressed in many malignancies (melanoma, lung carcinoma and multiple other histotypes) and iii) is considered to have high immunogenic potential.34,35 In this study, Ad and MG1 viral vectors encoding human (h) MAGE-A3 (Ad-MAGEA3 and MG1-MAGEA3, respectively) were manufactured and administered to non-human primates (NHPs).
The Ad-MAGEA3:MG1-MAGEA3 prime-boost strategy was well tolerated with no severe treatment-related adverse events. Immune responses of both CD4+ and CD8+ T-cell subsets as well as humoral immunogenicity were detected against hMAGE-A3 in all macaques. Circulating CD8+ T-cell populations specific for multiple MAGE-A3 epitopes expanded remarkably following MG1-MAGEA3 and remained detectable until necropsy. Reactivity against MAGE-A3 epitopes fully conserved between the human antigen expressed by the vaccine vectors and the macaque homolog of the host were pinpointed. Such observation validated the ability of the Ad:MG1 approach to induce auto-reactive MAGE-A3 T-cells; a critical propensity for anticancer vaccines targeting “self antigens”. Multi-parameter immune analyses and comprehensive safety profiling data reported here have informed clinical trial design for the treatment of various advanced solid neoplasms with oncolytic MAGE-A3 vaccination (NCT02285816 and NCT02879760).
Results
Ad-MAGEA3:MG1-MAGEA3 vaccination was well tolerated by non-human primates
Eight macaques were randomized to receive MG1-MAGEA3 alone, the remaining 20 were treated with MG1-MAGEA3 following Ad-MAGEA3 prime (Ad-MAGEA3:MG1-MAGEA3) at three different intervals (Figure 1A). Animals were also assigned to a low (1x1010 PFU) or high (1x1011 PFU) dose of MG1-MAGEA3, infused twice at 3-day intervals. No adverse events were observed during the administration of Ad-MAGEA3 or MG1-MAGEA3. Of the 8 primates treated with MG1-MAGEA3 alone, 87.5% experienced weight loss (5 x grade 1/2, 2 x grade 3/4; Table S1). Conversely, only 25% of the 20 macaques treated with Ad-MAGEA3:MG1-MAGEA3 experienced weight loss (all grade 1/2; Table S1). The degree and duration of weight loss was greater in animals treated with MG1-MAGEA3 alone compared to Ad-MAGEA3:MG1-MAGEA3 (p = 0.006; Figure 1B). Weight loss was not associated with the dose of MG1-MAGEA3 (p = 0.729; Figure 1C). Eighty-seven percent (n = 7/8) of the primates administered with MG1-MAGEA3 alone experienced fever shortly after virus delivery (Figure 1D). To a lesser extent, pyrexia was observed in 35% (7/20) of the macaques treated with Ad-MAGEA3:MG1-MAGEA3 (Figure 1D). The dose of MG1-MAGEA3 did not impact pyrexia (Figure 1E). Only 3 primates out of the 28 experienced fever at both MG1-MAGEA3 infusions. Episodes of pyrexia remained acute with only two events still present at 24 hours post-MG1-MAGEA3 delivery. Some animals experienced emesis, diarrhea, and constipation (Grade 1/2) within the first 3 days after MG1-MAGEA3 administration (Table S1). Two animals, M-hi 2 (macaque #2 of the cohort that received high-dose MG1-MAGEA3 – Figure 1A) and AM2w-hi 1 (macaque #1 of the cohort infused with high-dose MG1-MAGEA3 two weeks after delivery of Ad-MAGEA3 – Figure 1A), presented with paresis of one of their upper extremities at day 39 and 59 post-MG1-MAGEA3, respectively, with no other associated pathology (Table S1). The neurologic deficits improved over time and completely resolved in one animal. These were attributed to trauma from restraint during venipuncture. No neurologic alterations were observed in other animals. The summary of constitutional adverse events is presented in Table S1. Across both treatment arms, there was a probable MG1-MAGEA3-related decline in circulating neutrophils and lymphocytes (Fig. S1A-1D). Six animals developed transient neutropenia. However, circulating neutrophils decreased in all animals following the first infusion of MG1-MAGEA3, regardless of the dose (Fig. S1A and S1B). A transient decrease in circulating lymphocytes was noted in only 2 animals after the first MG1-MAGEA3 dose. The reduction of the lymphoid population was less pronounced than the neutrophils (Fig. S1C and S1D). Total leukocyte counts also tended to diminish following MG1-MAGEA3 treatment, being most apparent in Ad-MAGEA3-pre-treated animals, independently of MG1-MAGEA3 dose (Fig. S1E and S1F). Transient and clinically silent grade 1/2 elevations of ALT were documented in both groups (data not shown). One primate had a chronic high-grade elevation of ALT that pre-dated vaccination, this animal was diagnosed histologically with intrahepatic hemosiderosis. Overall, MG1-MAGEA3 was well tolerated by primates with only low-grade adverse events ascribed to animals receiving Ad-MAGEA3:MG1-MAGEA3.
Figure 1.
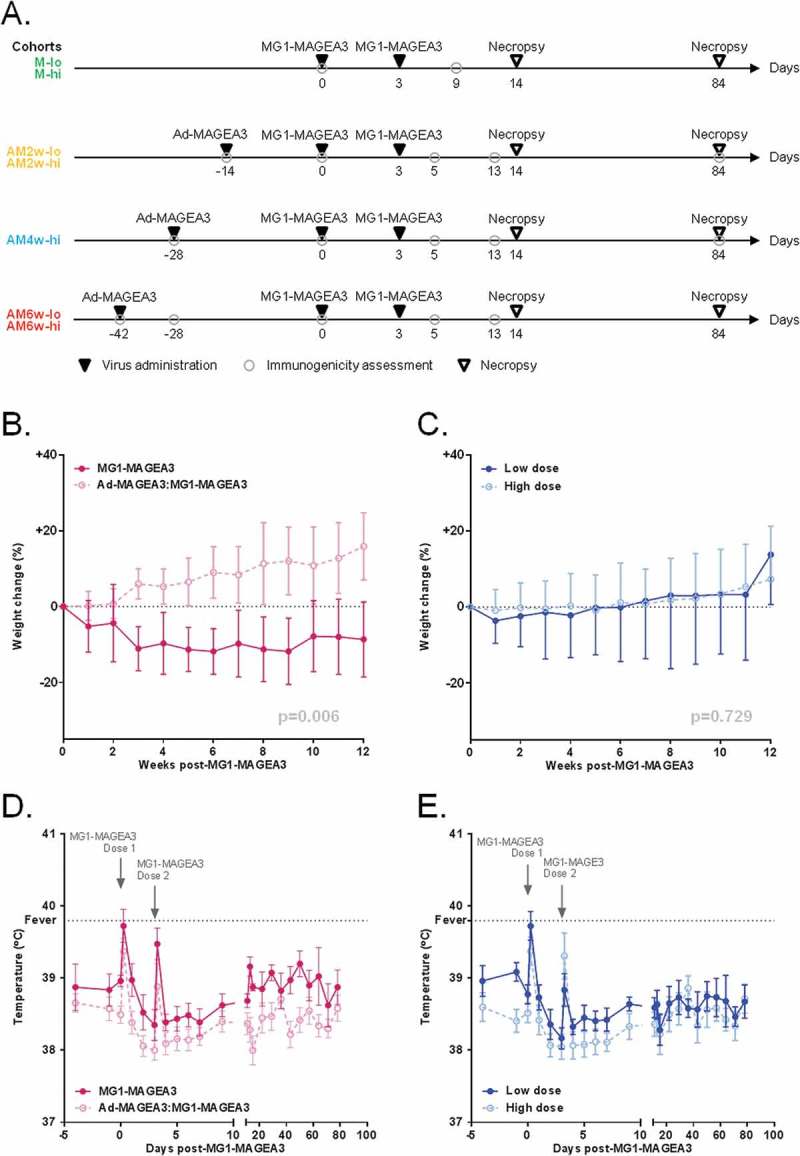
Ad-MAGEA3 and MG1-MAGEA3 are well tolerated by non-human primates.
(A) Study timeline in cynomolgus macaques. MG1-MAGEA3 was infused twice (3 days apart) at low (1 x 1010 PFU; ‘lo”) or high (1 x 1011 PFU; “hi”) doses, either as a standalone treatment (cohorts M-lo and M-hi; n = 4 each) or sequentially after receiving one intramuscular injection of 1 × 1010 PFU Ad-MAGEA3. For the latter setting, three intervals between Ad-MAGEA3 and MG1-MAGEA3 injections were tested: 2 weeks (cohorts AM2w-lo and AM2w-hi; n = 4 each), 4 weeks (cohort AM4w-hi; n = 4) or 6 weeks (cohorts AM6w-lo and AM6w-hi; n = 4 each). Blood samples were collected at the indicated time-points for immunogenicity assessments. Of note, the AM6w cohorts were dedicated to assessing the immunogenicity of the Ad-MAGEA3:MG1-MAGEA3 prime-boost and were not enrolled in the toxicology study. Body weight change relative to body weight at day 0 (cohorts M, AM2w and AM4w; n = 20) (B,C) and rectal temperatures (all macaques; n = 28) (D,E) by treatment arm (MG1-MAGEA3 alone versus Ad-MAGEA3:MG1-MAGEA3 regardless of the interval) or MG1-MAGEA3 dose (low dose versus high dose regardless of the treatment arm). Fever was classified as a rectal temperature ≥ 39.8°C (indicated by the horizontal dashed line). Body weight change is illustrated as mean ± SD and temperature as mean ± SEM. p-values (linear mixed models) are indicated and were considered significant when p < 0.05. MAGE-A3, human melanoma-associated antigen, family A, member 3; PFU, plaque-forming unit.
MG1-MAGEA3 bio-distribution and shedding
Multiple tissues from macaques were analyzed for viral bio-distribution (Table S2). All animals had Maraba RNA viral genomes detectable in the blood by RTqPCR (data not shown). Among tissue subtypes, immune-related organs (spleen and lymph nodes) were the most frequently positive for MG1-MAGEA3 genomes (Fig. S2A). No detectable viral plaques were identified in any of the tissues positive for MG1-MAGEA3 by RTqPCR, indicating that the genomes identified were non-replicating MG1-MAGEA3. No Ad-MAGEA3 genomes were detected by qPCR in any tissue. All fecal samples tested for MG1-MAGEA3 by RTqPCR were negative. MG1-MAGEA3 genomes were detected in the urine of 3 animals treated with high-dose MG1-MAGEA3, without replicating virus being present (data not shown). MG1-MAGEA3 genomes were detectable in the saliva of 43% (3/7) of the animals that received low-dose MG1 and of 67% (8/12) of those injected with high-dose Maraba (Fig. S2B). For all animals, histopathological analyses revealed no indication of virus infection or lesions of pathologic significance in any of the tissues evaluated. Of note, there was no evidence of pathology in the testes (MAGE-A3+ MHC– tissue) of any macaques, nor was there evidence of pathology in the peripheral nerves of the 2 animals that developed peripheral neuropathy. Overall, despite the widespread distribution of MG1-MAGEA3 genomes, no virally induced histologic pathology was detected.
Ad-MAGEA3 efficiently primed CD8+ T-cells against human MAGE-A3
Ad-MAGEA3 and MG1-MAGEA3 were evaluated for their ability to immunize naive macaques against hMAGE-A3. Cellular hMAGE-A3-specific immune responses were detected in the blood by quantifying CD4+ and CD8+ T-lymphocytes secreting interferon-γ (IFNγ) following ex vivo re-stimulation with pools of hMAGE-A3 peptides (Fig. S3A,B). Re-stimulation was performed without ex vivo expansion of peripheral blood mononuclear cells (PBMCs). Eight primates received only MG1-MAGEA3, either at low (n = 4, cohort M-lo) or high (n = 4, cohort M-hi) doses (Figure 1A). T-cell responses against hMAGE-A3 were measured before and 9 days after the first dose of the MG1 vaccine. As illustrated in the Figures 2A and 2B, no significant hMAGE-A3-specific CD8+ T-cell response was detected following MG1-MAGEA3 prime, regardless of the dose, when compared to pre-immune baseline (Figure 2A,B). Ad-MAGEA3 priming efficacy was evaluated in the 20 macaques enrolled in the five prime-boost cohorts: AM6w-lo, AM6w-hi, AM2w-lo, AM2w-hi, AM4w-hi (Figure 1A). hMAGE-A3-specific T-cell populations were quantified before and 14 days after intramuscular injection of Ad-MAGEA3. At day 14, mean Ad-primed response was 4.46 ± 1.49 IFNγ+ CD8+ T-cells/µl blood and accounted for more than 0.5% (0.62 ± 1.02%) of all total circulating CD8+ T-cells (Figure 2C,D). Additionally, the prime response was evaluated at day 28 post-Ad-MAGEA3 (AM4w-hi cohort (n = 4)) and at day 42 (AM6w-lo and AM6w-hi cohorts (n = 8)). At these later time-points, the reactive CD8+ T-cell population had expanded in only 25% of the primates assessed (n = 3/12, data not shown). Overall, the mean response remained stable over a month after Ad-MAGEA3 (Figure 2C,D). Out of the 20 macaques, 17 (85%) displayed responses above pre-immune baseline, ranging from 0.32 to 28.07 IFNγ+ CD8+ T-cells/µl blood (Figure 2C and S4A,B). Ad-MAGEA3 was able to prime specific CD8+ responses against multiple pools of hMAGE-A3 peptides (Fig. S4A-D). Primed CD8+ T-cells represented up to 4.25% of total circulating CD8+ T-lymphocytes (Figure 2D and S4D). In conclusion, Ad-MAGEA3 was able to prime significant CD8+ T-cell immunity in the majority of primates against broad-ranging hMAGE-A3 epitopes.
Figure 2.
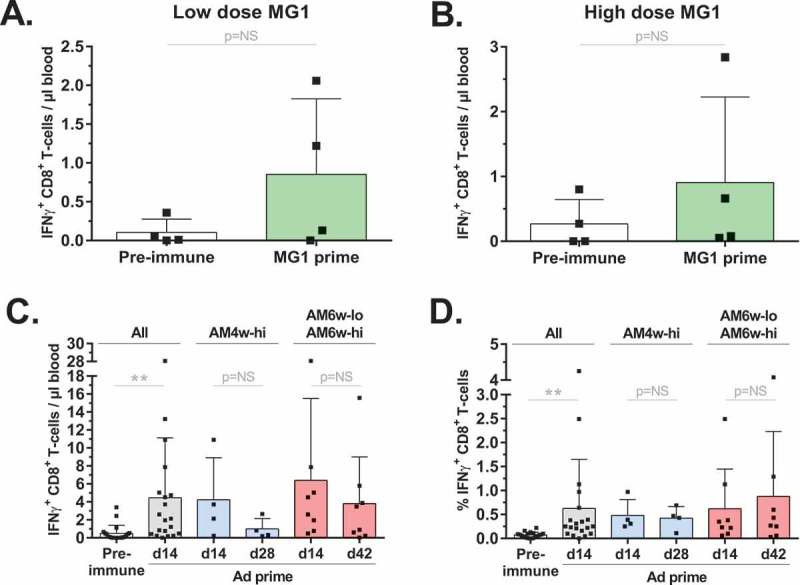
Ad-MAGEA3 primed CD8+ T-cell population against hMAGE-A3.
hMAGE-A3-specific CD8+ T-cell responses were detected by intracellular staining of IFNγ following ex vivo re-stimulation with pools of overlapping peptides covering the full length MAGE-A3 antigen. (A,B) Pre-immune and MG1-MAGEA3-mediated prime responses against hMAGE-A3 were measured before and 9 days after receiving MG1-MAGEA3. MG1-MAGEA3 was delivered systemically at two doses (3 days apart) of 1e10 PFU to the M-lo cohort (n = 4, A) or 1e11 PFU to the M-hi cohort (n = 4, B). (C,D) The Ad-MAGEA3 vaccine was administered intramuscularly at 1e10 PFU to a total of 20 macaques in the AM2w-lo, AM2w-hi, AM4w-hi, AM6w-lo and AM6w-hi cohorts. CD8+ T-cell responses against hMAGE-A3 were measured before and 14 days after Ad-MAGEA3 administration. Ad-MAGEA3-mediated prime response was also measured at day 28 in the AM4w-hi cohort (n = 4) and at day 42 in the AM6w-lo and AM6w-hi cohorts (n = 8). Mean hMAGE-A3-specific prime response is displayed both as the count (cells per µl of blood; C) and as the frequency (% among total circulating CD8+ T-cell population); D) of IFNγ-producing CD8+ T-lymphocytes. Histograms represent mean ± SD. p-value considered non significant (NS) when > 0.05; **p < 0.01 (Wilcoxon’s paired test for A and B; Dunn’s test for C and D). hMAGE-A3, human melanoma-associated antigen, family A, member 3; IFNγ, interferon gamma; PFU, plaque-forming unit.
MG1-MAGEA3 boosts hMAGE-A3 specific CD4+ T-cells
Specific hMAGE-A3 CD4+ T-cell immunity was also evaluated. MG1-MAGEA3 alone failed to prime detectable CD4+ responses (data not shown). Mean priming of the CD4+ T-lymphocyte compartment following Ad-MAGEA3 injection was of lower magnitude than the CD8+ subset (Figure 3A,B). Of the 20 immunized macaques, 40% (n = 8) displayed reactivity above the pre-immune baseline with a maximum of 0.29 IFNγ+ CD4+ T-cells/µl blood observed in AM6w-lo 4 (Fig. S5A). All 20 macaques that received the Ad-MAGEA3 prime were boosted with MG1-MAGEA3 either at a 6-week interval in the AM6w-lo and AM6w-hi cohorts, 4-week interval in the AM4w-hi cohort, or 2-week interval in the AM2w-lo and AM2w-hi cohorts (Figure 1A). MAGE-A3-specific T-cell populations were quantified at day 5 (n = 20), 13 (n = 20) or 84 (n = 6) after intravenous MG1-MAGEA3. Ad-MAGEA3:MG1-MAGEA3 boosted responses to a mean of 0.35 ± 0.06 reactive CD4+ T-cells/µl blood (approximately 0.1% of total CD4+ T-lymphocytes) at day 5 post-MG1 (0.08 ± 0.06%; Figure 3A,B). The mean boost response eventually contracted, with a ~ 2-fold decrease observed by the following week: 0.16 ± 0.03 hMAGE-A3-activated CD4+ T-cells/µl blood (0.04 ± 0.03% of total CD4+ T-lymphocytes) at day 13 (Figure 3A,B). CD4+ T-cell responses were undetectable at d84 in samples analyzed from the AM2w-lo/hi (n = 4) and AM4w-hi (n = 2) cohorts (Figure 3A). Overall, hMAGE-A3-specifc CD4+ T-cells were detected in 85% of the primates post-MG1 boost (17/20; Figure 3C). AM2w-lo 1 was the strongest responder, with 1.26 IFNγ+ CD4+ T-lymphocytes/µl blood specific for hMAGE-A3 accounting for 0.31% of total circulating CD4+ T-cells (Figure 3C,Dand Fig. S5B).
Figure 3.
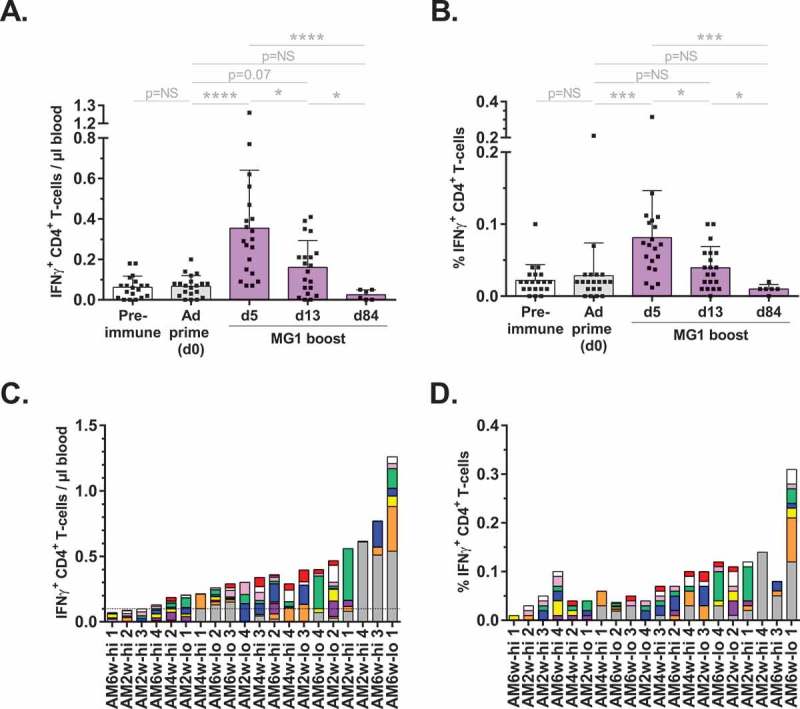
Ad-MAGEA3:MG1-MAGEA3 induced CD4+ T-cell reactivity against hMAGE-A3.
hMAGE-A3-educated CD4+ T-cells were quantified by intracellular staining of IFNγ following ex vivo (re-)stimulation with pools of overlapping peptides covering the full length MAGE-A3 antigen. (A, B) Kinetics of CD4+ T-cell responses against hMAGE-A3 in the 20 macaques that received the Ad-MAGEA3:MG1-MAGEA3. Response is displayed both as the count (cells per µl of blood; A) and as the frequency (% among total CD4+ T-lymphocytes; B) of IFNγ-producing CD4+ T-lymphocytes. Histograms show mean ± SD. p-value (Dunn’s test) considered non significant (NS) when > 0.05; *p < 0.05, ***p < 0.001, ****p < 0.0001. (C, D) Peak CD4+ T-cell boost responses against hMAGE-A3 in each individual macaque, ranked according to their magnitude expressed as absolute count per µl of blood (C). Corresponding MG1-MAGEA3 boost responses expressed as the frequency (%) of reactive cells inside the total CD4+ T-cell population (D). Bars illustrate the number of IFNγ-producing CD4+ T-cells per µl blood, with the dashed line indicating pre-immune baseline (set to 0.1 reactive CD4+ T-cells/µl blood). Sub-responses to each of the nine peptide pools used for antigenic (re-)challenge are color-coded: pool 1–10 in red, pool 11–20 in white, pool 21–30 in pink, pool 31–40 in green, pool 41–50 in blue, pool 51–60 in yellow, pool 61–70 in purple, pool 71–80 in orange and pool 81–87 in grey. hMAGE-A3, human melanoma-associated antigen, family A, member 3; IFNγ, interferon gamma.
MG1-MAGEA3 stimulates humoral immunity
Humoral immunogenicity was assessed on serum samples. None of the primates vaccinated with MG1-MAGEA3 alone had detectable hMAGE-A3 antibodies (Fig. S6A). In contrast, all macaques immunized with Ad-MAGEA3:MG1-MAGEA3 produced hMAGE-A3-specific immunoglobulins (Fig. S6A). Ad-MAGEA3 priming was required to evoke anti-hMAGE-A3 B-cell immunity (Fig. S6A,B). Although Ad-MAGEA3 vaccination did not uniformly induce detectable Th1 CD4+ T-cell prime responses (Figure 3A and Fig. S5A), it was capable of driving some detectable humoral responses. Similarly to cellular immunogenicity, consecutive administration of MG1-MAGEA3 significantly boosted production of antibodies targeting hMAGE-A3 (Fig. S6B). There was no effect of gender, age, prime-boost interval or dose of MG1-MAGEA3 on the magnitude of circulating hMAGE-A3 antibodies (Fig. S6A). However, the magnitude of B-cell responses corresponded to the magnitude of hMAGE-A3-specific CD8+ T-cell responses (Fig. S6A). Taken together, Ad-MAGEA3:MG1-MAGEA3 vaccination was able to engage Th1 and humoral effector cell populations alongside activation of CD8+ T-cells from the endogenous immune system of outbred primates.
MG1-MAGEA3 boosting leads to marked expansion of MAGE-A3-specific CD8+ T-cells
Kinetics of the hMAGE-A3-specific CD8+ T-cell expansion in the prime-boost cohorts were evaluated. Prior to MG1-MAGEA3 boost (day 0), Ad prime responses were documented at a mean of 2.77 ± 0.97 IFNγ+ CD8+ T-lymphocytes/µl blood which represented slightly more than 0.5% (0.72 ± 1.22%) of circulating CD8+ T-cells (Figure 4A,B). Five days after the first infusion of MG1-MAGEA3, the mean reactive CD8+ T-cell population underwent a 4-fold increase, reaching 10.94 ± 3.78 cells/µl blood and represented more than 2% of total circulating CD8+ T-cells (2.4 ± 3.26%; Figure 4A,B). One week later (day 13), the mean response had contracted (Figure 4A). However, hMAGE-A3-specific CD8+ T-lymphocytes persisted at a magnitude comparable to pre-boost until necropsy at almost three months (d84) after MG1-MAGEA3 boost (Figure 4A). At these later time-points, the reactive cell population accounted for approximately 0.5% of total circulating CD8+ T-lymphocytes (Figure 4B). At the individual level, this acute expansion-contraction occurred in 70% (14/20) of the macaques as illustrated by AM6w-lo 4 (Figure 4C). In the remaining 30% primates, we recorded either a slower expansion of the hMAGE-A3-educated CD8+ T-cell population, peaking later than 5 days post-boost (3/20 = 15%; Figure 4D), or no significant expansion post-boost (3/20 = 15%; Figure 4E). Overall, Ad-MAGEA3:MG1-MAGEA3 vaccination induced specific immunity in all macaques with some variations in the kinetics. Observed responses ranged between 0.48 and 76.24 IFNγ+ CD8+ T-cells/µl blood (median = 5.67; Figure 4F,G). In the 10 weakest responders, median reactivity against hMAGE-A3 comprised 0.68% of all circulating CD8+ T-cells (Figure 4H). In the 10 strongest responders, median reactive lymphocyte population represented 3.55% of all circulating CD8+ T-cells, ranging between 1.26% and a remarkable maximum of 13.47% in AM6w-lo 4 (Figure 4I). In the 17 macaques that exhibited definitive boosting, reactive CD8+ T-cells expanded by a mean fold of 26.83 ± 57.55 post-boost (Figure 4J). Boost fold magnitude varied significantly, ranging between 1.45 and 200, with a median at 7.35 (Figure 4J). In conclusion, Ad-MAGEA3:MG1-MAGEA3 vaccination was 100% immunogenic, inducing remarkable CD8+ T-cell expansions against the tumor antigen with durability observed over several months.
Figure 4.
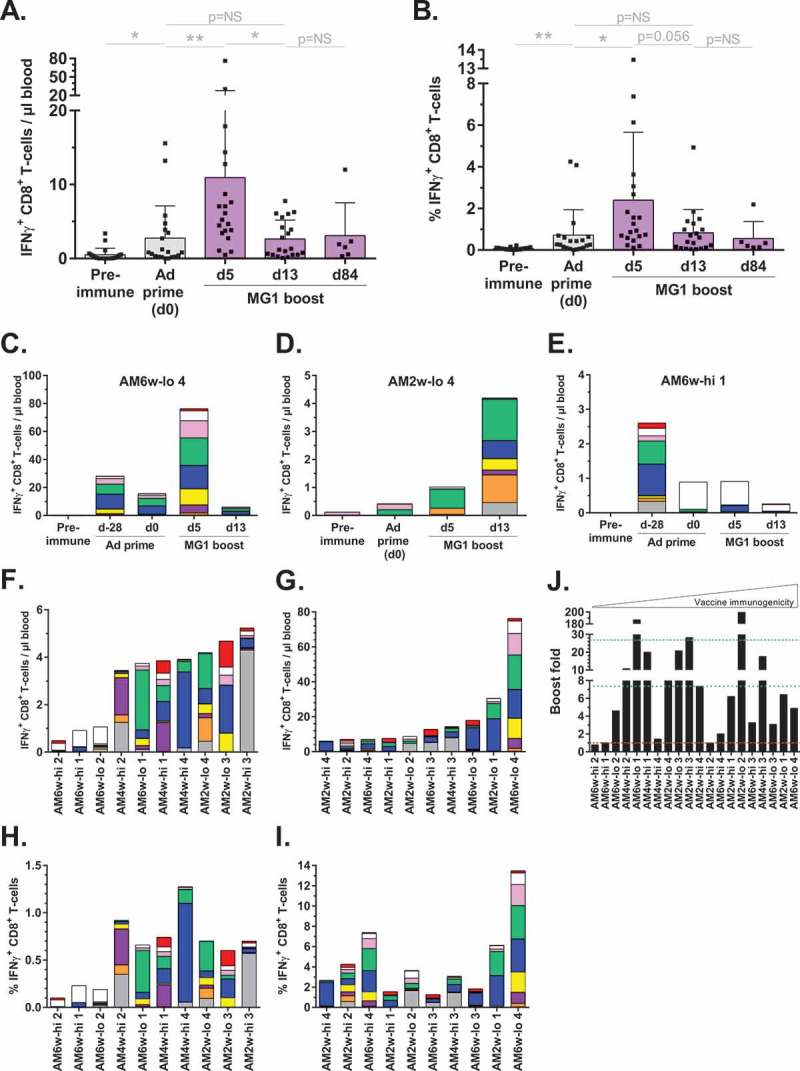
Marked expansion of the hMAGE-A3-specific CD8+ T-cell population following MG1-MAGEA3 boost.
(A,B) Kinetics of CD8+ T-cell responses against hMAGE-A3 in 20 macaques treated with Ad-MAGEA3:MG1-MAGEA3. Response is displayed both as the count (cells per µl of blood; A) and as the frequency (% among total CD8+ T-lymphocytes; B) of IFNγ-producing CD8+ T-lymphocytes. Histograms represent mean ± SD. p-value (Dunn’s test) considered non significant (NS) when > 0.05; *p < 0.05, **p < 0.01. (C-E) Illustrations of the three different kinetics of the boost response witnessed across the vaccinated macaques. (F, G) Peak CD8+ T-cell boost responses against hMAGE-A3 in each individual macaque, ranked according to their magnitude with bottom 10 and top 10 responders respectively displayed in the panels F and G. (H, I) Corresponding MG1-MAGEA3 boost responses expressed as the frequency (%) of reactive cells inside the total CD8+ T-cell population. (C-I) Sub-responses to each of the nine pools of hMAGE-A3 peptides used for antigenic re-stimulation are color-coded: pool of peptides 1–10 in red, 11–20 in white, 21–30 in pink, 31–40 in green, 41–50 in blue, 51–60 in yellow, 61–70 in purple, 71–80 in orange and 81–87 in grey. (J) Fold by which hMAGE-A3-reactive CD8+ T-cell population expanded between day 0 and the peak response day following MG1-MAGEA3 boost. In the absence of any detectable prime response in AM2w-lo 2, its boost fold was arbitrarily set to 200. Red dashed line illustrates boost fold of 1 (i.e. failure of MG1 boost). Green dotted line and dashed line illustrate median and mean boost folds, respectively. hMAGE-A3, human melanoma-associated antigen, family A, member 3; IFNγ, interferon gamma.
Factors impacting the magnitude and quality of vaccine-induced cellular immunity
Additional analyses were performed to interrogate the effects of experimental variables and individual characteristics of macaques on the induced hMAGE-A3 cellular immunity. Variations in gender, age, interval between Ad-MAGEA3 and MG1-MAGEA3 administration and the dose of MG1 did not affect vaccine immunogenicity (data not shown). However, predominant reactivity against MHC-I-restricted epitope(s) located in the N-terminal region covered by the pool of peptides 11–20 was associated with weaker CD8+ T-cell boost responses (macaques AM6w-lo2, AM6w-hi1 and AM6w-hi2, Figure 5A). Thus, individual variables affecting antigen presentation and/or recognition were implicated as determinants for the magnitude of cellular immunity. Additionally, the quality of the CD8+ T-cell immunity induced by Ad-MAGEA3:MG1-MAGEA3 vaccination was evaluated by the ability of reactive immune cells to produce not only IFNγ but also TNFα (Figure 5B). As illustrated in the Figure 5C, the quality of the mounted hMAGE-A3 boost response was remarkable with an average of 62 ± 21% of the IFNγ+ reactive CD8+ T-cells co-secreting TNFα. In 65% of macaques (13/20), the majority of MAGE-3-specific circulating CD8+ T-cells were dual cytokine positive. Interestingly, T-cell quality was independent of the magnitude of the immune response (Figure 5C). Additionally, the quality of the induced cellular immunity was independent of gender, MG1 dose and the prime-boost interval (data not shown). However, the frequency of IFNγ+ TNFα+ double positive CD8+ T-lymphocyte population was lower in the youngest primates (Figure 5D). Taken together, Ad-MAGEA3:MG1-MAGEA3 vaccination was broadly immunogenic in an NHP population with varied biologic and genotypic characteristics.
Figure 5.
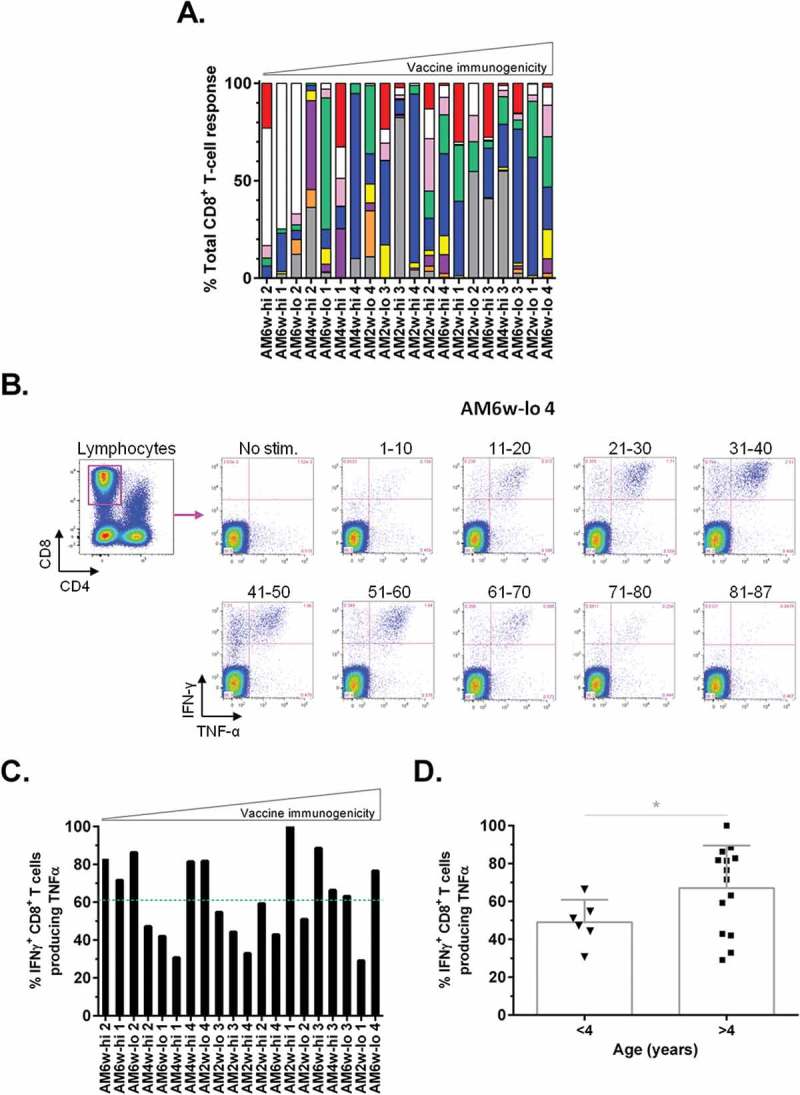
Genotypic and biological traits impacting on the profile of Ad:MG1 vaccination.
(A) Distribution of the CD8+ T-cell reactivity against the different regions of the hMAGE-A3 antigen following Ad-MAGEA3:MG1-MAGEA3 immunization. Each region was color-coded according to the peptide pool covering it, as follows: the N-terminal extremity was encompassed within the pool of peptides 1–10 in red, the inner regions were covered by the pools of peptides 11–20 in white, 21–30 in pink, 31–40 in green, 41–50 in blue, 51–60 in yellow, 61–70 in purple and 71–80 in orange while the C-terminal extremity was covered by the peptides 81–87 in grey. (B-D) Quality of vaccine immunogenicity was assessed by measuring the proportion of IFNγ+ reactive CD8+ T-cells that co-produced TNFα. (B) Flow cytometry gating strategy. CD8+ lymphocytes were first gated (top left plot – magenta frame). In the child gates (right), IFNγ TNFα double positive CD8+ lymphocytes were counted following each (re-)stimulation with one of the nine peptide pools from the hMAGE-A3 library (pool 1–10 to pool 81–87). Illustrated dot plots were recorded in the AM6w-lo 4 macaque who best responded to Ad-MAGEA3 prime:MG1-MAGEA3 boost. (C) Quality of the CD8+ T-lymphocyte reactivity illustrated as the percentage of IFNγ+ T cells co-producing TNFα upon ex vivo (re-)exposure to hMAGE-A3 peptides in each macaque immunized with Ad-MAGEA3:MG1-MAGEA3. (A, C) Primates are ranked according to the magnitude of the CD8+ T-cell boost response illustrated in the Figure 5E-F, from the weakest (on the left) to strongest (on the right). Green dashed line illustrates both mean (62%) and median (61%) TNFα positivity values. (D) Quality of vaccine immunogenicity according to the age of the macaque (3 year-old versus 4 to 6 year-old subjects). Histograms represent mean ± SD. p-value (Dunn’s test) considered non significant (NS) when > 0.05; *p < 0.05. hMAGE-A3, human melanoma-associated antigen, family A, member 3; IFNγ, interferon gamma; TNFα, tumor necrosis factor alpha.
Ad-MAGEA3:MG1-MAGEA3 induces auto-reactive T-cells
To further appraise the potential clinical relevance of oncolytic vaccination, the ability to invoke populations of auto-reactive T-cells was tested. For this purpose, T-cell epitope mapping of the hMAGE-A3 was performed. Blood from 16 out of the 20 macaques that received Ad-MAGEA3 plus MG1-MAGEA3 was collected (the four weakest responders were excluded). Circulating simian T-cells were re-stimulated for 24 hours with each individual peptide of the hMAGE-A3 peptide library. Immunogenicity and relative immune dominance of every peptide was characterized by ELISpot (Figure 6A). Subsequently, peptides with sequence homology between human and macaque were identified by blasting the H. sapiens MAGE-A3 reference sequence (accession number: P43357) against the NCBI protein database and allowed identification of three M. fascicularis MAGE-A3 sequences (accession numbers: XP_005594921, XP_015299818, XP_005594918). Sequence alignment revealed that the 314 amino acids of the human and cynomolgus MAGE-A3 were 89% identical (Fig. S3B). Highly conserved regions were encompassed within 17 of the 87 peptides. Among them, eight peptides shared sequence identity (#31–36, 42, 61) while the other nine differed by only one amino acid located at the N- or C-terminal extremity (#6, 26, 27, 30, 53, 60, 73, 77, 78; Fig. S3A). Fourteen of 16 macaques (88%) that received Ad-MAGEA3:MG1-MAGEA3 demonstrated T-cell reactivity against highly conserved MAGE-A3 peptides (Figure 6B). With the exception of peptide #32, cellular immunogenicity was observed at least once against each of the highly conserved peptides (Figure 6B). T-cell responses against conserved peptides were mostly subdominant compared to non-conserved peptides. Although such an observation was predictable, strong subdominant immunogenicity against conserved regions (magnitude ≥ 50% compared to the immunodominant peptide response) was documented in 7 animals (44%). Remarkably, immunodominant reactivity against the fully conserved peptide #42 was observed at day 84 post-MG1-MAGEA3 in AM6w-hi4 (Figures 6A and B). Peptides #42, #36 and #78 were the most frequently immunogenic sequences amongst highly conserved peptides, with specific T-cell responses detected for each peptide in 5 macaques (31%). These data illustrate the ability of the Ad-MAGEA3:MG1-MAGEA3 vaccination to induce populations of auto-reactive T-cells in primates.
Figure 6.
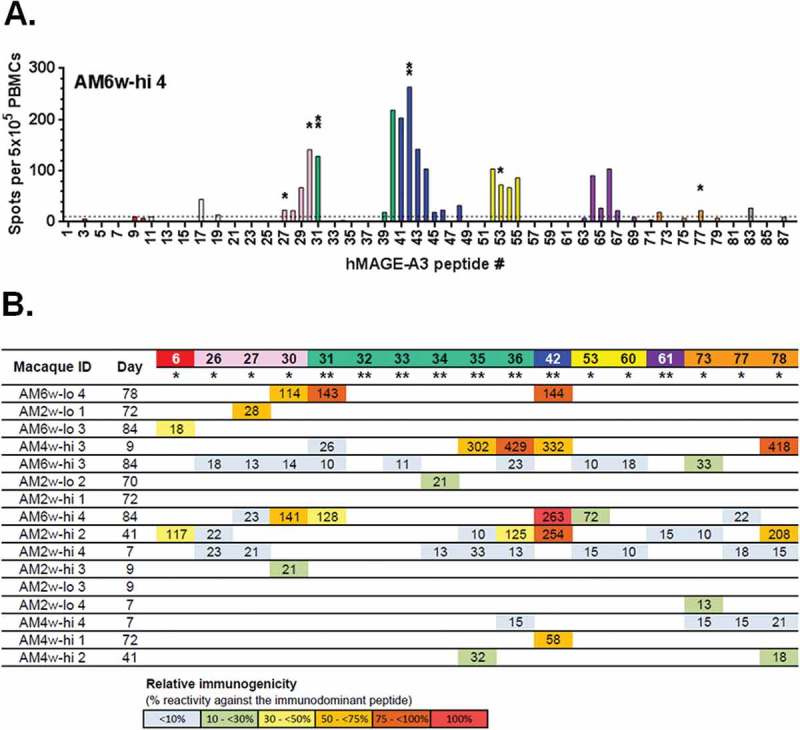
Ad-MAGEA3:MG1-MAGEA3 induces auto-reactive T-cells.
5x105 PBMCs were seeded and re-stimulated for 24 hours with one of the 87 overlapping peptides that cover the complete sequence of the hMAGE-A3 antigen. Reactivity against each peptide was detected by ELISpot. (A) Representative epitope mapping from the AM6w-hi4 macaque. Spots of T-lymphocytes secreting IFNγ following exposure to each peptide were counted and displayed in a bar graph. Cut-off was set to 10 spots (dashed line). Peptides with highly conserved sequences between human and simian MAGE-A3 were labeled as follows: ** = sequences identical or * = sequences diverging by one amino acid located at an extremity. (B) Reactivity against each of the highly conserved MAGE-A3 peptides was measured in 16 macaques at the indicated time-point post-MG1-MAGEA3 injection and reported as the number of spots detected. For each macaque, highlighting illustrates the relative immunogenicity of each peptide expressed as the percentage of the response measured against the immunodominant peptide. The immunodominant peptide was determined as the peptide leading to the highest number of IFNγ+ T-cell spots and consisted, except in AM6w-hi4, of a poorly conserved epitope not displayed in the table. For instance, in AM6w-hi4, as indicated by the red highlighting, the conserved peptide #42 turned out immunodominant with 263 spots detected (100% immunogenicity) while the conserved peptide #31 was subdominant with 128 spots detected (49% immunogenicity of the immunodominant peptide #42 illustrated by the yellow highlight). MAGE-A3, human melanoma-associated antigen, family A, member 3; IFNγ, interferon gamma; PBMCs, peripheral blood mononuclear cells.
Discussion
Due to their multi-faceted mechanisms of action and ability to engage the endogenous immune system, various oncolytic viruses (OVs) are undergoing clinical assessment.36 Two phase I/II clinical trials using the MAGE-A3 oncolytic vaccination platform are underway. The first utilizes the oncolytic Maraba MG1-MAGEA3 vaccine, either alone or following Ad-MAGEA3, for the treatment of patients with advanced MAGE-A3-positive solid malignancies (NCT02285816). The second is evaluating the treatment of MAGE-A3-positive non-small cell lung cancer (NSCLC) in combination with pembrolizumab (NCT02879760). The comprehensive safety profile and potent immunogenic properties documented herein were key facilitators to enable the launch of these first-in-man studies.
Following systemic administration of 1 × 1010 PFU (low dose) or 1 × 1011 PFU (high dose) MG1-MAGEA3, only mild and transient constitutional adverse events were recorded. Anorexia, fever, vomiting, diarrhea and constipation were amongst the most commonly encountered side effects, however no apparent association with the dose of MG1-MAGEA3 and frequency or severity of adverse events was noted. Pyrexia was more frequently observed in animals receiving MG1-MAGEA3 alone but remained short-lived in all cases. Weight loss was also more frequent in primates receiving MG1-MAGEA3 alone with the only grade III/IV events recorded in this cohort. One potential explanation for the beneficial effect of priming lies in the host’s immune response to the OV. When Ad is used to prime a response against an antigen that is also encoded within the OV, cellular immunity is skewed towards the antigen and away from the oncolytic vector. Conversely, when the OV encoding the antigen is administered alone, immunity is dominated by a vector-centric response.17 In light of this information, more marked physiologic inflammatory sequelae may be anticipated when MG1-MAGEA3 is administered without priming. Mild and self-limiting increases in circulating ALT (with the exception of one animal that was co-incidentally diagnosed with an unrelated hepatopathy) were also noted. No morphologic hepatic pathology was observed and the reasons for increases in transaminase activity are unclear. Nonetheless, such biochemical changes will be closely monitored within the clinic. Temporary neutropenia was documented in some of the animals and appeared independent of MG1-MAGEA3 dose. In all cases, neutrophils returned to within normal reference range. Clinically, viral infections are considered a common differential for neutropenia,37 so transient iatrogenic neutropenia following MG1-MAGEA3 is expected. Oncolytic vaccination was well tolerated and priming enhanced the safety profile thus highlighting the possibility of clinically escalating Maraba MG1 doses in the prime:boost setting.
All primates were carefully evaluated for any signs of neurotoxicity. Neurovirulence of the prototypical rhabdoviral family member, vesicular stomatitis virus (VSV) has been documented in NHPs.38,39 In our study, two animals developed peripheral nerve paralysis that was deemed attributable to trauma from restraint and not secondary to vaccination. No pathology was noted within the examined peripheral nerve tissue sections of any of the macaques. Severe neurological toxicity of human cancer patients following treatment with anti-MAGE-A3 TCR gene therapy has been reported. Cross-reaction of the high affinity TCR with neurologically expressed MAGE-A12 was presumably the cause. Indeed, engineered T-cells demonstrated higher affinity for the HLA-A*0201-restricted epitope KMAELVHFL of MAGE-A12 than for the targeted KVAELVHFL epitope of MAGE-A3.40 Peptide #31 (used to re-stimulate PBMCs in our ELISpot) contained the sequence KVAELVHFL. In our study, four primates had specific immune responses to the fully conserved peptide 31. Central nervous system pathology was not observed in any of the treated animals despite sequence homology with the cynomolgus MAGE-A12 epitope KVTELVHFL (putative M. fascicularis sequence NP_001270578.1). In preclinical murine studies splenic tropism of MG1-Maraba has been found to be fundamentally important to the mechanism by which MG1-Maraba potently boosts primed CD8+ T-cell responses thus the detection of intrasplenic MG1-Maraba genomes in primates supports common mammalian bio-distribution properties.31 Although genome was recovered from some tissues primarily comprising secondary lymphoid organs, no replicating virus was detected. This is consistent with the premise that the attenuated Maraba MG1 virus is extremely sensitive to type I IFN and selectively replicates in tumor cells that have defective type I responses but not in normal cells.25 Non-human primates are considered to be ideal models for the assessment of potential neurotoxicity resulting from live viral vaccines 41 thus instilling confidence in the preclinical safety profile established here.
Boosting with MG1-MAGEA3 significantly expanded specific CD8+ T-lymphocytes as part of a broad endogenous immune response. Transfusion of engineered anti-MAGE-A3 CD8+ T-cells resulted in clinical regressions of tumors, but this treatment was deemed unsafe due to the aforementioned neurotoxocity.40 Anti-MAGE-A3 vaccination has also been evaluated clinically using a recombinant protein strategy and, although this treatment was safe, no survival advantage was conferred. However, the induction of T-cell responses was not reported, precluding full assessment of this platform’s immunogenicity.42 Objective tumor responses have revealed the potential value of generating CD4+ T-cells against MAGE-A3 as demonstrated in patients treated with adoptive cellular therapy.43 MAGE-A3 antibodies have a potential role in increasing cellular immunity against the antigen by forming immune complexes; such complexes enhance the cross-presentation of the CT antigen, NY-ESO-1, in the context of vaccination.44 More broadly, tumor-infiltrating B-cells have been associated with favorable prognoses in various malignant indications, including lung cancer.45 We predict that the ability of oncolytic vaccination to engage multiple different effector arms of the immune system will be advantageous compared to more polarized approaches. By encoding the entire MAGE-A3 sequence, the immune system is exposed to all potential MAGE-A3 epitopes, irrespective of the genetic haplotype of the host.
A number of variables were interrogated to ascertain their possible impact on vaccine immunogenicity. Neither gender nor age affected the immune response. We did observe a reduction in the number of multi-functional T-cells in the youngest cohort of primates. Further investigation is needed to discern the cause and relevance of this finding but, as cancer is primarily associated with an aging population, this should not be overtly detrimental.46 Overall, the majority of MAGE-A3-specific CD8+ T-cells were capable of producing multiple cytokines and these multifunctional cells are important for efficacy of cancer immunotherapeutics.47 Clinically, Maraba MG1 can be administered rapidly after priming, as the interval between prime and boost did not alter immunogenicity. Mechanistically, MG1-Maraba is able to rapidly boost central memory T cells (TCM) induced by priming as previously demonstrated.31 As this minimizes any delay in treatment schedule, this versatility is also potentially advantageous. Considerable variation of the magnitude of immune responses and the apparent dominance of CD8+ responses to a specific pool of peptides in the lowest responding animals suggests that there are individual factors responsible for the magnitude of immunity invoked. Although the definitive cause of these variations was not established, it is likely that individual genotype is the main determinant of the size and specificity of response as seen in other vaccines.48 MG1 genomes were found predominantly in secondary lymphoid organs. This distribution supports the ability of MG1-MAGEA3 to directly boost TCM resulting in massive expansion of effector T-cells as previously elucidated in mice.31 MG1-MAGEA3 was capable of inducing very large and specific anti-MAGE-A3 immunity comprising up to 13.47% of all circulating CD8+ T cells in primates.
Some limitations were inherent in this study. In order to avoid loss of cellular viability secondary to freeze/ thawing, flow cytometry was performed on fresh samples hence there may be some variability between differing runs. The same personnel performed sample processing and data acquisition following identical protocols to minimize operator and procedural variation. As all animals were tumor free, no detectable viral replication occurred. So, the impact of functional oncolysis on the safety and immunogenicity has not been extended beyond previously published murine data.14,20,23–25 Toxicity has been associated with acute, massive cytokine release in some immunotherapeutics, such as CAR T-cells.49 Cytokines were not monitored in this study as their importance in a tumor-free animal is questionable. In most primates, cellular immunity rapidly contracted after boosting. Whether such kinetics will be reflected in the case of ongoing antigenic stimulation by the tumor is unknown. Finally, when using healthy hosts, studying the effect of oncolytic vaccination on the TME was not possible and, although we clearly demonstrate the induction of circulating immunity, how the oncolytic Maraba MG1 virus and effector immune populations will interact within the TME of spontaneous tumors remains to be determined. Data gathered during our clinical trials will address these areas of intrigue.
Based on the lack of viral replication, the absence of MG1-MAGEA3-related lesions and clinically significant hematologic or serum biochemical abnormalities, we conclude that the intravenous administration of MG1-MAGEA3 following Ad-MAGEA3 is safe. Previous rodent-based studies have highlighted the remarkable immunogenicity of this platform.14,20 The ability of Maraba MG1 to boost multiple arms of the endogenous immune system against a relevant tumor-associated antigen in a genetically outbred NHP population has now been confirmed. In light of these findings, the safety, immunogenicity and efficacy of oncolytic MAGE-A3 vaccination are currently being assessed for patients diagnosed with advanced malignancies refractory to established standard-of-care treatment.
Materials and methods
Macaques
Eighteen female and ten male cynomolgus macaques (Macaca fascicularis), aged 3–6 years and weighing 3–6 kg, were studied. Primates were obtained from Charles River Laboratories Preclinical Services (QC, Canada) and Primus Bio-Resources (QC, Canada). Animals were in groups of up to four when possible, with normal day-night cycles and an approved diet. Animals were identified by tattoos and neck collars. NHP study protocols were approved by the Animal Resource Centre, University Health Network, Toronto, ON, Canada.
Recombinant viruses
Ad-MAGEA3 is a human serotype 5 replication-deficient adenovirus (E1/E3-deleted).50 It contains a transgene encoding the full-length human melanoma-associated antigen, family A, member 3 (hMAGE-A3). Ad-MAGEA3 was injected in both quadriceps at a total dose of 1 × 1010 plaque forming units (PFU) (Figure 1A). MG1-MAGEA3 is a replication-competent oncolytic rhabdovirus.14,20,23–27 It was generated by inserting the hMAGE-A3 transgene between the G and L genes of the attenuated MG1 strain of Maraba. Macaques received two doses of 1 × 1010 or 1 × 1011 PFU MG1-MAGEA3, delivered intravenously three days apart (Figure 1A).
Peptides
A library of 87 overlapping 12 to 17-mer peptides (Pepscan, Netherlands) covering the full-length hMAGE-A3 antigen (314 amino acids) was used for re-stimulating T-cells ex vivo. Peptides 1 to 87 sequentially cover hMAGE-A3 from N- to C-terminal. T-lymphocytes were re-stimulated either with individual peptides or with the following pools: “red” pool = peptides 1 to 10, “white” pool = peptides 11 to 20, “pink” pool = peptides 21 to 30, “green” pool = peptides 31 to 40, “blue” pool = peptides 41 to 50, “yellow” pool = peptides 51 to 60, “purple” pool = peptides 61 to 70, “orange” pool = peptides 71 to 80, “grey” pool = peptides 81 to 87.
Antibodies
Monoclonal antibodies used in flow cytometry were purchased from BD Biosciences (ON, Canada): anti-human CD8α-PerCP-Cy5.5 (dilution 1:100, clone RPA-T8, ref. 560662) and anti-human CD4-FITC (dilution 1:25, clone L200, ref. 550628) for cell surface staining, anti-human IFNγ-APC (dilution 1:40, clone B27, ref. 562017) and anti-human TNFα-PE (dilution 1:4, clone Mab11, ref. 557068) for intracellular cytokine staining (ICS).
Animal testing procedures and necropsy
For blood draws, injections and minor procedures, macaques were restrained on a purpose-designed chair. Animals were anesthetized for surgery by intramuscular administration of 50 μM/kg of Domitor for induction (Medetomidine hydrochloride, Novartis) then maintained with 1.0–2.5% isoflurane during the surgery. Buprenorphine (0.01–0.03 mg/kg) was administered intramuscularly every 12 hours, and acetaminophen (Tylenol®, 5–15 mg/kg) administered orally every 8–12 hours as needed for analgesia. A subcutaneous vascular access port (VAP) connected to a central venous catheter through the femoral vein was surgically implanted to facilitate MG1-MAGEA3 administration and blood sampling (CP4/PCP-4, Access Technologies, Norfolk Medical Products Inc., IL, USA). Primates were treated with the antibiotic enrofloxacin (Baytril®, Bayer Healthcare, ON, Canada) for 24 hours after the procedure. Animals were euthanized by exsanguination under deep anesthesia while being perfused with 1 L of normal saline. Craniotomy was conducted to expose the brain, and the brain was removed within minutes of death. Samples from specific regions were taken from one hemisphere and while the contralateral hemisphere was preserved intact and fixed in 10% neutral-buffered formalin.
Clinical assessment for MG1-MAGEA3 safety profile
Animals were monitored daily throughout the experiment by clinical assessment and were weighed weekly. Vital signs and rectal temperature were measured prior to MG1-MAGEA3, 6 hours after administration, and thereafter following the sample collection schedule (Figure 1A). Fever was designated as temperature ≥ 39.8°C.
Cellular immunogenicity of the Ad-MAGEA3 and MG1-MAGEA3 vaccines
MAGE-A3-specific CD8+ and CD4+ T-cell responses were measured by detecting interferon-γ (IFNγ) production using ICS. Responses were evaluated before and at varying times after vaccination (Figure 1A). 2 ml of blood was diluted in four volumes of ammonium-chloride-potassium buffer (ACK) for 5 minutes at room temperature to lyse erythrocytes. Cells were washed twice in Hanks medium and the ACK lysis procedure was repeated. One tenth of the peripheral leukocytes were incubated in complete RPMI (RPMI-1640 medium supplemented with 10% fetal calf serum, 1% penicillin-streptomycin and 1% L-glutamine) either without (un-stimulated) or with one of the nine pools of hMAGE-A3 peptides (15 µg/ml) for re-stimulation of MAGE-A3-specific T-lymphocytes. Incubation was performed at 37°C, 5% CO2 and 95% humidity for 5 hours, with brefeldin A (1 μg/ml, GolgiPlug, BD Biosciences, ON, Canada) during the last 4 hours. Leukocytes were first incubated for 10 minutes in pH7.4 phosphate-buffered saline (PBS) with the LIVE/DEAD fixable near-IR dye (dilution 1:500; Life Technologies, CA, USA) to further discriminate live lymphocytes from dead entities; viability of the lymphogate events exceeded 80% in all samples collected (data not shown). Leukocytes were stained with the above listed surface antibodies for 25 minutes at 4°C in FACS buffer (PBS containing 0.5% bovine serum albumin). Cells were permeabilized and fixed with Cytofix/Cytoperm (BD Biosciences, ON, Canada) for 20 minutes at 4°C in FACS buffer and finally stained for intracellular cytokines for 25 minutes at 4°C in FACS buffer. Data were acquired using a FACSCanto cytometer with FACSDiva software (BD Biosciences, ON, Canada) and analyzed with FlowJo Mac software (Treestar, OR, USA). Absolute counts of reactive lymphocytes were obtained taking into account the following parameters: i) the proportion of the whole blood cell suspension processed and stained; typically 200 µl of whole blood for each peptide re-stimulation condition out of the 2 ml drawn per macaque (normalization factor #1) and ii) the proportion of the stained cell suspension ran through the flow cytometer; typically 300 out of 400 µl of the stained lymphocyte suspension (normalization factor #2). CD4+ and CD8+ lymphocyte counts quantitated by flow cytometry analysis (~ 1x105 events for each lymphocyte subset) were finally normalized per µl of blood initially collected by considering the two normalization factors. Background signal measured in un-stimulated leukocytes was subtracted from the responses detected in the re-stimulated samples. Mean pre-immune reactivity against hMAGE-A3 was established at 0.25 and 0.1 IFNγ+ CD8+ and CD4+ T-cells/µl blood, respectively.
T-cell epitope mapping of human MAGE-A3
Human MAGE-A3 T-cell epitopes were mapped by ELISpot measuring IFNγ release (Monkey IFNγ ELISpot PLUS kit from Mabtech Inc., OH, USA). T-cell reactivity was evaluated at various time-points after MG1-MAGEA3 administration in the five cohorts that received Ad-MAGEA3 prime-MG1-MAGEA3 boost. Briefly, 10 to 15 ml of blood were drawn and processed as described for ICS. 500,000 peripheral leukocytes were incubated in complete RPMI without (un-stimulated) or with each individual peptide of the hMAGE-A3 peptide library (20 µg/ml) for (re-)stimulation of MAGE-A3-specific T-lymphocytes. Incubation was performed at 37°C, 5% CO2 and 95% humidity for 24 hours. Spots of IFNγ-secreting T-cells were revealed following manufacturer’s recommendations and quantified using CTL-ImmunoSpot S6 Micro Analyzer (CTL, OH, USA). Background signal produced by un-stimulated leukocytes is deducted from the responses detected in the re-stimulated samples. Ten IFNγ-positive cells per well were used as minimum cut-off.
Bioinformatics
M. fascicularis MAGE-A3 sequences were identified in the NCBI protein database using the BLASTP program (blast.ncbi.nlm.nih.gov/) and the H. sapiens MAGE-A3 reference sequence (NCBI accession number: P43357) as protein query. Sequence alignment was performed using Clustal Omega (www.ebi.ac.uk/Tools/msa/clustalo/).
Statistical analyses
Linear mixed effect models were fitted to analyze weekly weight variation from baseline between experimental arms (MG1-MAGEA3 alone versus Ad-MAGEA3 + MG1-MAGEA3) and dose groups (low versus high dose) considering the time points as fixed effects. Statistical analyses were performed using SAS version 9.4 (SAS Institute Inc., NC, USA). For vaccine immunogenicity, GraphPad Prism version 6 for Windows (GraphPad Software, San Diego, CA) was used for data graphing and the R software for statistical analysis.51 Immune data are displayed as histograms or scatter plots displaying the mean value ± standard deviation (SD). For testing data normality, we applied the Shapiro-Wilk test and considered that data distribution was not normal when p-value < 0.05. For comparing two conditions, we applied the Wilcoxon test when data distribution was not normal while the Welch t-test was used for normally distributed data sets. For multiple pairwise comparisons (> 2 conditions), Dunn’s test (Kruskal-Wallis multiple comparisons) was applied, followed by a p-value adjustment using the Benjamini-Hochberg method, using the FSA R package.52 Differences between conditions were considered statistically significant when adjusted p ≤ 0.05 (*p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Funding Statement
B.D.L was supported by the Terry Fox Foundation (COVCo team grant) and the Ontario Institute for Cancer Research (ORBIT team grant). J.A.M was supported by the Ontario Institute for Cancer Research (ORBIT team grant);Ontario Institute for Cancer Research (CA) [ORBIT team grant];
Author contributions
B.D.L, J.A.M, D.S, J.C.B, Y.W, J.L.B and N.B designed the study. J.G.P and S.A.A jointly carried out most of the experiments/data analysis and statistical analysis and did the final data representations. J.G.P performed immunogenicity assessment with the assistance of K.B.S, C.E, G.D, R.P and J.M. Animal surgical procedures and anesthesia were performed by J.A.M, S.A.A, D.H, A.E, A.G, B.M, and S.L. The safety assessment, hematology and blood chemistry analyses were performed by S.A.A, N.T, D.H, A.E, A.G, B.M, S.L and R.L. P.V.T did the histopathological processing and analysis. C.L and J.R standardized various methods/protocols and contributed to coordinate the study. G.S contributed to statistical analysis. J.G.P, S.A.A, B.Y, C.J.B, M.J.A, J.A.M and B.D.L wrote the manuscript/made the figures. B.D.L, J.A.M and D.F.S jointly supervised the study and participated in the experimental design, critical reading and revision.
Acknowledgments
We would like to express many thanks to U. Sankar, A. del Rocio Ortega Rolando and M.F.C. Medina (Robert E. Fitzhenry Vector Laboratory, McMaster Immunology Research Centre, Hamilton, ON, Canada) for the production of both Ad-MAGEA3 and MG1-MAGEA3 viruses. Our appreciation also goes to S. Avery and M. D’Souza (UHN Animal Resource Centre, Toronto, ON, Canada) for assisting with animal housing and experiments. Finally, we want to express our gratitude to A.C. Smith, A. Laporte, J. Hamill, M. Becker and X. Gao (Ottawa Health Research Institute, Ottawa, ON, Canada) for their technical assistance in screening tissues for Maraba virus detection.
Conflict of Interest
Jonathan Pol, John Bell, Yonghong Wan, David Stojdl and Brian Lichty are named as inventors on patents covering Maraba virus as an oncolytic vaccine.
Material availability
Ad-MAGEA3 and MG1-MAGEA3 must be obtained through an MTA.
Supplemental material
Supplemental data for this article can be accessed here.
References
- 1.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies. Eur J Cancer. 2016;53:125–134. doi: 10.1016/j.ejca.2015.09.013. [DOI] [PubMed] [Google Scholar]
- 2.Ugurel S, Rohmel J, Ascierto PA, Flaherty KT, Grob JJ, Hauschild A, Larkin J, Long GV, Lorigan P, McArthur GA, et al. Survival of patients with advanced metastatic melanoma: the impact of novel therapies-update 2017. Eur J Cancer. 2017;83:247–257. doi: 10.1016/j.ejca.2017.06.028. [DOI] [PubMed] [Google Scholar]
- 3.Daud A, Blank CU, Robert C, Puzanov I, Richtig E, Margolin KA, O’Day S, Nyakas M, Lutzky J, Tarhini AA, et al. KEYNOTE-006 study of pembrolizumab (pembro) versus ipilimumab (ipi) for advanced melanoma: efficacy by PD-L1 expression and line of therapy. J Clin Oncol. 2016;34:9513. [Google Scholar]
- 4.Hodi FS, Chesney J, Pavlick AC, Robert C, Grossmann KF, McDermott DF, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Combined nivolumab and ipilimumab versus ipilimumab alone in patients with advanced melanoma: 2-year overall survival outcomes in a multicentre, randomised, controlled, phase 2 trial. Lancet Oncol. 2016;17:1558–1568. doi: 10.1016/S1470-2045(16)30366-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- 7.Buque A, Bloy N, Aranda F, Castoldi F, Eggermont A, Cremer I, Starmann J, Tjwa M, Plate KH, Sültmann H, et al. Trial Watch: immunomodulatory monoclonal antibodies for oncological indications. Oncoimmunology. 2015;4:e1008814. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vacchelli E, Aranda F, Bloy N, Buque A, Cremer I, Eggermont A, Fridman WH, Fucikova J, Galon J, Spisek R, et al. Trial Watch-Immunostimulation with cytokines in cancer therapy. Oncoimmunology. 2016;5:e1115942. doi: 10.1080/2162402X.2015.1115942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vacchelli E, Pol J, Bloy N, Eggermont A, Cremer I, Fridman WH, Starmann J, Tjwa M, Plate KH, Sültmann H, et al. Trial watch: tumor-targeting monoclonal antibodies for oncological indications. Oncoimmunology. 2015;4:e985940. doi: 10.1080/2162402X.2015.1008371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bloy N, Pol J, Aranda F, Eggermont A, Cremer I, Fridman WH, Fučíková J, Galon J, Tartour E, Spisek R, et al. Trial watch: dendritic cell-based anticancer therapy. Oncoimmunology. 2014;3:e963424. doi: 10.4161/21624011.2014.963424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pol J, Buque A, Aranda F, Bloy N, Cremer I, Eggermont A, Erbs P, Fucikova J, Galon J, Limacher J-M, et al. Trial Watch-Oncolytic viruses and cancer therapy. Oncoimmunology. 2016;5:e1117740. doi: 10.1080/2162402X.2015.1117740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pol J, Kroemer G, Galluzzi L.. First oncolytic virus approved for melanoma immunotherapy. Oncoimmunology. 2016;5:e1115641. doi: 10.1080/2162402X.2015.1115641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.First-Ever CAR T-cell Therapy Approved in U.S. Cancer Discov. 2017;7:OF1. doi: 10.1158/2159-8290.CD-NB2017-126. [DOI] [PubMed] [Google Scholar]
- 14.Atherton MJ, Stephenson KB, Pol J, Wang F, Lefebvre C, Stojdl DF, Nikota JK, Dvorkin-Gheva A, Nguyen A, Chen L, et al. Customized Viral Immunotherapy for HPV-Associated Cancer. Cancer Immunol Res. 2017;5:847–859. doi: 10.1158/2326-6066.CIR-17-0102. [DOI] [PubMed] [Google Scholar]
- 15.Boisgerault N, Kottke T, Pulido J, Thompson J, Diaz RM, Rommelfanger-Konkol D, Embry A, Saenz D, Poeschla E, Pandha H, et al. Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse. Mol Ther. 2013;21:1507–1516. doi: 10.1038/mt.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bridle BW, Clouthier D, Zhang L, Pol J, Chen L, Lichty BD, Bramson JL, Wan Y. Oncolytic vesicular stomatitis virus quantitatively and qualitatively improves primary CD8 T-cell responses to anticancer vaccines. Oncoimmunology. 2013;2:e26013. doi: 10.4161/onci.26013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, Brunellière J, Bramson JL, Lichty BD, Wan Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18:1430–1439. doi: 10.1038/mt.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P, Diaz RM, Chong H, Ilett E, Chester J, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Naslund TI, Uyttenhove C, Nordstrom EK, Colau D, Warnier G, Jondal M, Van Den Eynde BJ, Liljeström P. Comparative prime-boost vaccinations using Semliki Forest virus, adenovirus, and ALVAC vectors demonstrate differences in the generation of a protective central memory CTL response against the P815 tumor. J Immunol. 2007;178:6761–6769. [DOI] [PubMed] [Google Scholar]
- 20.Pol JG, Zhang L, Bridle BW, Stephenson KB, Resseguier J, Hanson S, Chen L, Kazdhan N, Bramson JL, Stojdl DF, et al. Maraba virus as a potent oncolytic vaccine vector. Mol Ther. 2014;22:420–429. doi: 10.1038/mt.2013.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM, Rommelfanger D, Ilett E, Pease L, Pandha H, et al. Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma. Nat Biotechnol. 2012;30:337–343. doi: 10.1038/nbt.2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rommelfanger DM, Wongthida P, Diaz RM, Kaluza KM, Thompson JM, Kottke TJ, Vile RG. Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer. Cancer Res. 2012;72:4753–4764. doi: 10.1158/0008-5472.CAN-12-0600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Le Boeuf F, Selman M, Son HH, Bergeron A, Chen A, Tsang J, Butterwick D, Arulanandam R, Forbes NE, Tzelepis F, et al. Oncolytic Maraba Virus MG1 as a treatment for sarcoma. Int J Cancer. 2017;141:1257–1264. doi: 10.1002/ijc.30813. [DOI] [PubMed] [Google Scholar]
- 24.Bourgeois-Daigneault MC, St-Germain LE, Roy DG, Pelin A, Aitken AS, Arulanandam R, Falls T, Garcia V, Diallo J-S, Bell JC. Combination of Paclitaxel and MG1 oncolytic virus as a successful strategy for breast cancer treatment. Breast Cancer Res. 2016;18:83. doi: 10.1186/s13058-016-0744-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brun J, McManus D, Lefebvre C, Hu K, Falls T, Atkins H, Bell JC, McCart JA, Mahoney D, Stojdl DF. Identification of genetically modified Maraba virus as an oncolytic rhabdovirus. Mol Ther. 2010;18:1440–1449. doi: 10.1038/mt.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mahoney DJ, Lefebvre C, Allan K, Brun J, Sanaei CA, Baird S, Pearce N, Grönberg S, Wilson B, Prakesh M, et al. Virus-tumor interactome screen reveals ER stress response can reprogram resistant cancers for oncolytic virus-triggered caspase-2 cell death. Cancer Cell. 2011;20:443–456. doi: 10.1016/j.ccr.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 27.Mahoney DJ, Stojdl DF. Molecular pathways: multimodal cancer-killing mechanisms employed by oncolytic vesiculoviruses. Clin Cancer Res. 2013;19:758–763. doi: 10.1158/1078-0432.CCR-11-3149. [DOI] [PubMed] [Google Scholar]
- 28.Bourgeois-Daigneault MC, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, Falls T, St-Germain LE, Pelin A, Lichty BD, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. 2018;10. [DOI] [PubMed] [Google Scholar]
- 29.Alkayyal AA, Tai LH, Kennedy MA, De Souza CT, Zhang J, Lefebvre C, Sahi S, Ananth AA, Mahmoud AB, Makrigiannis AP, et al. NK-cell recruitment is necessary for eradication of peritoneal carcinomatosis with an IL12-expressing maraba virus cellular vaccine. Cancer Immunol Res. 2017;5:211–221. doi: 10.1158/2326-6066.CIR-16-0162. [DOI] [PubMed] [Google Scholar]
- 30.Zhang J, Tai LH, Ilkow CS, Alkayyal AA, Ananth AA, De Souza CT, Wang J, Sahi S, Ly L, Lefebvre C, et al. Maraba MG1 virus enhances natural killer cell function via conventional dendritic cells to reduce postoperative metastatic disease. Mol Ther. 2014;22:1320–1332. doi: 10.1038/mt.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bridle BW, Nguyen A, Salem O, Zhang L, Koshy S, Clouthier D, Chen L, Pol J, Swift SL, Bowdish DME, et al. Privileged antigen presentation in Splenic B cell follicles maximizes T cell responses in prime-boost vaccination. J Immunol. 2016;196:4587–4595. doi: 10.4049/jimmunol.1600106. [DOI] [PubMed] [Google Scholar]
- 32.Tong JG, Valdes YR, Sivapragasam M, Barrett JW, Bell JC, Stojdl D, DiMattia GE, Shepherd TG. Spatial and temporal epithelial ovarian cancer cell heterogeneity impacts Maraba virus oncolytic potential. BMC Cancer. 2017;17:594. doi: 10.1186/s12885-017-3600-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tong JG, Valdes YR, Barrett JW, Bell JC, Stojdl D, McFadden G, McCart JA, DiMattia GE, Shepherd TG. Evidence for differential viral oncolytic efficacy in an in vitro model of epithelial ovarian cancer metastasis. Mol Ther Oncolytics. 2015;2:15013. doi: 10.1038/mto.2015.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Drake CG, Lipson EJ, Brahmer JR. Breathing new life into immunotherapy: review of melanoma, lung and kidney cancer. Nat Rev Clin Oncol. 2014;11:24–37. doi: 10.1038/nrclinonc.2013.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gjerstorff MF, Andersen MH, Ditzel HJ. Oncogenic cancer/testis antigens: prime candidates for immunotherapy. Oncotarget. 2015;6:15772–15787. doi: 10.18632/oncotarget.4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017;3:841–849. doi: 10.1001/jamaoncol.2016.2064. [DOI] [PubMed] [Google Scholar]
- 37.Boxer L, Dale DC. Neutropenia: causes and consequences. Semin Hematol. 2002;39:75–81. [DOI] [PubMed] [Google Scholar]
- 38.Mire CE, Miller AD, Carville A, Westmoreland SV, Geisbert JB, Mansfield KG, Feldmann H, Hensley LE, Geisbert TW, Bausch DG. Recombinant vesicular stomatitis virus vaccine vectors expressing filovirus glycoproteins lack neurovirulence in nonhuman primates. PLoS Negl Trop Dis. 2012;6:e1567. doi: 10.1371/journal.pntd.0001567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, et al. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology. 2007;360:36–49. doi: 10.1016/j.virol.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, Dudley ME, Feldman SA, Yang JC, Sherry RM, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013;36:133–151. doi: 10.1097/CJI.0b013e3182829903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Levenbook I. The role of non-human primates in the neurological safety of live viral vaccines (review). Biologicals. 2011;39:1–8. doi: 10.1016/j.biologicals.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Vansteenkiste JF, Cho BC, Vanakesa T, De Pas T, Zielinski M, Kim MS, Jassem J, Yoshimura M, Dahabreh J, Nakayama H, et al. Efficacy of the MAGE-A3 cancer immunotherapeutic as adjuvant therapy in patients with resected MAGE-A3-positive non-small-cell lung cancer (MAGRIT): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol. 2016;17:822–835. doi: 10.1016/S1470-2045(16)00099-1. [DOI] [PubMed] [Google Scholar]
- 43.Lu YC, Parker LL, Lu T, Zheng Z, Toomey MA, White DE, Yao X, Li YF, Robbins PF, Feldman SA, et al. Treatment of Patients With Metastatic Cancer Using a Major Histocompatibility Complex Class II-Restricted T-Cell Receptor Targeting the Cancer Germline Antigen MAGE-A3. J Clin Oncol. 2017;35:3322–3329. doi: 10.1200/JCO.2017.74.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagata Y, Ono S, Matsuo M, Gnjatic S, Valmori D, Ritter G, Garrett W, Old LJ, Mellman I. Differential presentation of a soluble exogenous tumor antigen, NY-ESO-1, by distinct human dendritic cell populations. Proc Natl Acad Sci U S A. 2002;99:10629–10634. doi: 10.1073/pnas.112331099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nielsen JS, Nelson BH. Tumor-infiltrating B cells and T cells: working together to promote patient survival. Oncoimmunology. 2012;1:1623–1625. doi: 10.4161/onci.21650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 47.Klebanoff CA, Gattinoni L, Restifo NP. CD8+ T-cell memory in tumor immunology and immunotherapy. Immunol Rev. 2006;211:214–224. doi: 10.1111/j.0105-2896.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Poland GA, Ovsyannikova IG, Jacobson RM. Personalized vaccines: the emerging field of vaccinomics. Expert Opin Biol Ther. 2008;8:1659–1667. doi: 10.1517/14712598.8.11.1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Davila ML, Riviere I, Wang X, Bartido S, Park J, Curran K, Chung SS, Stefanski J, Borquez-Ojeda O, Olszewska M, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ng P, Beauchamp C, Evelegh C, Parks R, Graham FL. Development of a FLP/frt system for generating helper-dependent adenoviral vectors. Mol Ther. 2001;3:809–815. doi: 10.1006/mthe.2001.0323. [DOI] [PubMed] [Google Scholar]
- 51.Team RDC R: A language and environment for statistical computing. R Foundation for Statistical Computing; 2008. [Google Scholar]
- 52.Ogle DH. FSA: fisheries Stock Analysis. 2018. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.