

SUMMARY
Beige adipocytes are present in white adipose tissue (WAT) and have thermogenic capacity to orchestrate substantial energy metabolism and counteract obesity. However, adipocyte-derived signals that act on progenitor cells to control beige adipogenesis remain poorly defined. Here, we show that adipose-specific depletion of Raptor, a key component of mTORC1, promoted beige adipogenesis through prostaglandins (PGs) synthesized by cyclooxygenase-2 (COX-2). Moreover, Raptor-deficient mice were resistant to diet-induced obesity and COX-2 downregulation. Mechanistically, mTORC1 suppressed COX-2 by phosphorylation of CREB-regulated transcription coactivator 2 (CRTC2) and subsequent dissociation of CREB to cox-2 promoter in adipocytes. PG treatment stimulated PKA and promoted differentiation of progenitor cells to beige adipocytes in culture. Ultimately, we show that pharmacological inhibition or suppression of COX-2 attenuated mTORC1 inhibition-induced thermogenic gene expression in inguinal WAT in vivo and in vitro. Our study identifies adipocyte-derived PGs as key regulators of white adipocyte browning, which occurs through mTORC1 and CRTC2.
In Brief
Beige adipocytes, which develop in white adipose tissue (WAT), have become a promising avenue to counteract obesity. However, the repertoire of extracellular signals that control beige adipogenesis remains largely unknown. Here, Zhang et al. show that COX-2-mediated prostaglandins act as paracrine signals that orchestrate beige adipogenesis and are controlled by the mTORC1/CRTC2 pathway.
Graphical Abstract
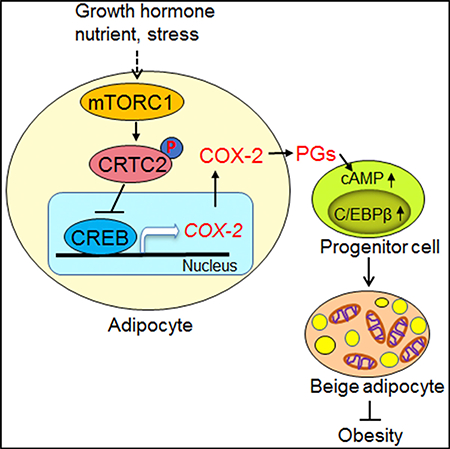
INTRODUCTION
Recruitment and activation of beige adipocytes (the “browning” effect) may have potential therapeutic implications for the treatment of obesity and related disorders such as insulin resistance, type 2 diabetes, and cardiovascular diseases (Boström et al., 2012; Cohen et al., 2014; Harms and Seale, 2013; Seale et al., 2011; Vegiopoulos et al., 2010). However, while activation of the β3-adrenoceptor signaling pathway promotes thermogenic gene expression in brown and beige adipocytes in rodents, this approach is clinically not feasible due to the low efficacy of the β3-adrenoceptor signaling agonists in humans (Boström et al., 2012; Cristancho and Lazar, 2011; Farmer, 2008; Fisher et al., 2012; White and Stephens, 2010). Thus, identifying alternative drug targets that specifically and effectively promote beige adipogenesis is critical for the treatment of obesity and associated metabolic diseases.
Cyclooxygenase (COX), a rate-limiting enzyme responsible for the biosynthesis of prostaglandins (PGs), exists in two isoforms: COX-1, the constitutive form, and COX-2, the inducible form (Marnett et al., 1999). COX-2 oxygenates arachidonic acid and converts it into a number of PGs, including PGD2, PGE2, PGF2α and prostacyclin (PGI2), all of which exert diverse hormone-like effects via autocrine or paracrine mechanisms (Marnett et al., 1999). In adipose tissue (AT), the COX-2/PG pathway is induced by cold exposure and plays a critical role in coldinduced beige/brite adipocyte formation and browning of white adipose tissue (WAT) (Bayindir et al., 2015; Vegiopoulos et al., 2010). In agreement with this, PGs shift the differentiation of progenitors toward a brown adipocyte phenotype (Bayindir et al., 2015; Vegiopoulos et al., 2010). The COX-2/PG axis also plays a critical role in regulating adipose tissue inflammation and obesity-induced insulin resistance (Chan et al., 2016; Gartung et al., 2016; Hsieh et al., 2010; Hu et al., 2016; Vegiopoulos et al., 2010). However, whether or not the COX-2/PG pathway is involved in the development of obesity remains an open question given that a 25-week high-fat diet (HFD) feeding had no significant effect on COX-2 expression in adipose tissue (Vegiopoulos et al., 2010). In addition, COX-2 is expressed constitutively in adipocytes as well as a variety of immune cells residing in adipose tissue such as macrophages, neutrophils, and T cells (Iñiguez et al., 1999; Kim et al., 2001; Pablos et al., 1999; Pfannkuche et al., 1986). This raises important questions as to whether adipocyte-derived PGs play a role in regulating adipose thermogenesis and maintaining energy homeostasis, and how PG biosynthesis is regulated in adipocytes.
The mechanistic targets of rapamycin (mTOR), an intracellular energy sensor that integrates distinct signals such as hormones, nutrients, and stress, is a vital regulator of multiple cellular processes such as protein translation, lipid metabolism, cell growth, and survival (Wullschleger et al., 2006). mTOR exists in two distinct complexes, mTORC1 and mTORC2, which differ in subunit compositions and biological function (Loewith et al., 2002). mTORC1 contains the rapamycin-associated TOR protein (Raptor), while mTORC2 contains the rapamycin-insensitive companion of mTOR (Rictor) (Loewith et al., 2002). Raptor is also a regulatory-associated protein of mTOR that binds to and mediates mTOR action (Hara et al., 2002). Loss of Raptor eliminates mTORC1 activity irrespective of feeding status (Sengupta et al., 2010). mTORC1 is understood to regulate multiple metabolic processes including protein synthesis, lipogenesis, energy expenditure, and autophagy (Laplante and Sabatini, 2012; Wullschleger et al., 2006) and is highly active in the adipose tissue of obese and HFD-fed rodents (Khamzina et al., 2005; Tremblay et al., 2005). Furthermore, over-activation of mTORC1 results in the development of adiposity and obesity (Carnevalli et al., 2010; Laplante and Sabatini, 2012; Polak et al., 2008; Um et al., 2004; Zhang et al., 2009). However, accumulating evidence has raised questions regarding its role in adipose tissue thermogenesis (Labbé et al., 2016; Lee et al., 2016; Liu et al., 2014, 2016; Polak et al., 2008; Shan et al., 2016; Tran et al., 2016; Um et al., 2004; Wada et al., 2016). Several studies agree that adipose inhibition of mTORC1 leads to browning of WAT and that overactivation of mTORC1 in adipose tissue suppresses thermogenesis and exacerbates diet-induced obesity and insulin resistance in mice (Liu et al., 2014; Polak et al., 2008; Um et al., 2004; Wada et al., 2016). However, other studies report that inactivation of mTORC1 in adipose tissue results in impaired thermogenesis (Liu et al., 2016; Tran et al., 2016). While the work from Lee, Shan, and colleagues suggests an inhibitory effect of mTOR signaling on WAT browning, both studies argue that adipose-specific inhibition of mTOR or mTORC1 results in lipodystrophy and systemic insulin resistance during postnatal growth (Labbé et al., 2016; Lee et al., 2016; Shan et al., 2016). In comparison to the well-studied physiological role of mTORC1 in adipose tissue, the mechanisms by which mTORC1 regulates adipose thermogenesis remain largely unknown.
Here, we show that adipose-specific knockout of Raptor induces COX-2 expression and PG biosynthesis, leading to beige adipogenesis in WAT. In support of this, activation of the mTORC1 signaling pathway inhibits COX-2 expression and biosynthesis of PGs including PGI2, PGD2, and PGE2. In adipocytes, mTORC1 inhibits transcription of COX-2 through phosphorylating cyclic-AMP-responsive element-binding protein (CREB)-regulated transcription coactivator 2 (CRTC2) at Ser136. Moreover, inhibition of COX-2 blunts the effect of mTORC1 on beige adipocyte differentiation in vivo. Collectively, our study reveals that adipocyte mTORC1 is a key negative regulator of beige adipocyte development via a CRTC2/COX-2/PGdependent paracrine mechanism and that activation of adipocyte COX-2/PG signaling protects against diet-induced obesity.
RESULTS
Raptor Deficiency Has Differential Effects on Thermogenic Capacity in WAT and Brown Adipose Tissue
To address the controversy regarding the role of mTORC1 in regulating thermogenesis, we generated adipose-specific Raptor knockout (KO) mice using adiponectin cre mice. Consistent with the previous finding that adipose-specific KO of Raptor impairs adipose tissue development during postnatal growth (Laplante and Sabatini, 2012; Lee et al., 2016; Liu et al., 2016; Tran et al., 2016), we observed that 10-week-old Raptor KO mice displayed less fat mass and exhibited lipodystrophy, liver steatosis, and insulin resistance under normal chow diet conditions despite similar body mass (Figures S1A– S1F). In agreement with the findings of others (Lee et al., 2016; Liu et al., 2016; Tran et al., 2016), Raptor deficiency significantly decreased levels of thermogenic markers UCP1, PGC1α, and C/EBPβ as well as adipogenic markers PPARγ and adiponectin in brown adipose tissue (BAT) accompanied by the occurrence of large lipid droplets in this tissue (Figures 1A and 1C). However, in inguinal WAT (iWAT) and epididymal WAT (eWAT), adipose-specific KO of Raptor led to a robust induction of UCP1 and C/EBPβ and development of multilocular lipid droplets despite suppression of adiponectin expression (Figures 1B, 1C–1E, and S1G). Consistent with this, the mRNA levels of ucp1 and C/EBPβ were significantly upregulated in iWAT of Raptor-deficient mice (Figure 1F). Although beige markers including CD137, TBX1, and TMEM26 as well as UCP1-independent thermogenesis markers SERCA2b and CKMT2 were not significantly affected despite downregulation of RyR2 and GATM, other brown markers including Coxt11, FGF21, and Kng2 were all upregulated in the iWAT of Raptor KO animals (Figure 1F), suggesting that mTORC1 signaling pathway plays distinct roles in regulating thermogenic gene expression in BAT and WAT. In agreement with this, inguinal fat of Raptor KO mice displayed a 2-fold greater O2 consumption compared to controls (Figure 1G). In contrast, the brown fat of the Raptor KO mice showed decreased O2 consumption (Figure 1G). Unexpectedly, Raptor deficiency had no significant effect on differentiation (Figure S1H), basal and β3-adrenegic signaling-induced UCP1, as well as the expression of PPARγ and adiponectin (Figure S1I), or O2 consumption (Figures S1J and S1K) in primary differentiated adipocytes from iWAT and BAT. Interestingly, despite no significant difference in Raptor expression, the stromal vascular fraction (SVF) of iWAT in Raptor KO animals displayed a notable upregulation of UCP1 compared to control samples (Figures 1H and S1L). Moreover, UCP1 levels were increased in the SVF to a markedly greater extent than in the adipocyte fraction (Figures 1H and S1L). Taken together, these results strongly suggest that the differential effects of mTORC1 inhibition on adipose tissue development and thermogenesis in WAT and BAT is mediated via a non-cell-autonomous mechanism.
Figure 1. Raptor Deficiency Has Differential Effects on Thermogenesis in WAT and BAT.

(A) The expression levels of the thermogenic andadipogenic markers UCP1, C/EBPβ, PGC1α, PPARγ, and adiponectin were suppressed by Raptor deficiency in interscapular BAT at room temperature.</P/>ADPN, adiponectin; TH, tyrosine hydroxylase.
(B) The expression levels of the thermogenic andadipogenic markers UCP1, C/EBPβ, but not PPARγ and PGC1α were induced in inguinal fat (iWAT) of Raptor-deficient mice.
(C) H&E staining of eWAT, iWAT, BAT, and liver from 10-week-old male Raptor-deficient and control mice.
(D) Quantification of adipocyte size in iWAT andeWAT of Raptor KO and control mice. Size of adipocytes were determined with ImageJ analysis of H&E-stained tissues.
(E) Immunohistochemistry (IHC) staining of UCP1 iniWAT and BAT from 10-week-old Raptor-deficient and control mice.
(F) The mRNA levels of thermogenic, beige, andbrown markers in inguinal fat of Raptor-deficient and control mice.
(G) O2 consumption of inguinal and brown fat from 10-week-old Raptor-deficient and control mice. Volume of O2/min/μg tissue was used as the unit for O2 consumption. The data are presented as mean ± SEM.
(H) The increase of UCP1 levels by Raptor deficiency was greater in the stromal vascular fraction than in adipocyte fraction in iWAT compared to the control samples. Adip, adipocyte fraction; AT, adipose tissue; SVF, stromal vascular fraction. The data in (D)–(F) are presented as the mean ± SEM. *p < 0.05, **p < 0.01.
Raptor Deficiency-Induced Browning of WAT Does Not Require Sympathetic Tone
It has previously been shown that impaired BAT function promotes the compensatory browning of WAT by enhancing sympathetic input to WAT (Schulz et al., 2013; Shin et al., 2017). To determine whether Raptor deficiency-induced beiging of iWAT is caused by cross talk between BAT and WAT, we examined the levels of tyrosine hydroxylase (TH), a key enzyme of catecholamine biosynthesis driven by sympathetic tone. The expression of TH was not significantly affected by Raptor deficiency in either BAT or iWAT (Figures 1A and 1B). Moreover, surgical denervation led to a marked reduction of TH but had no significant effect on Raptor deficiency-induced UCP1 and C/EBPβ expression in iWAT (Figure 2A), suggesting that Raptor deficiency-induced beige adipocyte development in WAT is not due to altered sympathetic pathetic tone. Furthermore, the increased levels of UCP1 and C/EBPβ in Raptor KO iWAT were also observed under thermoneutrality with lower sympathetic activity (Figure 2B). However, the inducing effect of Raptor deficiency on thermogenic gene expression in iWAT was blunted under cold stress, a condition associated with activation of sympathetic tone (Figure 2D). In addition, Raptor-deficient mice displayed decreased levels of UCP1 and C/EBPβ in BAT under cold stress but not thermoneutrality (Figures 2C and 2E). In accordance with this, Raptor KO mice displayed significantly lower O2 consumption under cold stress conditions with no significant effect on food intake and activity (Figures 2F, S2A, and S2B). Whereas there was no significant difference in systemic O2 consumption between Raptor KO and control mice at room temperature (Figure 2F), probably due to increased WAT browning but reduced BAT function under this condition (Figures 1A and 1B).
Figure 2. Raptor Deficiency-Induced WAT Browning Does Not Require the Sympathetic Nervous System.

(A) Surgical denervation led to significant downregulation of tyrosine hydroxylase (TH), while slightly decreasing Raptor deficiency-induced UCP1 expression in iWAT. n = 6/group. 10-weekold male Raptor KO and control mice were used for the following studies. For cold stress and thermoneutrality studies, animals were housed in an environmental chamber at 22_C for 72 hr followed by 30_C, or 6_C for an additional 48 hr.
(B–E) Raptor deficiency upregulated expression levels of thermogenic markers UCP1 and C/EBPb in iWAT (B), while slightly downregulating thermogenic gene expression in BAT (C) under thermoneutrality as well as room temperature. n = 5–7/group in (B) and (C). The Raptor deficiency-induced expression of thermogenic genes ucp1 and c/ebpb was blunted under cold exposure conditions (6_C, 2 days) in iWAT (D) despite persistent reduction of thermogenic gene expression in BAT of Raptor KO mice (E). n = 8/group in (D) and (E).
(F) Raptor mice displayed decreased O2 consumption under cold stress but not room temperature conditions. O2 consumption was normalized by body mass and analyzed using Student’s t test. The data in (A) and (F) are presented as the mean ± SEM.
*p < 0.05, **p < 0.01.
Raptor Deficiency in Adipocytes Induces COX-2 Expression and PG Production In Vivo and In Vitro
To elucidate the underlying mechanism by which mTORC1 signaling regulates beige adipocyte development, we performed RNA-sequencing experiments on inguinal fat from 3-month-old male Raptor KO and control mice. Principal-component analysis (PCA) showed that three KO samples (red) were separated from two control samples (black) (Figure S2C). A total of 11,278 genes remained for analysis after filtering out genes with little or no expression, and a total of 1,812 genes was differentially expressed as shown in the Volcano plot (Figure S2D). The results of the differential expression analysis are summarized in the heatmap (Figure 3A), which shows the gene expression signatures of the most differentially expressed genes in these samples (a larger version with the genes labeled is given in Figure S2E). Among the differentially expressed genes, a group of inflammatory cytokines or immune cell markers including Cxcl2, Tnfsf4, Pdcd1, IL-5, IL1rl1, IL-7r, and Gata3 were elevated by Raptor deficiency (Figure 3B), suggesting the activation of type 2 inflammation, a pathway that has been well documented to be linked to WAT browning. Given the depletion of adipocyte-specific Raptor in these animals, our data also imply that adipocyte-derived paracrine signals may mediate the type 2 immunity and WAT browning in Raptor KO WAT. Notably, Raptor deficiency significantly upregulated PG-endoperoxide synthase 2 (ptgs2), known as COX-2, which is responsible for biosynthesis of PGs, a group of cyclic fatty acid compounds with varying hormone-like effects (Figure 3B). As the COX-2/PG pathway plays a key role in cold-induced browning of WAT as well as inflammation (Hu et al., 2016; Vegiopoulos et al., 2010), we asked whether COX-2 could mediate the promoting effect of mTORC1 deficiency on WAT browning. In support of this, the upregulation of ptgs2, cxcl2, pdcd1, Il-5, and gata3 by Raptor deficiency was validated by RT-PCR in inguinal fat (Figure 3C). Both protein and mRNA levels of COX-2 were higher in WAT compared to BAT (Figures 3D and 3E). Moreover, Raptor deficiency significantly upregulated protein levels of COX-2 but not COX-1 in iWAT and eWAT (Figures 3F and 3G), suggesting that the COX-2 pathway is downstream of mTORC1 in WAT, whereas expression levels of COX-1 and COX-2 were only minimally affected by Raptor deficiency in BAT (Figure S2F). Consistent with this, levels of several PGs including PGD2, PGE2, and PGI2 were increased in inguinal fat of Raptor KO mice, and the increase of PGI2 level by Raptor deficiency was greater than the increase of PGD2 and PGE2 in iWAT (Figure 3H). In support of our in vivo data, protein levels of COX-2 were upregulated in primary adipocytes from iWAT of Raptor KO compared to control mice (Figure 3I). COX-2 mRNA levels and production of PGD2, PGE2, and PGI2 were also upregulated in Raptor KO adipocytes compared to control cells (Figures 3J and 3K). These results indicate that mTORC1 plays a critical role in regulating the COX-2/ PG pathway in a cell-autonomous manner.
Figure 3. Raptor Deficiency in Adipocytes Induces the Expression of COX-2 and Production of Prostaglandins In Vivo and In Vitro.

RNA-sequencing gene expression signatures of iWAT from 10-week-old male Raptor KO and control mice. n = 2–3/group.
(A) Heatmap summarizing differential gene expression in adenoid cystic carcinoma (ACC) tumors versus normal samples and illustrating gene expression signatures for ACC tumors. Blue and red shading indicates downregulation and upregulation, respectively, as shown in the color key at upper right. A larger version of the heatmap with genes labeled is provided in Supplemental Experimental Procedures (Figure S2E).
(B) The fold change of differential genes involved ininflammation in iWAT between 2-month-old Raptor KO and control mice.
(C) RT-PCR analysis for part of differential genes inRNA sequencing (RNA-seq) of the iWAT in Raptor KO and control mice.
(D) The protein levels of COX-2 in various fat depots. n = 4/group.
(E) The mRNA levels of COX-2 in various fat depots. n = 3/group. The data are presented as mean ± SEM.
(F and G) The expression of COX-2 was induced in iWAT (F) and eWAT (G) of Raptor KO mice.
(H) The secretion of PGE2, PGD2, and PGI2 was elevated by Raptor deficiency in iWAT. The levels of PGs were determined by ELISA with the kits.
(I) The expression levels of COX2 but not COX-1 were suppressed by Raptor depletion in primary differentiated adipocytes from iWAT.
(J) mRNA levels of COX-2 were induced by Raptordeficiency in primary adipocytes from iWAT. n = 4/ group.
(K) Secretion of PGE2, PGD2, and PGI2 in primaryRaptor KO and control adipocytes. n = 4/group. The data in (I) was the representative data from three individual experiments. The data in (C), (E), (H), (J), and (K) are presented as the mean ± SEM. *p < 0.05, **p < 0.01.
Inhibition or Suppression of COX-2 Diminishes Raptor Deficiency-Induced Development of Beige Adipocytes in WAT
To gain insight into the role of COX-2 in Raptor deficiency-induced beige fat development, we administered 15 μg/kg/ day celecoxib, a specific inhibitor of COX-2, to Raptor KO and control mice for 10 days. Inhibition of COX-2 reversed the Raptor deficiency-induced increase in protein (Figures 4A and S2G) and mRNA (Figure 4B) levels of UCP1 in iWAT but had little effect on thermogenic gene expression in BAT (Figure S2H), suggesting that COX-2 mediates the inducing effect of Raptor deficiency on the browning of WAT. In addition, COX-2 inhibition diminished Raptor deficiency-induced development of multilocular adipocytes as well as large adipocytes in both iWAT and eWAT (Figures 4C and 4D). The increased O2 consumption was reversed by COX-2 inhibition in iWAT but not BAT of Raptor KO mice (Figure 4E). In addition, factors from Raptor KO adipocytes significantly induced expression of UCP1 and C/EBPβ during differentiation of primary preadipocytes from iWAT (Figure 4F). However, this inducing effect was suppressed by RNAi-mediated knockdown of COX-2 in Raptor KO adipocytes (Figures S2I and 4F). These data suggest that COX-2 mediates the inhibitory effect of mTORC1 on beige adipocyte differentiation.
Figure 4. Inhibiting COX-2 Suppresses Raptor Deficiency-Induced Development of Beige Adipocytes in White Adipose Tissue.

10-week-old male Raptor-deficient and control mice (n = 8/group) were treated with or without COX-2 inhibitor celecoxib by i.p. injection for 10 days (15 μg/kg).
(A) Administration of celecoxib blunted the inducing effect of Raptor deficiency on UCP1 and C/EBPβ in iWAT. n = 4/group.
(B) The increase in mRNA levels of thermogenic markers UCP1 was diminished by treatment of celecoxib.
(C) Representative image for H&E staining of iWAT,eWAT, interscapular BAT, and liver.
(D) Quantification of adipocyte size in iWAT andeWAT of Raptor KO and control mice from (C). Size of adipocytes were determined with ImageJ analysis of H&E-stained tissues. n = 4/group.
(E) O2 consumption of inguinal and brown fat from Raptor-deficient and control mice treated with or without celecoxib. Volume of O2/min/mg tissue was used as the unit for O2 consumption.
(F) The media from Raptor KO primary differentiated adipocytes induced expression of UCP1 and C/EBPβ during differentiation of primary preadipocytes from iWAT, while suppressing COX-2 using RNAi attenuated the promoting effect of Raptor deficiency on thermogenic gene expression in vitro. n = 4/group. The data in (B) and (D)–(F) are presented as the mean ± SEM. *p < 0.05, **p < 0.01. For (D), *p < 0.05, **p < 0.01, control versus KO with vehicle; #p < 0.05, ##p < 0.01, KO with vehicle versus KO with celecoxib.
Blocking PG Signaling Alleviates Raptor Deficiency-Induced Beige Adipogenesis in WAT
To delineate the role of PG signaling in Raptor deficiencyinduced beige adipocyte development, we administered 100 μg/kg PGD2, PGE2, or PGI2 to C57BL/6 mice through subcutaneous injection for 2 days. Administration of PGD2, PGE2, or PGI2 significantly upregulated protein levels of thermogenic genes including ucp1 and C/EBPβ in inguinal fat but not in BAT (Figures 5A and S3A). In addition, mRNA levels of UCP1 and C/EBPβ were upregulated by administration of PGD2, PGE2, or PGI2 (Figure 5B), indicating the promoting effects of PGs on beige adipocytes. The injection of PGI2 and PGE2 induced thermogenesis to a greater extent than PGD2 treatment (Figures 5A and 5B). Although treatment with PGD2, PGE2, or PGI2 had no significant effect on the expression of ucp1 and C/EBPβ in differentiated primary adipocytes (Figure S3B), treatment of these PGs upregulated protein and mRNA levels of UCP1 and C/EBPβ during the differentiation of primary iWAT-derived preadipocytes (from day 1 to day 4) (Figures 5C, 5D, S3C, and S3D). In support of previous findings reporting that PGE2, PGD2, and PGI2 bind to their respective receptors and stimulate cyclic AMP (cAMP) signaling (Clasadonte et al., 2011; Mohan et al., 2012), we found that the receptors for PGE2 and PGI2 were enriched in the SVF, adipocytes and preadipocytes (Figure S3E), and that treatment of primary adipocytes with PGD2, PGE2, and PGI2 for 2 hr increased intracellular levels of cAMP along with PKA signaling (Figures 5E and 5F). Consistent with this, induction of UCP1 and C/EBPβ by PGI2 was suppressed by PKA inhibition following treatment with 10 μM H89 during adipocyte differentiation (Figure 5G).
Figure 5. Blocking PG Signaling Alleviates Raptor Deficiency-Induced Beige Adipogenesis in WAT.

(A) Administration of PGD2, PGE2, and PGI2 by subcutaneous injection (0.1 μg/kg body weight) for 2 days induced the expression of UCP1 and C/EBPβ in iWAT. D2, PGD2; E2, PGE2; I2, PGI2.
(B) The mRNA levels of thermogenic markers UCP1 and C/EBPβ were enhanced by treatment of PGD2, PGE2, and PGI2. n = 6/group.
(C) The expression levels of UCP1 and C/EBPβ but not PPARγ and PGC1α were induced by PGI2 treatment at different dose during the differentiation of primary preadipocytes.
(D) The mRNA levels of UCP1 were analyzed by RTPCR in the differentiated adipocytes treated with or without PGD2, PGE2, or PGI2. n = 4/group.
(E and F) The intracellular cAMP levels (E) were determined by ELISA, and phosphorylation of PKA substrates (F) were analyzed by western blot analysis in primary preadipocytes that were starved for 3 hr followed by treatment of PGE2, PGD2, or PGI2 for 2 hr and 20 min, respectively. n = 3/group.
(G) The induced expression levels of UCP1 and C/EBPβ by PGI2 were suppressed by PKA inhibition with H89 treatment during the differentiation of primary adipocytes from iWAT. n = 3/group.
(H) Administration of PGI antagonist CAY10441 partially reversed the promoting effect of Raptor deficiency on UCP1 in iWAT.
(I) The media from Raptor KO primary differentiatedadipocytes induced the expression of UCP1 and C/EBPβduringthedifferentiationofpreadipocytefrom iWAT, while this inducing effect by Raptor deficiency was diminished by the treatment of PGI antagonist CAY10441. n = 3/group. (A), (C), (F), and (H) were the representative data from three individual experiments. The data in (B), (D), (E), (G), and (I) are presented as the mean ± SEM. *p < 0.05, control versus treatment or two particular groups as indicated.
To further investigate the role of PG signaling in mTORC1 inhibition-induced beige adipogenesis, we administered 2 μg/kg of PGI2 receptor antagonist CAY10441 through intraperitoneal (i.p.) injection in Raptor KO and control littermates for 10 days. Inhibiting PGI2 signaling suppressed Raptor deficiency-induced upregulation of UCP1 and C/EBPβ in iWAT, suggesting that PGI2 mediates the browning effect induced by Raptor deficiency (Figure 5H). Moreover, the conditioned media of primary Raptor KO differentiated adipocytes induced the expression of UCP1 and C/EBPβ during the differentiation of primary preadipocytes, an effect that was partially reversed by treatment with the PGI2 antagonist CAY10441 during the differentiation (Figure 5I). These results suggest that mTORC1 inhibition promotes the development of beige adipocytes via a PG-mediated paracrine mechanism.
Raptor Deficiency Protects Mice from Diet-Induced Suppression of COX-2 and Loss of Beige Adipocytes
COX-2 has been shown to orchestrate beige/brite adipocyte formation (Bayindir et al., 2015; Vegiopoulos et al., 2010). However, its role in the development of obesity remains poorly defined. To examine the effect of HFD feeding on adipose tissue COX-2 as well as phosphorylation of S6K at Thr389, 6-week-old male C57BL/6 mice were fed with 45% HFD for 4, 16, or 24 weeks as we described previously (Liu et al., 2012, 2014). We found that 4-week HFD feeding significantly downregulated protein and mRNA levels of COX-2 but not COX-1 in iWAT (Figures 6A and 6B) and eWAT (Figure S4A), concurrent with increased phosphorylation of S6K at Thr389. In contrast, HFD feeding significantly upregulated COX-2 expression and suppressed phosphorylation of S6K in BAT (Figure S4B). While not statistically significant, trending COX-2 suppression in iWAT (Figures 6C, 6D, S4C, and S4D) and eWAT (Figures S4C–S4F) was also observed following 16 weeks of HFD feeding, with no effect seen at 24 weeks despite persistent activation of S6K. Along this line, mTORC1 was activated, and expression levels of COX-2 were suppressed in the iWAT and eWAT but not in the BAT of ob/ob mice compared to lean mice (Figures 6E and S4G). In addition, rapamycin treatment upregulated COX-2 in human adipocytes from neck adipose tissue (Figure 6F). Moreover, Raptor-deficient mice were resistant to diet-induced downregulation of COX-2 (Figure 6G). Consistent with this, basal and HFD-induced expression of UCP1 in inguinal fat was significantly upregulated in Raptor KO mice compared to control mice (Figure 6G). In addition, Raptor deficiency-induced downregulation of UCP1 in BAT was restored under HFD feeding (Figure S4H). In accordance with this, despite no significant effect on body weight after 4 weeks of HFD feeding, Raptor deficiency protected against HFD-induced decrease of O2 consumption with no change to food intake or activity (Figure 6H and data not shown). Along with this, Raptor-deficient mice were protected against dietinduced obesity and glucose intolerance despite no significant changes in triglyceride (TG) content in liver, hepatic steatosis, or insulin tolerance (Figures S1A–S1F). These results together suggest that adipocyte mTORC1 inhibition protects mice against HFD-induced downregulation of COX-2, energy imbalance, and obesity. However, we did not observe the induction of COX-2 by 48-hr cold exposure (6C) despite the upregulation of UCP1 in iWAT and BAT (Figure S6I). On the other hand, mTORC1 as well as food intake was stimulated by cold stress (Figure S6J), implying that cold stress activation of mTORC1 may be partially due to the increase of food intake, and that mTORC1/COX-2 pathway is dispensable for cold-induced thermogenesis.
Figure 6. Adipocyte mTORC1 Inhibition Protects Mice against HFD-Induced Downregulation of COX-2 and Energy Expenditure.

(A–D) 6-week-old male C57BL/6 mice were fed with normal chow diet (NCD) or HFD for 4, 16, or 24 weeks. Protein levels of COX-2 and COX-1 along with phosphorylation of S6K (A) and mRNA level of COX-2 and COX-1 (B) in iWAT after 4-week-HFD feeding. Phosphorylation of S6K at Thr389 in iWAT (C) were stimulated by HFD feeding for 4, 16, or 24 weeks. Conversely, the expression levels of COX-2 in iWAT (D) were suppressed by 4 or 16 weeks of HFD feeding, while not significantly altered by 24 weeks of HFD feeding. n = 4–8/group in (C) and (D).
(E) Phosphorylation of S6K at Thr389 was increased, and expression levels of COX-2 were suppressed in iWAT of ob/ob mice compared to lean mice. n = 5/group.
(F) Rapamycin treatment induced expression of COX-2 in human primary adipocytes from neck adipose tissue. n = 3/group.
(G)RaptordeficiencyprotectedagainstthesuppressionofCOX-2inducedby4-weekHFDandupregulatedbasalandHFD-inducedUCP1iniWAT in vivo.n=5–7/group.
(H) O2 consumption in Raptor KO and control mice throughout light and dark cycles on the day before HFD feeding and day 7 and day 30 after HFD feeding. The average O2 consumption was normalized to whole-body mass and analyzed using Student’s t test. (F) was the representative data from three individual experiments. The data in (B)–(H) are presented as the mean ± SEM. *p < 0.05.
mTORC1 Suppresses COX-2 Expression by Phosphorylating CRTC2 at Ser136 in Adipocytes
Given that β-adrenergic signaling drives lipolysis and COX-2 gene activation (Gartung et al., 2016; Klein et al., 2007; Vegiopoulos et al., 2010), we hypothesized that PKA signaling may mediate the inhibitory effect of mTORC1 on COX-2. We found that treatment of rapamycin or isoproterenol significantly induced COX-2 expression in primary preadipocytes (Figures S5A and S5B). Moreover, inhibition of PKA by H-89 treatment diminished the stimulatory effect of Raptor deficiency or rapamycin treatment on COX-2 expression in primary adipocytes (Figures 7A and 7B), suggesting a mediatory role of PKA signaling in mTORC1 regulation of COX-2. However, rapamycin and Raptor deficiency had no significant effect on the phosphorylation of PKA substrates (Figure 7B). Because mTORC1 has been shown to interact with PKA signaling through phosphorylation of CRTC2 at Ser136 in hepatocytes (Han et al., 2015), we examined the effect of mTORC1 on the localization of CRTC2 and CRTC3 in primary preadipocytes. We found that both CRTC2 and CRTC3 were expressed in preadipocytes (Figures 7C and S5C). CRTC2 is localized in both the nucleus and the cytoplasm and CRTC3 is predominantly localized in the nucleus under growth medium-culturing conditions (Figures 7C and S5C). Treatment with a cAMP analog promoted CRTC2 translocation to the nucleus, while having little effect on CRTC3 localization (Figures 7C and S5C). In addition, inhibition of mTORC1 by rapamycin had no effect on the nuclear translocation of CRTC2 (Figure 7C). However, leucine treatment induced and rapamycin treatment suppressed phosphorylation of CRTC2 at Ser136 in preadipocytes (Figures 7D and 7E). Moreover, Raptor deficiency suppressed leucine-induced P-CRTC2 at Ser136 in preadipocytes (Figures 7F and S5D), suggesting that mTORC1 may modulate CRTC2 activity through phosphorylation rather than cellular localization.
Figure 7. mTORC1 Signaling Inhibits CRTC2 Phosphorylation at Ser136 and Subsequently Suppresses COX-2 Transcription in Adipocytes.

(A) Raptor deficiency induced COX-2 expression, which was diminished by treatment of PKA inhibitor H89 in primary preadipocytes. n = 3/group.
(B) PKA inhibition attenuated the inducing effect of rapamycin treatment on COX-2 expression.
(C) Immunofluorescence staining of CRTC2 after treatment of 100 mM cAMP homolog dibutyryl cyclic AMP, 10 nM rapamycin, or co-treatment in primary preadipocytes.
(D) Leucine treatment stimulated the phosphorylation of CRTC2 at Ser136 in primary preadipocytes.
(E) Rapamycin treatment inhibited CRTC2 phosphorylation at Ser136 in primary preadipocytes.
(F) Raptor deficiency suppressed rapamycin-stimulated phosphorylation of CRTC2 at Ser136 in primary preadipocytes.
(G) (G) The effect of rapamycin treatment on COX-2 expression in CRTC2 suppressed and scrambled primary adipocytes.
(H–K) Overexpression of CRTC2 markedly upregulated expression levels of COX-2 (H), increased the interaction between CRTC2 and CREB (I), promoted CREB binding to the cox-2 promoter (J), and enhanced luciferase activity of the cox-2 promoter (K), while diminishing the inducing effects of rapamycin in 3T3-L1 preadipocytes. n = 3/group in (J) and (K). Meanwhile, the ability of CRTC2 to induce expression of COX-2, increase CREB binding to the cox-2 promoter, and activate cox-2 promoter was attenuated by phosphor-mimic (S136D) but not phosphor-defective (S136A) mutation of CRTC2 in 3T3-L1 preadipocytes. The interaction between CREB and CRTC2 was determined using immunoprecipitation. The binding of CREB to the cox-2 promoter was determined using ChIP assay. The data in (B)–(I) are representative from at least three individual experiments. The quantified data for (D) and (E) are presented in Figures S5D and S5E. The data in (A), (J), and (K) are presented as the mean ± SEM. *p < 0.05.
Next, we investigated the potential role of mTORC1-mediated CRTC2 phosphorylation in regulating COX-2 expression in preadipocytes given the promoting effect of CREB on cox-2 promoter activation (Reddy et al., 2000; Yang and Bleich, 2004). Suppressing CRTC2 by RNAi diminished rapamycin-induced COX-2 expression in preadipocytes (Figures 7G and S5E). In contrast, overexpression of CRTC2 markedly upregulated the expression levels of COX-2 and attenuated rapamycin-induced COX-2 expression in preadipocytes (Figure 7H). To dissect the mediatory role of CRTC2 in mTORC1 suppression of COX-2, we performed immunoprecipitation, chromatin immunoprecipitation (ChIP) analysis, and luciferase assays. We found that rapamycin treatment promoted the interaction between CRTC2 and CREB (Figure 7I), induced CREB binding to the cox-2 promoter (Figures 7J and S5F), and enhanced luciferase activity of the cox-2 promoter (Figure 7K) in preadipocytes. Moreover, overexpression of CRTC2 increased the association between CRTC2 and CREB as well as CREB binding to the cox-2 promoter, luciferase activity of the cox-2 promoter, and COX-2 expression, and diminished the inducing effect of rapamycin in preadipocytes (Figures 7H–7K). Furthermore, the ability of CRTC2 to interact with CREB, facilitate CREB binding to the cox-2 promoter, and activate the cox-2 gene to induce COX-2 expression was suppressed by a serine-to-aspartate mutation at 136 (S136D) but not by a serine-to-alanine mutation (S136A) (Figures 7H–7K). The results from this study indicate that mTORC1 suppresses COX-2 through phosphorylation of CRTC2 at Ser136 and subsequent inhibition of CREB binding to the cox-2 promoter in adipocytes.
DISCUSSION
Beige adipocytes, which develop in WAT, have become a promising avenue to counteract obesity (Harms and Seale, 2013). However, the adipocyte-derived signals that target progenitor cells and control beige adipogenesis in WAT have not yet been defined. Here, we show that COX-2-mediated PGs act as paracrine signals that target adipocyte progenitor cells and orchestrate beige adipogenesis. In addition, we propose that mTORC1 signaling is a key regulator of the COX-2/PG pathway through phosphorylation of CRTC2 in adipocytes and mediates obesity-induced suppression of COX-2 and beige adipogenesis in WAT. These results reveal that adipocyte mTORC1 controls beige adipogenesis via PG-mediated paracrine signaling, and establish a pathway underlying the suppression of COX-2 as well as energy imbalance during the development of obesity.
COX-2 plays a pivotal role in regulating adipose inflammation including macrophage recruitment and subsequent immune response and insulin resistance (Chan et al., 2016; Gartung et al., 2016; Hsieh et al., 2010; Hu et al., 2016). Additionally, COX-2 promotes energy metabolism such as beige adipocyte formation as well as browning of WAT (Davis et al., 2004; Fain et al., 2001; Lundholm et al., 2004; Vegiopoulos et al., 2010). However, whether COX-2 is critically involved in the development of obesity remains an open question. In addition, the physiological signals that control the COX-2 pathway during the development of obesity are unclear. We found that HFD feeding suppresses COX-2, which is accompanied by activation of mTORC1 in WAT, while BAT is protected from these alterations under HFD conditions (Figures 6A–6D, S4A, and S4B). In support of this, both protein and mRNA levels of COX-2 are more enriched in WAT compared to BAT (Figures 3D and 3E). Notably, mTORC1 activation and COX-2 suppression in iWAT by HFD feeding occurs within 4 weeks, even before UCP1 suppression (Figures 6A and 6B). In addition, inhibition of mTORC1 in adipocytes restores HFD-induced suppression of COX-2 (Figure 6G). Despite previous findings that 25 weeks of HFD (60% kcal from fat; Research Diet) feeding has no significant effect on COX-2 expression (Vegiopoulos et al., 2010), our data suggest that COX-2 expression is indeed suppressed in WAT in the earlier stage of obesity, as indicated by 4- to 16-week HFD feeding (Figures 6C, 6D, S4C, and S4D). However, diet-induced COX-2 suppression disappears at the later stage of obesity as indicated by 24-week HFD feeding. Given that COX-2 plays a critical role in obesity-induced adipose inflammation and insulin resistance, the mechanisms underlying the regulation of COX-2 expression at the later stage of obesity may be more complicated than in the earlier stage.
Previous studies have reported that β3-adrenergic receptors are required for the recruitment and activation of beige adipocytes in response to cold stress in WAT (Barbatelli et al., 2010; Cannon and Nedergaard, 2004; Jimenez et al., 2003; Nahmias et al., 1991). However, these findings were challenged by evidence showing that, in the absence of β3-adrenergic receptors, cold-induced activation of thermogenic programming in both BAT and WAT remains fully intact (de Jong et al., 2017). These findings also indicate the possibility of an alternative β3-adrenergic receptor-independent signaling pathway that may control beige adipogenesis and activation. Adipocyte-derived PGs facilitates the activation of PKA-C/EBPβ pathway, which induces beige adipogenesis in adipocyte progenitor cells (Figures 5E–5G). Therefore, treatment with the PG receptor antagonist diminishes the ability of Raptor deficiency to induce progenitor cell differentiation into beige adipocytes (Figures 5H and 5I). The results of this study indicate that PG receptors in progenitor cells may be possible therapeutic targets for the treatment of obesity and its related disorders.
Despite accumulating evidence indicating that COX-2 is induced by lipolysis in adipocytes (Gartung et al., 2016; Klein et al., 2007; Vegiopoulos et al., 2010), the mechanisms underlying modulation of COX-2 transcription in adipocytes remain elusive. Our data suggest that overactivation of mTORC1 by obesity suppresses COX-2 expression and PG production through phosphorylation of CRTC2 at Ser136 and subsequent blocking of CREB binding to the cox-2 promoter (Figure 7). CRTCs facilitate hepatic gluconeogenesis and lipid homeostasis at the transcriptional level (Han et al., 2015; Wang et al., 2009); however, the physiological role of CRTCs in adipose tissue is incompletely understood. CRTC2 and CRTC3 are present in adipose tissue as well as in adipocytes and are involved in the regulation of energy balance and glucose uptake in adipose tissue (Henriksson et al., 2015; Park et al., 2014; Song et al., 2010). However, little is known about CRTCs modulation and target genes in adipocytes. Our study established that mTORC1 inactivates the cox-2 gene by phosphorylating CRTC2 (Figure 7). Although mTORC1 signaling has little effect on cytoplasm-nucleus shuttling of CRTC2 in adipocytes, mTORC1-mediated phosphorylation of CRTC2 at Ser136 inhibits association between CREB and CRTC2, CREB binding to the cox-2 gene, and the promoter activity of the cox-2 gene (Figures 7I–7K). Our study establishes a pivotal role for mTORC1 in regulating COX-2 transcription and PG production/secretion in adipocytes through phosphorylation of CRTC2. Consistent with the findings of Paschos et al. (2018), mTORC1/COX-2 pathway is not required for cold-induced thermogenesis. Whereas adipocyte-derived PGs stimulate accumulation of cAMP in adipocyte progenitor cells and subsequently promote beige adipogenesis, favoring calorie restriction or weight loss induced WAT browning.
The role of adipocyte mTORC1 signaling in the regulation of thermogenesis remains controversial. Early studies show that inhibition of mTORC1 by S6K1 ablation as well as adipose-specific deficiency of Raptor enhances thermogenesis and energy expenditure and protects against diet-induced obesity and glucose intolerance (Polak et al., 2008; Um et al., 2004). In support of this, the basal expression levels of thermogenic genes are elevated in the white fat of Raptor- or mTOR-deficient mice (Lee et al., 2016; Meng et al., 2017; Shan et al., 2016; Tran et al., 2016). However, other studies show that adipose inactivation of mTORC1 by rapamycin or Raptor deficiency leads to lipodystrophy, fatty liver, and insulin resistance (Labbé et al., 2016; Lee et al., 2016; Shan et al., 2016; Tran et al., 2016). In addition, inhibition of mTORC1 results in impaired thermogenesis by interacting with PKA signaling in either BAT or both BAT and WAT (Liu et al., 2016; Tran et al., 2016). Therefore, addressing this controversy regarding mTORC1 in adipose tissue is urgently needed. Our present study shows that adipocyte mTORC1 differentially modulates thermogenic programming of BAT and WAT. Raptor deficiency promotes browning of white adipose tissue by regulating beige adipocyte differentiation in a PG-dependent manner, while suppressing thermogenesis in BAT through impairment of brown fat development. Interestingly, while increased thermogenesis and O2 consumption in iWAT occurs under thermoneutrality and at room temperature, experimental conditions in mice resemble human conditions, an effect that disappears under cold stress conditions. Our study demonstrates that, while mTORC1 inhibition does not intrinsically increase thermogenic capacity, it activates PKA signaling in adipocyte progenitor cells via CRTC2-COX-2 axis-dependent PG production. Our study uncovers PG as a paracrine signal that promotes beige adipogenesis and mediates adipocyte mTORC1 inhibition-induced browning of WAT. Given the activation of type 2 inflammation by Raptor deficiency in iWAT (Figures S2E, 3B, and 3C), further investigation is needed to address whether COX-2 mediates the promoting effect of mTORC1 inhibition on immune response in the context of thermogenic regulation. In addition, the type and function of immune cells controlled by adiposespecific mTORC1/COX-2 pathway remain to be clarified.
In summary, our data show that adipocyte mTORC1 signaling impacts adipocyte progenitor cells and controls beige adipocyte differentiation via a PG-dependent paracrine mechanism. Moreover, the mTORC1 pathway inhibits the production and secretion of PG through the phosphorylation of CRTC2 and the suppression of COX-2 in adipocytes. In addition, mTORC1mediated suppression of COX-2 is critically involved in the development of obesity. Our study strongly suggests that adipocyte mTORC1 signaling plays a key role in regulating the development of beige adipocytes in WAT.
EXPERIMENTAL PROCEDURES
ChIP Assay
3T3-L1 preadipocytes or CRTC2-overexpressed preadipocytes were treated with or without 5 nM rapamycin for 10, 30, 60, or 120 min, and then fixed in 1% formaldehyde for 10 min at 37°C. Cells were washed twice with cold PBS containing protease inhibitor cocktail, and subsequently lysed in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1). Extracts were sonicated until DNA fragments of 2001,000 bp were achieved. Sonicated solution was diluted in ChIP dilution buffer (TE buffer, 1.1% Triton X-100, 167 mM NaCl, 1.13 cocktail). The immuno-complexes were precipitated using an anti-CREB (Millipore) antibody or IgG as the control. Precipitated complexes were reverse cross-linked at 65°C. The amount of precipitated DNA was detected by semiquantitative PCR using the primers 5ʼ-CAGAGAGGGGGAAAA GTTGG-3ʼ and 5ʼ-GAGCAGAGTCCTGACTGACTC-3ʼ.
Luciferase Assay
The luciferase construct of the cox-2 promoter was generated by subcloning Xhol-HindIII fragment (−74 to −727) of the cox-2 promoter into pGL3-basic with the primers 5ʼ-CCCTCGAGAACCCGGAGGGTAGTTCCAT-3ʼ and 5ʼ-CCAAGCTTGAGCAGAGTCCTGACTGACTC-3ʼ. 3T3-L1 preadipocytes were grown in a 24-well plate to 70% confluency. The cox-2 promoter luciferase construct was co-transfected with pSV-β-Galactosidase Control Vector (Promega, Madison, WI, USA) in combination with pcDNA 3.1, pcDNA/CRTC2, pcDNA/CRTC2-S136A, or pcDNA/CRTC2-S136D. 24 hr post-transfection, cells were treated with or without 5 nM rapamycin for 4 hr and then lysed for the measurement of the luciferase with the Luciferase Assay Kit (Promega) and β-galactosidase activities using a Turner TD20/20 luminometer.
Human Study
Patients were identified either before undergoing anterior cervical spine surgery, before undergoing parathyroidectomy or before undergoing thyroidectomy, and then were recruited in the ENT clinic at time of consent for primary surgery. This study has been reviewed and approved by Human Research Review Committee at UNMHSC. 5–10 μg neck fat was harvested during anterior cervical spine surgery, parathyroidectomy, or thyroidectomy. Primary adipocytes were isolated and treated with or without 5 nM Rapamycin for 20 hr.
Animals
The ob/ob mice were obtained from Jackson Laboratory (stock number 000632). The raptor floxed mice (raptorfl/fl) (Jackson Laboratory, stock number 13188) were crossed with adiponectin cre mice (Jackson Laboratory, stock number 10803) to generate raptor fl+//adiponectin cre mice. The Raptor ad+/ mice were then crossed back with raptorfl/fl mice to generate adipose-specific raptor knockout (KO) and raptor-floxed control mice. The knockout efficiency was confirmed in adipose tissue and other tissues by western blot analysis using anti-raptor antibodies. Unless otherwise noted, 10-week-old male mice were used for all experiments. Animals were housed in a specific pathogen-free barrier facility with a 12-hr light/12-hr dark cycle with free access to food and water. For cold stress study, animals were housed in the temperature-controlled chamber at 22°C for three days followed by either 30°C or 6°C for another 48 hr. For the high-fat diet challenge study, 6-week-old male mice were fed with 45% of normal chow diet provided by the animal facility at the University of New Mexico Health Sciences Center or high-fat diet (45% kcal from fat) from Research Diets (D12451; New Brunswick, NJ, USA) for 4, 16, or 24 weeks. For surgical denervation, an incision was made at the dorsal hindlimb of the animal and lateral to the spinal column that continued rostrally and then ventrally to the ventral hindlimb as described in our previous study (Luo et al., 2017). Two weeks post-surgery, mice were euthanized and inguinal fat was collected for western blot. All animal experimental protocols were reviewed and approved by the Animal Care Committee of the University of New Mexico Health Sciences Center.
RNA Isolation and Sequencing
Total RNA was isolated from single 10-mm slide-mounted FFPE sections using the RNeasy FFPE Kit from Qiagen. After isolation, total RNA was mixed with ERCC Spike-In Mix 1 control RNA (Life Technologies) and converted to cDNA using the SMARTer Universal Low Input RNA Kit for Sequencing (Clontech), which uses random primers for the conversion. The Ion Plus Fragment Library Kit (Life Technologies) was used to add barcodes, and amplify and size-select the final library. Four individually barcoded samples were sequenced together on Ion Proton P1v2 chips by the Analytical and Translational Genomics Shared Resource at the University of New Mexico Cancer Center (Brayer et al., 2016). RNA-sequencing data are available for download from the NCBI Sequence Read Archive using SRA study accession number SRP059557.
Statistics
Statistical analysis of the data was performed using a two-tailed Student’s t test between two groups or one-way ANOVA among three different groups. All of the results were presented as the mean ± SEM, and p value of <0.05 was considered to be statistically significant.
Supplementary Material
Highlights.
mTORC1 is a potent regulator of white to beige adipogenesis
mTORC1 inhibition leads to WAT browning and occurs despite sympathetic denervation
COX-2 and prostaglandins are essential for browning of WAT in Raptor-deficient mice
mTORC1 suppresses COX-2/PG pathway via a CRTC2-dependent mechanism
ACKNOWLEDGMENTS
This work is supported by an R01 Award (DK110439; PI, M.L.) from NIDDK; a P20 Award (GM121176; PD, Vojo Deretic; mPI, M.L.) from NIGMS; an Innovative Basic Science Award (1–17-IBS-261; PI, M.L.) from the American Diabetes Association; a Grant in Aid Award (15GRNT2490018; PI, M.L.); a National Basic Research Program of China (2014CB910501; PI, Feng Liu); a CTSC pilot award (PI, M.L.); a CoBRE pilot award associated with P30 (P30GM103400 [PD, J. Liu]; mPI, M.L.); RAC pilot awards (PI, M.L.); and a UNMCCC pilot award (PI, M.L.) at the University of New Mexico Health Sciences Center (UNMHSC).
We would like to acknowledge the National Institute of Diabetes and Digestive and Kidney Diseases, the National Institute of General Medical Science, the American Diabetes Association, the American Heart Association, the National Basic Research Program of China, and the University of New Mexico Health Sciences Center (UNMHSC) for funding support. We thank Dr. Yiguo Wang at Tsinghua University for providing us with the CRTC2 constructs and the p-CRTC2 (S136) antibody. We thank the Autophagy, Inflammation and Metabolism (ATM) Center at UNMHSC for providing the Seahorse XF Analyzer (Agilent) and the Cellomics HCS scanner for our present study and technical support from Dr. Michael Mandell and John Michael Weaver. The AIM center is supported by NIH grant P20GM121176 from NIGMS. We thank Dr. Feng Liu at the University of Texas Health at San Antonio for providing us with the adiponectin antibody. We thank the Clinical & Translational Science Center (CTSC) at UNM for the recruitment of human subjects and the coordination of human study. We also thank Dr. Curt Hines at UNMHSC for providing EVOS FL Cell Imaging System for the immunofluorescence staining. In addition, we thank Dr. Jesse Denson for editing this manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information includes Supplemental Experimental Procedures and five figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.08.055.
REFERENCES
- Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, Giacobino JP, De Matteis R, and Cinti S (2010). The emergence of coldinduced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab 298, E1244–E1253. [DOI] [PubMed] [Google Scholar]
- Bayindir I, Babaeikelishomi R, Kocanova S, Sousa IS, Lerch S, Hardt O, Wild S, Bosio A, Bystricky K, Herzig S, and Vegiopoulos A (2015). Transcriptional pathways in cPGI2-induced adipocyte progenitor activation for browning. Front. Endocrinol. (Lausanne) 6, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boström P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostro¨ m EA, Choi JH, Long JZ, et al. (2012). A PGC1-α-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brayer KJ, Frerich CA, Kang H, and Ness SA (2016). Recurrent fusions in MYB and MYBL1 define a common, transcription factor-driven oncogenic pathway in salivary gland adenoid cystic carcinoma. Cancer Discov. 6, 176–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, and Nedergaard J (2004). Brown adipose tissue: function and physiological significance. Physiol. Rev 84, 277–359. [DOI] [PubMed] [Google Scholar]
- Carnevalli LS, Masuda K, Frigerio F, Le Bacquer O, Um SH, Gandin V, Topisirovic I, Sonenberg N, Thomas G, and Kozma SC (2010). S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 18, 763–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan PC, Hsiao FC, Chang HM, Wabitsch M, and Hsieh PS (2016). Importance of adipocyte cyclooxygenase-2 and prostaglandin E2-prostaglandin E receptor 3 signaling in the development of obesity-induced adipose tissue inflammation and insulin resistance. FASEB J. 30, 2282–2297. [DOI] [PubMed] [Google Scholar]
- Clasadonte J, Sharif A, Baroncini M, and Prevot V (2011). Gliotransmission by prostaglandin e(2): a prerequisite for GnRH neuronal function? Front. Endocrinol. (Lausanne) 2, 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen P, Levy JD, Zhang Y, Frontini A, Kolodin DP, Svensson KJ, Lo JC, Zeng X, Ye L, Khandekar MJ, et al. (2014). Ablation of PRDM16 and beige adipose causes metabolic dysfunction and a subcutaneous to visceral fat switch. Cell 156, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cristancho AG, and Lazar MA (2011). Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol 12, 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TW, Zweifel BS, O’Neal JM, Heuvelman DM, Abegg AL, Hendrich TO, and Masferrer JL (2004). Inhibition of cyclooxygenase-2 by celecoxib reverses tumor-induced wasting. J. Pharmacol. Exp. Ther 308, 929–934. [DOI] [PubMed] [Google Scholar]
- de Jong JMA, Wouters RTF, Boulet N, Cannon B, Nedergaard J, and Petrovic N (2017). The β3-adrenergic receptor is dispensable for browning of adipose tissues. Am. J. Physiol. Endocrinol. Metab 312, E508–E518. [DOI] [PubMed] [Google Scholar]
- Fain JN, Ballou LR, and Bahouth SW (2001). Obesity is induced in mice heterozygous for cyclooxygenase-2. Prostaglandins Other Lipid Mediat. 65, 199–209. [DOI] [PubMed] [Google Scholar]
- Farmer SR (2008). Molecular determinants of brown adipocyte formation and function. Genes Dev. 22, 1269–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher FM, Kleiner S, Douris N, Fox EC, Mepani RJ, Verdeguer F, Wu J, Kharitonenkov A, Flier JS, Maratos-Flier E, and Spiegelman BM (2012). FGF21 regulates PGC-1a and browning of white adipose tissues in adaptive thermogenesis. Genes Dev. 26, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartung A, Zhao J, Chen S, Mottillo E, VanHecke GC, Ahn YH, Maddipati KR, Sorokin A, Granneman J, and Lee MJ (2016). Characterization of eicosanoids produced by adipocyte lipolysis: implication of cyclooxygenase-2 in adipose inflammation. J. Biol. Chem 291, 16001–16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Li E, Chen L, Zhang Y, Wei F, Liu J, Deng H, and Wang Y (2015). The CREB coactivator CRTC2 controls hepatic lipid metabolism by regulating SREBP1. Nature 524, 243–246. [DOI] [PubMed] [Google Scholar]
- Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, and Yonezawa K (2002). Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110, 177–189. [DOI] [PubMed] [Google Scholar]
- Harms M, and Seale P (2013). Brown and beige fat: development, function and therapeutic potential. Nat. Med 19, 1252–1263. [DOI] [PubMed] [Google Scholar]
- Henriksson E, Säll J, Gormand A, Wasserstrom S, Morrice NA, Fritzen AM, Foretz M, Campbell DG, Sakamoto K, Ekelund M, et al. (2015). SIK2 regulates CRTCs, HDAC4 and glucose uptake in adipocytes. J. Cell Sci 128, 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh PS, Lu KC, Chiang CF, and Chen CH (2010). Suppressive effect of COX2 inhibitor on the progression of adipose inflammation in high-fatinduced obese rats. Eur. J. Clin. Invest 40, 164–171. [DOI] [PubMed] [Google Scholar]
- Hu X, Cifarelli V, Sun S, Kuda O, Abumrad NA, and Su X (2016). Major role of adipocyte prostaglandin E2 in lipolysis-induced macrophage recruitment. J. Lipid Res 57, 663–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iñiguez MA, Punzó n C, and Fresno M (1999). Induction of cyclooxygenase-2 on activated T lymphocytes: regulation of T cell activation by cyclooxygenase-2 inhibitors. J. Immunol 163, 111–119. [PubMed] [Google Scholar]
- Jimenez M, Barbatelli G, Allevi R, Cinti S, Seydoux J, Giacobino JP, Muzzin P, and Preitner F (2003). Beta 3-adrenoceptor knockout in C57BL/ 6J mice depresses the occurrence of brown adipocytes in white fat. Eur. J. Biochem 270, 699–705. [DOI] [PubMed] [Google Scholar]
- Khamzina L, Veilleux A, Bergeron S, and Marette A (2005). Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146, 1473–1481. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kim JM, Jung HC, and Song IS (2001). Expression of cyclooxygenase-2 in human neutrophils activated by Helicobacter pylori water-soluble proteins: possible involvement of NF-kappaB and MAP kinase signaling pathway. Dig. Dis. Sci 46, 2277–2284. [DOI] [PubMed] [Google Scholar]
- Klein T, Shephard P, Kleinert H, and Kö mhoff M (2007). Regulation of cyclooxygenase-2 expression by cyclic AMP. Biochim. Biophys. Acta 1773, 1605–1618. [DOI] [PubMed] [Google Scholar]
- Labbé SM, Mouchiroud M, Caron A, Secco B, Freinkman E, Lamoureux G, Gélinas Y, Lecomte R, Bossé Y, Chimin P, et al. (2016). mTORC1 is required for brown adipose tissue recruitment and metabolic adaptation to cold. Sci. Rep 6, 37223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laplante M, and Sabatini DM (2012). mTOR signaling in growth control and disease. Cell 149, 274–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PL, Tang Y, Li H, and Guertin DA (2016). Raptor/mTORC1 loss in adipocytes causes progressive lipodystrophy and fatty liver disease. Mol. Metab 5, 422–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Xiang R, Wilk SA, Zhang N, Sloane LB, Azarnoush K, Zhou L, Chen H, Xiang G, Walter CA, et al. (2012). Fat-specific DsbA-L overexpression promotes adiponectin multimerization and protects mice from dietinduced obesity and insulin resistance. Diabetes 61, 2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Bai J, He S, Villarreal R, Hu D, Zhang C, Yang X, Liang H, Slaga TJ, Yu Y, et al. (2014). Grb10 promotes lipolysis and thermogenesis by phosphorylation-dependent feedback inhibition of mTORC1. Cell Metab. 19, 967–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Bordicchia M, Zhang C, Fang H, Wei W, Li JL, Guilherme A, Guntur K, Czech MP, and Collins S (2016). Activation of mTORC1 is essential for β-adrenergic stimulation of adipose browning. J. Clin. Invest 126, 1704–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, and Hall MN (2002). Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 10, 457–468. [DOI] [PubMed] [Google Scholar]
- Lundholm K, Daneryd P, Kö rner U, Hyltander A, and Bosaeus I (2004). Evidence that long-term COX-treatment improves energy homeostasis and body composition in cancer patients with progressive cachexia. Int. J. Oncol 24, 505–512. [PubMed] [Google Scholar]
- Luo Y, Liu B, Yang X, Ma X, Zhang X, Bragin DE, Yang XO, Huang W, and Liu M (2017). Myeloid adrenergic signaling via CaMKII forms a feedforward loop of catecholamine biosynthesis. J. Mol. Cell Biol 9, 422–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marnett LJ, Rowlinson SW, Goodwin DC, Kalgutkar AS, and Lanzo CA (1999). Arachidonic acid oxygenation by COX-1 and COX-2. Mechanisms of catalysis and inhibition. J. Biol. Chem 274, 22903–22906. [DOI] [PubMed] [Google Scholar]
- Meng W, Liang X, Chen H, Luo H, Bai J, Li G, Zhang Q, Xiao T, He S, Zhang Y, et al. (2017). Rheb inhibits beiging of white adipose tissue via PDE4D5-dependent downregulation of the cAMP-PKA signaling pathway. Diabetes 66, 1198–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan S, Ahmad AS, Glushakov AV, Chambers C, and Dore´ S (2012). Putative role of prostaglandin receptor in intracerebral hemorrhage. Front. Neurol 3, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahmias C, Blin N, Elalouf JM, Mattei MG, Strosberg AD, and Emorine LJ (1991). Molecular characterization of the mouse beta 3-adrenergic receptor: relationship with the atypical receptor of adipocytes. EMBO J. 10, 3721–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablos JL, Santiago B, Carreira PE, Galindo M, and Gomez-Reino JJ (1999). Cyclooxygenase-1 and −2 are expressed by human T cells. Clin. Exp. Immunol 115, 86–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Yoon YS, Han HS, Kim YH, Ogawa Y, Park KG, Lee CH, Kim ST, and Koo SH (2014). SIK2 is critical in the regulation of lipid homeostasis and adipogenesis in vivo. Diabetes 63, 3659–3673. [DOI] [PubMed] [Google Scholar]
- Paschos GK, Tang SY, Theken KN, Li X, Verginadis I, Lekkas D, Herman L, Yan W, Lawson J, and FitzGerald GA (2018). Cold-induced browning of inguinal white adipose tissue is independent of adipose tissue cyclooxygenase-2. Cell Rep. 24, 809–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfannkuche HJ, Kaever V, and Resch K (1986). A possible role of protein kinase C in regulating prostaglandin synthesis of mouse peritoneal macrophages. Biochem. Biophys. Res. Commun 139, 604–611. [DOI] [PubMed] [Google Scholar]
- Polak P, Cybulski N, Feige JN, Auwerx J, Rüegg MA, and Hall MN (2008). Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab. 8, 399–410. [DOI] [PubMed] [Google Scholar]
- Reddy ST, Wadleigh DJ, and Herschman HR (2000). Transcriptional regulation of the cyclooxygenase-2 gene in activated mast cells. J. Biol. Chem 275, 3107–3113. [DOI] [PubMed] [Google Scholar]
- Schulz TJ, Huang P, Huang TL, Xue R, McDougall LE, Townsend KL, Cypess AM, Mishina Y, Gussoni E, and Tseng YH (2013). Brown-fat paucity due to impaired BMP signalling induces compensatory browning of white fat. Nature 495, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J, Cohen P, Cinti S, and Spiegelman BM (2011). Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J. Clin. Invest 121, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sengupta S, Peterson TR, Laplante M, Oh S, and Sabatini DM (2010). mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468, 1100–1104. [DOI] [PubMed] [Google Scholar]
- Shan T, Zhang P, Jiang Q, Xiong Y, Wang Y, and Kuang S (2016). Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia 59, 1995–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin H, Ma Y, Chanturiya T, Cao Q, Wang Y, Kadegowda AKG, Jackson R, Rumore D, Xue B, Shi H, et al. (2017). Lipolysis in brown adipocytes is not essential for cold-induced thermogenesis in mice. Cell Metab. 26, 764–777.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song Y, Altarejos J, Goodarzi MO, Inoue H, Guo X, Berdeaux R, Kim JH, Goode J, Igata M, Paz JC, et al. ; CHARGE Consortium; GIANT Consortium (2010). CRTC3 links catecholamine signalling to energy balance. Nature 468, 933–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CM, Mukherjee S, Ye L, Frederick DW, Kissig M, Davis JG, Lamming DW, Seale P, and Baur JA (2016). Rapamycin blocks induction of the thermogenic program in white adipose tissue. Diabetes 65, 927–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F, Jacques H, and Marette A (2005). Modulation of insulin action by dietary proteins and amino acids: role of the mammalian target of rapamycin nutrient sensing pathway. Curr. Opin. Clin. Nutr. Metab. Care 8, 457–462. [DOI] [PubMed] [Google Scholar]
- Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, and Thomas G (2004). Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431, 200–205. [DOI] [PubMed] [Google Scholar]
- Vegiopoulos A, Mu€ller-Decker K, Strzoda D, Schmitt I, Chichelnitskiy E, Ostertag A, Berriel Diaz, Rozman J, Hrabe de Angelis M, Nu€sing RM, et al. (2010). Cyclooxygenase-2 controls energy homeostasis in mice by de novo recruitment of brown adipocytes. Science 328, 1158–1161. [DOI] [PubMed] [Google Scholar]
- Wada S, Neinast M, Jang C, Ibrahim YH, Lee G, Babu A, Li J, Hoshino A, Rowe GC, Rhee J, et al. (2016). The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Vera L, Fischer WH, and Montminy M (2009). The CREB coactivator CRTC2 links hepatic ER stress and fasting gluconeogenesis. Nature 460, 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White UA, and Stephens JM (2010). Transcriptional factors that promote formation of white adipose tissue. Mol. Cell. Endocrinol 318, 10–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, and Hall MN (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. [DOI] [PubMed] [Google Scholar]
- Yang F, and Bleich D (2004). Transcriptional regulation of cyclooxygenase-2 gene in pancreatic beta-cells. J. Biol. Chem 279, 35403–35411. [DOI] [PubMed] [Google Scholar]
- Zhang HH, Huang J, Du€vel K, Boback B, Wu S, Squillace RM, Wu CL, and Manning BD (2009). Insulin stimulates adipogenesis through the Akt-TSC2-mTORC1 pathway. PLoS One 4, e6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.