

ABSTRACT
Immune checkpoint inhibitors have shown great potential in treating solid tumors, inducing durable remission and prolonged survival time in responders. Despite their promise, a large fraction of patients remains unresponsive to these treatments highlighting the need for biomarkers that can predict patient sensitivity. Pre-treatment gene expression profiles for patients receiving immune checkpoint inhibitors have recently become available, establishing a new medium by which to discover biomarkers that predict therapy response. In this study, we mined for transcriptomic correlates of response by applying immune cell-derived gene expression signatures to publicly available datasets containing matched gene expression and response efficacy information. These datasets were comprised of urothelial carcinoma patients receiving anti-PD-L1 (n = 25), melanoma patients receiving anti-PD-1 (n = 28), and melanoma patients receiving anti-CTLA-4 (n = 42). We identified one signature, derived from a subpopulation of B cells, with scores that were significantly and reproducibly elevated in patients experiencing clinical benefit following therapy targeting the PD-1/PD-L1 axis and were additionally elevated in patients responsive to anti-CTLA-4 therapy. Multivariate models revealed that this signature was associated with response independent of other response-predictive biomarkers, including tumor mutation burden. Functional annotation of the signature revealed it to be associated with features indicative of an immunologically active microenvironment, including B and T cell activation as well as antigen presentation activity. The preliminary findings presented detail a transcriptomic signature associated with response to multiple checkpoint inhibitors and suggest novel biological associations that warrant further investigation.
Keywords: Immunotherapy, genomics, tumor immunology, B cells, immune checkpoint blockade, gene signatures, biomarkers
Introduction
Immune checkpoint blockade therapy is a revolutionary cancer treatment modality that relies on the inhibition of tumor-mediated immunosuppressive mechanisms to stimulate an anti-cancer immune response. Monoclonal antibodies targeting the proteins programmed cell death protein 1 (PD-1), PD-1 ligand (PD-L1), and cytotoxic T-lymphocyte antigen-4 (CTLA-4) have now been approved as first-line cancer management options for a variety of tumor types, with treatment-sensitive patients showing reduced tumor burdens and prolonged survival times.1 Several studies have shown that anti-PD-1/PD-L1 and anti-CTLA-4 act through distinct mechanisms.2,3 CTLA-4 interferes in the early stages of T cell activation by inhibiting stimulatory signaling that is required following antigen-T cell receptor (TCR) binding. Conversely, PD-1 is involved later in T cell activation and acts by attenuating TCR signaling following engagement with its ligands, PD-L1 or PD-L2.2 Blocking these proteins has led to increased immune activity in the tumor microenvironments of responsive patients, resulting in long-term remission and clinical benefit.
Despite these early successes, the response rate to these therapies remains around 20–30% in unselected patient populations, creating a need for biomarkers that can predict clinical benefit.1,4,5 The T cell-centric mechanisms behind each immune checkpoint protein suggest that correlates of lymphocytic infiltration in the tumor microenvironment may better inform patient response.6 Early immune-related biomarkers that have been identified for anti-PD-1/anti-PD-L1 include CD8 + T cell density7 and intratumoral PD-L1 expression,8 while the expression of immune-related genes has been associated with response to anti-CTLA-4.9,10 In addition to markers of immune infiltration, a higher tumor mutation burden (TMB) has been associated with response to both therapies, implicating the neoantigen-driven immune reaction as a common factor involved in immune checkpoint blockade response.10–14 Interestingly, somatic copy number alterations (SCNAs) have been shown to be more strongly correlated with tumor immune activity and response to anti-CTLA-4 than TMB, with lower SCNA levels associated with higher immune activity and improved response rates.15 The pre-treatment transcriptome of patients receiving immune checkpoint inhibitors is one area that has yet to be fully explored. Recent studies have released transcriptomic data and identified expression-based correlates of response for small cohorts of patients receiving immune checkpoint blockade therapy.10,12,13 However, no studies have examined whether common and reproducible correlates exist that are associated with response to drugs targeting the PD-1/PD-L1 axis and CTLA-4.
In this study, we applied an expression-based, immune signature-driven framework16 to identify correlates of response to the immune checkpoint inhibitors anti-PD-L1, anti-PD-1, and anti-CTLA-4. This analysis encompassed three available datasets containing gene expression data matched to immune checkpoint response information. One signature in our analysis, derived from a series of B cell expression profiles, was reproducibly associated with response to therapies targeting the PD-1/PD-L1 axis and was additionally associated with anti-CTLA-4 response. These associations were highly significant in a meta-analysis pooling these datasets and were also independent of other known response-predictive biomarkers. Our results provide preliminary evidence of a transcriptomic signature that can be used to predict response to multiple immune checkpoint inhibitors.
Results
A memory B cell-like score is associated with response to checkpoint blockade therapies
We applied an expression-based method16 to identify immunological correlates of response to immune checkpoint blockade therapy. For this study, we used six immune cell gene expression signatures that had been validated to represent one of naive B cells, memory B cells, CD8 + T cells, CD4 + T cells, natural killer cells, or myeloid cells in peripheral blood mononuclear cell (PBMC) mixtures and correlate with the flow cytometry fraction of related cell types in NSCLC tumors.17 We began our study by applying this method to a discovery dataset that included gene expression and treatment response information for 21 pre-treatment urothelial tumor biopsies from patients receiving anti-PD-L1 therapy. Of the six immune cell signatures used by this method, only the memory B cell-like (MBL) signature produced scores that significantly differed between patients experiencing clinical benefit and those that did not (P = 0.01; Figure 1A, Supplementary Figure S1). These improved response rates also translated to post-treatment survival, where patients with MBL scores in the top quartile had significantly longer progression-free survival (median 398 days) compared to patients with scores in the bottom quartile (median 64 days, P = 0.02; Figure 1B). This trend mirrored findings in the TCGA bladder urothelial carcinoma dataset (P = 0.02; Supplementary Figure S2A).
Figure 1.
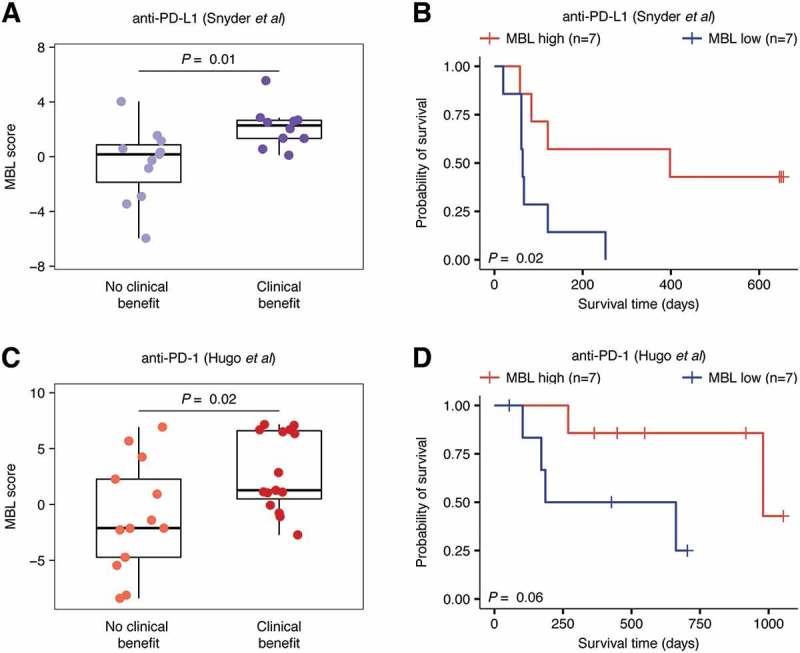
MBL score predicts clinical benefit and improved survival in patients receiving checkpoint inhibitors targeting the PD-1/PD-L1 axis. A, Distribution of MBL scores in patients that experienced no clinical benefit (n = 11) or clinical benefit (n = 10) in response to anti-PD-L1 therapy. B, Progression-free survival of patients with either high or low MBL scores in the Snyder et al anti-PD-L1 dataset. C, Distribution of MBL scores in patients that experienced no clinical benefit (n = 13) or clinical benefit (n = 15) to anti-PD-1 therapy. D, Overall survival of patients with either high or low MBL scores in the Hugo et al anti-PD-1 dataset. In figures A, and C, boxes span quartiles, with the lines representing the median score in each group. In figures B and D, high/low MBL designation was made using the top and bottom MBL quartiles in each dataset and vertical hash marks indicate censored data. P-values were calculated using the two-tailed Wilcoxon sum-rank test in A and C, and the log-rank test in B and D.
While the MBL signature was associated with response to anti-PD-L1 therapy, this analysis was performed in a small dataset where the findings could be due to chance. Thus, we next applied the MBL signature to a validation dataset consisting of 28 pre-treatment melanoma biopsies from patients receiving anti-PD-1 checkpoint blockade therapy.12 As was the case for the anti-PD-L1 dataset, patients experiencing clinical benefit in response to anti-PD-1 therapy had significantly higher MBL scores compared to patients that did not experience clinical benefit (P = 0.02; Figure 1C). Furthermore, patients with MBL scores in the top quartile had a median overall survival time of 548 days compared to 186 days for patients with MBL scores in the bottom quartile, a difference that trended toward significance (P = 0.06; Figure 1D). Unlike in urothelial carcinoma, the MBL score was not associated with prolonged survival in the metastatic samples of the TCGA melanoma dataset (P = 0.57; Supplementary Figure 2B).
Our success in the two datasets targeting the PD-1/PD-L1 axis led us to examine whether the MBL score could predict response to anti-CTLA-4, an immune checkpoint inhibitor that acts on a pathway distinct from PD-1. To test this, we applied our method to a dataset consisting of 42 pre-treatment melanoma biopsies from patients that received anti-CTLA-4 therapy.10 As in the PD-1/PD-L1 data, we observed significantly higher MBL scores in patients experiencing clinical benefit within 6 months (P = 0.05) and long-term survival with no clinical benefit (P = 0.05) relative to patients experiencing no clinical benefit following treatment (Figure 2A). Furthermore, high-MBL patients exhibited significantly prolonged overall survival compared to low-MBL patients, with high-MBL patients having a median overall survival time of 853 days compared to 160 days in low-MBL patients (P = 0.01; Figure 2B).
Figure 2.
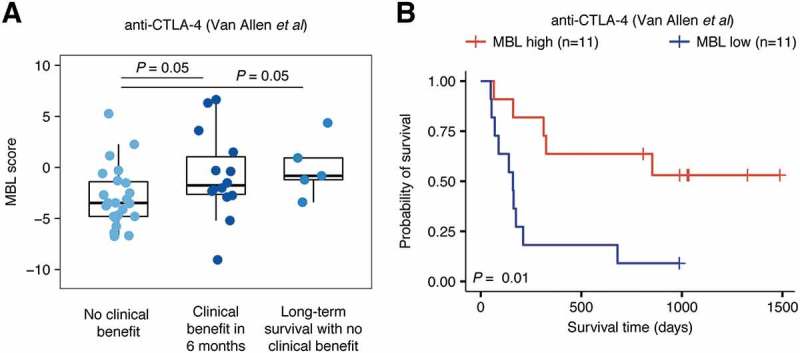
MBL score predicts clinical benefit and improved survival in patients receiving anti-CTLA-4 therapy. A, Distribution of MBL scores in patients that experienced no clinical benefit (n = 23), clinical benefit within 6 months (n = 14), and long-term survival with no clinical benefit (n = 5) from anti-CTLA-4 therapy. Boxes span quartiles, with the lines representing the median score in each group. P-values were calculated using the two-tailed Wilcoxon sum-rank test. B, Overall survival of patients with either high or low MBL scores. High/low MBL designation was made using the top and bottom MBL quartiles in each dataset and vertical hash marks indicate censored data. P-values were calculated using the log-rank test.
To provide context to these findings, we performed a series of analyses to compare the response and survival associations of the MBL signature to that of other genomic measurements of immune infiltration, including the five other immune signatures calculated using our method, a series of single cell RNAseq-derived immune signatures calculated using ssGSEA18, and the inferred immune cell fractions of 22 lymphocytes calculated using CIBERSORT.19 The MBL signature was the only signature that exhibited a significant association with response to immune checkpoint blockade therapy and post-treatment survival in each of the datasets tested (Supplementary Table S1). To increase the power of these analyses, we then pooled the datasets into two cohorts, one combining the anti-PD-1 and anti-PD-L1 datasets and one combining the datasets for all three immune checkpoint inhibitors (Supplementary Table S1). Compared to the other signatures, the MBL score exhibited the strongest associations with immune checkpoint blockade response in both the anti-PD-1/anti-PD-L1 cohort (meta-P = 6e-4) and the cohort containing all three datasets (meta-P = 2e-4; Figure 3A). This result was also reflected in the two-class survival analyses, where patients with MBL scores in the top quartile of each dataset exhibited significantly prolonged survival time in the combined anti-PD-1/anti-PD-L1 cohort (meta-P = 3e-3) and the combined checkpoint inhibitor cohort (meta-P = 6e-5; Figure 3B).
Figure 3.

Meta-analyses of the MBL signature’s association with response and survival in three immune checkpoint inhibitor datasets. Z-score absolute values indicating significance of the association between A, MBL score and clinical benefit in response to immune checkpoint inhibitor therapy using the two-tailed Wilcoxon sum-rank test and B, MBL score and either progression-free (anti-PD-L1) or overall survival (anti-PD-1, anti-CTLA-4) using the log-rank test. Light gray line indicates a z-score equal to 1.96 (P < 0.05). Z-scores from the PD-1 and PD-L1 cohort were combined using Stouffer’s method to get the PD-1/PD-L1 meta-z-score. Z-scores from all three datasets were combined using Stouffer’s method to get the meta-z-score. All p-values correspond to their respective z-scores.
The MBL score outperforms other response-associated biomarkers
Many factors have been shown to associate with response to immune checkpoint blockade therapy, including TMB and the level of immune checkpoint proteins in the tumor microenvironment.6 To rule out that the MBL signature was simply a proxy for other known markers of immunotherapy response, we first performed univariate logistic regression and Cox proportional hazards models to identify each biomarker’s dataset-specific association with response and survival. We then combined each variable, including our score, to predict treatment response and patient survival in a series of multivariate logistic regression and Cox proportional hazards models. Models from all three datasets included TMB, and expression of the genes encoding CTLA-4 (CTLA4), PD-1 (PDCD1), and PD-L1 (CD274) as covariates. Additionally, the anti-CTLA-4 dataset models included SCNA level which has been shown to associate with treatment response in a previous study.15 In both the anti-PD-L1 and anti-PD-1 datasets, the MBL score was the only biomarker predictive of response at the univariate level (Supplementary Table S2) and remained so even after adjusting for the covariates in the multivariate model (P = 0.04 and 0.03, respectively; Table 1). Similarly, in the anti-CTLA-4 dataset, the MBL score was the only variable predictive of response in the univariate model and remained predictive in the multivariate model (P = 0.02; Table 1). These results differed from the study that showed SCNA level was associated with response to anti-CTLA-4 therapy using many of the same samples.15 However, this study was performed on a larger cohort of samples for which genomic data was available (n = 110), making it more highly powered to observe these associations.
Table 1.
Multivariate regression models to predict clinical benefit and survival following immune checkpoint blockade therapy. Multivariate logistic and Cox proportional hazards regression models for the anti-PD-L1, anti-PD-1, and anti-CTLA-4 datasets. Logistic regression in the anti-PD-L1 and anti-PD-1 datasets classified patients experiencing clinical benefit or no clinical benefit. Logistic regression in anti-CTLA-4 dataset classified patients with no clinical benefit from those with either clinical benefit within 6 months or long-term survival with no clinical benefit. Sample sizes for the anti-PD-L1, anti-PD-1, and anti-CTLA-4 datasets vary between the logistic regression (n = 21, 28, and 40, respectively) and Cox proportional hazards models (n = 25, 28, and 40, respectively) depending on available variables, with some samples having survival data with no response data.
| Logistic regression |
Cox proportional hazards regression |
|||
|---|---|---|---|---|
| Covariate | Estimate (SE) | P-value | Hazard ratio (95% CI) | P-value |
| anti-PD-L1 | ||||
| Intercept | −3.89 (2.98) | 0.19 | ||
| MBL score | 1.12 (0.55) | 0.04 | 0.79 (0.66–0.94) | 0.01 |
| TMB | 0.00 (0.00) | 0.94 | 1.00 (1.00–1.00) | 0.85 |
| CTLA-4 | −5.72 (3.29) | 0.08 | 2.10 (0.41–10.79) | 0.38 |
| PD-1 | 1.95 (1.76) | 0.27 | 0.72 (0.27–1.91) | 0.51 |
| PD-L1 | 4.45 (3.10) | 0.15 | 0.35 (0.05–2.30) | 0.28 |
| anti-PD-1 | ||||
| Intercept | 0.53 (1.49) | 0.72 | ||
| MBL score | 0.26 (0.12) | 0.03 | 0.91 (0.81 – 1.03) | 0.14 |
| TMB | 0.00 (0.00) | 0.41 | 1.00 (1.00 – 1.00) | 0.14 |
| CTLA-4 | −0.79 (1.20) | 0.51 | 0.55 (0.10 – 2.94) | 0.48 |
| PD-1 | −0.99 (1.51) | 0.51 | 1.66 (0.21 – 13.29) | 0.63 |
| PD-L1 | −0.13 (1.70) | 0.94 | 1.56 (0.08 – 30.92) | 0.77 |
| anti-CTLA-4 | ||||
| Intercept | −0.84 (1.28) | 0.51 | ||
| MBL score | 0.33 (0.14) | 0.02 | 0.82 (0.70 – 0.96) | 0.01 |
| TMB | 0.00 (0.00) | 0.08 | 1.00 (1.00 – 1.00) | 0.73 |
| SCNA level | −0.46 (0.65) | 0.48 | 1.30 (0.68 – 2.48) | 0.43 |
| CTLA-4 | 1.77 (1.10) | 0.11 | 0.27 (0.07 – 1.03) | 0.06 |
| PD-1 | −1.54 (1.44) | 0.28 | 1.56 (0.40 – 6.04) | 0.52 |
| PD-L1 | −0.55 (1.70) | 0.75 | 0.92 (0.15 – 5.58) | 0.93 |
Univariate Cox proportional hazards models treating MBL score as a continuous variable showed similar results to the logistic regression analyses in two of the datasets, with higher MBL scores significantly associated with prolonged survival in the anti-PD-L1 and anti-CTLA-4 datasets (P = 0.01, HR = 0.80 and P = 0.01, HR = 0.83, respectively; Supplementary Table S2). In the anti-PD-L1 dataset, no other covariates were significantly associated with progression-free survival, while in the anti-CTLA-4 dataset, CTLA4 expression was the only other significant covariate. When including all covariates in multivariate Cox proportional hazards models, the MBL score was the only marker significantly associated with patient prognosis in either the PD-L1 or CTLA-4 dataset (P = 0.01, HR = 0.79; P = 0.01, HR = 0.82; Table 1). In the anti-PD-1 dataset none of the covariates were significantly associated with survival at the univariate level, including the MBL score, though there was a protective directionality (P = 0.27, HR = 0.93; Supplementary Table S2). The survival associations remained insignificant in the multivariate model, though the MBL score showed a protective trend (P = 0.14, HR = 0.91; Table 1). These results notably differed from the original anti-PD-1 study, which reported that somatic mutation burden was associated with improved survival following treatment.12 However, similar to the SCNA study in the anti-CTLA-4 dataset, the anti-PD-1 TMB analysis was performed on a larger cohort of samples for which whole exome sequencing was available (n = 38).
In addition to these genomic correlates of immunotherapy response, the original anti-PD-1 study identified a group of 26 transcriptomic signatures that were co-enriched in non-responders compared to responders. These signatures, referred to collectively as the innate PD-1 resistance (IPRES) signature, were associated with functions including mesenchymal transition, angiogenesis, hypoxia, and wound healing.12 In a univariate setting, the IPRES signature was associated with both patient response to anti-PD-1 in a logistic regression model (P = 3e-3; Supplementary Table S2) and poor patient survival in a Cox proportional hazards model (P = 0.04, HR = 3.46; Supplementary Table S2). Interestingly, we found that IPRES-enriched samples in the anti-PD-1 dataset had significantly lower MBL scores compared to non-IPRES-enriched samples (P = 0.03; Supplementary Figure S3). This result was not surprising given that both signatures were associated with response to anti-PD-1 therapy. However, while the MBL signature was associated with response to anti-CTLA-4 therapy, the IPRES signature has been reported to not associate with anti-CTLA-4 therapy response.12
We concluded our multivariate and univariate modeling by examining how the MBL score’s performance compared to that of our five other immune signatures as well as CD8A and CD4 gene expression. In univariate logistic regression models, none of the immune cell signatures and genes we tested exhibited significant associations with immune checkpoint blockade response. However, in the anti-CTLA-4 dataset, CD8A and CD4 gene expression were significantly associated with prolonged patient survival (P = 0.03 and 0.03, respectively) while the CD8 + T cell infiltration score showed an insignificant trend in the same direction (P = 0.08), mirroring findings from the previous study (Supplementary Table S2).10 Notably, across datasets, none of these genes and immune signatures exhibited significant associations when adjusting for other markers of immunotherapy response (Supplementary Table S3). These results together provide evidence that the MBL signature is detecting immunological features distinct from other correlates of immune infiltration.
The MBL score is robustly associated with immune checkpoint blockade response
We next tested the MBL score’s accuracy in predicting response to immune checkpoint blockade therapy. For each dataset, we ranked the patients from low to high based on their respective MBL score. We then performed an iterative procedure where we used each patient’s score as a threshold by which to classify the dataset. Patients with a score less than or equal to this threshold were classified as non-responders and patients with a score greater than this threshold were classified as responders. For each iteration, we then calculated the sensitivity and specificity of these classifications and used these results to determine the overall area under the receiver operating characteristic curve (AUC) for that dataset. Performing this procedure in each dataset gave us AUCs of 0.74, 0.68, and 0.72 for the anti-PD-L1, anti-PD-1 and anti-CTLA-4 datasets, respectively (Supplementary Table S2). Notably, these AUCs were higher than those calculated using TMB in all three datasets (AUC = 0.55, 0.57, and 0.63 for anti-PD-L1, anti-PD-1, and anti-CTLA-4, respectively) and this remained true when using SCNA level to predict anti-CTLA-4 response (AUC = 0.66; Supplementary Table S2). To test the robustness of the MBL score’s prediction, we next performed a resampling procedure on each of the datasets, where we created simulated versions of the datasets that consisted of 75% of the samples from the original datasets with the proportion of responders to non-responders held constant. We performed 1,000 simulations for each dataset and then recalculated the AUCs for each set of simulations. In all three datasets, the AUCs from the simulated datasets remained high (AUC = 0.70, 0.66, and 0.71 for anti-PD-L1, anti-PD-1, and anti-CTLA-4, respectively), with the lower-bound of the 95% confidence interval never crossing the 0.5 random classification threshold in any case (Supplementary Table S2).
The MBL score associates with B cell activity
The findings using the MBL score were intriguing, as B cells have not previously been implicated in response to checkpoint blockade therapy. We thus examined the underlying signature to better understand the signals it was capturing from the tumor microenvironment. In the flow cytometry benchmark datasets originally used to define the signature, the MBL score was directly associated with memory B cell fraction (R = 0.68) in PBMC mixtures and CD19 + B cell fraction in non-small cell lung cancer (R = 0.57), while showing weak associations with the flow cytometry fractions of non-B cells in both contexts (Supplementary Figure S4 and S5). Each gene in the MBL signature was weighted based on its differential expression level in the underlying B cell expression profile compared to all other immune cell profiles in the reference gene expression dataset.17 To functionally characterize our signature, we examined how the genes from an independent set of previously published immune signatures19 were weighted throughout the MBL signature. We found that the most highly weighted genes in the signature were those specific to naive B cells, memory B cells, and activated dendritic cells, while genes specific to other cell types were not weighted significantly higher than background (Supplementary Figure S6).
We hypothesized based on these analyses that the signature was detecting B cell activity in the tumor microenvironment. To evaluate this hypothesis, we examined the association between the MBL score and B cell receptor (BCR) heavy chain expression in patient tumor gene expression data. For this analysis, we used the anti-PD-1, anti-CTLA-4, and TCGA datasets, as they all included the raw RNAseq reads required to measure BCR levels from bulk expression data. In all three datasets, the MBL score was positively associated with BCR expression (R = 0.37, 0.53, and 0.36, respectively) and samples with high MBL scores, defined as above the median, exhibited significantly higher levels of BCR heavy chain abundance (P = 0.02, 4e-4, and 2e-11, respectively; Figure 4A). BCR abundance has previously been associated with melanoma patient survival in TCGA.20 Thus, we tested this association, and how well BCR abundance associated with immune checkpoint inhibitor response, in the anti-PD-1 and anti-CTLA-4 datasets. While no significant associations were observed in the anti-PD-1 dataset (Supplementary Figure S7), BCR heavy chain abundance was associated with clinical benefit (P = 0.02; Figure 4B) and prolonged patient survival (P = 0.05; Figure 4C) in the anti-CTLA-4 dataset.
Figure 4.
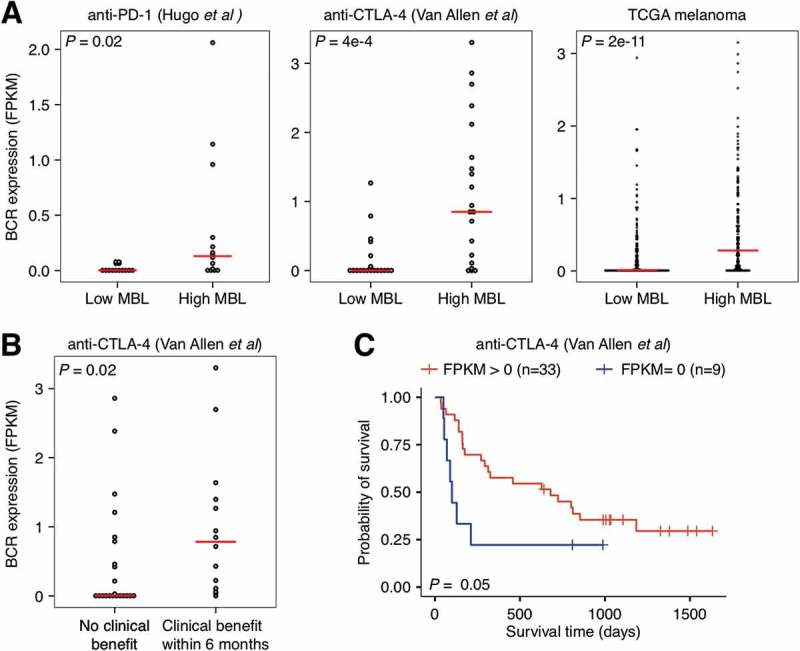
Relationship between B cell receptor expression, MBL score, and response and survival following anti-CTLA-4 therapy in melanoma. A, Comparison of B cell receptor heavy chain expression in high MBL patients versus low MBL patients for the anti-PD-1 (n = 28), anti-CTLA-4 (n = 42), and TCGA melanoma datasets (n = 469). High/low MBL designation was made using the median MBL score in each dataset. B, Comparison of B cell receptor heavy chain expression in patients that did not exhibit a clinical benefit to anti-CTLA-4 therapy (n = 14) versus those that did (n = 23). C, Overall survival of patients in patients that exhibited heavy chain B cell receptor expression relative to those that did not in the anti-CTLA-4 dataset. Vertical hash marks indicate censored data. For figure B, patients in the long-term survival with no clinical benefit group were excluded. P-values were calculated using the two-tailed Wilcoxon sum-rank test in A and B and the log-rank test in C. Boxes span quartiles, with the lines representing the median score in each group.
The MBL score associates with immune activation in the tumor microenvironment
While the BCR analyses indicated that the MBL signature was associated with B cell signals, it was possible that the MBL signature was capturing additional immune-associated functions. To provide a more thorough annotation of the signals detected by the MBL signature, we identified the genes it correlated with in each dataset (R > 0.3) and created a 55-gene core MBL signature that represented the intersection of these signatures (Figure 5A; Supplementary Table S4). The core MBL signature included genes associated with T cell function (CCL19, CXCL9, CXCR3) and MHC class II antigen presentation (HLA-DMA, HLA-DPA1, HLA-DQB1), in addition to genes specific to B cells (MS4A1, TNFRSF17, ADAM28). We then performed a gene ontology (GO) enrichment analysis to identify the functions that were most over-represented in the core and dataset-specific signatures (Figure 5B, Supplementary Table S5). The most enriched terms in the core signature included functions relating to antigen receptor-mediated signaling (GO:0050857; 53.68 fold-enrichment, false discovery rate (FDR) = 0.02), leukocyte and T cell activation (GO:2000516; 34.16 fold-enrichment; FDR = 0.05), leukocyte and T cell proliferation (GO:0042102; 18.98 fold-enrichment; FDR = 0.01), and cell-cell adhesion (GO:1903039; 12.53 fold-enrichment; FDR = 4e-3). Notably, GO terms related to B cell function, such as B cell proliferation (GO:0042100; 30.47 fold-enrichment, FDR = 0.06) and B cell activation (GO:0042113; 10.58 fold-enrichment, FDR = 0.14) were enriched, but at a lower significance threshold.
Figure 5.
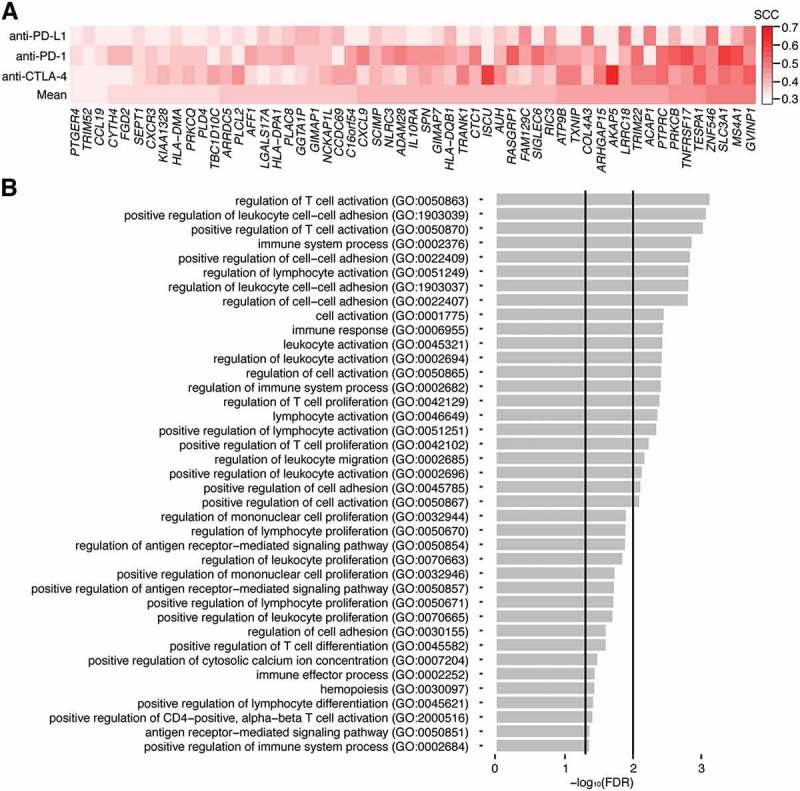
Gene ontology enrichment analysis for the core MBL signature. A, Spearman coefficients representing the correlation between a dataset’s MBL scores and expression values for the 55 genes of the core MBL signature. Each row represents a different immunotherapy dataset and the mean correlation. Genes are arranged from lowest to highest using the mean correlation. B, False discovery rates (FDR) for the gene ontology (GO) terms most overrepresented in the core MBL gene set. Vertical lines indicate where FDR = 0.05 and 0.01. FDRs were derived from p-values calculated using a Fisher’s exact test.
GO enrichment analyses of the MBL-correlated genes within the individual datasets were largely in agreement with those from the core dataset, though there were subtle differences between them. The anti-PD-L1 signature, which had the fewest MBL-correlated genes, uniquely exhibited enrichment in genes associated with dendritic cell apoptosis regulation (GO:2000669). In contrast, the anti-PD-1 genes were most associated with antigen presentation (GO:0002475, GO:0019883), T cell activity (GO:0001916) and B cell activity (GO:0050855), while the anti-CTLA-4 genes were most associated with T-helper cell function (GO:0002295, GO:0042088) and B cell activity (GO:0002923, GO:0045579) (Supplementary Table S5). Together, these results suggested that the MBL signature was primarily capturing signals indicative of immune activation in the tumor microenvironment.
Discussion
This study presents intriguing findings linking the activity of a B cell-derived expression signature to immune checkpoint blockade therapy response. In all three datasets, patients experiencing clinical benefit had significantly higher MBL scores compared to those that did not experience clinical benefit. This makes the MBL signature unique as it is associated with response to immune checkpoint blockade therapies targeting the PD-1/PD-L1 axis as well as therapies targeting CTLA-4. Interestingly, scores from this signature were predictive of therapy response and post-treatment survival even after adjusting for other known biomarkers, suggesting that a novel transcriptomic process exists that can distinguish responders from non-responders for each treatment.
The MBL signature was derived from B cell gene expression profiles and exhibited strong correlations with memory B cell fractions in PBMC mixtures and B cell infiltration in NSCLC tumors.17 Furthermore, when applied to TCGA and the immunotherapy response datasets used in this study, the MBL score exhibited significant associations with BCR expression. Together, these analyses suggested that the MBL signature was well-suited to serve as a proxy for B cell activity in the tumor microenvironment. However, functional analyses of the genes that were positively correlated with the MBL score across the three immunotherapy datasets indicated that the signature was more broadly capturing signals of immune activity in the tumor microenvironment. In addition to B cell activity, the genes that were correlated with MBL score in all three datasets were enriched in functions related to T cell activation and antigen presentation. Furthermore, when examining the MBL-correlated genes specific to each dataset we identified differences regarding the most enriched functions in each signature, with the anti-PD-L1 signature uniquely enriched in dendritic cell-based functions, the anti-PD-1 signature exhibiting broader lymphocyte activation and antigen presentation signals and the anti-CTLA-4 signature most associated with T-helper cell and B cell activity.
The diverse immunological functions captured by the MBL signature may explain why it was the only immune cell-derived signature tested in this study to exhibit significant associations with immune checkpoint inhibitor response in each of the three immunotherapy datasets. However, the interaction of these functions in the context of immunotherapy remains unclear. Interestingly, the core MBL signature included a series of MHC-class II genes, indicating the importance of antigen presentation with regards to immune checkpoint blockade response. MHC-class II genes are expressed on a variety of cell types including B cells, dendritic cells, and macrophages.21 Furthermore, melanoma-specific expression of MHC-class II genes has been shown to be associated with response to anti-PD-1 therapy.22 MHC-class II antigen presentation is primarily involved in the activation of CD4 + T cell-based immune responses. Given the role of neoantigens in eliciting a tumor immune response,23 and the B cell, T cell and antigen presentation functions associated with the MBL signature, it is possible that the MBL signature was detecting an MHC-class II driven immune response. This process may have been driven by different cell populations in each dataset, with B cells providing the signal in the anti-CTLA-4 and anti-PD-1 melanoma datasets, which exhibited higher degrees of B cell function in responders, while MHC-class II-expressing dendritic cells providing the signal in the anti-PD-L1 dataset. Understanding the extent to which this cross-presentation is taking place in each context will be necessary to further characterize the underlying mechanisms of immune checkpoint blockade response.
A major limitation of this study is that all associations were observed in three datasets with limited numbers of patients (n = 25, n = 28, and n = 42). As of now, there are few publicly available gene expression datasets for patients receiving immune checkpoint blockade therapy. Despite the small sample sizes in our study, our signature was reproducibly associated with response in both datasets targeting the PD-1/PD-L1 axis and a meta-analysis pooling the datasets revealed highly significant associations. Furthermore, while our signature was associated with response in all three datasets, many previously reported genomic-based biomarkers were not robust to the smaller sample size, including SCNA level in anti-CTLA-415 and mutation burden in anti-PD-1.12 These findings suggest that the MBL signature would perform well in more highly powered studies. As additional data is released, it will be important to further test this signature to demonstrate its reproducibility and validate it as a response-predictive biomarker.
In conclusion, we present a B cell-derived expression signature that can predict patient response to immune checkpoint inhibitors in two different cancer types. Despite the preliminary stage of these findings, this study provides evidence of a shared biology driving anti-PD-1/PD-L1 and anti-CTLA-4 response and details a versatile signature that is reported to be predictive of response to both therapies. While it will be important to validate these findings both mechanistically and in large patient cohorts, we believe the associations presented will be useful going forward in the efforts to personally tailor immunotherapy treatment regimens.
Methods
Datasets
Raw gene expression data (.fastq) and clinical information for the anti-CTLA-4 dataset were obtained from the Database of Genotypes and Phenotypes (dbGaP) under accession number phs000452. Raw read files were aligned to the GRCh37 human genome assembly using TopHat v2.1.0.24 From the aligned reads, transcript assembly and abundance estimation were performed using Cufflinks v2.2.1.25 Raw and processed gene expression data for the anti-PD-1 dataset were obtained from the gene expression omnibus (GEO) under accession number GSE78220. Clinical information for this dataset was downloaded from a supplement in the original publication.12 Processed gene expression data and clinical information for the anti-PD-L1 dataset in urothelial carcinoma were downloaded from Zenodo (https://zenodo.org/record/546110) and GitHub (https://github.com/hammerlab/multi-omic-urothelial-anti-pdl1), respectively. In all datasets, clinical benefit designations were defined using the anti-CTLA-4 study’s criteria,10 where clinical benefit was defined as complete response, partial response, or stable disease with overall survival > 1 year by RECIST criteria, while no clinical benefit was defined using RECIST criteria for progressive disease or stable disease with overall survival less than 1 year. The PBMC and non-small cell lung cancer gene expression used for validation were obtained from GEO under accession numbers GSE65133 and GSE84797 and their associated flow cytometry data were obtained from their respective publications.19,26 Raw TCGA gene expression data (.fastq) for skin cutaneous melanoma (SKCM) was downloaded from the National Cancer Institute Genomic Data Commons Legacy Archive (https://portal.gdc.cancer.gov/legacy-archive/), while processed TCGA RNASeqV2 data for SKCM and bladder urothelial carcinoma (BLCA) were downloaded from the Broad Institute TCGA GDAC Firehose repository (http://firebrowse.org/). In cases where samples lacked response information or survival, they were excluded from the respective analyses. Sample sizes for each immunotherapy dataset and the associated covariates used in each analysis are available in Supplementary Table S6.
Calculation of immune cell signature scores
To calculate immune cell signature scores from patient gene expression data, we used a previously established framework,16,17 that calculates scores for six distinct cell types from patient gene expression data using specificity weights that capture how uniquely a gene is expressed in a given immune cell. The algorithm driving this procedure, BASE,27 calculates these scores by summarizing how the weights are distributed through a patient’s ranked gene expression profile. This distribution is captured by calculating a Kolmogorov-Smirnov statistic-like metric comparing two functions. The first function (foreground) represents the cumulative sum of a patient’s ranked gene expression profile weighted by a given immune cell’s specificity weight, while the second function (background) represents the cumulative sum of a patient’s ranked gene expression profile weighted by the complement (1-weight) of the immune cell’s specificity weight. The maximal deviation between these two functions represents the patient’s signature score for that cell type, with a high score representing high expression of immune-specific genes in that patient and a low score indicating the opposite. Full details of this procedure and code for both immune cell specificity weight calculation and immune cell signature score calculation have been published in prior work.16 Details on the calculation of the weights and the benchmarking analyses used to characterize the signatures used in this study, as well as the signatures themselves, are available in a prior publication.17 The signature scores for each immunotherapy dataset are available in this article (Supplementary Table S7).
Additional infiltration scores were calculated using gene expression signatures derived from single cell RNAseq data18 as well as the LM22 matrix used by CIBERSORT.19 Infiltration scores using the single cell RNAseq signatures were calculated using single sample gene set enrichment analysis (ssGSEA) from the R GSVA package.28 Cellular fractions were inferred from the LM22 matrix using the CIBERSORT webserver (https://cibersort.stanford.edu/runcibersort.php) run in relative mode using the following parameters: 100 permutations and quantile normalization disabled (as is recommended for RNAseq data). Prior to immune signature score calculation using BASE and ssGSEA, all genes with a transcript abundance of 0 across all samples were removed from each dataset and all expression data was then multiplied by 100 to minimize unwanted noise from the addition of a pseudocount before being log10-transformed. For CIBERSORT, all immune cell fractions were calculated from gene expression data in non-log space.
B cell receptor analyses
BCR heavy chain abundance was inferred using V’DJer. FASTQ files for each dataset used in BCR repertoire analyses were aligned using STAR (v2.5.2b).29 V’DJer with default settings was then run on the aligned files to infer each patient’s BCR heavy chain (IGH) repertoire.20 V’DJer was initially unable to process some samples in the anti-PD-1 dataset as they contained reads of variable length. To address this, each sample in the anti-PD-1 dataset was filtered to only contain reads of 100 base pairs before being input into V’DJer. Transcript abundance of the resulting BCR contigs output by V’DJer were quantified using RSEM.30 FPKM for each BCR contig was calculated by dividing the expected counts output by RSEM by the total number of fragments in the file divided by one million.
Statistical analyses
Signature score and BCR expression comparisons were made using the two-tailed Wilcoxon-sum rank test. Correlation coefficients represent Spearman correlations unless stated otherwise. Response classification was performed using the “glm()” function in R with family “binomial.” For survival analyses, samples were stratified into high and low groups based on whether they were above or below the dataset’s third and first score quartile, respectively, unless stated otherwise. Survival distributions between the two groups were compared using the log-rank test through the “survdiff()” function from the R survival package. Cox proportional hazards regression was used to model the relationship between signature scores and patient survival and was performed using the “coxph()” function from the R survival package. For meta-analyses, p-values from Wilcoxon sum-rank tests or log-rank tests, for response and survival associations respectively, were converted to z-scores. The z-scores were then collapsed into meta-z-scores by applying a weighted version of Stouffer’s method.31
GO enrichment analyses were performed using the GO Enrichment Analysis webserver (http://geneontology.org/page/go-enrichment-analysis). The analysis type run was the PANTHER Overrepresentation Test, with the Homo sapiens reference list, the GO biological process annotation dataset, and with FDRs calculated using the Fisher’s exact test.
Funding Statement
This study was supported by the American Cancer Society (IRG-82-003-30) and the National Center for Advancing Translational Sciences of the National Institutes of Health (KL2TR001088) (C. Cheng). F.S. Varn was additionally supported in part by the National Institute of General Medical Sciences of the National Institutes of Health (T32GM008704);American Cancer Society (ACS) [IRG-82-003-30];HHS | NIH | National Institute of General Medical Sciences (NIGMS) [T32GM008704];HHS | NIH | National Center for Advancing Translational Sciences (NCATS) [KL2TR001088];
Disclosure of Potential Conflicts of Interest
The authors declare no potential conflicts of interest
Supplementary data
Supplemental data for this article can be accessed here
References
- 1.Nishino M, Ramaiya NH, Hatabu H, Hodi FS.. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol 2017;14:655–668. doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252–264. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, et al. Distinct cellular mechanisms underlie anti-CTLA-4 and anti-PD-1 checkpoint blockade. Cell 2017;170:1120–33 e17. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, Patt D, Chen -T-T, Berman DM, Wolchok JD. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol 2015;33:1889–1894. doi: 10.1200/JCO.2014.56.2736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, Garbe C, Chiarion-Sileni V, Testori A, Chen -T-T, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol 2015;33:1191–1196. doi: 10.1200/JCO.2014.56.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–287. doi: 10.1038/nrc.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568–571. doi: 10.1038/nature13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med 2015;372:2018–2028. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 9.Ji RR, Chasalow SD, Wang L, Hamid O, Schmidt H, Cogswell J, Alaparthy S, Berman D, Jure-Kunkel M, Siemers NO, et al. An immune-active tumor microenvironment favors clinical response to ipilimumab. Cancer Immunol Immunother 2012;61:1019–1031. doi: 10.1007/s00262-011-1172-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350:207–211. doi: 10.1126/science.aad0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–128. doi: 10.1126/science.aaa1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. Genomic and transcriptomic features of response to anti-PD-1 therapy in metastatic melanoma. Cell 2016;165:35–44. doi: 10.1016/j.cell.2016.02.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–2199. doi: 10.1056/NEJMoa1406498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Turajlic S, Litchfield K, Xu H, Rosenthal R, McGranahan N, Reading JL, Wong YNS, Rowan A, Kanu N, Al Bakir M, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol 2017;18:1009–1021. doi: 10.1016/S1470-2045(17)30516-8. [DOI] [PubMed] [Google Scholar]
- 15.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355:eaaf8399. doi: 10.1126/science.aaf8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varn FS, Wang Y, Mullins DW, Fiering S, Cheng C. Systematic pan-cancer analysis reveals immune cell interactions in the tumor microenvironment. Cancer Res 2017;77:1271–1282. doi: 10.1158/0008-5472.CAN-16-2490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varn FS, Tafe LJ, Amos CI, Cheng C. Computational immune profiling in lung adenocarcinoma reveals reproducible prognostic associations with implications for immunotherapy. OncoImmunology 2018;7:e1431084. doi: 10.1080/2162402X.2018.1431084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 2016;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mose LE, Selitsky SR, Bixby LM, Marron DL, Iglesia MD, Serody JS, Perou CM, Vincent BG, Parker JS. Assembly-based inference of B-cell receptor repertoires from short read RNA sequencing data with V’DJer. Bioinformatics 2016;32:3729–3734. doi: 10.1093/bioinformatics/btw526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Roche PA, Furuta K. The ins and outs of MHC class II-mediated antigen processing and presentation. Nat Rev Immunol 2015;15:203–216. doi: 10.1038/nri3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Johnson DB, Estrada MV, Salgado R, Sanchez V, Doxie DB, Opalenik SR, Vilgelm AE, Feld E, Johnson AS, Greenplate AR, et al. Melanoma-specific MHC-II expression represents a tumour-autonomous phenotype and predicts response to anti-PD-1/PD-L1 therapy. Nat Commun 2016;7:10582. doi: 10.1038/ncomms10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science 2015;348:69–74. doi: 10.1126/science.aaa4971. [DOI] [PubMed] [Google Scholar]
- 24.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 2012;7:562–578. doi: 10.1038/nprot.2012.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lizotte PH, Ivanova EV, Awad MM, Jones RE, Keogh L, Liu H, Dries R, Almonte C, Herter-Sprie GS, Santos A, et al. Multiparametric profiling of non-small-cell lung cancers reveals distinct immunophenotypes. JCI Insight 2016;1:e89014. doi: 10.1172/jci.insight.89014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng C, Yan X, Sun F, Li LM. Inferring activity changes of transcription factors by binding association with sorted expression profiles. BMC Bioinformat 2007;8:452. doi: 10.1186/1471-2105-8-452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformat 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformat 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med 2015;21:938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.