

Abstract
Objective
Insulin-like growth factor 1 (IGF-1) is a major autocrine/paracrine growth factor, which promotes cell proliferation, migration, and survival. We have shown previously that IGF-1 reduced atherosclerosis and promoted features of stable atherosclerotic plaque in Apoe−/− mice, an animal model of atherosclerosis. The aim of this study was to assess effects of smooth muscle cell (SMC) IGF-1 signaling on the atherosclerotic plaque.
Approach and Results
We generated Apoe−/− mice with IGF-1 receptor (IGF-1R) deficiency in SMC and fibroblasts (SM22α-CreKI/IGF1R-flox mice). IGF-1R was decreased in the aorta and adventitia of SM22α-CreKI/IGF1R-flox mice and also in aortic SMC, embryonic, skin and lung fibroblasts (FB) isolated from SM22α-CreKI/IGF1R-flox mice. IGF-1R deficiency downregulated collagen mRNA-binding protein LARP6 and vascular collagen, and mice exhibited growth retardation. The high-fat diet fed SM22α-CreKI/IGF1R-flox mice had increased atherosclerotic burden and inflammatory responses. Alpha smooth muscle actin-positive plaque cells had reduced proliferation and elevated apoptosis. SMC/FB-targeted decline in IGF-1 signaling decreased atherosclerotic plaque SMC, markedly depleted collagen, reduced plaque fibrous cap, and increased plaque necrotic cores. Aortic SMC isolated from SM22α-CreKI/IGF1R-flox mice had decreased cell proliferation, migration, increased sensitivity to apoptosis and these effects were associated with disruption of IGF-1-induced Akt signaling.
Conclusions
IGF-1 signaling in SMC and in FB is a critical determinant of normal vascular wall development and atheroprotection.
Keywords: atherosclerosis, cardiovascular disease, smooth muscle cells, fibroblasts, atherosclerotic plaque stability, Animal Models of Human Disease, Vascular Biology, Atherosclerosis, Vascular Disease
Introduction
Insulin-like growth factor 1 (IGF-1) mediates somatic development and growth, and IGF-1 deficiency delays puberty and induces multiple growth abnormalities1. Circulating IGF-1 levels are maximal during early childhood; however, they progressively decline with age2 and reduced IGF-I in the elderly may contribute to the increased risk of cardiovascular diseases (CVD)3. CVD including atherosclerotic heart disease remain the primary cause of morbidity and mortality in the USA4. Most acute coronary events are related to rupture or erosion of atherosclerotic plaques that are not hemodynamically significant5. Thus, plaque stability is a critical determinant of clinical events. We have shown that systemic IGF-1 administration in Apoe−/− mice, an animal model of atherosclerosis, reduced atherosclerosis, decreased inflammatory responses and promoted features of stable atherosclerotic plaque6,7. Consistent with our results, another group reported that administration of long R3 IGF-1 increased plaque smooth muscle cells (SMC), cap thickness and reduced the rate of intraplaque hemorrhage, indicating that IGF-1 promoted plaque stabilization8. These findings are in line with most (but not all9) clinical studies, which have suggested that lower circulating IGF-1 levels and higher IGF-1 binding protein-3 (IGFBP-3) are associated with an increased risk of atherosclerotic disease10,11.
Most actions of IGF-1 are mediated via its cognate receptor tyrosine kinase, IGF-1 receptor (IGF-1R)12. IGF-1R signaling activates multiple downstream signaling pathways, including PI3-kinase, Akt, and mitogen-activated protein kinase (MAPK). The activation of these pathways mediates differential biologic actions of IGF-1, including cell growth, differentiation, migration, and survival12. Reduced levels of IGF-1 and IGF-1R were associated with apoptotic SMC in advanced human atherosclerotic plaque13 suggesting a link between downregulation of IGF-1 signaling, SMC apoptosis, and atherosclerotic burden. IGF-1R is ubiquitously expressed; therefore multiple cell types can mediate IGF-1-induced effects on atherosclerotic plaque development. An animal model with cell-specific IGF-1R deficiency is thus an indispensable tool to delineate the contribution of intracellular IGF-1 signaling to net IGF-1 effects. We have demonstrated recently that IGF-1R deficiency in macrophages decreased expression of cholesterol transporters, reduced lipid efflux and increased foam cell accumulation in lesions, which in turn resulted in elevated atherosclerotic burden and reduced plaque vulnerability14.
SMC and adventitial fibroblasts (FB) are major vascular cell types determining normal vessel wall function and development, repair and response to atherogenesis15. IGF-1 promotes vascular SMC migration, proliferation and survival, however the specific contribution of SMC in IGF-1-induced atheroprotective effects is unknown. In the current study we aimed to assess the role of SMC IGF-1 signaling on the atherosclerotic plaque. For this purpose we crossbred Igf1r-floxed mice with SM22α-Cre knockin (SM22α-CreKI) mice, both nullizygous for the Apoe gene (Apoe−/−), anticipating to induce IGF-1R deficiency in SM22α-expressing cell types, i.e. smooth muscle cells. Cre recombinase expression and reduced IGF-1R level were detected in SM22α-CreKI/IGF1R-flox mice in SMC-rich organs and also, unexpectedly, in FB-rich vascular adventitia. We assessed the effect of SMC and FB-targeted IGF-1R deficiency on vascular wall morphology, atherosclerotic plaque composition, atherosclerotic burden and plaque stability. We found that IGF-1R deficiency reduced vessel size, depleted vascular collagen, and increased atherosclerosis. Our results establish the critical importance of IGF-1 signaling for normal vascular wall development and for atheroprotection.
Materials and Methods
Mice
All animal experiments were approved by the Institutional Committee for Use and Care of Laboratory Animals of University of Missouri at Columbia. Mice with SM22α Cre recombinase knockin (SM22α-CreKI+/+ mice) were obtained from Dr. Eugene Chen (Medical School, University of Michigan)16. Mice with LoxP-flanked IGF-1R were a generous gift from Dr. Jens Brüning (University of Leipzig, Germany)17. Apoe−/− mice (C57BL/6 background) were from Jackson Lab. SM22α Cre knockin mice and mice with LoxP-flanked IGF-1R gene were crossed with Apoe−/− mice for 10 generations to generate SM22α-Cre+/+/Apoe−/− and LoxP+/+-IGF-1R/Apoe−/− mice (FIR), respectively. SM22α-Cre+/+/Apoe−/− and FIR mice were crossed with each other to obtain LoxP+/+-IGF-1R/SM22α-Cre+/−/Apoe−/− mice (SM22α-CreKI/IGF1R-flox mice or IGF1R/FIR mice). We were not able to generate mice homozygous for both LoxP and SM22α-Cre genes because of fetal mortality. Six week-old IGF1R/FIR (n=28, males, 13; females, 15) and FIR mice (control, n=55, males, 26; females, 29) were fed with a high-fat Western type diet (WD, Harland-Teklad) for 12 weeks. A smaller group of 6-week-old IGF1R/FIR (n=14, males, 7; females, 7) and FIR mice (control, n=14, males, 7; females, 7) were fed with WD for 4 weeks. The body weight and the blood pressure were measured every week.
Cells
Aortic SMC were isolated by attaching the adventitial fat-cleaned, opened aorta to the culture dishes containing DMEM/F-12 media (Thermo Fisher), supplemented with 10% fetal bovine serum (FBS), 4 mmol/L L-glutamine, 100 U/mL penicillin and 100 ug/mL streptomycin. Cell identity was confirmed by staining for calponin and α-smooth muscle actin (α-SMA), SMC markers. More then 90% cells were double positive for calponin and α-SMA (Suppl.Fig.I). Mouse embryonic fibroblasts (MEF) were harvested according to the standard protocol18. Mouse adult skin and lung fibroblasts were isolated as per Seluanov et al.19. Briefly, mouse lung and skin tissue were minced and treated with 0.14 U/mL Liberase Blendzyme collagenase mixture (Roche) in EMEM/F-12 media for 60 min and tissue fragments were left to attach to the culture dish for 48h. Fibroblast identity was confirmed by immunostaining for ER-TR7, a fibroblast marker20. We used male mice for cell isolation.
Atherosclerosis quantification
Under anesthesia, mice were perfused with saline then 4% buffered paraformaldehyde plus 5% sucrose and the heart and the entire aorta were dissected. The heart was separated from the aortic arch and fixed overnight followed by embedding into paraffin. Three aortic valve equally spaced sections were stained with H&E and lesions were manually outlined with CellSense Dimension software (Olympus) by two investigators blinded to the group assignment. For the current study, the average internal variability (section-to-section coefficient of variation per mouse) was 9.5±0.7%, external variability (mice-to-mice variability within group) was 8.7±1.7% and the average discrepancy between investigators was less then 10%. To quantify aortic lipid accumulation we performed en face analysis. The entire aorta was split longitudinally, pinned flat in a black-waxed dish, and stained with neutral lipid-sensitive stain Oil Red to visualize lesions.
Vessel morphometry
The aorta length was quantified from the top of the left ventricle until the iliac bifurcation. Aortic valve, thoracic aorta and brachiocephalic artery (BCA) circumference were quantified at the external elastic lamina using H&E-stained cross-sections. Vessel medial area was calculated by subtracting the area within the internal elastic lamina from the area within the external elastic lamina.
Immunohistochemistry
Information about antibodies is included in the Major Resources Table in the Supplemental Material. Antibody specificity was verified by staining of serial section with “normal” IgG (obtained from an unimmunized animal of the same species as primary antibody). For IHC with anti-N-tyrosine antibody, sections stained with “normal” rabbit IgG or with anti-N-tyrosine antibody blocked by incubation with 3-nitro-L-tyrosine (Millipore-Sigma, 200 uM, 1h) served as the negative control and sections pre-treated with peroxynitrite (Millipore-Sigma) and stained with anti-N-tyrosine antibody served as the positive control (Suppl.Fig.II).
Collagen level in the vascular wall was quantified using Trichrome-stained aortic rings. Each image was converted into the multichannel mode and “green” signal (average pixels value) was assessed from region-of-interests (ROI) manually placed within the image. We kept the same ROI size to quantify collagen in images obtained for IGF-1R-deficient and control mice.
Analysis of lesions in the BCA
BCA was dissected from SM22α-CreKI/IGF1R-flox (n=10) and FIR (n=8) mice fed with WD for 12 weeks. One hundred 5 um sections per artery segment were cut and 10 equally spaced sections were immunostained for calponin, fibrinogen, collagen (Trichrome) and H&E. In addition, serial sections were stained by Carstairs method to detect fibrin (orange-red), SMC (dark red), and collagen (bright blue). Data obtained by IHC were consistent with results generated by Carstairs (data not shown).
Laser capture microdissection (LCM)
The distal end of the small intestine (terminal ileum) was dissected, embedded in OCT and intestinal SMC were isolated by LCM. Cross-section was immunostained for calponin to identify SMC, and used as a reference for LCM (Suppl.Fig.III). Eight to ten serial sections were stained with Histogene staining solution (Thermo Fisher) as per kit’s instructions. LCM was performed with the Applied Biosystems ArcturusXT LCM System. LCM dissected tissue was used for RNA isolation with PicoPure Frozen RNA Isolation Kit followed by cDNA synthesis with RiboAmp HS Plus cDNA kit (Thermo Fisher) according to the manufacturer’s instructions.
Cytokine assay
Serum cytokine levels were quantified by Inflammatory Cytokines Multi-Analyte ELISArray (Qiagen) according to manufacturer’s instructions. IGF-1 levels were quantified with IGF-1 ELISA (R&D Systems), IGFBP-1 levels with IGFBP-1 DuoSet ELISA (R&D Systems), IGFBP-2 levels with IGFBP-2 ELISA (Thermo Scientific), IGFBP-3 levels with IGFBP-3 Quantikine ELISA (R&D Systems), IGFBP-5 levels with IGFBP-5 ELISA (Sigma-Aldrich) and IGF-1 acid-labile subunit (ALS) levels with ALS ELISA (Biomatek).
Cell proliferation, migration and cell apoptosis
Cell apoptosis was quantified with TUNEL-Fluorescein kit (Roche) as per manufacturer’s instructions. Total cell apoptosis was defined as TUNEL+ cell number per 1000 plaque cells and α-SMA-positive cell apoptosis was measured as the number of TUNEL+/α-SMA+ cells per 1000 α-SMA+ cells. Sections treated with DNAse I and sections stained with dUTP-omitted TUNEL mixture served as positive and negative controls for TUNEL assay, respectively. To assess cell proliferation, in vivo labeling of DNA with 5-ethynil-2′-deoxyuridine (EdU) was performed according to Salic et al21. EdU (5 mg/kg/day) was administered i.p. for 4 days before sacrifice and labeled DNA was detected by AlexaFluor488-azide (Click-It Imaging Kit, Invitrogen). A section of small intestine was the positive control for the EdU assay.
Cell growth curves were built by counting cell number with CyQuant Cell Proliferation kit (Life Technologies). Cell proliferation was measured with BrdU Cell proliferation ELISA (Roche). Cell migration was quantified using a wound-healing (“scratch”) assay as described22. Chemoattractant-stimulated cell migration was measured with FluoroBlock plate inserts (BD Biosciences). Cells were seeded on inserts and migrated toward 20% FBS for 6h followed by labeling with calcein-AM (Molecular Probes). Migrated cell number was quantified on the bottom part of the insert with a Bio-Tek Synergy microplate reader.
Data analysis
Authors declare that the design, execution, and reporting of the current study adhere to the guidelines for experimental atherosclerosis studies described in the scientific statement from the American Heart Association23. Authors considered sex as biological variable in accordance with AHA ATVB Council statement24.
All numeric data are expressed as mean ± SEM. Statistical analyses were performed with GraphPad Prism 6.07. Unless otherwise specified, two-tailed unpaired Student t tests were performed to determine statistical significance. For data that were not normally distributed, Mann-Whitney U test was performed. ANOVA was used to compare multiple groups followed by post hoc analysis with Dunnett’s multiple comparison test. We used Grubbs’ test to check data sets for potential outliers. Differences were considered significant at P<0.05.
Results
SM22α promoter-driven Cre recombinase decreased IGF-1R expression in SMC, and in fibroblasts (FB).
To assess the effects of SMC IGF-1 signaling on the atherosclerotic plaque we generated Apoe−/− mice with SM22α-CreKI mediated IGF-1R deficiency (SM22α-CreKI/IGF1R-flox mice, IGF1R/FIR mice). To generate SM22α-CreKI/IGF1R-flox mice we used SM22α-CreKI+/+ mice16, which are widely used for generation of models with SMC-specific knockout25,26. Mice with a LoxP-flanked IGF-1R gene (FIR mice) were used as control.
To characterize the phenotype of SM22α-CreKI/IGF1R-flox mice, Cre recombinase and IGF-1R expression were quantified in skeletal muscles, spleen, heart, brain, adventitia-free aortas, adventitia and urinary bladder (Fig.1). Cre recombinase was not found in any tissue dissected from FIR mice. Cre recombinase was detected in SMC-rich tissues (aortas and urinary bladder), and in the heart in SM22α-CreKI/IGF1R-flox mice. Cre expression in the heart of SM22α-CreKI/IGF1R-flox mice is consistent with the report from Zhang et al. showing high Cre expression in aorta and low Cre levels in the heart of SM22α-CreKI+/+ mice16. As expected SM22α-CreKI/IGF1R-flox mice have reduced IGF-1R expression in the aorta and in the urinary bladder (85±4% and 47±4% decrease, respectively) and also in the heart (51±3% decrease compared to FIR controls) without changes in IGF-1R levels in brain, skeletal muscles and spleen. IGF-1R expression was also reduced (78±7% decrease compared to FIR) in non-vascular (intestinal) SMC: cells were identified by immunostaining with calponin (SMC marker) and isolated by laser capture microdissection14 (Suppl.Fig.III).
Figure 1. SM22α promoter-driven Cre recombinase decreased IGF-1R expression in SMC, and in fibroblasts (FB).

Skeletal muscles, brain, spleen, adventitia, urinary bladder, heart, and adventitia-free aortas were dissected from FIR and SM22α-CreKI/IGF1R-flox (IGF1R/FIR) mice (n=4/group) and used for immunoblotting (A, B). Aortic SMC were isolated from mice and cells were exposed to 100 ng/ml IGF-1 (n=6/group). Expression of IGF-1R and IGF-1R downstream molecules was quantified by immunoblotting (C). IGF-1R and Cre expression in FB isolated from FIR and IGF1R/FIR mice (n=4/group) (D, E). MW, molecular weight, *P<0.05.
To our surprise, we found detectable Cre expression and reduced IGF-1R levels in fibroblast-rich adventitia (43±8% decrease compared to FIR) (Fig.1) suggesting leakage of Cre in non-SMC tissue. We isolated aortic SMC and also embryonic, skin and lung FB from SM22α-CreKI/IGF1R-flox and control mice. More than 90% aortic cells isolated from SM22α-CreKI/IGF1R-flox or FIR mice were double positive for calponin and α-smooth muscle actin (α-SMA), SMC markers (Suppl.Fig.I). IGF-1R levels were virtually undetectable in SMC isolated from SM22α-CreKI/IGF1R-flox mice (Fig.1C). IGF-1 induced Ser473 Akt and also p38 MAPK and ERK1/2 phosphorylation in control cells, however no Akt and weak p38 MAPK and ERK1/2 activation was found in SM22α-CreKI/IGF1R-flox aortic SMC suggesting inhibition of IGF-1R-downstream signaling in these cells. IGF-1R expression was downregulated by approximately 85% in fibroblasts (Fig.1D, E). Thus, SM22α promoter-driven Cre recombinase induced IGF-1R deficiency in SMC and in FB in SM22α-CreKI/IGF1R-flox mice.
Serum IGF-1 levels were reduced by 24–28% in SM22α-CreKI/IGF1R-flox mice compared to control (Supp.Fig.IVA) and this effect was observed in both males and females (data not shown). IGF-1 is mainly secreted by the liver as a result of stimulation by growth hormone (GH)12. Besides GH, a group of IGF-binding proteins (IGFBPs), and an acid-labile subunit (ALS) serve to regulate circulating IGF-1 concentration and action12. We measured expression levels of IGF-1, GH, ALS and IGFBPs in SM22α-CreKI/IGF1R-flox and FIR mice after 12 weeks of feeding with a high-fat Western type diet (WD). IGF-1 gene expression in liver and aortas in SM22α-CreKI/IGF1R-flox mice was not different from controls (data not shown). GH level was slightly increased in SM22α-CreKI/IGF1R-flox mice potentially due to a feedback mechanism (Supp.Fig.IVB). IGF-1R deficiency did not change circulating level of IGFBP-3 (494.4±33.2 ng/ml, SM22α-CreKI/IGF1R-flox vs. 406.3 ± 32.8 ng/ml, FIR, P=NS) and ALS (9.65 ± 0.56 ug/ml, SM22α-CreKI/IGF1R-flox vs. 8.53±0.56 ug/ml, FIR, P=NS), primary components of the IGF-1/IGFBP-3/ALS ternary complex. We found that IGFBP-5 level was downregulated in SM22α-CreKI/IGF1R-flox mice (1.05±0.09 ng/ml, SM22α-CreKI/ IGF1R-flox vs. 1.50±0.18 ng/ml, FIR) without changes in IGFBP-1 (31.57 ± 5.74 ng/ml, SM22α-CreKI/IGF1R-flox vs. 28.34 ± 0.88 ng/ml, FIR, P=NS) and IGFBP-2 level (74.05±18.35 ng/ml, SM22α-CreKI/IGF1R-flox vs. 89.1±17.6ng/ml, FIR, P=NS) (Suppl.Fig.IV).
IGF-1R deficiency induced growth retardation, decrease of vascular collagen and downregulation of LARP6, a collagen mRNA-binding protein.
SM22α-CreKI/IGF1R-flox mice had a basal reduction in body weight compared with FIR mice: 25.8±1.9% decrease at 6 weeks age when fed with normal chow (P<0.05). We observed a similar reduction in body weight for males (SM22α-CreKI/IGF1R-flox: 14.46±0.68g, FIR: 19.81±0.35g, 27.0±2.2% decrease, P<0.05) and for females (SM22α-CreKI/IGF1R-flox: 12.60±0.47g, FIR: 16.65±0.35g, 24.3±1.6% decrease, P<0.05). IGF-1R-deficient mice had shorter aortas with decreased lumen area, cross-sectional vessel circumference and reduced area of tunica media compared to control mice. The brachiocephalic artery (BCA) had also decreased circumference and reduced medial area (Suppl.Table 1). SM22α-CreKI/IGF1R-flox mice had slightly higher weight gain during 12 weeks feeding with WD, however this effect was not statistically significant (Suppl.Fig.V). SM22α-CreKI/IGF1R-flox mice had food intake similar to control mice (SM22α-CreKI/IGF1R-flox: 140.1±15.3g/kg/day, FIR: 152.6±5.23g/kg/day, P=NS). Feeding mice with a WD for 12 weeks increased body weight in both groups of mice but SM22α-CreKI/IGF1R-flox mice still had a significant 15.8±1.2% decrease in body weight compared to controls.
In order to obtain further insights into vascular effects of SM22α-Cre mediated IGF-1R deficiency we identified SMC and FB in the vascular wall by immunostaining of cross-sections of ascending aorta with α-SMA antibody or with TR7 antibody (FB marker27), respectively. The adventitial layer was TR7-immunopositive but α-SMA-negative and vice versa, the medial layer was positive for α-SMA and negative for TR7 (Suppl.Fig.VI) indicating that these antibodies are suitable to distinguish SMC and FB in the vascular wall. SM22α-CreKI/IGF1R-flox mice had a thinner aortic wall (39.1±7.5% decrease compared to FIR mice, P<0.05) due to reduced thickness of both medial and adventitial layers (37.0±3.8% and 43.2±8.6% decrease compared to FIR mice, respectively, both are P<0.05). Although IGF-1R deficiency reduced medial area, it did not change either the number of layers in the media (data not shown) or SMC number (SM22α-CreKI/IGF1R-flox: 244.8±13.5 cells, FIR: 252.6±9.2 cells, P=NS) suggesting that SMC size might be smaller in SM22α-CreKI/IGF1R-flox mice. SM22α-CreKI/IGF1R-flox mice had reduced collagen in both media and in vascular adventitia (Fig.2A, B). We have shown that IGF-1 increased collagen in human SMC and in mouse FB via upregulation of collagen mRNA-binding La ribonucleoprotein domain family, member 6 (LARP6)28. LARP6 expression was significantly decreased in aortas and in adventitia in SM22α-CreKI/IGF1R-flox mice (>80% decrease compared to FIR mice) (Fig.2CE). LARP6 levels were also downregulated by 65–75% in embryonic, skin and lung FB isolated from SM22α-CreKI/IGF1R-flox mice (Fig.2DF). Neither IGF-1 receptor deficiency nor mouse feeding with a WD changed systolic blood pressure (Supp.Fig.IVD).
Figure 2. IGF-1R deficiency decreased vascular collagen (A, B) and downregulated LARP6 (C-F).
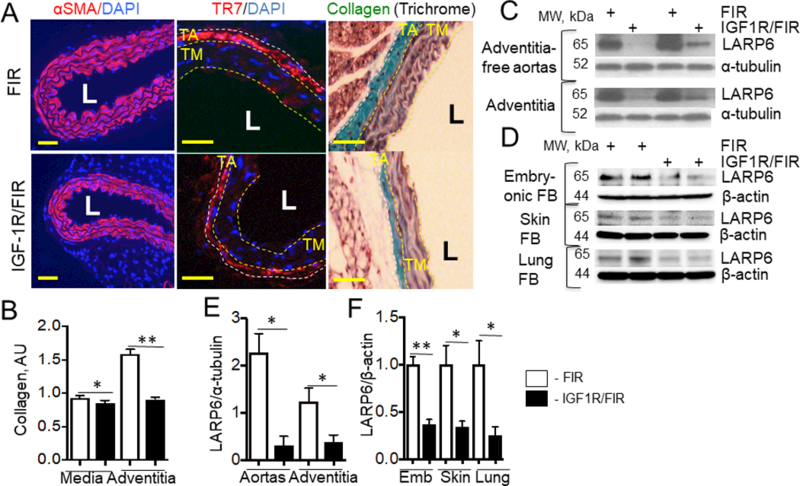
A, Cross-sections of descending aorta were stained for αSMA (SMC marker), TR7 (FB marker) and DAPI or stained for collagen (Trichrome) (n=6/group). LARP6 expression was quantified in adventitia-free aortas, adventitia (C, E) and in embryonic, skin and lung fibroblasts isolated from mouse tissues (D, F) (n=4/group). Yellow line, tunica media (TM), white line, tunica adventitia (TA). L – lumen, *P<0.05, **P<0.01. Scale bar, 10um.
IGF-1R deficiency increased atherosclerotic burden, upregulated plaque macrophages and decreased SMC and collagen.
We tested the effect of IGF-1R deficiency on atherosclerosis after 4 weeks and after 12 weeks of mice feeding with a WD. SM22α-CreKI/IGF1R-flox mice had a 3.2-fold increase in lipid accumulation in the aorta (en face analysis, 12 weeks feeding with WD) and increased atherosclerotic plaque cross-sectional area (CSA) after 4 and after 12 weeks of WD compared to control (Fig.3) indicating that IGF-1R deficiency promoted atherosclerosis development. We found no gender effect on atherosclerotic burden in SM22α-CreKI/IGF1R-flox or in FIR mice. Atherosclerotic plaques in SM22α-CreKI/IGF1R-flox mice fed for 12 weeks with WD were markedly less cellular than plaques in FIR mice (insert in Fig.3C); they contained large cholesterol crystals and had increased necrotic cores compared to control. SMC levels were decreased in atherosclerotic plaques in SM22α-CreKI/IGF1R-flox mice fed with WD for either 4 weeks (IHC for α-SMA: 3.1-fold decrease) or 12 weeks (IHC for α-SMA, 2.9-fold decrease; calponin, 2.8-fold decrease; smooth muscle myosin heavy chain, SM-MHC, 2.7-fold decrease compared to control) (Fig.4 and Suppl.Fig.VII). Atherosclerotic plaques in SM22α-CreKI/IGF1R-flox mice had a marked reduction in collagen (Fig.4A).
Figure 3. SM22α-creKI mediated IGF-1R deficiency increased atherosclerosis.
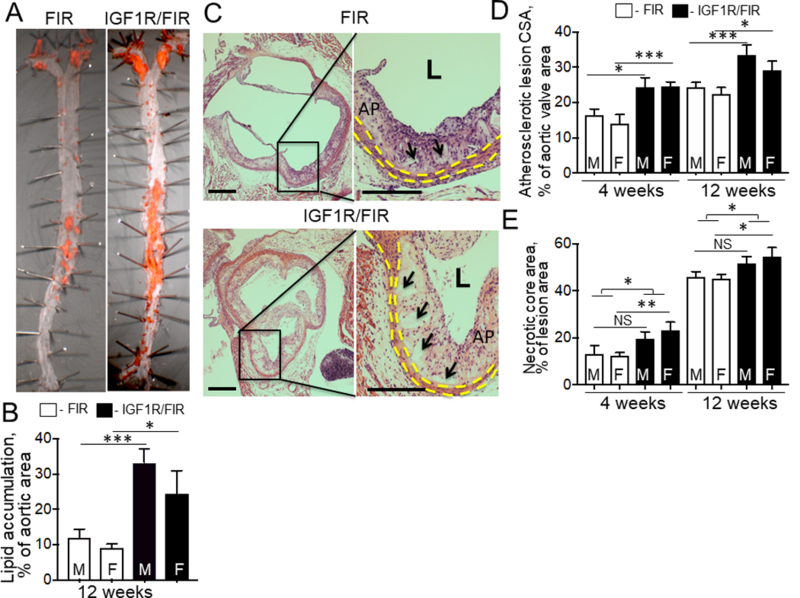
Mice were fed with a Western-type diet for 4 and 12 weeks. Lipid accumulation was measured by en face analysis (n=12/group) (A, B). Lesion cross-sectional area (CSA) was measured in H&E-stained aortic valve sections (n=14/group) (C, D). The necrotic core (C, insert) was identified as the acellular (hematoxylin-negative) part of the atherosclerotic plaque. Black arrows, necrotic cores. E, necrotic core area, quantitative data. Yellow dashed line, tunica media, AP, atherosclerotic plaque, L, lumen. *P<0.05, **P<0.01, ***P<0.005. M, males, F, females. There was no statistically significant difference between males and females within each group. Scale bar, 50um
Figure 4. IGF-1R deficiency reduced plaque SMC, collagen, increased macrophages, promoted cell apoptosis and decreased cell proliferation.
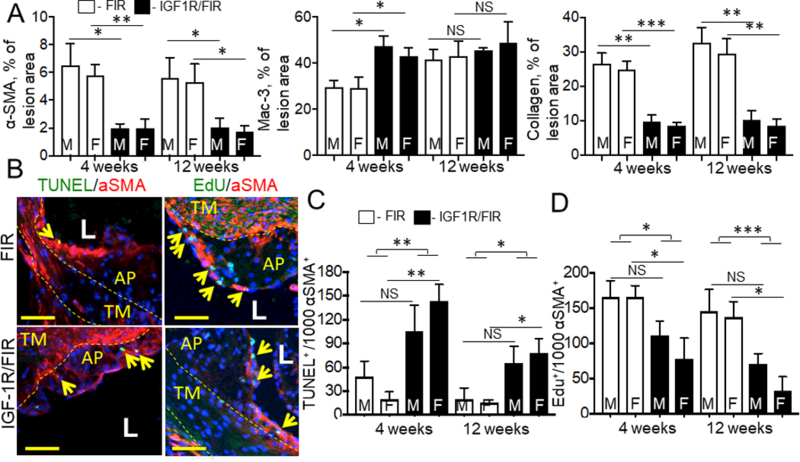
Aortic valve sections were stained for αSMA, Mac-3 (macrophage marker) or collagen (n=14/group) (A). B-D, Cell apoptosis and proliferation were assessed in αSMA-immunopositive plaque area and data shown per 103 αSMA+ plaque cells. B, representative images for mice fed with WD for 12 weeks. Yellow arrows, apoptotic (TUNEL+) or proliferating (Edu+) cells. Yellow dashed line, tunica media (TM), AP, atherosclerotic plaque, L, lumen. M, males, F, females. There was no statistically significant difference between males and females within each group *P<0.05, **P<0.01, ***P<0.005. Scale bar, 10 um.
SMC/FB-targeted IGF-1R deficiency significantly increased plaque macrophages after 4 weeks of feeding with WD (Fig.4A and Suppl.Fig.VIII) suggesting crosstalk between SMC and macrophages. To further study molecular mechanism we quantified expression of 86 chemokines and chemokine receptors in aortic SMC isolated from SM22α-CreKI/IGF1R-flox and FIR mice using real-time RT PCR array (Qiagen). We found the strong trend toward upregulation of Ackr4, Cxcr3, Cxcl12 (SDF-1α) and Xcl1 in IGF-1R-deficient SMC compared to FIR cells (Suppl.Table 3). We confirmed that Ackr4 and Cxcl12 were significantly upregulated in IGF-1R deficient SMC (10.8- and 6.0-fold overexpression, respectively) using real-time PCR (Suppl.Table 4). Cxcl12 (SDF-1α) was involved in regulating of recruitment of inflammatory cells29 and activation of macrophage phagocytosis30. We found that Cxcl12 (SDF-1α) expression was upregulated in αSMA-positive plaque cells in SM22α-CreKI/IGF1R-flox compared to FIR mice fed with WD for 4 weeks (2.1-fold increase, P<0.01) (Suppl.Fig.IX).
IGF-1R deficiency stimulated cell apoptosis, reduced cell proliferation and promoted inflammatory responses.
α-SMA is expressed by SMC and myofibroblasts in mouse atherosclerotic plaque31. Cell apoptosis and cell proliferation of α-SMA-positive cells were quantified by staining with TUNEL assay or with 5-ethynyl-2’-deoxyuridine assay (EdU), respectively. SM22α-CreKI/IGF1R-flox mice fed with WD either for 4 or 12 weeks had increased rates of apoptosis and reduced proliferation of α-SMA-positive cells in the atherosclerotic plaque compared to control (Fig.4B-D). Apoptosis and proliferation of α-SMA-negative cells were not changed in mice after 12 weeks of feeding with WD (data not shown). Serum levels of cytokines were quantified in mice fed with WD for 12 weeks using Multi-Analyte ELISA array. Expression of major pro-inflammatory cytokines IL-1β, IL-6 and IL-12 was upregulated in SM22α-CreKI/IGF1R-flox mice and levels of IL-4 and IL-10 (these cytokines can act as pro- and also as anti-inflammatory molecules32) were also increased (Suppl. Table 2). IGF-1R deficiency had no effect on circulating 8-isoprostane levels, a marker of systemic oxidative stress33 and did not change serum glucose and total cholesterol levels (Suppl.Fig.X).
IGF-1R deficiency reduced plaque fibrous cap, and increased necrotic cores.
The reduction in plaque SMC and collagen levels, and increase in necrotic cores in IGF-1R-deficient mice are consistent with an unstable plaque phenotype34. We and others have observed signs of former plaque rupture (such as intraplaque hemorrhage and elastin breaks) in the brachiocephalic artery (BCA)14,35,36. BCAs were dissected from SM22α-CreKI/IGF1R-flox and FIR mice and equally spaced cross-sections were stained for calponin to identify SMC-positive plaque fibrous cap (Fig.5). SM22α-CreKI/IGF1R-flox mice had reduced plaque fibrous cap area (53±14% decrease) and increased necrotic cores compared to control mice. Fibrous cap in IGF-1R-deficient mice was markedly thinner and contained multiple disruptions (Fig.5A, C). BCA plaques in IGF-1R-deficient mice contained reduced collagen levels compared to controls (SM22α-CreKI/IGF1R-flox: 13.1±6.4%, FIR, 48.5±9.3%, P<0.001). We detected minor fibrin deposition (2–3% of lesion area) in BCA plaques and IGF-1R-deficiency did not change fibrin levels (Suppl.Fig.XI). Oxidative stress in BCA plaques was quantified by immunostaining for N-tyrosine, a marker of protein oxidation37. IGF-1R deficiency did not change plaque N-tyrosine levels (Suppl.Fig.II). Consistent with data obtained for BCAs, SM22α-CreKI/IGF1R-flox mice had markedly thinner SMC-positive fibrous caps in the aortic valve plaques (SM22α-CreKI/IGF1R-flox: 3.98±0.55um, FIR, 9.48±0.81um, P<0.001, n=14/group).
Figure 5. IGF-1R deficiency reduced plaque fibrous cap, and increased necrotic cores in the brachiocephalic artery (BCA).
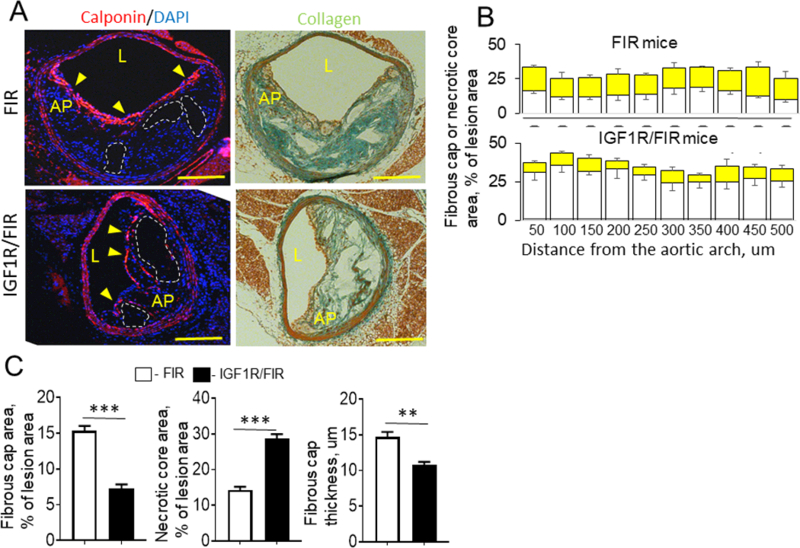
BCA were dissected from IGF-1R-deficient (n=10) and control mice (n=8) and 10 equally spaced sections were stained for calponin to identify SMC-positive plaque fibrous cap and serial sections were stained for collagen. A, Representative images (sections cut at 250 um from the aortic arch). B, Quantitative data for fibrous cap (yellow square) and necrotic core area (empty square). Data obtained for each BCA were averaged and shown as mean value ± SEM (C). AP, atherosclerotic plaque, L, lumen, yellow arrows, fibrous cap, white dashed line, necrotic cores, **P<0.01, ***P<0.005, Scale bar, 50um.
Aortic SMC isolated from SM22α-CreKI/IGF1R-flox mice have reduced proliferation, migration and increased apoptotic rates.
SM22α-CreKI/IGF1R-flox- and FIR-derived aortic SMC had similar basal viability as assessed by calcein-AM viability assay (data not shown). SM22α-CreKI/IGF1R-flox cells had a longer cell-doubling period (SM22α-CreKI/IGF1R-flox, 79±6h vs. FIR, 61±5h) and less SM22α-CreKI/IGF1R-flox cells were labeled with BrdU (BrdU ELISA) and with EdU (in vitro labeling) (Fig.6AB and Suppl.Fig.XII) suggesting reduced cell proliferation. SM22α-CreKI/IGF1R-flox cells had decreased ability to migrate spontaneously or toward chemoattractant (20% FBS) as established by a wound-healing (“scratch”) assay or by transmembrane migration assay with FluoroBlock inserts, respectively (Fig.6C). SM22α-CreKI/IGF1R-flox-derived SMC had increased sensitivity to apoptosis induced either by cell incubation in serum-free media or by exposure to the oxidant H2O2 (Fig.6D and Suppl.Fig.XII).
Figure 6. IGF-1R deficiency reduced SMC proliferation, decreased cell migration and increased apoptotic rates.
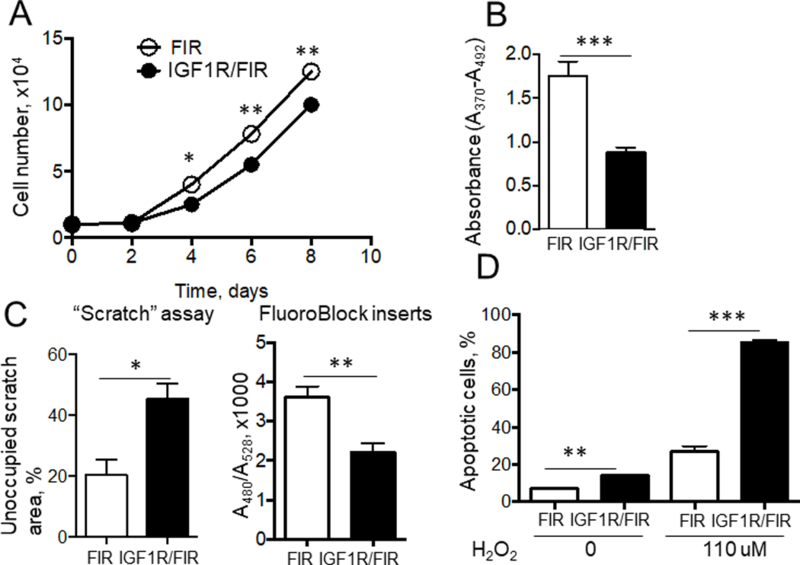
SMC were isolated from mouse aortas. Cell growth curves were obtained by counting cell number (A) and cell proliferation was assessed by DNA labeling with BrdU ELISA (B) (n=6/group). Cell migration was measured using a wound-healing (“scratch”) assay as the percentage of scratch area, which was left unoccupied (n=6/group). Chemo attractant-stimulated cell migration was quantified toward 20% FBS using FluoroBlock plate inserts (C). To quantify apoptosis, aortic SMC were incubated with/without H2O2 and stained with TUNEL assay (n=4/group) (D). *P<0.05, **P<0.01, ***P<0.005
Discussion
Here we report that SMC- and FB-targeted IGF-1R deficiency reduced vascular size and vascular collagen content, and induced growth retardation. IGF-1R deficiency increased atherosclerotic plaque burden and circulating inflammatory cytokines and these effects were associated with reduced proliferation and elevated apoptosis of α-SMA-positive plaque cells. SMC/FB-targeted decline in IGF-1 signaling decreased plaque SMC, increased necrotic core area and markedly depleted collagen. Consistent with these in vivo findings we found that aortic SMC isolated from mice with IGF-1R deficiency had disrupted IGF-1R-dependent downstream signaling, decreased cell proliferation, migration and increased sensitivity to apoptosis.
IGF-1 is a potent mitogen and pro-survival factor for vascular SMC and FB, and it also stimulates migration of SMC38 and FB39. Thus, a reduction in IGF-1 effects might be beneficial in certain pathologic conditions, such as restenosis, hypertension and the early stages of atherosclerotic plaque formation characterized by hypertrophy/hyperplasia of vascular SMC, but detrimental in other conditions in which loss of SMC contributes to the disease process, such as destabilization of atherosclerotic plaques40. To assess the role of SMC IGF-1 signaling on the atherosclerotic plaque we generated mice with IGF-1R deficiency induced by Cre recombinase under control of the SM22α promoter (SM22α-CreKI/IGF1R-flox mice). Cre recombinase expression and corresponding reduction in IGF-1R levels were detected in aorta and vascular adventitia in SM22α-CreKI/IGF1R-flox mice, indicating strong activity of the SM22α promoter in SMC- and in FB-rich tissues and this is consistent with reports showing SM22α expression in vascular SMC41 and also in adventitial FB42. Further, we found that embryonic, lung and skin FB isolated from SM22α-CreKI/IGF1R-flox mice expressed Cre and these cells also had reduced IGF-1R levels. SMC and FB play important roles in atherosclerosis via proliferation and migration to the subendothelial region where these cells undergo phenotypic switching: FB acquire additional SMC markers (including α-SMA) and SMC loose “classical” smooth muscle markers31. We have shown that SM22α immunopositivity was detected in plaque cells in WD-fed Apoe−/− mice and SM22α was co-localized with other markers of SMC43 indicating that the SM22α promoter is active in plaque SMC. Thus, SM22α-CreKI/IGF1R-flox mice are a valuable tool to investigate the effect of Cre recombinase-driven IGF-1R deficiency in both normal vascular wall SMC and FB and in SM22α expressing plaque cells. SM22α Cre knockin mice are widely used for the generation of murine models with vascular SMC-specific knockout25,26. Our finding that SM22α-CreKI/IGF1R-flox mice (which were derived from SM22α-CreKI+/+ mice16) expressed Cre in both SMC and in FB indicates the limited feasibility of using SM22α-CreKI+/+ mice for generation of SMC-specific knockouts. Some prior studies exploring SM22α-CreKI+/+ mice might be potentially re-evaluated in view of our current results.
The liver is the major source of circulating IGF-1, however almost every organ also produces IGF-1, thus there are endocrine (liver-derived) and autocrine/paracrine forms of IGF-144. There is debate about the relative role of endocrine vs. autocrine/paracrine IGF-1 in body development. Mice with conditional liver-specific IGF-1-knockout (with intact Igf1 gene locus in non-hepatic tissues) had 75% reduction in circulating IGF-1 without changes in body weight45 showing a dominant role of non-hepatic IGF-1 for normal growth and development. However, Stratikopoulos et al. have demonstrated that bi-transgenic mice lacking Igf1 gene expression in non-hepatic tissues and partially retaining liver IGF-1, had decreased circulating IGF-1 levels as well as markedly reduced body weight suggesting that liver-derived and locally produced IGF-1 have at least nearly equal importance for organismal growth46. We have shown that SMC isolated from SM22α-CreKI/IGF1R-flox mice had markedly reduced Akt, p38 MAPK and ERK1/2 phosphorylation indicating that autocrine IGF-1 signaling was inhibited in these cells. Our data are consistent with a critical role of local tissue-specific responses to IGF-1 for entire body development. To our knowledge this is the first report demonstrating the importance of autocrine/paracrine SMC/FB IGF-1 signaling for organismal growth. Of note, SM22α transcripts are expressed transiently in the embryonic heart (between embryonic day (E) 8.0 and E12.5), in skeletal muscle cells in the somites (E9.5-E12.5) and in vascular SMC beginning E9.5 and thereafter they continue to be expressed in all SMC into adulthood47,48. One can speculate that activation of the SM22α promoter during embryogenesis in SM22α-CreKI/IGF1R-flox mice contributed to mechanisms inducing growth retardation. Our data show that IGF-1R deficiency reduced overall vascular size, and in particular medial area, although the number of nuclei/media was unchanged, potentially suggesting a reduction in individual cell size. SM22α-CreKI/IGF1R-flox-derived aortic SMC had reduced proliferation rate and increased sensitivity to apoptosis and these mechanisms could contribute to growth retardation. Furthermore, intriguingly we found that SM22α promoter-driven deficiency of IGF-1 receptor reduced circulating IGF-1 levels. IGF-1R deficiency had no effect on IGF-1 production by the liver. We found no changes in expression of IGFBP-3 and ALS (these molecules form the ternary IGF-1/IGFBP-3/ALS complex, the primary stabilizer of circulating IGF-112), however IGFBP-5 levels were decreased in SM22α-CreKI/IGF1R-flox mice. IGFBP-5 is known to form an alternative complex with ALS in the presence of IGF-1, competing with IGFBP-349. IGFBP-5 overexpressing mice have increased circulating IGF-1 levels suggesting the ability of IGFBP-5 to regulate circulating IGF-1 levels50 and it is known that IGF-1 can also regulate IGFBP-5 levels51. It is thus difficult to know whether the reduction in circulating IGF-1 resulted in a decrease in IGFBP-5, or vice versa.
We have shown that IGF-1 exerts anti-atherogenic effects in Apoe−/− mice and IGF-1-induced reduction in atherosclerotic burden was associated with suppression of inflammatory responses, reduced cell apoptosis, decrease in plaque macrophages, increase in SMC, and vascular collagen6, 7, 52. In the current study we found that SM22α-CreKI-mediated loss of IGF-1 signaling increased atherosclerosis and cell apoptosis, reduced plaque SMC and collagen, increased plaque macrophages and potentiated inflammation. Since these effects are attributed to SMC and FB, our results demonstrate a key role of these cells in mediating IGF-1-induced atheroprotection. It is important to outline that increased sensitivity to apoptosis of SM22α-CreKI/IGF1R-flox-derived aortic SMC was associated with inhibition of IGF-1-induced Akt signaling. We have demonstrated previously that the anti-apoptotic function of the IGF-1 receptor was mediated by PI3-kinase/Akt signaling in human aortic SMC53. These in vitro data taken together with results of the current study are consistent with an important role of IGF-1/IGF-1R/Akt signaling for SMC resistance to apoptosis.
SMC-derived cytokines/chemokines have been reported to modulate macrophage recruitment and phenotype54,29. We have found that Cxcl12 (SDF-1α) chemokine was upregulated in aortic SMC isolated from SM22α-CreKI/IGF1R-flox mice and in αSMA+ plaque cells in these mice. Different cell types, including vascular SMCs and monocytes express Cxcl12 and its main receptor Cxcr455. Cxcl12/Cxcr4 axis has been implicated in recruitment of inflammatory cells29 and formation of macrophage-derived foam cells56. Plaque macrophages were identified in this study by immunohistochemistry for Mac-3 (CD107b), general macrophage marker57. Mac-3 is also expressed by some dendritic cells (DC)58. DC are known to express Cxcr4 and respond to Cxcl1259,60. DC play the important role in all stages of atherosclerosis including lipid uptake, antigen presentation, efferocytosis, and inflammation resolution61. We speculate that IGF-1R deficiency stimulated Cxcl12/Cxcr4 signaling pathway and this mechanism promoted recruitment of inflammatory cells and increased the number of Mac-3-positive plaque cells in SM22α-CreKI/IGF1R-flox mice. We have found that IGF-1R deficiency induced a relatively large increase in the number of apoptotic aSMA+ plaque cells (from 0.4% to 1.2% at 4 weeks of WD). It has been shown that an increase in SMC apoptosis (from 1.2% to 2.3% at 10 weeks of WD) was sufficient to increase plaque size in Apoe-null mice62. Therefore, our data suggest that elevated SMC apoptosis and an increase in plaque macrophages are responsible for increased atherosclerotic plaque burden seen in SM22α-CreKI/IGF1R-flox mice.
In advanced atherosclerotic plaques, a balance between cell death and survival of cells within the fibrous cap, primarily composed of vascular SMC and extracellular matrix, appears to correlate with plaque instability or stability62. We have shown that SMC-specific IGF-1 overexpression did not change atherosclerotic burden, however plaques in IGF-1 overexpressing mice had increased SMC levels, collagen content, and reduced necrotic cores suggesting that SMC-derived IGF-1 promoted plaque stability43. SMC-derived IGF-1 may act in an autocrine manner (on SMC) and in a paracrine manner on neighboring fibroblasts, monocytes/macrophages, endothelial cells and others. Our current study demonstrates that SMC and FB IGF-1 signaling plays a critical role in anti-atherosclerotic and plaque stabilizing effect of IGF-1.
IGF-1 upregulated collagen mRNA-binding protein LARP6 in cultured aortic SMC, mouse FB and also in vivo in Apoe−/− mice and LARP6 upregulation mediated IGF-1-induced increase in collagen28. In the current study we found that IGF-1R deficiency reduced α-SMA positive cells, increased necrotic cores and depleted collagen in atherosclerotic plaque suggesting decreased plaque stability. We found that SM22α-CreKI/IGF1R-flox mice had a reduced fibrous cap area, increased necrotic cores and thinner fibrous caps consistent with the unstable plaque phenotype. It is important to note that these effects correlated with marked downregulation of LARP6 in aortas, vascular adventitia and also in embryonic, skin and lung FB. Taken together these findings established the potential link between SMC/FB LARP6 and plaque stability and justify further investigation into the role of LARP6 in promoting plaque stabilization.
In summary, we assessed effects of SM22α-CreKI mediated IGF-1R deficiency on vascular wall morphology and atherosclerotic plaque development. A decline in SMC and FB IGF-1 signaling reduced overall vascular size, decreased SMC proliferation, increased apoptotic rates and furthermore resulted in mouse growth retardation. The high-fat diet fed SM22α-CreKI/IGF1R-flox mice had increased inflammatory responses and atherosclerotic burden. Atherosclerotic plaques in SM22α-CreKI/IGF1R-flox mice had larger necrotic cores, increased macrophages, decreased SMC levels, markedly depleted collagen inducating that IGF-1R downregulation potentially compromised plaque stability. Our data demonstrate the important role of IGF-1 signaling in SMC and in FB for normal vascular wall development and for prevention of atherogenesis.
Supplementary Material
Highlights.
IGF-1R deficiency in smooth muscle cells and fibroblasts reduced overall vascular size, downregulated vascular collagen, and induced growth retardation.
Mice with SMC/FB-targeted IGF-1R deficiency had increased atherosclerotic plaque burden and inflammatory responses.
SMC/FB-targeted decline in IGF-1 signaling decreased plaque SMC, and markedly depleted collagen.
Aortic SMC isolated from IGF-1R-deficient mice had decreased cell proliferation, migration, increased sensitivity to apoptosis and these effects were associated with disruption of IGF-1-induced Akt signaling.
Acknowledgements
We thank Dr. Eugene Chen (Medical School, University of Michigan) for providing SM22α-CreKI+/+ mice and Dr. Jens Brüning (University of Leipzig, Germany) for mice with LoxP-flanked IGF-1 receptor. We thank to Patricia Lobelle-Rich (Tulane University, New Orleans) for our lab management and support. The authors are grateful for the support provided by the Molecular Cytology Core of University of Missouri.
Sources of Funding
This work was supported by grants from the NIH R21-HL113705 (SS), R01-HL070241 (PD), RO1-HL080682 (PD), 1-U54 GM104940 (PD), American Heart Association 13GRNT17230069 (SS), American Diabetes Association 1–13-BS-210 (CW), the Japan Society for the Promotion of Science S#25221204 (SIT) and Veterans Affairs VA-I01-BX002255 and Research Career Scientist grants (BC).
Abbreviations:
- SMC
smooth muscle cells
- IGF-1
insulin-like growth factor 1
- IGF-1R
insulin-like growth factor 1 receptor
- CVD
cardiovascular disease
- IRS
insulin receptor substrate
- MAPK
mitogen-activated protein kinase
- IGFBP
insulin-like growth factor binding protein
- FB
fibroblasts
- SM22α
smooth muscle protein 22 α
- GH
growth hormone
- ALS
acid-labile subunit
- WD
Western-type diet
- α-SMA
α-smooth muscle cell actin
- LARP6
La ribonucleoprotein domain family, member 6
- CSA
cross-sectional area
- TUNEL
Terminal deoxynucleotidyl transferase dUTP Nick-End Labeling
- EdU
5-ethynyl-2’-deoxyuridine
Footnotes
Disclosures
None
References
- 1.Puche JE and Castilla-Cortazar I. Human conditions of insulin-like growth factor-I (IGF-I) deficiency. J Transl Med 2012;10:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamberts SW, van den Beld AW and van der Lely AJ. The endocrinology of aging. Science 1997;278:419–24. [DOI] [PubMed] [Google Scholar]
- 3.Higashi Y, Sukhanov S, Anwar A, Shai SY and Delafontaine P. Aging, atherosclerosis, and IGF-1. J Gerontol A Biol Sci Med Sci 2012;67:626–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C and Stroke Statistics S. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 5.Ambrose JA, Tannenbaum MA, Alexopoulos D, Hjemdahl-Monsen CE, Leavy J, Weiss M, Borrico S, Gorlin R and Fuster V. Angiographic progression of coronary artery disease and the development of myocardial infarction. J Am Coll Cardiol 1988;12:56–62. [DOI] [PubMed] [Google Scholar]
- 6.Sukhanov S, Higashi Y, Shai SY, Vaughn C, Mohler J, Li Y, Song YH, Titterington J and Delafontaine P. IGF-1 reduces inflammatory responses, suppresses oxidative stress, and decreases atherosclerosis progression in ApoE-deficient mice. Arterioscler Thromb Vasc Biol 2007;27:2684–90. [DOI] [PubMed] [Google Scholar]
- 7.Higashi Y, Sukhanov S, Anwar A, Shai SY and Delafontaine P. IGF-1, oxidative stress and atheroprotection. Trends Endocrinol Metab 2010;21:245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.von der Thusen JH, Borensztajn KS, Moimas S, van Heiningen S, Teeling P, van Berkel TJ and Biessen EA. IGF-1 has plaque-stabilizing effects in atherosclerosis by altering vascular smooth muscle cell phenotype. Am J Pathol 2011;178:924–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawachi S, Takeda N, Sasaki A, Kokubo Y, Takami K, Sarui H, Hayashi M, Yamakita N and Yasuda K. Circulating insulin-like growth factor-1 and insulin-like growth factor binding protein-3 are associated with early carotid atherosclerosis. Arterioscler Thromb Vasc Biol 2005;25:617–21. [DOI] [PubMed] [Google Scholar]
- 10.Kaplan RC, Strickler HD, Rohan TE, Muzumdar R and Brown DL. Insulin-like growth factors and coronary heart disease. Cardiol Rev 2005;13:35–9. [DOI] [PubMed] [Google Scholar]
- 11.Juul A, Scheike T, Davidsen M, Gyllenborg J and Jorgensen T. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation 2002;106:939–44. [DOI] [PubMed] [Google Scholar]
- 12.Delafontaine P, Song YH and Li Y. Expression, regulation, and function of IGF-1, IGF-1R, and IGF-1 binding proteins in blood vessels. Arterioscler Thromb Vasc Biol 2004;24:435–44. [DOI] [PubMed] [Google Scholar]
- 13.Okura Y, Brink M, Itabe H, Scheidegger KJ, Kalangos A and Delafontaine P. Oxidized low-density lipoprotein is associated with apoptosis of vascular smooth muscle cells in human atherosclerotic plaques. Circulation 2000;102:2680–6. [DOI] [PubMed] [Google Scholar]
- 14.Higashi Y, Sukhanov S, Shai SY, Danchuk S, Tang R, Snarski P, Li Z, Lobelle-Rich P, Wang M, Wang D, Yu H, Korthuis R and Delafontaine P. Insulin-Like Growth Factor-1 Receptor Deficiency in Macrophages Accelerates Atherosclerosis and Induces an Unstable Plaque Phenotype in Apolipoprotein E-Deficient Mice. Circulation 2016;133:2263–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Patel S, Shi Y, Niculescu R, Chung EH, Martin JL and Zalewski A. Characteristics of coronary smooth muscle cells and adventitial fibroblasts. Circulation 2000;101:524–32. [DOI] [PubMed] [Google Scholar]
- 16.Zhang J, Zhong W, Cui T, Yang M, Hu X, Xu K, Xie C, Xue C, Gibbons GH, Liu C, Li L and Chen YE. Generation of an adult smooth muscle cell-targeted Cre recombinase mouse model. Arterioscler Thromb Vasc Biol 2006;26:e23–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stachelscheid H, Ibrahim H, Koch L, Schmitz A, Tscharntke M, Wunderlich FT, Scott J, Michels C, Wickenhauser C, Haase I, Bruning JC and Niessen CM. Epidermal insulin/IGF-1 signalling control interfollicular morphogenesis and proliferative potential through Rac activation. EMBO J 2008;27:2091–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saeed H, Taipaleenmaki H, Aldahmash AM, Abdallah BM and Kassem M. Mouse embryonic fibroblasts (MEF) exhibit a similar but not identical phenotype to bone marrow stromal stem cells (BMSC). Stem Cell Rev 2012;8:318–28. [DOI] [PubMed] [Google Scholar]
- 19.Seluanov A, Vaidya A and Gorbunova V. Establishing primary adult fibroblast cultures from rodents. J Vis Exp 2010. [DOI] [PMC free article] [PubMed]
- 20.Sorensen I, Susnik N, Inhester T, Degen JL, Melk A, Haller H and Schmitt R. Fibrinogen, acting as a mitogen for tubulointerstitial fibroblasts, promotes renal fibrosis. Kidney Int 2011;80:1035–44. [DOI] [PubMed] [Google Scholar]
- 21.Salic A and Mitchison TJ. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc Natl Acad Sci U S A 2008;105:2415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liang CC, Park AY and Guan JL. In vitro scratch assay: a convenient and inexpensive method for analysis of cell migration in vitro. Nat Protoc 2007;2:329–33. [DOI] [PubMed] [Google Scholar]
- 23.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B and Council on Basic Cardiovascular S. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 24.Robinet P, Milewicz DM, Cassis LA, Leeper NJ, Lu HS and Smith JD. Consideration of Sex Differences in Design and Reporting of Experimental Arterial Pathology Studies-Statement From ATVB Council. Arterioscler Thromb Vasc Biol 2018;38:292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Holtwick R, Gotthardt M, Skryabin B, Steinmetz M, Potthast R, Zetsche B, Hammer RE, Herz J and Kuhn M. Smooth muscle-selective deletion of guanylyl cyclase-A prevents the acute but not chronic effects of ANP on blood pressure. Proc Natl Acad Sci U S A 2002;99:7142–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hassane S, Claij N, Jodar M, Dedman A, Lauritzen I, Duprat F, Koenderman JS, van der Wal A, Breuning MH, de Heer E, Honore E, DeRuiter MC and Peters DJ. Pkd1-inactivation in vascular smooth muscle cells and adaptation to hypertension. Lab Invest 2011;91:24–32. [DOI] [PubMed] [Google Scholar]
- 27.Mathur A, Kumar A, Babu B and Chandna S. In vitro mesenchymal-epithelial transition in NIH3T3 fibroblasts results in onset of low-dose radiation hypersensitivity coupled with attenuated connexin-43 response. Biochim Biophys Acta 2018;1862:414–426. [DOI] [PubMed] [Google Scholar]
- 28.Blackstock CD, Higashi Y, Sukhanov S, Shai SY, Stefanovic B, Tabony AM, Yoshida T and Delafontaine P. Insulin-like growth factor-1 increases synthesis of collagen type I via induction of the mRNA-binding protein LARP6 expression and binding to the 5’ stem-loop of COL1a1 and COL1a2 mRNA. J Biol Chem 2014;289:7264–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S and Weiser-Evans MC. SDF-1alpha induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol 2011;31:1300–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ma W, Liu Y, Ellison N and Shen J. Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J Biol Chem 2013;288:15481–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gomez D and Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res 2012;95:156–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhao XN, Li YN and Wang YT. Interleukin-4 regulates macrophage polarization via the MAPK signaling pathway to protect against atherosclerosis. Genet Mol Res 2016;15. [DOI] [PubMed] [Google Scholar]
- 33.Montuschi P, Barnes PJ and Roberts LJ 2nd. Isoprostanes: markers and mediators of oxidative stress. Faseb J 2004;18:1791–800. [DOI] [PubMed] [Google Scholar]
- 34.Finn AV, Nakano M, Narula J, Kolodgie FD and Virmani R. Concept of vulnerable/unstable plaque. Arterioscler Thromb Vasc Biol 2010;30:1282–92. [DOI] [PubMed] [Google Scholar]
- 35.Gough PJ, Gomez IG, Wille PT and Raines EW. Macrophage expression of active MMP-9 induces acute plaque disruption in apoE-deficient mice. J Clin Invest 2006;116:59–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matoba T, Sato K and Egashira K. Mouse models of plaque rupture. Curr Opin Lipidol 2013;24:419–25. [DOI] [PubMed] [Google Scholar]
- 37.Darwish RS, Amiridze N and Aarabi B. Nitrotyrosine as an oxidative stress marker: evidence for involvement in neurologic outcome in human traumatic brain injury. J Trauma 2007;63:439–42. [DOI] [PubMed] [Google Scholar]
- 38.Arnqvist HJ, Bornfeldt KE, Chen Y and Lindstrom T. The insulin-like growth factor system in vascular smooth muscle: interaction with insulin and growth factors. Metabolism 1995;44:58–66. [DOI] [PubMed] [Google Scholar]
- 39.Gross SM and Rotwein P. Unraveling Growth Factor Signaling and Cell Cycle Progression in Individual Fibroblasts. J Biol Chem 2016;291:14628–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Libby P and Aikawa M. Stabilization of atherosclerotic plaques: new mechanisms and clinical targets. Nat Med 2002;8:1257–62. [DOI] [PubMed] [Google Scholar]
- 41.Shanahan CM, Weissberg PL and Metcalfe JC. Isolation of gene markers of differentiated and proliferating vascular smooth muscle cells. Circ Res 1993;73:193–204. [DOI] [PubMed] [Google Scholar]
- 42.Faggin E, Puato M, Zardo L, Franch R, Millino C, Sarinella F, Pauletto P, Sartore S and Chiavegato A. Smooth muscle-specific SM22 protein is expressed in the adventitial cells of balloon-injured rabbit carotid artery. Arterioscler Thromb Vasc Biol 1999;19:1393–404. [DOI] [PubMed] [Google Scholar]
- 43.Shai SY, Sukhanov S, Higashi Y, Vaughn C, Kelly J and Delafontaine P. Smooth muscle cell-specific insulin-like growth factor-1 overexpression in Apoe−/− mice does not alter atherosclerotic plaque burden but increases features of plaque stability. Arterioscler Thromb Vasc Biol 2010;30:1916–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Delafontaine P Insulin-like growth factor I and its binding proteins in the cardiovascular system. Cardiovasc Res 1995;30:825–34. [PubMed] [Google Scholar]
- 45.Yakar S, Liu JL, Stannard B, Butler A, Accili D, Sauer B and LeRoith D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc Natl Acad Sci U S A 1999;96:7324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stratikopoulos E, Szabolcs M, Dragatsis I, Klinakis A and Efstratiadis A. The hormonal action of IGF1 in postnatal mouse growth. Proc Natl Acad Sci U S A 2008;105:19378–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li L, Miano JM, Cserjesi P and Olson EN. SM22 alpha, a marker of adult smooth muscle, is expressed in multiple myogenic lineages during embryogenesis. Circ Res 1996;78:188–95. [DOI] [PubMed] [Google Scholar]
- 48.Zhang JC, Kim S, Helmke BP, Yu WW, Du KL, Lu MM, Strobeck M, Yu Q and Parmacek MS. Analysis of SM22alpha-deficient mice reveals unanticipated insights into smooth muscle cell differentiation and function. Mol Cell Biol 2001;21:1336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Twigg SM and Baxter RC. Insulin-like growth factor (IGF)-binding protein 5 forms an alternative ternary complex with IGFs and the acid-labile subunit. J Biol Chem 1998;273:6074–9. [DOI] [PubMed] [Google Scholar]
- 50.Salih DA, Tripathi G, Holding C, Szestak TA, Gonzalez MI, Carter EJ, Cobb LJ, Eisemann JE and Pell JM. Insulin-like growth factor-binding protein 5 (Igfbp5) compromises survival, growth, muscle development, and fertility in mice. Proc Natl Acad Sci U S A 2004;101:4314–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Beattie J, Allan GJ, Lochrie JD and Flint DJ. Insulin-like growth factor-binding protein-5 (IGFBP-5): a critical member of the IGF axis. Biochem J 2006;395:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sukhanov S, Higashi Y, Shai SY, Blackstock C, Galvez S, Vaughn C, Titterington J and Delafontaine P. Differential requirement for nitric oxide in IGF-1-induced anti-apoptotic, anti-oxidant and anti-atherosclerotic effects. FEBS Lett 2011;585:3065–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Y, Higashi Y, Itabe H, Song YH, Du J and Delafontaine P. Insulin-like growth factor-1 receptor activation inhibits oxidized LDL-induced cytochrome C release and apoptosis via the phosphatidylinositol 3 kinase/Akt signaling pathway. Arterioscler Thromb Vasc Biol 2003;23:2178–84. [DOI] [PubMed] [Google Scholar]
- 54.Ostriker A, Horita HN, Poczobutt J, Weiser-Evans MC and Nemenoff RA. Vascular smooth muscle cell-derived transforming growth factor-beta promotes maturation of activated, neointima lesion-like macrophages. Arterioscler Thromb Vasc Biol 2014;34:877–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van der Vorst EP, Doring Y and Weber C. Chemokines and their receptors in Atherosclerosis. J Mol Med (Berl) 2015;93:963–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tanaka G, Nakase I, Fukuda Y, Masuda R, Oishi S, Shimura K, Kawaguchi Y, Takatani-Nakase T, Langel U, Graslund A, Okawa K, Matsuoka M, Fujii N, Hatanaka Y and Futaki S. CXCR4 stimulates macropinocytosis: implications for cellular uptake of arginine-rich cell-penetrating peptides and HIV. Chem Biol 2012;19:1437–46. [DOI] [PubMed] [Google Scholar]
- 57.Ho MK and Springer TA. Tissue distribution, structural characterization, and biosynthesis of Mac-3, a macrophage surface glycoprotein exhibiting molecular weight heterogeneity. J Biol Chem 1983;258:636–42. [PubMed] [Google Scholar]
- 58.Flotte TJ, Springer TA and Thorbecke GJ. Dendritic cell and macrophage staining by monoclonal antibodies in tissue sections and epidermal sheets. Am J Pathol 1983;111:112–24. [PMC free article] [PubMed] [Google Scholar]
- 59.Kabashima K, Shiraishi N, Sugita K, Mori T, Onoue A, Kobayashi M, Sakabe J, Yoshiki R, Tamamura H, Fujii N, Inaba K and Tokura Y. CXCL12-CXCR4 engagement is required for migration of cutaneous dendritic cells. Am J Pathol 2007;171:1249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Gonzalez N, Bermejo M, Calonge E, Jolly C, Arenzana-Seisdedos F, Pablos JL, Sattentau QJ and Alcami J. SDF-1/CXCL12 production by mature dendritic cells inhibits the propagation of X4-tropic HIV-1 isolates at the dendritic cell-T-cell infectious synapse. J Virol 2010;84:4341–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Subramanian M and Tabas I. Dendritic cells in atherosclerosis. Semin Immunopathol 2014;36:93–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clarke MC, Figg N, Maguire JJ, Davenport AP, Goddard M, Littlewood TD and Bennett MR. Apoptosis of vascular smooth muscle cells induces features of plaque vulnerability in atherosclerosis. Nat Med 2006;12:1075–80. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.