

ABSTRACT
In non-small cell lung carcinoma (NSCLC), stimulation of toll-like receptor 7 (TLR7), a receptor for single stranded RNA, is linked to tumor progression and resistance to anticancer chemotherapy. However, the mechanism of this effect has been elusive. Here, using a murine model of lung adenocarcinoma, we demonstrate a key role for TLR7 expressed by malignant (rather than by stromal and immune) cells, in the recruitment of myeloid derived suppressor cells (MDSCs), induced after TLR7 stimulation, resulting in accelerated tumor growth and metastasis. In adenocarcinoma patients, high TLR7 expression on malignant cells was associated with poor clinical outcome, as well as with a gene expression signature linked to aggressiveness and metastastic dissemination with high abundance of mRNA encoding intercellular adhesion molecule 1 (ICAM-1), cytokeratins 7 and 19 (KRT-7 and 19), syndecan 4 (SDC4), and p53. In addition, lung tumors expressing high levels of TLR7 have a phenotype of epithelial mesenchymal transition with high expression of vimentin and low abundance of E-cadherin. These data reveal a crucial role for cancer cell-intrinsic TLR7 expression in lung adenocarcinoma progression.
KEYWORDS: TLR7, lung adenocarcinoma, metastasis, EMT, MDSC
Introduction
Tumors constitute a complex microenvironment, which is composed of genetically altered malignant and non-mutated stromal cells, the latter including fibroblasts, vascular and lymphatic vessels, as well as immune cells 1,2. Many tumor-infiltrating immune cells including CD4+ and CD8+ T cells, B lymphocytes, or type 1 macrophages (M1) participate in immunosurveillance, meaning that they are commonly associated with good clinical outcome. Other leukocyte subtypes including regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs) and type 2 macrophages (M2) have immunosuppressive functions, meaning that their local presence has a negative clinical impact 1. The composition of tumor infiltrating immune cells strongly influences the tumor microenvironment and the behavior of the tumor in terms of progression, metastasis and treatment resistance.
Toll-like receptors (TLRs), which recognize ligands from pathogens (pathogen-associated molecular patterns, PAMPs) or dying cells (damaged-associated molecular patterns, DAMPs), are involved in the immune response against pathogens 3, in thus far that they induce the secretion of inflammatory cytokines and chemokines upon their activation 4. These receptors are largely expressed by immune cells and epithelial cells but also by several types of tumors cells 5. The role of TLRs in tumor microenvironment is complex. On one hand, TLRs expressed by immune cells can favor immunosurveillance, because their stimulation favors the maturation and activation of innate and adaptive immune effectors. On the other hand, TLRs expressed by cancer cells may receive stimuli that favor tumor progression 6,7.
We have previously demonstrated that TLR7, a single stranded RNA receptor that is usually expressed in endosomes of immune cells including plasmacytoid and conventional dendritic cells (pDCs and cDCs), macrophages and B lymphocytes 8,9, is expressed by malignant cells from NSCLC patients 10. High expression by cancer cells was associated with poor prognosis, both in early stages that were treated with surgery alone and more advanced stages treated by neo-adjuvant chemotherapy. Moreover, in a mouse model of lung cancer, TLR7 stimulation favored tumor growth 11. The present study has been designed to decipher the mechanisms involved in the pro-tumorigenic and possible pro-metastatic effects of TLR7 stimulation.
Here, we report that the stimulation of TLR7 expressed by malignant cells favors tumor progression and metastasis, increases CCL2 and GM-CSF secretion in the tumor microenvironment, and elicits the recruitment of MDSCs into the tumor. In vivo depletion of MDSCs abolished the TLR7-dependent pro-tumorigenic and pro-metastatic effect. Finally, we observed in lung adenocarcinoma patients, that high expression of TLR7 by tumor cells is associated to a pro-metastatic gene expression signature as well as to epithelial–mesenchymal transition (EMT).
Results
Intratumoral TLR7 agonist injection favors tumor progression through a direct effect on carcinoma cells
Murine lung adenocarcinoma LLC-luc cells express several TLRs including TLR4, TLR7 and TLR9 (Figure 1A-C). Expression of TLR7 was also validated by quantitative PCR (data not shown). We have previously demonstrated that intratumoral injection of TLR7 agonist results in increased tumor progression in immunocompetent as well as in immunodeficient mice 11. However, the precise mechanisms involved in such pro-tumorigenic effects are elusive. To determine if this effect was specific, we analyzed the growth of subcutaneous grafted LLC-luc tumors in wild-type (WT) mice locally injected with TLR4, TLR7 or TLR9 agonists. Bacterial lipopolysaccharide (LPS), an agonist of TLR4, failed to affect tumor growth (Figure 1A), while CpG, an agonist of TLR9, actually inhibited tumor growth (Figure 1B). On the contrary, intratumoral injection of the TLR7 agonist CL264 induced a pro-tumorigenic effect (Figure 1C), in accordance to our previous observations 11.
Figure 1.
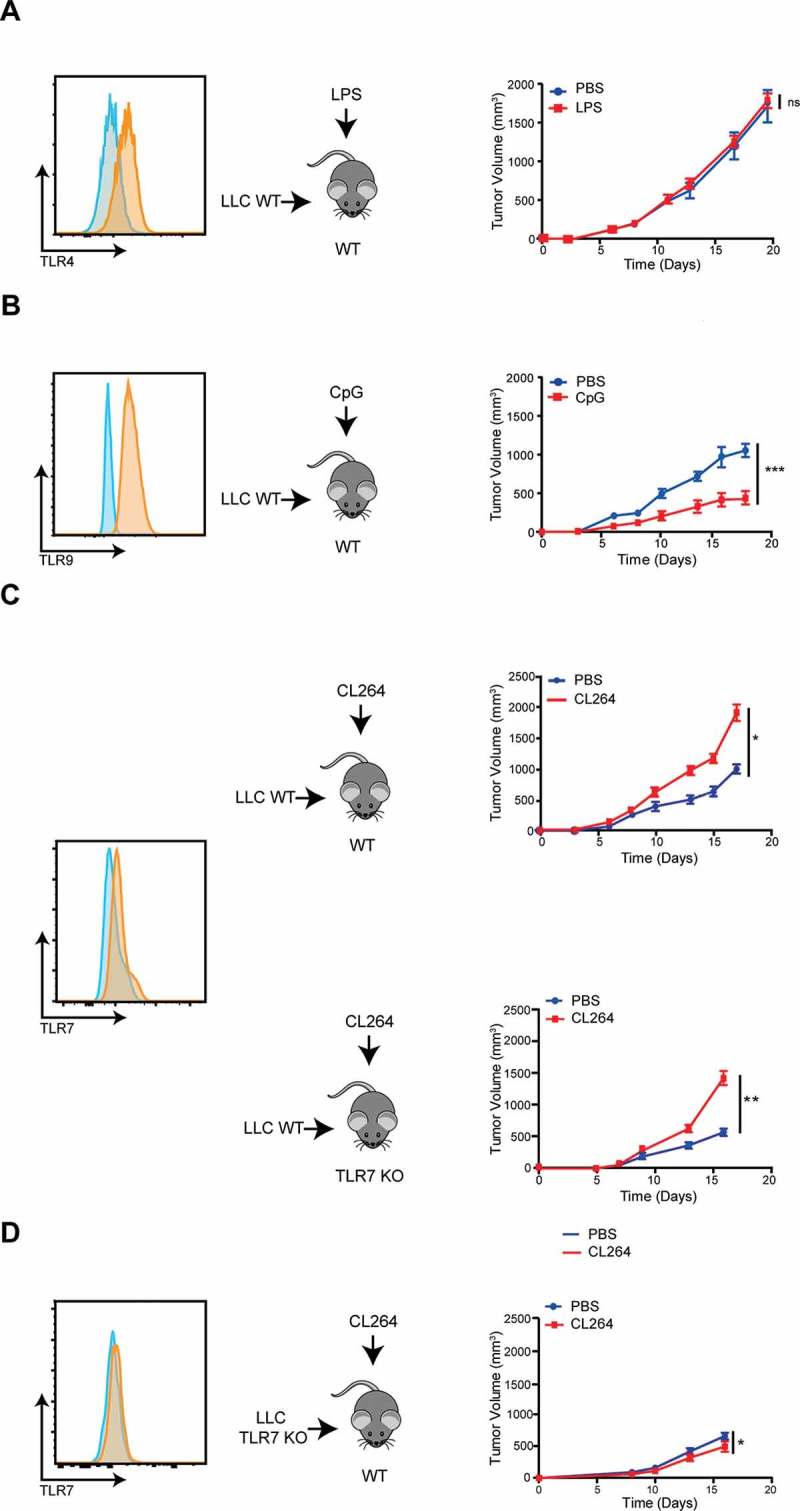
Pro- or anti-tumoral effects of different TLRs stimulation.
Left panel: TLR4 (A), TLR9 (B) and TLR7 (C) expression by LLC-luc cells and TLR7 expression by LLC-luc cells deficient for TLR7 (obtained by CRISPR/Cas9 technology) (D). Control isotype is shown in blue and stained cells in orange.Right panel: Tumor progression in WT C57Bl/6 mice grafted with LLC-luc cells, after LPS (A) or CpG injection (B). Tumor progression in WT C57Bl/6 or in TLR7 KO mice grafted with LLC-luc cells after CL264 injection (C). Tumor progression in WT C57Bl/6 mice grafted with TLR7-deficient LLC-luc cells (LLC TLR7 KO) after CL264 injection (D). Data are mean ± SEM (5 mice/group). *: p < 0,05, **: p < 0,01, ***: p < 0,001. ns = not significant. Each experiment was repeated three times.
The pro-tumorigenic effect of TLR7 stimulation could result from a direct stimulation of TLR7 on tumor cells and/or from stimulation of immune cells that express TLR7. We determined the level of TLR7 expression of immune cells in the tumor, the spleen and the blood of tumor-bearing mice by flow cytometry after optimization of the gating strategy (Fig. S1). TLR7 was highly expressed by macrophages, pDCs, and to a less extent by cDCs in all compartments including the tumor (Fig. S2). Thus, TLR7 agonists may act both on malignant cells and on tumor-infiltrating myeloid cells to favor tumor progression.
To discriminate the target population of TLR7 agonist, we took advantage of TLR7-deficient mice (TLR7 KO), and generated TLR7-deficient LLC-luc cells (LLC TLR7 KO) by CRISPR/Cas9-mediated gene editing (Figure 1D). We injected WT (TLR7 expressing) LLC-luc cells into TLR7 KO mice or alternatively TLR7-deficient LLC-luc cells into WT mice, and then evaluated the effects of the TLR7 agonist CL264 on tumor progression. We observed a growth accelerating effect of CL264 in TLR7 KO mice, similar to that observed in WT mice (Figure 1C). In sharp contrast, the TLR7 agonist failed to stimulate the growth of TLR7-deficient LLC-luc tumors (Figure 1D). These results clearly demonstrate that the pro-tumorigenic effect of CL264 effect is mediated by an effect on TLR7 expressed by malignant (rather than immune) cells.
Stimulation of TLR7 alterates the intratumoral immune infiltrates
TLR stimulation is known to modify the inflammatory microenvironment of tumors. We analyzed whether the local injection of TLR7 agonist would impact the composition of the immune infiltrate. Immune phenotyping by flow cytometry (for gating strategy see Fig. S1) revealed the percentages of T, B and NK cells, Tregs, macrophages, cDCs, pDCs and MDSCs, including granulocytic and monocytic subpopulations. The immune infiltrate of vehicle-only treated tumors was composed by a majority of myeloid cells including 49% of MDSCs, 15% of cDCs and 5% of macrophages, as well as a by a minority of adaptive immune cells including 10% T cells, 5% NK cells and 3% Tregs (Figure 2A). The local injection of CL264 induced a modification of percentages and absolute cells numbers of MDSCs, with a significant (unpaired Student T test, p < 0.05) increase from 49 ± 7% to 66 ± 10% (Figure 2A-C). CL264 also induced a decrease of cDCs (from 15% ± 1% to 9%± 2%) and of pDCs (from 2% to 1.5%) (Figure 2D-E). In control tumors, MDSCs were mostly monocytic, and CL264 treatment resulted in the increase of granulocytic MDSCs (Figure 2F-G). In contrast, CL264 did not affect the frequency of macrophages, NK, T, Tregs, and B cells (Figure 2A, Fig. S3A).
Figure 2.
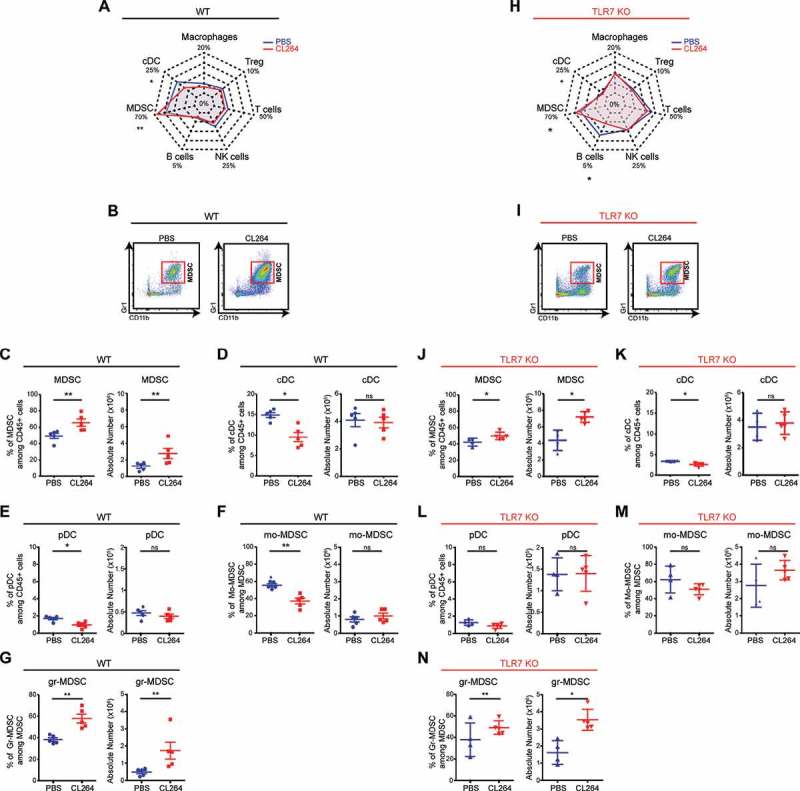
TLR7 stimulation alterates intratumoral immune infiltrates.
WT (A-G) or TLR7 KO (H-N) C57BL/6 mice received subcutaneous injection of LLC-luc cells, followed by 4 injections of CL264 (red) or PBS (blue), on days 0, 3, 6 and 9. The percentages and absolute numbers of immune cells were analyzed 10 days after. Radar plot representation for immune cell subpopulations is shown in (A) and (H). Representative flow cytometry image of Gr1+CD11b+ MDSC cells in WT (B) and in TLR7 KO mice (I), having received PBS or CL264. Percentages and absolute numbers of each cell population in WT (C-G) and in TLR7 KO mice (J-N). Data are mean ± SEM (5 mice/group). *: p < 0,05, **: p < 0,01, ***: p < 0,0001. ns = not significant. Each experiment was repeated three times.
CL264 stimulates MDSC recruitment by acting on TLR7 expressed by malignant cells
We investigated if the increase of MDSCs within the tumor was a consequence of TLR7 stimulation on cancer or immune cells. The TLR7 agonist CL264 was injected into LLC-luc tumors that were established in WT or TLR7 KO mice.
At baseline levels (without CL264 treatment), WT and TLR7KO mice showed low frequencies of MDSCs, cDCs and macrophages in their blood and spleens (Fig. S4A, B). TLR7 KO mice exhibited a lower proportion of MDSCs, cDCs, NK and T cells and increased proportion of B cells in the spleen and the blood compared to WT mice (Fig. S4A, B). The tumor immune infiltrate was characterized by high frequencies of MDSCs, both in TLR7 KO and in WT mice. Higher proportions of macrophages and T cells and lower proportions of cDCs were detected in the tumors of TLR7 KO compared to WT mice (Fig. S4C).
In tumors growing on TLR7 KO mice (Figure 2H-N), compared to WT mice (Figure 2A-G) the injection of TLR7 agonist increased the proportion and absolute number of MDSCs (Figure 2H-J), with a marked shift towards a granulocytic MDSC phenotype (Figure 2N), concomitant with a slight decreased proportion of cDCs (Figure 2K), but did not modify the frequency of pDCs (Figure 2L), macrophages, NK, T, Tregs and B cells (Fig. S3B).
Collectively, these data indicate that CL264 maintains its local immunomodulatory function resulting in an increased presence of MDSCs in WT and TLR7 KO mice.
Mdscs are involved in the pro-tumorigenic effect mediated by TLR7 stimulation
To decipher the possible role of MDSCs in the tumor growth stimulatory effect of CL264, we experimentally depleted MDSCs from the system. Tumor bearing mice that were treated with CL264 or vehicle only received intraperitoneal injections of anti-Gr1 monoclonal antibody (mAb) or control isotype every 48 hours (Figure 3A). During the experiment, less than 0.4% of MDSCs were detectable in the blood of mice, confirming the efficacy of MDSC depletion (Figure 3B-C). MDSC depletion resulted in decreased tumor growth, both in WT and in TLR7 KO mice (Figure 3D, G). The TLR7 agonist CL264 stimulated tumor growth (Figure 3E, H), and this pro-tumorigenic effect was completely lost in the absence of MDSCs, both in WT mice (Figure 3F) and in TLR7 KO mice (Figure 3I).
Figure 3.
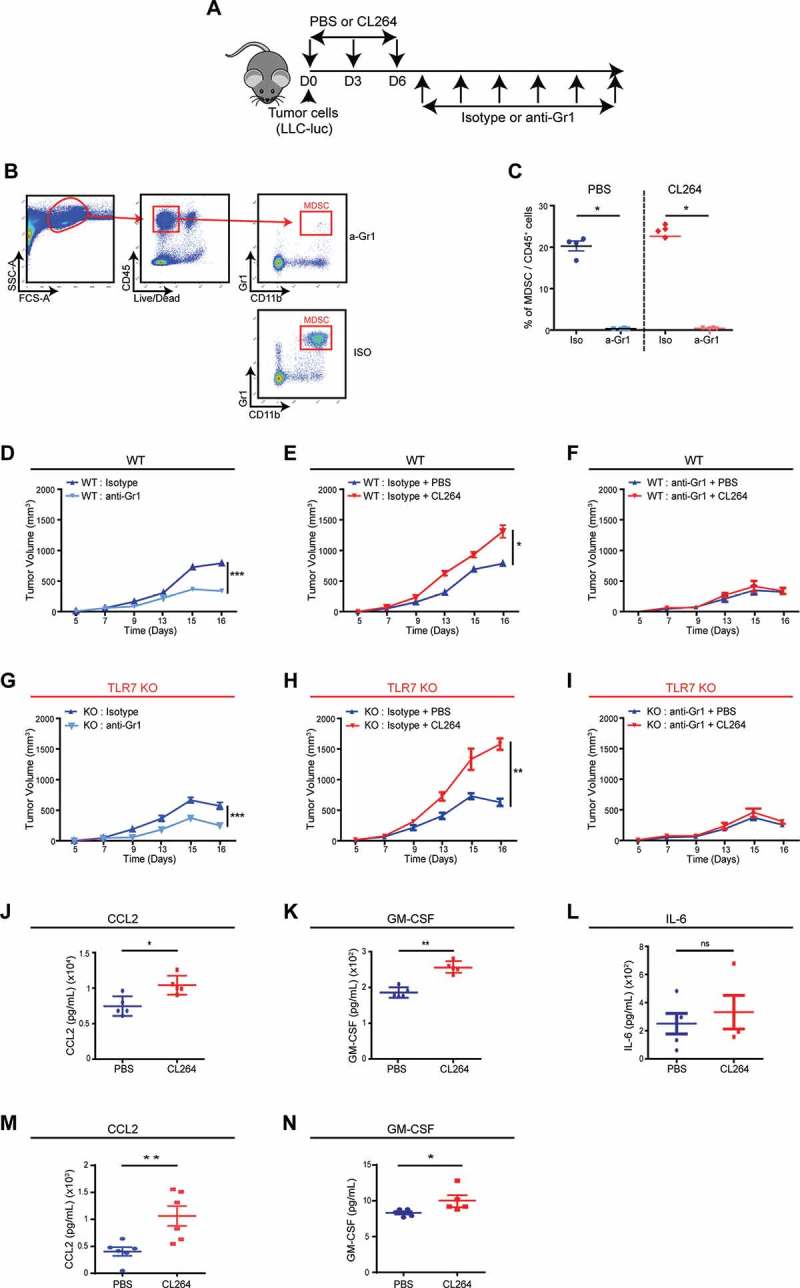
MDSCs are involved in the pro-tumorigenic effect mediated by TLR7 stimulation.
WT C57BL/6 and TLR7 KO mice received subcutaneous injection of LLC-luc cells, followed by intratumoral injections of CL264 or PBS, and intraperitoneal injections of anti-Gr1 or control antibody every 48 hours (A). Gating strategy for MDSC staining and representative FACS image showing the efficacy of MDSCs depletion in the blood (B). Percentage of MDSCs among total CD45+ cells in the blood is shown for all treated mice (C).Tumor progression in WT C57Bl/6 (D) or in TLR7 KO (G) mice grafted with LLC-luc cells, after isotype or anti-Gr1 antibody injections. Tumor progression in WT C57Bl/6 (E) or in TLR7 KO (H) mice grafted with LLC-luc cells, after CL264 or PBS injection and control isotype antibody injections. Tumor progression in WT C57Bl/6 (F) or in TLR7 KO (I) mice grafted with LLC-luc cells, after CL264 or PBS injection and anti-Gr1 antibody injections. CCL2 (J), GM-CSF (K) and IL-6 (L) titration by ELISA in supernatants from tumor cells from PBS or CL264-treated mice. CCL2 (M) and GM-CSF (N) titration by ELISA in supernatants from in vitro PBS or CL264-stimulated tumor cells.*: p < 0,05, **: p < 0,01, ***: p < 0,0001. Data are mean ± SEM (5 mice/group). ns = not significant. Each experiment was repeated three times.
These results demonstrate that the tumor growth exacerbating effect of TLR7 stimulation is mediated by the increased number of MDSCs within the tumor microenvironment.
TLR7 stimulation induced an increased production of CCL2 and GM-CSF within the tumor microenvironment
Given the involvement of MDSCs in the pro-tumorigenic effect of TLR7 stimulation, we investigated the mechanisms that might be involved in the local CL264-induced MDSCs expansion. Several cytokines such as IL-6, GM-CSF or CCL2 stimulate the recruitment or proliferation of MDSCs and may be produced by tumor cells. Local injection of the TLR7 agonist CL264 enhanced the ELISA-detectable concentration of CCL2 and GM-CSF within the tumor. The concentrations of CCL2 and GM-CSF were 10407 ± 1326 and 256 ± 15 pg/ml, respectively, in mice treated with CL264, and 7453 ± 1380 and 186 ± 15 pg/ml, respectively, in control mice receiving PBS (Figure 3J, K). In contrast, CL264 failed to significantly modulate the amount of IL-6 found in the tumor (Figure 3L). The secretion of CCL2 and GM-CSF by tumor cells was confirmed in vitro, which was found increased in supernatants of CL264-stimulated cells (Figure 3M, N).
These results suggest that CL264 may stimulate the recruitment of MDSCs via the induction of CCL2 and GM-CSF.
TLR7 agonist does not modify the immunosuppressive capacities of mdscs
To unravel the possible impact of TLR7 stimulation on MDSCs functions, we measured the production of molecules involved in MDSCs-mediated immunosuppressive circuitries. The production of nitrate, H2O2, and arginase 1 by MDSCs was not affected by CL264 injection. Similarly, the proportion of ROS producing cells was not altered by CL264 (Fig. S5A). In addition, the capacity of MDSCs to suppress T cell proliferation was similar for MDSCs purified from PBS-treated and CL264-treated tumors (Fig. S5B-C). These results demonstrate that intratumoral injection of TLR7 agonist does not modify the immunosuppressive properties of intratumoral MDSCs.
MDSC are involved in the pro-metastatic effect of TLR7 stimulation
Next, we analyzed the capacity of CL264 to stimulate lung metastasis. NOD/SCID or C57BL/6 mice were subcutaneously grafted with LLC-luc cells, and the apparition of metastasis was imaged over time, by bioluminescence or precisely quantified at necropsy following perfusion with Indina Ink and labelling of the lungs with Feket solution. Either of these methods revealed the capacity of CL264 to stimulate lung metastasis, both in immunodeficient NOD/SCID mice (Figure 4A) and in immunocompetent C57Bl/6 mice (Figure 4B).
Figure 4.
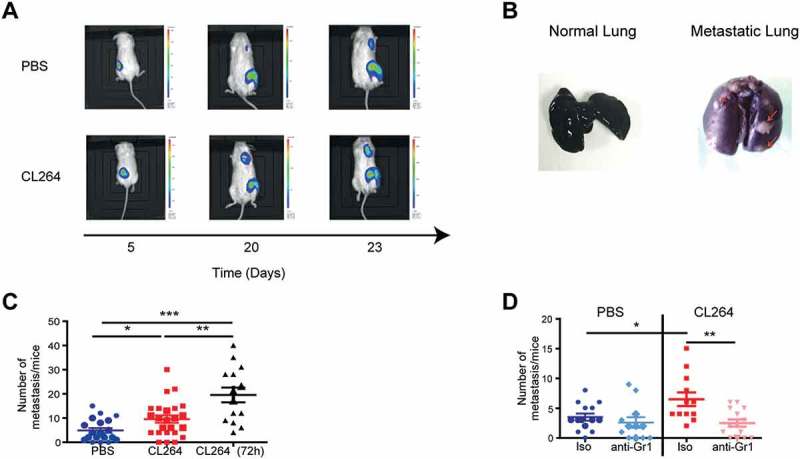
MDSCs are involved in the pro-metastatic effect of TLR7 stimulation.
NOD/SCID (A, C) or C57Bl/6 mice (B, D) received subcutaneous injection of LLC-luc cells, followed by 4 injections of CL264 or PBS, on days 0, 3, 6 and 9. Tumor progression was analyzed by the measurement of D-luciferine bioluminescence (A) or by staining with indian ink and Feket solution at day 25 (B). The number of metastasis per lungs was quantified in NOD/SCID mice treated by PBS or CL264 (3 injections or continuous injections every 72h) (C) and in C57Bl/6 mice having also received control isotype or anti-Gr1 antibody (D). *: p < 0,05, **: p < 0,01, ***: p < 0,0001. Data are mean ± SEM (10–25 mice/group).
The number of metastasis was quantified in PBS- and CL264-treated mice. We observed the presence of lung metastases in both groups of mice, but the mice having received three injections of TLR7 agonist had a significantly higher number of metastases with a mean of 9.5 (± 7.5) metastases per lungs for CL264-treated mice, compared to 4.9 (± 4.5) metastases per lungs for the control group (Figure 4C). Repeated injections of CL264 (every three days throughout the duration of the experiment) led to even higher number of lung metastasis (19.5 (± 11.5) metastasis per lungs) (Figure 4C). These results indicate that TLR7 stimulation has a marked pro-metastatic effect.
Given the implication of MDSC in metastatic dissemination 12, we investigated the potential role of MDSCs in the pro-metastatic effects of TLR7 stimulation. MDSC depletion by injection of anti-Gr1 mAbs led to a significant decrease in the number of metastases (mean of 2.5 (± 2) metastases per lungs) in CL264-treated mice compared to control mice that did not receive depleting antibody (mean of 6.5 (± 4) metastases per lungs) (Figure 4D), showing that TLR7 stimulation increases the number of lung metastases through a mechanism involving MDSCs.
High expression of TLR7 in human lung adenocarcinoma is associated with a metastatic gene expression signature
We further characterized the in vitro impact of CL264 on metastatic properties of malignant cells. The stimulation of LLC cells with CL264 resulted in epithelio-mesenchymal transition gene expression signature, with an increased gene expression of N-cadherin, vimentin, snail1, snail 2 and zeb, and a decreased expression of E-cadherin (Figure 5A, B).
Figure 5.
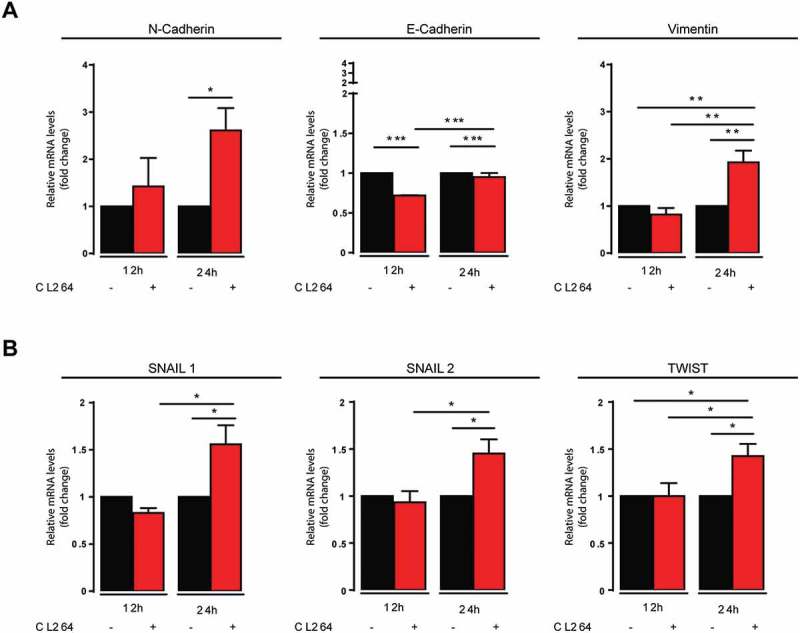
TLR7 stimulation of LLC cells leads to epithelio-mesenchymal signature.
Gene expression of membrane molecules (A) and transcription factors (B) involved in epithelio-mesenchymal transition, in LLC cells that were stimulated or not with CL264, determined 12 and 24h after stimulation.
Immunohistochemistry was then used to determine the expression of E-cadherin, associated to epithelial cell phenotype, and that of vimentin, expressed by mesenchymal cells, on human malignant cells expressing high or low levels of TLR7. Forty-five adenocarcinoma patients were analyzed for the expression of TLR7, E-cadherin and vimentin (for representative staining patterns see Figure 6A). Patients were divided into two groups that were expressing TLR7 or not on malignant cells. The expression of E-cadherin and vimentin by tumor cells was determined in a semiquantitative fashion. In TLR7hi patients, the expression of E-cadherin was significantly lower (25.6%± 3.7% of cancer cells expressing E-Cadherin) than in TLR7low patients (49.3%± 7% of tumor cells expressing E-cadherin) (Figure 6B). Concomitantly, in TLR7hi patients, the expression of vimentin was higher (34.6%± 6.7% of tumor cells expressed vimentin), than in TLR7low patients (15.3%± 3.6% of tumor cells expressed vimentin) (Figure 6C). In conclusion, in patients with lung adenocarcinoma, high expression of TLR7 on cancer cells is associated with signs of EMT.
Figure 6.
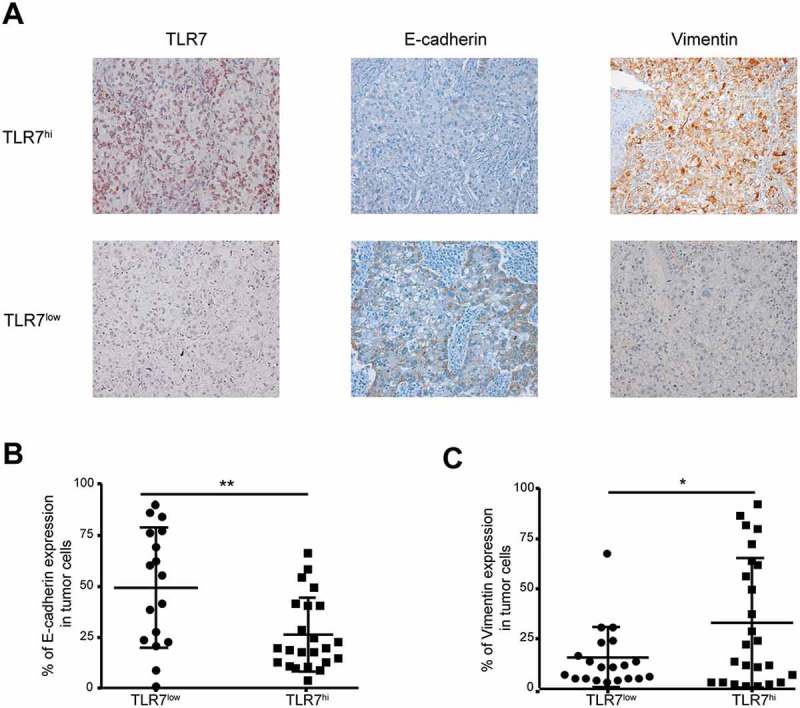
Strong TLR7 expression by malignant cells is link to signs of EMT.
Representative immunohistochemistry staining of TLR7, E-cadherin and vimentin on malignant cells of adenocarcinoma patients. Images were taken at x20 magnification (A). Percentage of malignant cells that express E-cadherin (B) and vimentin (C) in TLR7hi and TLR7low patients (n = 43). *: p < 0,05, **: p < 0,01, ***: p < 0,001. Data are mean ± SEM.
We then determined the physiopathology impact of TLR7 expression on the metastasis process in a large cohort of adenocarcinoma patients. We first determined the prognostic value of TLR7 on a cohort of 154 adenocarcinoma patients (sup Table 1), the malignant cells of whom displayed TLR7 either at a rather low level (< 5%) or were almost completely positive (> 82%, cut off determined in 11). We found that high TLR7 expression by tumor cells was associated with poor clinical outcome compared to low TLR7 expression, with a median OS of 45 months, compared to 90 months, respectively (Figure 7A). Multivariate analysis revealed TLR7 expression as an independent prognostic marker (Table 1). We then selected twelve patients from this cohort, including six patients having low (< 5%, TLR7low) and six having high (100%, TLR7hi) TLR7 expression, and performed gene expression analysis using nanostring technology and the pan cancer progression panel. Volcano plot analysis revealed that among 770 genes analyzed, six were significantly overexpressed and five were underexpressed in TLR7hi compared to TLR7low patients (Figure 7B). Unsupervised clustering of differentially expressed genes, revealed a small cluster of three genes including ICAM1, keratin 7 (KRT-7) and keratin 19 (KRT-19) that were overexpressed in TLR7hi patients. Additionally, NME1 (encoding nucleoside diphosphate kinase 1) and syndecan 4 (SDC4) (heparin sulfate proteoglycan molecule) were found overexpressed in the TLR7hi group of patients (Figure 7C). More detailed box-plot analysis of these genes confirmed their overexpression in TLR7hi patients (Figure 7D). Interestingly, TP53 was also found highly expressed in TLR7hi patients (Figure 7D). We then searched for correlation between the level of gene expression and survival of adenocarcinoma lung cancer patients, using the public software KM plotter 13. High expression of KRT-7 and KRT-19, as well as that of NME1 and TP53, significantly correlated with poor clinical outcome (Figure 7E).
Figure 7.
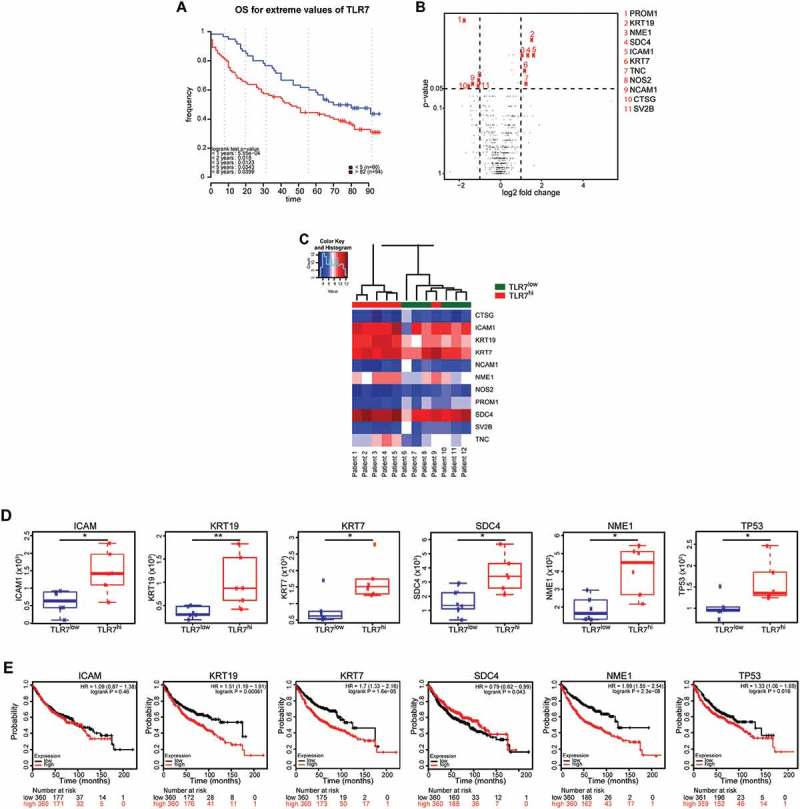
High expression of TLR7 by malignant adenocarcinoma cells is associated with metastatic gene expression signature.
Kaplan-Meier survival curve for overall survival (OS) for the 154 lung adenocarcinoma patients, divided in two groups according to the stratification of TLR7 expression. Optimal cutoff of 82% was previously determined 11. Patients are divided into TLR7hi (> 82% expression of TLR7) and TLR7low group (< 5% TLR7 expression). p-value was determined by log-rank test (A). Gene expression analysis was performed in 12 adenocarcinoma patients including 6 TLR7hi and 6 TLR7low, using nanostring technology. Volcano plot comparison of differentially expressed genes between TLR7hi and TLR7low group of patients. The horizontal dimension is the variation of median gene expression (log2 (ratio)) between the 2 groups, and the vertical axis represents the Mann-Whitney test p-value between TLR7hi and TLR7low patients. In red are represented genes that are over-or under-expressed in TLR7hi patients, with a fold-change superior to 2 (11 genes among 770 studied) (B). Hierarchical clustering of the 11 differentially expressed genes in TLR7hi compared to TLR7low groups. Individual patients are oriented in columns and expression level for each gene is oriented in rows (C). Box plot representation of ICAM1, KRT-19, KRT-7, SCD4, NME1, and P53 gene expression in TLR7hi and in TLR7low patients (D). Kaplan-Meier survival curve for overall survival (OS) for adenocarcinoma patients divided in two groups according to the stratification of ICAM1, KRT-19, KRT-7, SCD4, NME1, and P53 gene expression, using online survival analysis software KM plotter. p value was determined by log-rank test (E).
Table 1.
Univariate and multivariate analysis for 154 patients with lung adenocarcinoma not treated with neoadjuvant chemotherapy.
| Variable | P | HR (95% CI) |
|---|---|---|
| Univariate analysis | ||
| TLR7 (> 82% vs < 5%) | 0,027 | 1.56 (1.1 – 2.3) |
| TLR7 (medium vs < 5%) | 0,49 | 1.15 (0.78 – 1.7) |
| Gender (male vs female) | 0,17 | 1.19 (0.93 – 1.5) |
| Smoking status (smoker vs non smoker) | 0,72 | 1.05 (0.79 – 1.4) |
| Pathologic stage | ||
| IB vs IA | 0,11 | 1.37 (0.93 – 2) |
| IIA vs IA | 0,42 | 1.22 (0.76 – 2) |
| IIB vs IA | 0,0078 | 1.81 (1.2 – 2.8) |
| IIIA vs IA | < 0.0001 | 2.86 (2 – 4.1) |
| IIIB vs IA | 0,00012 | 3.42 (1.8 – 6.4) |
| IV vs IA | 0,0045 | 2.74 (1.4 – 5.5) |
| Multivariate analysis | ||
| TLR7 (> 82% vs < 5%) | 0,02 | 1.62 (1.1 – 2.4) |
| TLR7 (medium vs < 5%) | 0,6 | 1.11 (0.74 – 1.7) |
| Pathologic stage | ||
| IB vs IA | 0,42 | 1.21 (0.75 – 2) |
| IIA vs IA | 0,5 | 1.21 (0.69 – 2.1) |
| IIB vs IA | 0,022 | 1.93 (1.1 – 3.4) |
| IIIA vs IA | < 0.0001 | 2.60 (1.6 – 4.1) |
| IIIB vs IA | 0,011 | 1.99 (1.4 – 12) |
| IV vs IA | 0,062 | 6.92 (0.91 – 53) |
Collectively, these data indicate the existence of a gene signature that is associated to TLR7 expression by malignant cells as well as with poor prognosis.
Discussion
Our present data indicate that TLR7 stimulation induces pro-tumorigenic and pro-metastatic effects in a preclinical model of lung cancer while providing two salient insights. First, our data indicate that TLR7 must be expressed by the cancer cells (rather than by immune cells) so that the local presence of a TLR7 agonist can stimulate tumor growth. Second, we reveal the direct involvement of MDSCs in both the tumor growth-promoting and pro-metastatic effects of TLR7 stimulation. The presence of excess amount of a TLR7 agonist within the tumor causes an expansion of the number of MDSCs yet fails to enhance their immunosuppressive capacity. Our results also suggest that MDSC recruitment is mediated via a higher CCL2 and GM-CSF production in the tumor microenvironment after TLR7 stimulation. In addition, we demonstrated that high TLR7 expression by tumor cells from adenocarcinoma patients is associated to signs of EMT, as well as to a pro-metastatic gene expression signature. In particular, we observed a higher expression of KRT-7, KRT-19 and TP53, which are associated to the metastasis process and to a poor prognosis.
Previous studies have reported pro-tumorigenic effects for different TLRs, such as TLR2 14–18, TLR4 19–23, TLR5 24, TLR7 10,11,25 and TLR9 26–30. In prostate and breast cancer, the stimulation of TLR9 induced an over-expression of matrix metalloproteinase 13 as well as an increase of cell invasion 31,32. Finally, TLR4 stimulation increased tumor cells invasion in breast cancer 19 and in lung adenocarcinoma 33.
MDSC, a cell type with marked immunosuppressive properties 34, has been linked to tumor progression in various murine models of cancer 35–37. In cancer patients, the number of circulating MDSCs significantly increases compared to healthy subjects 38. Our study, however, is the first demonstration in a cancer model that TLR stimulation leads to local MDSCs expansion, accounting for enhanced tumor growth and metastasis.
TLR7 stimulation has previously mostly reported to stimulate anticancer immunosurveillance, for instance through direct stimulation of TLRs on antigens presenting cells, leading to their maturation 39,40. However, in recent years, TLR7 expression by tumor cells has been associated with a pro-tumoral effect in pancreatic adenocarcinoma 25 and in NSCLC 10,11. We found that stimulation of TLR7 expressed by adenocarcinoma cells modulated the immune infiltrate, leading to a significant expansion in MDSCs, associated with an increased secretion of CCL2 and GM-CSF in the tumor microenvironment. Both these cytokines are known to play a key role in the recruitment of MDSCs 38,41,42. Indeed, the absence of CCL2 production by cancer cells considerably reduces the recruitment of MDSCs into tumors 42.
Importantly, TLR7 stimulation had a pro-metastatic effect, while MDSC depletion drastically reduced this number of metastasis. In melanoma, MDSCs are associated with tumor progression, signs of EMT such as decreased E-Cadherin expression and increased migratory capacities of malignant cells 36. Similarly, MDSCs may contribute to metastatic dissemination of different types of cancer, including NSCLC 43. Finally, Srivastava’s team demonstrated that when MDSCs were depleted, the size of tumors decreased and cell migration was reduced 44.
In line with the involvement of EMT in the metastatic process, we found that TLR7 expression in lung adenocarcinoma was associated with EMT. We observed that a high expression of TLR7 was associated with low expression of E-cadherin and overexpression of vimentin. EMT is a process in which cells lose adhesive properties because of down-regulating E-cadherin expression and rearrange their cytoskeleton through enhanced vimentin expression 45. These changes in cancer cells play a critical role in metastasis development.
In adenocarcinoma patients, high TLR7 expression was associated with gene expression signature possibly linked to metastasis, with overexpression of KRT-7, KRT-19 and SDC4. These three genes have already been ascribed with pro-metastatic properties in different types of cancers. Indeed, KRT-7 is established as a diagnostic marker in a variety of malignancies including biliary duct, bladder, breast, cervix, kidney, liver, lung adenocarcinoma, ovary and pancreas cancer 46. In patients with lung adenocarcinoma, strong expression of KRT-7 was associated with a poor overall survival 47. Keratin 19 is a diagnostic marker in some tumors, such as in biliary duct, bladder, breast, cervix, colon, lung adenocarcinoma, ovary, pancreas, pleura, prostate, stomach and uterus cancer 46. In addition, KRT-19-positive hepatocellular carcinomas highly express invasion-related/metastasis-related markers 48. Finally, SDC4 plays a role in the development and metastatic occurrence in renal cell carcinoma 49.
Strong expression of TLR7 in patients with lung cancer is associated with poor survival prognosis but also with a pro-metastatic gene signature. As a result, it would be interesting to explore the molecular etiology of this pathogenic TLR7 overexpression. Ligation of TLR7 by agonists can stimulate TLR7 expression in a feedforward loop 50, which implies that high TLR7 abundance might be a sign of the natural stimulation of this receptor by ligands present in the tumor microenvironment. The molecular nature and source of such natural TLR7 agonists remains to be clarified. Moreover, it will be important to develop and characterize TLR7 antagonists or inhibitors of TLR7-elicited signals that might have immunostimulatory and anticancer effects.
Materials and methods
Cell lines
LLC-Luc cells (gift from Vincenzo di Bartolo, Institut Pasteur, Paris) were obtained from LLC cells (Lewis Lung Carcinoma, ATCC CRL-1642) that were stably transfected with firefly luciferase. LLC-Luc cells were cultured in DMEM+ GlutaMax (Gibco) with 10% heat-inactivated FCS, 1% penicilin/streptomycin mixture, and 0.5mg/ml hygromycin B. The cumulative culture length of the cells was fewer than 6 months after resuscitation. Early passage cells were used for all experiments and they were not reauthenticated.
Mice
Wild-type C57Bl/6J mice, mice deficient in TLR7 (B6.129S1-Tlr7tm1Flv/J strain), and NOD/SCID mice were purchased from Charles River Laboratories. All the animals were house kept at CEF (Centre d’Explorations Fonctionnelles of Centre de Recherche des Cordeliers, Agreement no. A75-06–12). They were maintained in a constant temperature and humidity in light controlled room with a 12 hours light cycle. They had free access to food (SAFE Laboratory) and tap water. All experiments were conducted in 6–8 week-old female mice in accordance with the institutional guidelines and the recommendations for the care and use of laboratory animals put forward by the Directive 2010/63/EU revising Directive 86/609/EEC on the protection of animals used for scientific purposes (project has been approved by a user establishment’s ethics committee and the Project Authorization: number Ce5/2010/057 and 6629).
Tumor growth and metastasis
Cancer cells (1 × 106 in 100μl PBS/injection) were injected into the flank of recipient mice. Mice then received intra-tumor injections of LPS (40 μg/injection), CL264 (40 μg/injection) or CpG (30 μg/injection) (Invivogen) at day 0, 3 and 6 after tumor grafting. Tumor volume (length (l), breath (b), depth (d)) was measured every 2–3 days using caliper, and was calculated taking into account the three values according to the ellipsoid volume calculation formula: tumor volume = 4/3 × π × l/2 × b/2 = 0.5236 × l × b × d. For metastasis detection, mice were injected (i.p.) with 225mg/kg body weight D-luciférine K+ (Interchim), then were anaesthetized using isofluran, 10 minutes before imaging for bioluminescence using an IVIS imaging system (Lumina II). Mice were sacrificed between days 22 and 24. The rib cage was excised and 1 ml of India ink (7.5% solution) was injected into the trachea. The lungs were harvested and washed with 1X PBS. Separation of lobes and connectives tissues elimination was performed. The lungs were immersed and stored in 1ml of Feket solution (300ml of 70% ethanol, 30ml of 37% formaldehyde and 5 ml of glacial acetic acid). The number of metastatic foci was counted.
Mdscs depletion
When tumors were palpable, mice were injected (i.p.) with anti-Gr1 (clone RB6-8C5, BioXCell) or control antibody (clone LTF-2, BioXCell) (200µg/injection, in 100µl PBS) every 48 hours. MDSC depletion was confirmed in the peripheral blood of recipient mice. Blood sample was recovered in heparin containing tubes. After red blood cell lysis by ACK, nucleated cells were incubated for 20 minutes at 4°C with blocking antibody (24G2, 1mg/ml, 1/100). Cells were stained for 30 minutes at 4°C with the following antibodies: PECy7 anti-CD45 (Clone 30-F11, BD Pharmingen, 1/800), eF450 anti-CD11b (Clone M1/70, eBioscience, 1/100), PerCPCy5.5 anti-Gr1 (Clone RB6-8C5, BD Pharmingen, 1/100), PE anti-CD11c (Clone N418, eBioscience, 1/100) and LIVE/DEAD® Fixable Yellow Dead Cell Stain Kit (Life Technologies, L34959, 1/100). Staining was assessed with a FACS Fortessa cytometer, and flow cytometry data were analyzed using FACSDiva (BD Biosciences) or FlowJO (Tree Star) software.
Preparation of single cell suspensions and flow cytometry analyzes
Mice were sacrificed 10 days after cancer cell injection, and tumors and spleens were excised, mechanically dissociated, and incubated at 37°C for 30 minutes with DNase I (1mg/ml, Roche) and liberase TM (2,5mg/ml, Roche). Resulting single-cell suspensions were filtered through 70µm cell strainers (BD), and were incubated for 20 minutes with blocking antibody (24G2, 1mg/ml, 1/100). Cells were then stained for 30 minutes with LIVE/DEAD® Fixable Yellow Dead Cell Stain Kit (Life Technologies, L34959) and the following antibodies: PECy7 anti-CD45 (clone 30-F11, BD Pharmingen, 1/800), APC-eF780 anti-CD3 (clone UCHT1, eBioscience, 1/100), PETR anti-CD4 (clone GK1.5, Caltag, 1/100), AF700 anti-CD8 (clone 53–6.7, eBioscience1/100), PE anti-CD19 (clone 1D3, BD Pharmingen, 1/100), BV605 anti-NK1.1 (clone PK136, Biolegend, 1/50), FITC anti-CD11b (clone M1/70, BD Pharmingen, 1/100), eF450 anti-CD11b (clone M1/70, eBioscience, 1/100), BV786 anti-CD11c (clone HL3, BD Pharmingen, 1/50), BV605 anti-Gr1 (clone RB6-5C8, BD Pharmingen, 1/100), AF700 anti-F4/80 (clone CI:A3-1, AbD serotec). For the FoxP3 and TLR7 labeling, the cells were first incubated with fixation/permabilisation reagent (eBiosciences), as recommended by the manufacturer and then stained during 30 minutes with AF647 anti-FoxP3 antibody (Biolegend) or PE anti-TLR7 antibody (BD Pharmingen).
Staining was assessed with a FACS Fortessa cytometer, and flow cytometry data were analyzed using FACSDiva or FlowJO software.
Cytokine detection by ELISA
Mice were sacrificed 10 days after cancer cell injection and tumors were excised, cut into pieces of 1 mm3 and placed in RPMI medium supplemented with 1% penicilin/streptomycin and 1% Glutamine for 24h at 37°C. Supernatants were then collected and stored at −80°C.
100000 LL/2 cells were cultured in a 12 well plate for 12 or 24 hours along with TLR7 agonist (2.5 µg/mL CL264), supernatants were collected and stored at −80°C.
Cytokine production was quantified by using GM-CSF ELISA ready-SET-Go! (88–7334-22), mouse CCL2 (MCP-1) ELISA ready-SET-Go! (88–7391-22) and mouse IL-6 ELISA ready-SET-Go! (88–7064-22) from eBiosciences, using the protocols provided by the manufacturer.
Mdscs sorting and biological activity
Mice were sacrificed 10 days after cancer cell injection and tumors were excised. Single cell suspension was prepared and incubated in blocking solution (24G2, 1mg/ml, 1/100), during 20 minutes, then stained for 30 minutes at 4°C with PECy7 anti-CD45, eF450 anti-CD11b, PerCPCy5.5 anti-Gr1, and LIVE/DEAD® Fixable Yellow Dead Cell Stain Kit. After cell sorting using a FACS Aria cytometer, sorted cells were centrifuged and cultured overnight in RPMI medium with 10% FBS, 2mM L-glutanime, 50U/ml penicilin, 50µg/ml streptomycine, 5% NaPyruvate, 0.1% β-mercapto-ethanol and 10% HEPES. Supernatants were used to perform Nitric oxide assay, (kit KGE001, R&D Systems), H2O2 assay (Kit A22188, Invitrogen, Molecular Probes), and ROS assay (Cell ROX Oxidative Stress Reagent, ThermoFisher Scientific), using protocols provided by the manufacturers.
T cells were purified from spleens using the Pan T cell Isolation Kit II (Miltenyi Biotech). Following centrifugation, purified T cells were labeled with CFSE (106 cells/ml in 5µM CFSE PBS BSA 0.1% solution for 10 minutes, CellTrace™ CFSE Cell Proliferation Kit, Invitrogen). The reaction was terminated by the addition of 10 volumes of cold RPMI 1640 with 10% FBS. Labeled cells were then incubated with anti-biotin MACSiBead particles coupled with CD3ϵ-Biotin and CD28-Biotin (T Cell Activation/Expansion Kit, mouse, Miltenyi) for 72 h, in the presence or not or sorted Gr1+ CD11b+ cells. T cell proliferation was determined by flow cytometry.
Patients
A retrospective series of 154 lung adenocarcinoma stage I-III patients who underwent primary surgery (without neoadjuvant chemotherapy) and who were operated between 2001 and 2005 was obtained from Hotel-Dieu hospital (Paris, France). Lung tumor samples were analyzed with the agreement of the French ethic committee (agreements 2008–133 and 201206–12) in accordance with article L.1121–1 of French law.
Immunohistochemistry
Human tumor samples were fixed in neutral-buffered 10% formalin solution and paraffin embedded. TLR7 staining and quantification was performed as previously described 11 using TLR7-specific polyclonal antibody (ENZO Lifesciences) at 10 mg/mL.
The expression of E-cadherin (Dako M3613, 1/100 dilution) and vimentin (Cells Signaling 5741, 1/100) by tumor cells was performed as follows. Serial 5-μm tissue sections were deparaffinized, rehydrated, and pretreated in 10 mM citrate buffer, pH 6, for antigen retrieval. Sections were incubated with hydrogen peroxide for 15 minutes, then blocked in 5% human serum for 30 minutes at room temperature. The slides were then incubated with a primary antibody (diluted in Dako real solution, antibody diluent) anti-E-cadherin (Dako M3613, 1/100 dilution) for one hour at room temperature or anti-vimentin (Cells Signaling 5741, 1/100) overnight. Slides were then incubated for 30 minutes at room temperature with the secondary antibody (anti-rabbit coupled to biotin, 1/200, JIR 715–066-150), followed by 30 minutes incubation with streptavidin-HRP (Dako, 1/300). After each incubation, the slides were washed 5 minutes with 1x TBS+ 0.04% Tween. Revelation was performed with the DAB kit (Dako, K3468) and stopped by placing slides in 1X TBS and distillated water. Counter coloration was performed with hematoxylin.
Gene expression analysis
RNA extraction was performed on LCC-Luc cells stimulated or not with 2.5 µg/mL CL264 using the Maxwell®16 LEV simplyRNA cells kit manufacturer procedure. 200 µg of RNA were used for RT-PCR with High-Capacity cDNA Reverse Transcription Kit with RNase Inhibitor kit from (Applied Biosystems) and the pPCR was performed using Taqman probes (FAM- Applied Biosystems: E-Cadherin (Mm01247357_m1), N-Cadherin (Mm01162497_m1), Keratin 19 (Mm00492980_m1), Keratin 7 (Mm00466676_m1), Vimentin (Mm01333430_m1), NME 1(Mm01612215_m1), snail 1 (Mm00441533_g1), snail 2(Mm00441531_m1), twist1 (Mm00442036_m1) and the housekeeping gene, GAPDH (Mm99999915_g1).
Twenty µm of FFPE sections were obtained from paraffin blocks. Deparaffination was performed by adding 1 mL 100% clearene for 5 min at room temperature. After centrifugation the pellet was washed twice with 1 mL 100% ethanol. Protease digestion was performed by adding digestion buffer and protease form RecoverAll™ Total Nucleic Acid Isolation Kit for FFPE (AM1975), then samples were incubated 3h at 50°C and for 15 min at 80°C for RNA isolation. Gene expression analysis was performed on 100ng RNA, using nCounter XT gene expression assays from nanosting (pancancer progression panel), following the recommendations of the manufacturer.
CRISPR/CAS9 mediated tlr7 gene editing
LLC-luc cells were transiently transfected with a plasmid coding for Tlr7-targeting gRNA and Cas9 protein fused to a red fluorescent protein (RFP) (Sigma Aldrich, St. Louis, MO, US) by means of lipofectamin 2000 (Thermo Fisher scientific, Waltham, MA, US) according to the manufacturer recommendation. The following day, RFP expressing cells were selected by single cell sort using a BD FACS Aria Fusion (Becton Dickinson, Franklin Lakes, NJ, US). Following TLR7-deficient clones were then selected.
Statistical analysis
Statistical analysis for in vivo experiments was performed using GraphPad Prism 5 (GraphPad Software) and using the R software (RFoundation for Statistical Computing; www.r-project.org) and the package survival. Significance threshold for p values was set to 0.05. All mice experiments were analyzed using two-way ANOVA method. Post hoc analyses with Bonferroni corrections were applied for pair-wise comparisons of tumor growth over time in different groups of mice. Tumor volumes at the endpoint of each experiment were compared using a nonparametric Mann–Whitney test with Bonferroni corrections for pair-wise comparisons. Mann–Whitney tests were also used to compare percentages and numbers of immune cells infiltrating the tumors in mice. Overall survival (OS) curves were estimated by the Kaplan – Meier method and compared by the log-rank test. The OS was defined from the date of the surgery until the date of death or the last day of the patient’s visit to the hospital. Unsupervised hierarchical clustering was performed with Euclidian distance and Ward linkage criterion. Comparisons of the expression of vimentin and E-cadherin in TLR7hi and TLR7low patients were performed using a Mann–Whitney test.
Funding Statement
This work was supported by the “Institut National de la Santé et de la Recherche Médicale” (INSERM), Sorbonne Universite, Universite Paris Descartes, the Cancer Research for Personalized Medicine (CARPEM), the LabEx Immuno-Oncology, and the Institut National du Cancer (2016-1-PLBIO-09-INSERM 6-1).
Abbreviations
- DAMP
damaged-associated molecular pattern
- EMT
epithelial–mesenchymal transition
- ICAM-1
intercellular adhesion molecule 1
- KRT
keratin
- NSCLC
non-small cell lung cancer
- MDSC
myeloid derived suppressor cells
- PAMP
pathogen-associated molecular patterns
- ssRNA
single-stranded RNA
- SDC4
syndecan 4
- TLR7
toll-like receptor 7
Technical support
We are most grateful for excellent technical assistance of the Centre d’’Explorations Fonctionnelles (CEF) crew for their support with the mice. Cell sorting was performed at CICC (Centre d’Imagerie Cellulaire et de Cytometrie, Centre de Recherche des Cordeliers UMR S 1138, Paris, France). CICC is a member of the UPMC Cell Imaging & Flow Cytometry (LUMIC) network and the UPD cell imaging network.
Supplemental Material
Supplemental data for this article can be accessed here.
Notes on contributions
MD, KI, SM, MG and HO performed experiments; FP performed statistical analysis; VN and VDB provided LLC-luciferase expressing cells; OK and GK performed CRISPR/cas9 TLR7 deletion on LLC-luc cells; AL and DD were responsible for clinical data; DD and MA were responsible for pathological data; IC, MD, KI and PEJ interpreted the data; IC designed and supervised the study; MD, KI, GK and IC wrote the manuscript.
References
- 1.Fridman WH, Pagès F, C S-F, Galon J.. The immune contexture in human tumours: impact on clinical outcome. Nat Rev Cancer. 2012; 12(4): 298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 2.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144(5): 646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 3.Newton K, Dixit VM. Signaling in innate immunity and inflammation. Cold Spring Harb Perspect Biol. 2012; 4(3): doi: 10.1101/cshperspect.a006049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basith S, Manavalan B, Lee G, Kim SG, Choi S. Toll-like receptor modulators: a patent review (2006-2010). Expert Opin Ther Pat. 2011; 21(6): 927–944. doi: 10.1517/13543776.2011.569494. [DOI] [PubMed] [Google Scholar]
- 5.Dajon M, Iribarren K, Cremer I. Toll-like receptor stimulation in cancer: A pro- and anti-tumor double-edged sword. Immunobiology. 2017; 222(1): 89–100. doi: 10.1016/j.imbio.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 6.Dajon M, Iribarren K, Cremer I. Dual roles of TLR7 in the lung cancer microenvironment. Oncoimmunology. 2015; 4(3): e991615. doi: 10.4161/2162402X.2014.991615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dranoff G. Coordinated tumor immunity. J Clin Invest. 2003; 111(8): 1116–1118. doi: 10.1172/JCI18359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hornung V, Rothenfusser S, Britsch S, Krug A, Jahrsdörfer B, Giese T, Endres S, Hartmann G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J Immunol Baltim Md. 1950 2002; 168(9): 4531–4537. [DOI] [PubMed] [Google Scholar]
- 9.Barr TA, Brown S, Ryan G, Zhao J, Gray D. TLR-mediated stimulation of APC: distinct cytokine responses of B cells and dendritic cells. Eur J Immunol. 2007; 37(11): 3040–3053. doi: 10.1002/eji.200636483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cherfils-Vicini J, Platonova S, Gillard M, Laurans L, Validire P, Caliandro R, Magdeleinat P, Mami-Chouaib F, M-C D-N, Fridman W-H, et al. Triggering of TLR7 and TLR8 expressed by human lung cancer cells induces cell survival and chemoresistance. J Clin Invest. 2010; 120(4): 1285–1297. doi: 10.1172/JCI36551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chatterjee S, Crozet L, Damotte D, Iribarren K, Schramm C, Alifano M, Lupo A, Cherfils-Vicini J, Goc J, Katsahian S, et al. TLR7 promotes tumor progression, chemotherapy resistance, and poor clinical outcomes in non-small cell lung cancer. Cancer Res. 2014; 74(18): 5008–5018. doi: 10.1158/0008-5472.CAN-13-2698. [DOI] [PubMed] [Google Scholar]
- 12.Kitamura T, Qian B-Z, Pollard JW. Immune cell promotion of metastasis. Nat Rev Immunol. 2015; 15(2): 73–86. doi: 10.1038/nri3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Győrffy B, Surowiak P, Budczies J, Lánczky A. Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PloS One. 2013; 8(12): e82241. doi: 10.1371/journal.pone.0082241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Farnebo L, Shahangian A, Lee Y, Shin JH, Scheeren FA, Sunwoo JB. Targeting Toll-like receptor 2 inhibits growth of head and neck squamous cell carcinoma. Oncotarget. 2015; 6(12): 9897–9907. doi: 10.18632/oncotarget.v6i12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang B, Zhao J, Shen S, Li H, He K-L, Shen G-X, Mayer L, Unkeless J, Li D, Yuan Y, et al. Listeria monocytogenes promotes tumor growth via tumor cell toll-like receptor 2 signaling. Cancer Res. 2007; 67(9): 4346–4352. doi: 10.1158/0008-5472.CAN-06-4067. [DOI] [PubMed] [Google Scholar]
- 16.Tye H, Kennedy CL, Najdovska M, McLeod L, McCormack W, Hughes N, Dev A, Sievert W, Ooi CH, Ishikawa T, et al. STAT3-driven upregulation of TLR2 promotes gastric tumorigenesis independent of tumor inflammation. Cancer Cell. 2012; 22(4): 466–478. doi: 10.1016/j.ccr.2012.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Scheeren FA, Kuo AH, Van Weele LJ, Cai S, Glykofridis I, Sikandar SS, Zabala M, Qian D, Lam JS, Johnston D, et al. A cell-intrinsic role for TLR2-MYD88 in intestinal and breast epithelia and oncogenesis. Nat Cell Biol. 2014; 16(12): 1238–1248. doi: 10.1038/ncb3058. [DOI] [PubMed] [Google Scholar]
- 18.Maruyama A, Shime H, Takeda Y, Azuma M, Matsumoto M, Pam2 ST. lipopeptides systemically increase myeloid-derived suppressor cells through TLR2 signaling. Biochem Biophys Res Commun. 2015; 457(3): 445–450. doi: 10.1016/j.bbrc.2015.01.011. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Wang B, Wang T, Xu L, He C, Wen H, Yan J, Su H, Zhu X. Toll-like receptor 4 prompts human breast cancer cells invasiveness via lipopolysaccharide stimulation and is overexpressed in patients with lymph node metastasis. PloS One. 2014; 9(10): e109980. doi: 10.1371/journal.pone.0109980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczepanski MJ, Czystowska M, Szajnik M, Harasymczuk M, Boyiadzis M, Kruk-Zagajewska A, Szyfter W, Zeromski J, Whiteside TL. Triggering of Toll-like receptor 4 expressed on human head and neck squamous cell carcinoma promotes tumor development and protects the tumor from immune attack. Cancer Res. 2009; 69(7): 3105–3113. doi: 10.1158/0008-5472.CAN-08-3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang B, Zhao J, Li H, He K-L, Chen Y, Chen S-H, Mayer L, Unkeless JC, Xiong H. Toll-like receptors on tumor cells facilitate evasion of immune surveillance. Cancer Res. 2005; 65(12): 5009–5014. doi: 10.1158/0008-5472.CAN-05-0784. [DOI] [PubMed] [Google Scholar]
- 22.Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, et al. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: role in proliferation and apoptosis in the intestine. Gastroenterology. 2006; 131(3): 862–877. doi: 10.1053/j.gastro.2006.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dapito DH, Mencin A, Gwak G-Y, Pradere J-P, Jang M-K, Mederacke I, Caviglia JM, Khiabanian H, Adeyemi A, Bataller R, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell. 2012; 21(4): 504–516. doi: 10.1016/j.ccr.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rutkowski MR, Stephen TL, Svoronos N, Allegrezza MJ, Tesone AJ, Perales-Puchalt A, Brencicova E, Escovar-Fadul X, Nguyen JM, Cadungog MG, et al. Microbially driven TLR5-dependent signaling governs distal malignant progression through tumor-promoting inflammation. Cancer Cell. 2015; 27(1): 27–40. doi: 10.1016/j.ccell.2014.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ochi A, Graffeo CS, Zambirinis CP, Rehman A, Hackman M, Fallon N, Barilla RM, Henning JR, Jamal M, Rao R, et al. Toll-like receptor 7 regulates pancreatic carcinogenesis in mice and humans. J Clin Invest. 2012; 122(11): 4118–4129. doi: 10.1172/JCI63606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jego G, Bataille R, Geffroy-Luseau A, Descamps G, Pellat-Deceunynck C. Pathogen-associated molecular patterns are growth and survival factors for human myeloma cells through Toll-like receptors. Leukemia. 2006; 20(6): 1130–1137. doi: 10.1038/sj.leu.2404226. [DOI] [PubMed] [Google Scholar]
- 27.Moreira D, Zhang Q, Hossain DMS, Nechaev S, Li H, Kowolik CM, D’Apuzzo M, Forman S, Jones J, Pal SK, et al. TLR9 signaling through NF-κB/RELA and STAT3 promotes tumor-propagating potential of prostate cancer cells. Oncotarget. 2015; 6(19): 17302–17313. doi: 10.18632/oncotarget.4029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zambirinis CP, Levie E, Nguy S, Avanzi A, Barilla R, Xu Y, Seifert L, Daley D, Greco SH, Deutsch M, et al. TLR9 ligation in pancreatic stellate cells promotes tumorigenesis. J Exp Med. 2015; 212(12): 2077–2094. doi: 10.1084/jem.20142162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Belmont L, Rabbe N, Antoine M, Cathelin D, Guignabert C, Kurie J, Cadranel J, Wislez M. Expression of TLR9 in tumor-infiltrating mononuclear cells enhances angiogenesis and is associated with a worse survival in lung cancer. Int J Cancer. 2014; 134(4): 765–777. doi: 10.1002/ijc.28413. [DOI] [PubMed] [Google Scholar]
- 30.Liu Y, Yan W, Tohme S, Chen M, Fu Y, Tian D, Lotze M, Tang D, Tsung A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J Hepatol. 2015; 63(1): 114–121. doi: 10.1016/j.jhep.2015.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ilvesaro JM, Merrell MA, Swain TM, Davidson J, Zayzafoon M, Harris KW, Selander KS. Toll like receptor-9 agonists stimulate prostate cancer invasion in vitro. Prostate. 2007; 67(7): 774–781. doi: 10.1002/pros.20562. [DOI] [PubMed] [Google Scholar]
- 32.Merrell MA, Ilvesaro JM, Lehtonen N, Sorsa T, Gehrs B, Rosenthal E, Chen D, Shackley B, Harris KW, Selander KS. Toll-like receptor 9 agonists promote cellular invasion by increasing matrix metalloproteinase activity. Mol Cancer Res MCR. 2006; 4(7): 437–447. doi: 10.1158/1541-7786.MCR-06-0007. [DOI] [PubMed] [Google Scholar]
- 33.Chow SC, Gowing SD, Cools-Lartigue JJ, Chen CB, Berube J, Yoon H-W, Chan CHF, Rousseau MC, Bourdeau F, Giannias B, et al. Gram negative bacteria increase non-small cell lung cancer metastasis via Toll-like receptor 4 activation and mitogen-activated protein kinase phosphorylation. Int J Cancer J Int Cancer. 2015; 136(6): 1341–1350. doi: 10.1002/ijc.29111. [DOI] [PubMed] [Google Scholar]
- 34.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012; 12(4): 253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu J, Wang H, Yu Q, Zheng S, Jiang Y, Liu Y, Yuan G, Qiu L. Aberrant frequency of IL-10-producing B cells and its association with Treg and MDSC cells in Non Small Cell Lung Carcinoma patients. Hum Immunol. 2016; 77(1): 84–89. doi: 10.1016/j.humimm.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 36.Toh B, Wang X, Keeble J, Sim WJ, Khoo K, Wong W-C, Kato M, Prevost-Blondel A, Thiery J-P, Abastado J-P. Mesenchymal transition and dissemination of cancer cells is driven by myeloid-derived suppressor cells infiltrating the primary tumor. PLoS Biol. 2011; 9(9): e1001162. doi: 10.1371/journal.pbio.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Feng P-H, Lee K-Y, Chang Y-L, Chan Y-F, Kuo L-W, Lin T-Y, Chung F-T, Kuo C-S, Yu C-T, Lin S-M, et al. CD14(+)S100A9(+) monocytic myeloid-derived suppressor cells and their clinical relevance in non-small cell lung cancer. Am J Respir Crit Care Med. 2012; 186(10): 1025–1036. doi: 10.1164/rccm.201204-0636OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Filipazzi P, Valenti R, Huber V, Pilla L, Canese P, Iero M, Castelli C, Mariani L, Parmiani G, Rivoltini L. Identification of a new subset of myeloid suppressor cells in peripheral blood of melanoma patients with modulation by a granulocyte-macrophage colony-stimulation factor-based antitumor vaccine. J Clin Oncol Off J Am Soc Clin Oncol. 2007; 25(18): 2546–2553. doi: 10.1200/JCO.2006.08.5829. [DOI] [PubMed] [Google Scholar]
- 39.Le Mercier I, Poujol D, Sanlaville A, Sisirak V, Gobert M, Durand I, Dubois B, Treilleux I, Marvel J, Vlach J, et al. Tumor promotion by intratumoral plasmacytoid dendritic cells is reversed by TLR7 ligand treatment. Cancer Res. 2013; 73(15): 4629–4640. doi: 10.1158/0008-5472.CAN-12-3058. [DOI] [PubMed] [Google Scholar]
- 40.Aspord C, Tramcourt L, Leloup C, Molens J-P, Leccia M-T, Charles J, Plumas J. Imiquimod inhibits melanoma development by promoting pDC cytotoxic functions and impeding tumor vascularization. J Invest Dermatol. 2014; 134(10): 2551–2561. doi: 10.1038/jid.2014.194. [DOI] [PubMed] [Google Scholar]
- 41.Serafini P, Carbley R, Noonan KA, Tan G, Bronte V, Borrello I. High-dose granulocyte-macrophage colony-stimulating factor-producing vaccines impair the immune response through the recruitment of myeloid suppressor cells. Cancer Res. 2004; 64(17): 6337–6343. doi: 10.1158/0008-5472.CAN-04-0757. [DOI] [PubMed] [Google Scholar]
- 42.Chun E, Lavoie S, Michaud M, Gallini CA, Kim J, Soucy G, Odze R, Glickman JN, Garrett WS. CCL2 Promotes Colorectal Carcinogenesis by Enhancing Polymorphonuclear Myeloid-Derived Suppressor Cell Population and Function. Cell Rep. 2015; 12(2): 244–257. doi: 10.1016/j.celrep.2015.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang A, Zhang B, Wang B, Zhang F, Fan K-X, Guo Y-J. Increased CD14(+)HLA-DR (-/low) myeloid-derived suppressor cells correlate with extrathoracic metastasis and poor response to chemotherapy in non-small cell lung cancer patients. Cancer Immunol Immunother CII. 2013; 62(9): 1439–1451. doi: 10.1007/s00262-013-1450-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mk S, Zhu L, Harris-White M, Uk K, Kar U, Huang M, Mf J, Jm L, Elashoff D, Strieter R, et al. Myeloid suppressor cell depletion augments antitumor activity in lung cancer. PloS One. 2012; 7(7): e40677. doi: 10.1371/journal.pone.0040677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139(5): 871–890. doi: 10.1016/j.cell.2009.11.007. [DOI] [PubMed] [Google Scholar]
- 46.Karantza V. Keratins in health and cancer: more than mere epithelial cell markers. Oncogene. 2011; 30(2): 127–138. doi: 10.1038/onc.2010.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pøhl M, Olsen KE, Holst R, Donnem T, Busund L-T, Bremnes RM, Al-Saad S, Andersen S, Richardsen E, Ditzel HJ, et al. Keratin 34betaE12/keratin7 expression is a prognostic factor of cancer-specific and overall survival in patients with early stage non-small cell lung cancer. Acta Oncol Stockh Swed. 2016; 55(2): 167–177. doi: 10.3109/0284186X.2015.1049291. [DOI] [PubMed] [Google Scholar]
- 48.Kawai T, Yasuchika K, Ishii T, Katayama H, Yoshitoshi EY, Ogiso S, Kita S, Yasuda K, Fukumitsu K, Mizumoto M, et al. Keratin 19, a Cancer Stem Cell Marker in Human Hepatocellular Carcinoma. Clin Cancer Res Off J Am Assoc Cancer Res. 2015; 21(13): 3081–3091. doi: 10.1158/1078-0432.CCR-14-1936. [DOI] [PubMed] [Google Scholar]
- 49.Erdem M, Erdem S, Sanli O, Sak H, Kilicaslan I, Sahin F, Telci D. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. Urol Oncol. 2014; 32(1): 25.e13–20. doi: 10.1016/j.urolonc.2012.08.022. [DOI] [PubMed] [Google Scholar]
- 50.Miettinen M, Sareneva T, Julkunen I, Matikainen S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2001; 2(6): 349–355. doi: 10.1038/sj.gene.6363791. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.