

The identification of HIV polymorphisms reproducibly selected under pressure by specific HLA alleles and the elucidation of their impact on viral function can help identify immunogenic viral regions where immune escape incurs a fitness cost. However, our knowledge of HLA-driven escape pathways and their functional costs is largely limited to HIV subtype B and, to a lesser extent, subtype C. Our study represents the first characterization of HLA-driven adaptation pathways in HIV subtypes A1 and D, which dominate in East Africa, and the first statistically rigorous characterization of differential HLA-driven escape across viral subtypes. The results support a considerable impact of viral genetic context on HIV adaptation to host HLA, where HIV subtype-specific sequence variation influences both epitope-HLA binding and the fitness costs of escape. Integrated bioinformatic and mechanistic characterization of these and other instances of differential escape could aid rational cytotoxic T-lymphocyte-based vaccine immunogen selection for both subtype-specific and universal HIV vaccines.
KEYWORDS: CTL, HLA, Uganda, adaptation, epitope, evolution, human immunodeficiency virus, immune escape, subtype
ABSTRACT
The extent to which viral genetic context influences HIV adaptation to human leukocyte antigen (HLA) class I-restricted immune pressures remains incompletely understood. The Ugandan HIV epidemic, where major pandemic group M subtypes A1 and D cocirculate in a single host population, provides an opportunity to investigate this question. We characterized plasma HIV RNA gag, pol, and nef sequences, along with host HLA genotypes, in 464 antiretroviral-naive individuals chronically infected with HIV subtype A1 or D. Using phylogenetically informed statistical approaches, we identified HLA-associated polymorphisms and formally compared their strengths of selection between viral subtypes. A substantial number (32%) of HLA-associated polymorphisms identified in subtype A1 and/or D had previously been reported in subtype B, C, and/or circulating recombinant form 01_AE (CRF01_AE), confirming the shared nature of many HLA-driven escape pathways regardless of viral genetic context. Nevertheless, 34% of the identified HLA-associated polymorphisms were significantly differentially selected between subtypes A1 and D. Experimental investigation of select examples of subtype-specific escape revealed distinct underlying mechanisms with important implications for vaccine design: whereas some were attributable to subtype-specific sequence variation that influenced epitope-HLA binding, others were attributable to differential mutational barriers to immune escape. Overall, our results confirm that HIV genetic context is a key modulator of viral adaptation to host cellular immunity and highlight the power of combined bioinformatic and mechanistic studies, paired with knowledge of epitope immunogenicity, to identify appropriate viral regions for inclusion in subtype-specific and universal HIV vaccine strategies.
IMPORTANCE The identification of HIV polymorphisms reproducibly selected under pressure by specific HLA alleles and the elucidation of their impact on viral function can help identify immunogenic viral regions where immune escape incurs a fitness cost. However, our knowledge of HLA-driven escape pathways and their functional costs is largely limited to HIV subtype B and, to a lesser extent, subtype C. Our study represents the first characterization of HLA-driven adaptation pathways in HIV subtypes A1 and D, which dominate in East Africa, and the first statistically rigorous characterization of differential HLA-driven escape across viral subtypes. The results support a considerable impact of viral genetic context on HIV adaptation to host HLA, where HIV subtype-specific sequence variation influences both epitope-HLA binding and the fitness costs of escape. Integrated bioinformatic and mechanistic characterization of these and other instances of differential escape could aid rational cytotoxic T-lymphocyte-based vaccine immunogen selection for both subtype-specific and universal HIV vaccines.
INTRODUCTION
Human leukocyte antigen (HLA) class I-restricted CD8+ cytotoxic T-lymphocyte (CTL) responses exert evolutionary pressure on an HIV-infected individual’s viral quasispecies, driving the selection of immune escape variants along reproducible mutational pathways (1–4). Using population-based approaches, HLA-associated polymorphisms in HIV, many of which have been experimentally confirmed to be immune escape mutations, have been comprehensively mapped across subtype B (5–9) and, to a lesser extent, subtype C and circulating recombinant form (CRF) 01_AE (CRF01_AE) (10–13). However, our knowledge of HLA-driven escape in other major HIV subtypes, as well as the extent to which viral genetic context modulates these pathways, remains limited.
It is also well established that the immune evasion benefits of some escape mutations are offset by costs to viral replication and/or protein function (14–23). A leading CTL-based HIV vaccine strategy aims to focus immune responses against such constrained regions (24); novel approaches to identify them within the diverse HIV subtypes (25) would thus be useful. Characterization of HLA-driven adaptation pathways across HIV subtypes is even more relevant, given the accumulating evidence that HIV subtype modulates viral evolution (26–29), pathogenesis (30–34), and immunity (35–40). Notably, the infecting HIV subtype influences the rate of clinical progression in untreated infection (30–37). Though the mechanisms underlying these effects are incompletely known, differential HLA-mediated viremia control as a result of sequence variation in key HIV CTL epitopes contributes at least in part (35–37). HLA-B*35 expression, for example, is associated with faster progression in subtype B, but not subtype C, infection in part because the subtype B consensus sequence of the key NY10 epitope (Gag codons 253 to 262) has a reduced affinity for B*35 (37). Instances of differential HLA-driven escape mutations across viral subtypes, attributable to viral backbone-specific fitness constraints (38–40), have also been described. For example, HLA-B*57/58:01-driven escape in KF11 (Gag codons 162 to 172) occurs via A163G in subtype C but S173T in subtype B, as the former compromises viral replication in a subtype B backbone (38, 39); similarly, M250I, located one codon downstream of TW10 (Gag codons 240 to 249), is readily selected in subtype C but only rarely in subtype B, where it attenuates viral replication (40). Differential HLA-B*57-associated escape within the KF11, IW9 (Gag 147 to 155), and TW10 epitopes has also been described between subtypes A1 and D, though the underlying mechanisms remain to be elucidated (41, 42). A framework to identify viral regions that differ in immunogenicity between HIV subtypes or where the functional costs of immune escape differ between HIV subtypes could thus aid in vaccine design.
We developed and applied such a framework here. We begin by characterizing the pathways of HLA-mediated adaptation in HIV subtypes A1 and D, two understudied viral subtypes that cocirculate in East Africa for which HLA-driven escape pathways have not yet been elucidated. Next, using a novel phylogenetically informed comparative statistical approach, we demonstrate that more than one-third of identified HLA-driven escape pathways are differentially selected between these subtypes. Finally, we mechanistically investigate two instances of statistically significant differential escape between subtypes and identify subtype-specific epitope-HLA binding and mutational constraints on immune escape as their likely causes. Our results confirm that viral genetic context modulates HIV adaptation to host cellular immunity and highlight the utility of integrated bioinformatic and molecular approaches to identify candidate epitopes for inclusion in subtype-specific and universal CTL-based HIV vaccines.
RESULTS
Subtype A1 and D cohort subsets exhibit distinct HIV, but concordant host, genetic profiles.
Host population HLA class I allele frequencies influence the prevalence of HLA-associated polymorphisms in circulating HIV strains (10, 43–45). To avoid confounding by this and other host population-related factors, studies attempting to identify differences in HLA-driven evolution across HIV subtypes must be undertaken in a single host population where two or more viral subtypes cocirculate at substantial frequencies. Uganda, where HIV subtypes A1 and D cocirculate, represents such a locale. We began by amplifying and bulk sequencing plasma HIV RNA gag, polymerase (pol), and nef from 513 chronically HIV-infected antiretroviral-naive participants in two established cohort studies (46–48). pol was amplified in two segments: protease-reverse transcriptase (RT) and integrase. Overall, 464 (90.4%) participants harbored a gag, pol, and/or nef sequence that could be classified as pure subtype A1 or D. This included 257 (50%) subtype A1-infected participants, 185 (36%) subtype D-infected participants, and 22 (4%) participants who were likely infected with a subtype A1/D recombinant but whose individual HIV genes could be classified as a single subtype (e.g., a participant infected with a virus whose 5′ and 3′ halves were A1 and D, respectively, would be classified as subtype A1 for Gag and subtype D for Nef) (Fig. 1). The remaining 49 (10%) participants were excluded because they harbored other viral subtypes and/or intersubtype recombinants where breakpoints occurred within the studied genes. In total, our analysis included 203 gag, 222 protease-RT, 200 integrase, and 210 nef subtype A1 sequences, as well as 134 gag, 154 protease-RT, 140 integrase, and 127 nef subtype D sequences.
FIG 1.
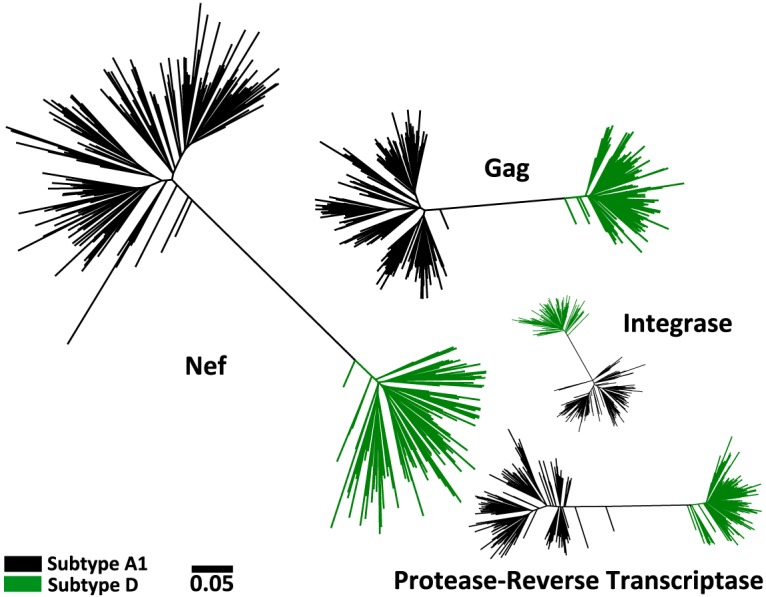
Maximum likelihood phylogenies inferred from subtype A1 and D gag, protease-RT, integrase, and nef sequence data sets. For subtype A1, 203 gag, 222 protease-RT, 200 integrase, and 210 nef sequences were collected. For subtype D, 134 gag, 154 protease-RT, 140 integrase, and 127 nef sequences were collected. All sequences cluster according to their classified subtype. All phylogenies are drawn on the same distance scale; units are in estimated numbers of nucleotide substitutions per site.
Critically, the HLA class I frequencies of the subtype A1 and D cohort subsets were highly comparable. Of the 106 total HLA-A, -B, and -C alleles observed, none differed significantly in frequency between cohort subsets at the predefined false discovery rate (FDR) threshold of a q value of <0.2 (P values for all comparisons were >0.05, except for HLA-B*47:01, for which P was 0.03) (Fig. 2). Cohort subsets were also generally well matched for sociodemographic and clinical characteristics (Table 1). They did not differ significantly with respect to plasma viral load (pVL) (median for subtype A1-infected participants, 5.2 log10 HIV RNA copies/ml [interquartile range {IQR}, 4.6 to 5.7 log10 HIV RNA copies/ml]; median for subtype D-infected participants, 5.1 log10 HIV RNA copies/ml [IQR, 4.6 to 5.5 log10 HIV RNA copies/ml]; P = 0.13), CD4+ T-lymphocyte count (median for subtype A1-infected participants, 135 cells/mm3 [IQR, 72 to 201 cells/mm3]; median for subtype D-infected participants, 120 cells/mm3 [IQR, 54 to 200 cells/mm3]; P = 0.17), or gender (proportion of females among subtype A1-infected participants, 67%; proportion of females among subtype D-infected participants, 75%; P = 0.15), though a modest age difference was noted (median age for subtype A1-infected participants, 35 years [IQR, 30 to 41 years]; median age for subtype D-infected participants, 34 years [IQR, 28 to 38 years]; P = 0.02). Together, these results are consistent with HIV subtypes A1 and D cocirculating in a single host population, thereby minimizing the potential for confounding by host-related differences between cohort subsets.
FIG 2.

HLA class I allele frequencies in subtype A1 and D cohort subsets. HLA alleles present at a >1% frequency in either cohort subset are shown. Cohort subsets did not differ in the frequency of any HLA allele at a q value of <0.2. The 22 (4%) individuals who were likely infected with recombinant subtype A1/D viruses but for whom individual HIV gene fragments could nevertheless be classified as pure subtypes were excluded from this analysis.
TABLE 1.
Sociodemographic and clinical characteristics of subtype A1- and D-infected cohort subsetsb
| Characteristic | Value(s) for patients infected with the following HIV subtype: |
P value | |
|---|---|---|---|
| A1 (n = 257) | D (n = 185) | ||
| % female | 67 | 75 | 0.15 |
| Median (IQR) age (yr) | 35 (30–41) | 34 (28–38) | 0.02 |
| Median (IQR) pVLa (log10 copies/ml) | 5.2 (4.6–5.7) | 5.1 (4.6–5.5) | 0.13 |
| Median (IQR) CD4+ T-cell count (no. of cells/mm3) | 135 (72–201) | 120 (54–200) | 0.17 |
pVL, plasma viral load.
Data for the 22 participants likely infected with a subtype A1/D recombinant but whose individual HIV genes could be classified as a single subtype are excluded from this table.
Identification of HLA-associated polymorphisms in HIV subtypes A1 and D.
Using phylogenetically informed approaches (9, 49) (see also Materials and Methods), we identified a total of 385 unique HLA-associated polymorphisms occurring at 183 unique HIV codons in subtype A1 and D Gag, protease-RT, integrase, and Nef at a q value of <0.2 (corresponding to P < 9 × 10−4 for subtype A1 and P < 8 × 10−4 for subtype D) (Fig. 3; see also Table S1 in the supplemental material). Specifically, 251 associations were identified at 123 codons in subtype A1 (55 [22%] in Gag, 11 [4%] in protease, 78 [31%] in RT, 52 [21%] in integrase, and 55 [22%] in Nef), while 180 HLA-associated polymorphisms were identified at 105 codons in subtype D (36 [20%] in Gag, 5 [3%] in protease, 47 [26%] in RT, 26 [14%] in integrase, and 66 [37%] in Nef). Consistent with previous observations in subtypes B, C, and CRF01_AE (8–13), Nef exhibited the highest density of HLA-associated polymorphisms. In subtype A1, 13% of Nef codons compared to 5% of Gag, 5% of protease, 7% of RT, and 10% of integrase codons harbored at least one HLA-associated polymorphism, while in subtype D, 17% of Nef codons compared to ≤5% of codons in other viral proteins harbored an HLA-associated polymorphism.
FIG 3.



Gag, Nef, protease, reverse transcriptase, and integrase immune escape maps of HLA-associated polymorphisms identified in subtypes A1 and D. HLA-associated polymorphisms identified in HIV subtype A1 (black consensus sequence) are displayed above; those identified in subtype D (green consensus sequence) are shown below. HIV subtype consensus sequences were obtained from the Los Alamos National Laboratory HIV sequence database, aligned to the HXB2 reference strain, and gap stripped to preserve HXB2 codon numbering. HIV residues that were overrepresented among individuals expressing the given HLA allele are shown in red; these represent the HLA adapted (inferred escaped) form. In contrast, HIV residues that are underrepresented in individuals expressing the given HLA allele are shown in blue; these represent the nonadapted (inferred susceptible) form for that HLA allele. Groups of polymorphisms in close proximity that are selected by the same HLA allele are boxed in yellow. HLA-associated HIV polymorphisms that do not survive correction for HIV codon covariation are shown in lowercase letters. Polymorphisms identified at a q value of <0.2 are shown.
In total, 32% of the 385 unique HLA-associated polymorphisms had been previously described in at least one other HIV subtype (9, 10, 12, 13, 45) (Table S1). HLA-A*03:01-associated Gag K28Q, for example, which is located within the HLA-A*03:01-restricted RY10 epitope (50), which ranks among the strongest HLA associations identified in subtype B (9) and which has also been described in subtype C (12), was among the strongest associations in subtype A1. We also observed numerous examples where the same HIV polymorphism was selected by more than one HLA allele. Gag K28Q in subtype A1, for example, is also strongly selected by HLA-A*30:01; this association has also been reported in subtypes B and C (9, 12). We also observed cases where HLA alleles exert opposing selection on HIV. S123D in RT in subtype A1, for example, represents the adapted (inferred escaped) form for HLA-B*57:03 but the nonadapted (inferred susceptible) form for HLA-B*49:01. Clusters of mutations selected by the same HLA allele, which often denote the presence of novel CTL epitopes (7), were also observed. Nef Y102H and E108D in subtype A1, for example, were selected by HLA-B*44:03, supporting the existence of a novel B*44:03-restricted epitope here.
Numerous HIV polymorphisms were selected by the same HLA in both subtypes A1 and D; this included, for example, HLA-C*07:01-associated Nef K105R, which is also selected by this allele in subtypes B, C, and CRF01_AE (9, 12, 13). Other HLA-associated polymorphisms were identified in only one subtype. Gag E161D, for example, located 1 residue upstream of the HLA-B*57-restricted Gag KF11 epitope, was associated with HLA-B*57:03 in subtype D, whereas no HLA associations were identified at a q value of <0.2 in subtype A1 at this site.
Substantial HIV subtype-specific adaptation to host HLA.
Though immune escape maps provide a convenient way to visualize HLA-associated HIV polymorphisms, they cannot be used to definitively identify viral sites under differential HLA-mediated selection between HIV subtypes. Even if the same association appears on multiple escape maps, its strength of selection may still differ between subtypes; conversely, the absence of a particular polymorphism on a given escape map may simply be because it narrowly missed the predefined statistical cutoff. We therefore employed a novel modification of a phylogenetically corrected logistic regression approach (9, 10, 49, 51) (see also Materials and Methods) to identify instances of statistically significant differential HLA-mediated selection between HIV subtypes A1 and D. Briefly, we took the union of all polymorphisms identified in either subtype where ≥3 individuals in both cohort subsets carried the relevant HLA allele, which, in total, was 289 HLA-associated polymorphisms. We then assessed whether, after controlling for other covariates, the viral subtype represented a significant independent predictor of each HLA-associated viral polymorphism. If so, it was classified as differentially selected between subtypes.
Overall, 34% (99 of 289) of polymorphisms were differentially selected between subtypes A1 and D at a q value of <0.1 and a P value of <0.05 (Fig. 4; Table S1). The proportion of HLA-associated polymorphisms that were differentially selected across subtypes A1 and D was not significantly different across all viral proteins investigated (P > 0.05 for all by Fisher’s exact test). In descending order these were protease, where 60% (6/10) of polymorphisms were differentially selected between subtypes, RT (37%, 34/92), integrase (37%, 13/35), Gag (34%, 22/64), and Nef (27%, 24/88).
FIG 4.

HLA-associated polymorphisms exhibiting differential selection between subtypes A1 and D. The natural logarithm of the odds ratio (lnOR) of selection of HLA-associated polymorphisms in Gag, Nef, protease, reverse transcriptase, and integrase that were differentially selected between subtypes A1 and D are shown. The lnOR of a specific association is shown only if it was independently statistically significant in a given HIV subtype; otherwise, it was set to 0. In most cases, differential selection of an HLA-associated polymorphism occurred because the polymorphism was selected in one subtype but not the other (e.g., B*57:03-Gag T242N was significantly selected in subtype D with an lnOR of 5.6 but not in subtype A1). A small filled circle denotes cases where the same HIV polymorphism was selected at significantly different strengths in the two subtypes. For example, HLA-C*07:01 selects for Nef K105R in subtype A1 (lnOR= 2.3) but more strongly in subtype D (lnOR= 6). Polymorphisms differentially selected at a false discovery rate threshold of a q value of <0.1 and a P value of <0.05 are shown. Infinite positive or negative lnORs were set to 6 or −6, respectively, for visualization purposes.
The vast majority of differentially selected viral polymorphisms (92 of 99; 93%) occurred because they were selected by the relevant HLA allele in one subtype but not the other (46 were selected in subtype A1 but not D, and 46 vice versa) (Fig. 4). Notably, HLA-B*57:03-associated Gag T242N, located within the immunodominant TW10 epitope (codons 240 to 249), was among the strongest instances of differential selection identified. In subtype D, the odds ratio (OR) of selection of Gag T242N by HLA-B*57:03 was 268 (corresponding to 5.6 when expressed as a natural logarithm [lnOR]), whereas in subtype A1, the OR of this association was not significantly different from 1. Applying our test for differential escape between HIV subtypes A1 and D for HLA-B*57:03-associated T242N thus yielded a highly statistically significant P value (P = 8 × 10−6, q = 3 × 10−4). HLA-A*03:01-associated 196R in Nef (intersubtype comparison, P = 7.3 × 10−6, q = 3 × 10−4), HLA-B*81:01-associated V77I in protease (P = 0.005, q = 0.02), and HLA-A*30:02-associated T218I in integrase (P = 1 × 10−4, q = 0.001), all of which were selected in subtype D but not subtype A1, represented the most significant instances of differential selection in these proteins, while HLA-A*30:01-associated K281R in RT, selected in subtype A1 alone, was the most significant instance of differential selection in this protein (P = 1.6 × 10−5; q = 4 × 10−4).
The remaining 7 instances of differential selection occurred when the same HLA allele selected the same polymorphism in both subtypes at significantly different strengths (3 were more strongly selected in subtype A1 than in subtype D, and 4 vice versa) (Fig. 4). For example, HLA-C*07:01 selected Nef K105R with an lnOR of ∞ in subtype D, whereas lnOR was only 2.3 in subtype A1 (intersubtype comparison, P = 0.02, q = 0.05). Similarly, HLA-A*66:01 selected alanine at integrase codon 97 with an lnOR of 4.2 in subtype A1 but an lnOR of 3.6 in subtype D (P = 0.03, q = 0.07). Our results therefore indicate that while HIV subtypes A1 and D display substantial overlap in their preferred mutational pathways for adaptation to host HLA (66% of identified polymorphisms were not differentially selected between the two subtypes), viral genetic context nevertheless plays a key role in defining these pathways.
Mechanisms underlying differential HLA-associated adaptation between HIV subtypes.
Understanding the mechanisms underlying differential escape is critical if we are to leverage this information for vaccine design. In particular, instances of differential escape attributable to epitope immunogenicity (e.g., because a particular epitope poorly binds its cognate HLA in a given subtype) might identify less useful vaccine epitopes for a given viral subtype, while those attributable to higher mutational barriers to escape in a particular HIV subtype might identify more useful vaccine epitopes. We thus interpreted our list of differentially selected viral polymorphisms (Fig. 4) in the context of subtype-specific HIV genetic variation, published CTL epitope immunogenicity data, protein domain structural predictions, and epitope/HLA binding predictions to identify candidates for mechanistic characterization. We identified HLA-B*57:03-associated Gag T242N to be a candidate for differential escape due to subtype-specific fitness costs and HLA-B*15:03-associated Nef K105R to be a candidate for differential escape due to subtype-specific differences in epitope/HLA binding.
HIV subtype-specific functional constraints underlie some instances of differential escape: B*57:03-Gag T242N.
Gag TW10 is strongly and frequently recognized by B*57-expressing individuals infected with various HIV subtypes (15, 50, 52), including A1 and D (41, 53). However, whereas Gag T242N is reproducibly selected by B*57:03 in subtype D (and others [9, 45]), it is not selected by this allele in subtype A1 (Fig. 4), despite our cohort being sufficiently powered to detect it (see Discussion). Notably, the subtype A1 consensus differs from that of all other major subtypes and CRFs at Gag codon 243, the residue immediately downstream of the mutation site: 75% of subtype A1 sequences harbor proline, whereas >97% of sequences belonging to subtypes B, C, D, G, and H as well as CRF01_AE and CRF02_AG harbor leucine (http://www.hiv.lanl.gov/). In fact, the TW10 consensus sequence in subtypes D and A1 differ only at this residue (indicated in boldface): 240TSTLQEQIGW249 in subtype D compared to 240TSTPQEQIGW249 in subtype A1.
Gag T242N can, however, occur in subtype A1 under some circumstances. We observed 8 instances of this mutation in subtype A1-infected individuals, 7 of whom expressed B*57:02 or B*58:01, alleles that belong to the same HLA supertype as B*57:03. In subtype A1, however, T242N never occurred in conjunction with the subtype consensus proline at position 243; rather, the adjacent residue was the HIV group M consensus L243 (3/8), V243 (2/8), T243 (1/8), I243 (1/8), or a mixture of the last two (1/8). We therefore hypothesized that consensus P243 prevents the selection of T242N in subtype A1 (41).
In silico structural models of Gag codons 232 to 254 in subtypes A1 and D, which encompass helix 6 of p24Gag and the region immediately upstream, supported this hypothesis (Fig. 5A). Atomic distances between aligned structures were expressed in terms of the root mean square deviation (RMSD) (54), where values greater than 2.5 to 3 Å in silico indicate substantial structural differences (55–57). The subtype A1 consensus proline at Gag codon 243 already notably alters the peptide structure compared to that of subtype D. Furthermore, whereas T242N is predicted to only minimally impact the helix 6 structure in subtype D (RMSD between predicted structures, 0.77 Å), T242N in subtype A1 is predicted to prevent helix 6 formation and to disrupt upstream protein folding (RMSD between predicted structures, 4.3 Å).
FIG 5.

Subtype-specific mutational constraints drive differential B*57:03-mediated selection of Gag T242N between subtypes A1 and D. (A) In silico modeling of the effect of Gag T242N (red, bottom models) on the structure of subtype D (green, left) and A1 (gray, right) consensus p24Gag codons 232 to 254 (top models). Differences in the subtype D and A1 consensus sequence at codon 243 are highlighted (light green, subtype D leucine; light gray, subtype A1 proline). While T242N is predicted to have minimal impact in subtype D (green, left, top versus bottom), its introduction into subtype A1 p24Gag is predicted to substantially alter the peptide structure (gray, right, top versus bottom) by disrupting helix formation (i.e., codon 241, blue) and upstream folding. (B) Representative flow cytometry plots confirming the infectivity (GFP) of VSVg-pseudotyped recombinant virus stocks generated from participant-derived plasma HIV RNA sequences in a CEM-derived LTR-driven GFP reporter T-cell line during experiments to determine the viral titer. Numbers indicate the percentage of GFP-positive (i.e., HIV-infected) cells. (C) Representative curves depicting the rate of spread of VSVg-pseudotyped subtype D (green, top) and subtype A1 (black, bottom) parental (circles; dark green, subtype D; black, subtype A1) and T242N mutant (squares; light green, subtype D; gray, subtype A1) viruses over multiple days in culture. The median and standard error of the percentage of GFP+ (infected) cells relative to that on day 2 are shown. Three independent experiments were performed. (Insets) Replication capacity of parental and T242N mutant viruses (where the value for the latter is normalized to that for the former) calculated over the exponential phase of viral replication. T242N had a modest, though not statistically significant, impact on viral replication in subtype D (median, 0.96 relative to parental subtype D; P = 0.1) but completely abrogated subtype A1 viral replication (P = 0.004).
To confirm mutational antagonism between T242N and P243, we engineered the former into a recombinant virus that harbored a participant-derived subtype A1 Gag-protease sequence and another that harbored a participant-derived subtype D Gag-protease sequence in an HIVNL4.3 reference strain backbone and generated virus stocks pseudotyped with the vesicular stomatitis virus G (VSVg) envelope (58). The titers of the virus stocks were determined by infecting a long-terminal repeat (LTR)-driven green fluorescent protein (GFP) reporter CD4+ T-cell line and measuring the percentage of GFP-expressing (i.e., infected) cells 48 h later. All viral stocks were confirmed to be infectious, though the titers of the T242N-containing subtype A1 mutant were lower than those of the others (Fig. 5B). We then assessed the ability of these viruses to replicate in the same GFP reporter T-cell line (59). Cells were infected with a standard viral inoculum (multiplicity of infection, 0.003), and viral spread was monitored by flow cytometry over up to 8 days postinfection. Dramatic differences in the impact of T242N on viral replication were observed between the two subtypes (Fig. 5C). Consistent with previous observations in subtypes B and C (19, 60), which also harbor leucine at codon 243, T242N conferred a modest impact on the viral replication capacity in subtype D (0.96 relative to parental subtype D; P = 0.1) but completely abrogated viral replication in subtype A1 (P = 0.004). Complete abrogation of T242N-containing subtype A1 viral replication continued to be observed even when cultures were infected at multiplicities of infection of up to 0.05 (data not shown). Viral replication was similarly abolished when T242N was engineered into a recombinant HIVNL4.3 strain harboring the consensus subtype A1 Gag-protease (Fig. 6).
FIG 6.

Gag T242N abrogates viral replication in the subtype A1 consensus backbone. (A) Representative flow cytometry plots demonstrating the infectivity (GFP) of subtype A1 consensus and T242N mutant recombinant viruses during experiments to determine the viral titer. The numbers indicate the percentage of GFP-positive (i.e., infected) cells. (B) Representative curves depicting the rate of spread of VSVg-pseudotyped subtype A1 consensus (black, circles) and T242N mutant (gray, squares) viruses over multiple days in culture. The median and standard error of the percentage of GFP+ (infected) cells relative to that on day 2 is shown. Two independent experiments were performed.
Subtype-specific epitope presentation underlies some instances of differential escape: B*15:03-Nef K105R.
While the result presented above confirms that subtype-specific mutational barriers underlie some instances of differential escape, other cases likely reflect a lack of immune pressure in a given subtype because the relevant CTL epitope is not efficiently presented by the restricting HLA in that viral genetic context. Nef K105R, which is selected by B*15:03 in subtype A1 but not subtype D (intersubtype comparison, P = 0.005, q = 0.02) (Fig. 4), represented a candidate for the latter mechanism. Of note, K105R is reproducibly selected by another allele, HLA-C*07:01, in both subtypes and, in fact, is more strongly selected in D than A1 (Fig. 4). This suggests that differential selection of K105R by B*15:03 is not due to subtype-specific fitness costs but, rather, is because B*15:03 exerts a greater selective pressure on this region of Nef in subtype A1 than in subtype D due to the higher-affinity binding of a putative viral epitope to this allele in subtype A1.
We first confirmed that K105R did not substantially impact Nef’s three most well-characterized functions—cell surface CD4, HLA class I, and SERINC5 downregulation (61–64)—in either subtype backbone (Fig. 7A). As hypothesized, K105R did not impact Nef-mediated CD4 downregulation capacity in subtype D (mean function of K105R relative to the subtype consensus, 0.99 [standard deviation = 0.004]; P = 0.25), nor did it majorly impair HLA class I (mean, 0.95 [standard deviation = 0.05]; P = 0.25) or SERINC5 (mean, 0.98 [standard deviation = 0.02]; P = 0.5) downregulation (Fig. 7B). Likewise, K105R had no impact on subtype A1 Nef function, where the CD4 (mean, 1.0 [standard deviation = 0.009]; P = 0.75), HLA (mean, 1.0 [standard deviation = 0.03]; P = 0.75), and SERINC5 (mean, 1.0 [standard deviation = 0.03]; P = 0.5) downregulation functions were not significantly different from the consensus downregulation function (Fig. 7B).
FIG 7.

Subtype-specific epitope presentation drives differential B*15:03-mediated selection of Nef K105R between subtypes A1 and D. (A) Representative flow cytometry plots depicting the ability of consensus and K105R Nef subtype D (top) and A1 (bottom) sequences to downregulate CD4, HLA class I, and SERINC5 in a transfection-based assay (dark green, consensus subtype D; black, consensus subtype A1; light green, K105R subtype D; gray, K105R subtype A1). The negative control (No Nef) corresponds to the empty pSelect-GFP vector. The numbers in the Nef-positive (GFP+) gate denote the median fluorescence intensity (MFI) of the cell surface molecule under investigation. (B) Histograms depicting the ability of the Nef K105R subtype D (top) and subtype A1 (bottom) mutant to downregulate CD4, HLA class I, and SERINC5, shown as the mean ± standard deviation from a minimum of three independent experiments, relative to that of their corresponding parental consensus sequences (shown as having a function of 1.0). The K105R polymorphism had no significant impact on either subtype D (P > 0.25 for all comparisons) or subtype A1 (P = 0.75 for all comparisons) CD4, HLA, or SERINC5 downregulation relative to that of the corresponding subtype consensus. (C) Epitopes used in the competitive binding assay, along with their predicted binding affinities to HLA-B*15:03 (derived from netMHC [version 4.0] software). (D) Titration (64-fold dilution) of the fluorescently labeled IY9 reference peptide over parental HLA-null K562-derived immortalized T cells and those retrovirally transduced to stably express HLA-B*15:03. The dose-dependent loss of fluorescence was observed only on B*15:03-expressing cells, indicating that the IY9 reference peptide is B*15:03 specific. (E) Epitope cellular competitive binding assay results. The median fluorescence relative to that with no competitor peptide is shown. As expected, the unlabeled reference peptide is able to compete with the labeled version of itself for B*15:03 binding in a dose-dependent manner. By comparison, the HLA-A*02-specific FS8 negative-control epitope was unable to compete with labeled IY9 for binding to B*15:03 at any concentration. Subtype A1 consensus epitope RL9 displaced more labeled reference peptide than the subtype D consensus epitope QL9 over all concentrations tested, confirming that RL9 (subtype A1) had the higher affinity of the two.
We then investigated whether differential B*15:03-mediated selection of Nef K105R is due to the presence of a B*15:03-restricted epitope in subtype A1 but not D. As no B*15:03 epitopes have been mapped in this region, we used netMHC (version 4.0) software (65) to identify a putative B*15:03-restricted epitope spanning Nef codons 104 to 112, where K105R sits at position 2. Consistent with our hypothesis, the subtype A1 consensus sequence (RL9; 104RKRQEILDL112; epitope amino acid differences are indicated in boldface) was predicted to bind nearly 7-fold more strongly to B*15:03 (predicted binding affinity, 131.2 nM) than the subtype D consensus (QL9; 104QKRKEILDL112; predicted binding affinity, 875.4 nM) (Fig. 7C).
Epitope binding to B*15:03 was experimentally confirmed using a competitive binding assay where a titration of the epitope of interest competes with a fixed dose of a fluorescently labeled reference peptide for B*15:03 binding on the surface of cells (66–68) (Fig. 7E). The reference peptide used was IY9 (IQQCFGIPY [IY9]), which was modified (as indicated in boldface) from the published B*15:03-restricted IQQEFGIPY epitope spanning HIV integrase codons 135 to 143 (69) to contain cysteine at position 4, on which the fluorophore was conjugated. The IY9 reference peptide, predicted to retain a high affinity for B*15:03 (3.3 nM; Fig. 7C), selectively bound K562-derived cells engineered to stably express B*15:03 across a 64-fold dilution range (Fig. 7D). It also behaved as expected in competition experiments, where titration of unlabeled IY9 consistently reduced the median fluorescence of target cells by up to 53% in a dose-dependent manner, while titration of the negative-control HLA-A*02:01-specific FS8 epitope, which spans Gag codons 433 to 440 (70) and which has a predicted B*15:03 binding affinity of 40,487 nM (Fig. 7C), did not reduce the fluorescence at any concentration tested (Fig. 7E).
Having validated our assay to be B*15:03 and peptide specific, we tested the predicted subtype A1 and D RL9/QL9 epitopes for their ability to compete with labeled IY9 for B*15:03 binding (Fig. 7E). While both RL9 and QL9 were able to compete with IY9 for binding to B*15:03 in a dose-dependent manner, subtype A1 RL9 consistently reduced the median fluorescence relative to that for no competitor peptide to a greater extent than subtype D QL9 over all concentrations tested (mean, 9% [standard deviation = 2.7%] greater reduction in subtype A1 than in subtype D). These results are consistent with netMHC binding predictions (Fig. 7C) for these epitopes and support the notion that the subtype A1 epitope sequence is more immunogenic. Therefore, the absence of the B*15:03-restricted K105R polymorphism in the context of subtype D infection is likely attributable to reduced or absent B*15:03-mediated selective pressure on this region of Nef in vivo.
DISCUSSION
This study represents the first systematic characterization of HLA-driven polymorphisms in major HIV group M subtypes A1 and D and the first rigorous statistical investigation of the extent to which mutational immune escape pathways differ across HIV subtypes in a cohort that was carefully selected to minimize confounding by host immunogenetic, sociodemographic, and other factors. Overall, our results indicate that while many immune escape pathways are shared across HIV subtypes—32% of HLA-associated polymorphisms identified in the present study had been previously described in at least one other HIV subtype—a substantial proportion are not. In particular, our observation that 34% of HLA-associated polymorphisms are statistically significantly differentially selected between subtypes A1 and D identifies the viral genetic backbone to be a key modulator of immune-driven HIV evolution.
The HLA-associated polymorphisms identified in the present study generally corroborate previous observations in subtypes A1 and D (41, 42), though some differences are noted. Gag T242N had previously been observed among individuals expressing B58 supertype alleles in subtype D, but not subtype A1, in a Kenyan cohort (41); our study, performed at subtype-level HLA resolution, further identifies B*57:03 to be the specific allele responsible for these differential effects. However, the Kenyan study also identified other B58 supertype-associated escape mutations in Gag that were not observed in the present study, at least not at the stringent cutoff of a q value of <0.2: these included P243T and I247X in the TW10 epitope in subtype A1, A163G/S165N in the KF11 epitope in both subtypes A1 and D, and A162X in subtype A1 (41). B*57-associated Gag I147L in the IW9 epitope, previously described in subtype D (41), was also not observed in the present study. While confounding by regional differences in HLA frequencies cannot be ruled out and issues relating to statistical power must be acknowledged, the observation that the Ugandan and Kenyan subtype A1 and D Gag consensus sequences differ at 10 and 7 codons, respectively, suggests that the influence of HIV genetic context on HLA-mediated adaptation may be even more nuanced, with differences possibly occurring at the sub-subtype or regional levels. The observation that 13% of polymorphisms restricted by a given HLA allele (where the frequency of each allele was verified to be comparable between cohorts) in subtype B were differentially selected in Japan compared to Canada, the United States, and Australia also supports regional HIV strain variation as a determinant of immune escape pathways, though the possibility of confounding by other host population factors, in particular, T-cell repertoire genetics, cannot be ruled out (10).
While our study supports a role of viral genetics in modulating immune-driven HIV evolution, our estimate that 34% of HLA-associated polymorphisms differ between viral subtypes is nevertheless lower than earlier estimates. In particular, a study comparing HLA-associated polymorphisms identified in subtype B-infected individuals in Canada and subtype C-infected individuals in South Africa estimated that HLA-mediated selection differed in 86% of shared epitopes, defined at the HLA allele level, between these HIV subtypes (11). This study, however, did not formally test for differential escape and was also likely confounded by differences in HLA subtype frequencies between cohorts (e.g., with B*57:01 versus B*57:03 being the most common B*57 allele in Canada and South Africa, respectively [39, 71]). Our observations that 32% of identified polymorphisms represented canonical escape mutations described in other subtypes and, furthermore, that 66% of identified HLA-associated polymorphisms did not significantly differ in the strength of selection across subtypes A1 and D underscore that, despite substantial differences, many immune escape pathways are nonetheless shared across HIV subtypes.
We further confirm that distinct mechanisms, namely, HIV subtype-specific fitness costs of escape and differential epitope binding, explain specific instances of differential HLA-driven adaptation. This is important, as the underlying mechanism has vaccine design implications. Gag TW10, for example, is highly immunogenic in multiple HIV subtypes (14, 15, 41, 52), and escape via T242N confers modest in vitro yet likely substantial in vivo fitness costs in most viral subtypes, as evidenced by its rapid and frequent reversion upon transmission to an HLA-mismatched host (15, 72). Here we demonstrate, through site-directed mutagenesis of one participant-derived sequence and one consensus sequence, that the T242N-P243 combination completely abrogates viral replication in subtype A1. Though we cannot completely rule out the possibility that compensatory mutational pathways could exist in some subtype A1 contexts, the severity of this defect demonstrated here and the lack of observation of T242N-P243 in natural sequences in this and other (41, 42) studies render this somewhat unlikely. We hypothesize that T242N reduces fitness by destabilizing the HIV capsid structure in the subtype A1 consensus viral backbone, a conclusion that is supported by our peptide modeling experiments and our observation that T242N/P243-containing subtype A1 viruses display lower infectivity, even when VSVg pseudotyped. While Gag T242N is reported to increase HIV capsid sensitivity to the cellular restriction factor TRIM5α (73) and we cannot rule out the contribution of TRIM5α to our results, the capsid of subtype A1 is less sensitive to TRIM5α than that of other subtypes (73) and the CEM-derived T-cell line used in our viral spread assay expresses relatively high levels of cyclophilin A (74), which counteracts the effect of TRIM5α on capsid stability (75). Regardless of the specific mechanism underlying the fitness cost, these observations support inclusion of this epitope in pan-subtype HIV vaccines and further identify it as a particularly useful immunogen in subtype A1.
Other cases of differential adaptation identify epitopes that are likely to represent poor immunogens. The putative B*15:03-restricted epitope RL9/QL9 (Nef codons 104 to 112), discovered via differential B*15:03-mediated escape via Nef K105R, is unlikely to be immunogenic in subtype D due to the weaker binding of its cognate sequence to this allele. Further evaluation of RL9’s immunogenicity in subtype A1 and other subtypes is merited, though K105R’s lack of an in vitro functional cost suggests a relatively low barrier to escape. Integrated bioinformatic and molecular approaches like those described in the present study, combined with information on epitope immunogenicity and HLA-mediated viremia control, could thus aid the rational selection of CTL-based vaccine immunogens.
The present study is not without limitations. Our subtype A1 and D cohort subsets were smaller and, thus, lower powered than those normally used to identify HLA-associated polymorphisms (9, 10, 12, 13, 45), and as such, these immune escape maps should not be considered comprehensive. Power limitations also precluded us from investigating certain associations more fully. Our observation that HLA-B*57:02 was significantly associated with Gag T242N in subtype A1 (P = 1.9 × 10−6, q = 0.006) (Fig. 3; see also Table S1 in the supplemental material) was particularly intriguing; however, we could not assess whether this association is differentially selected between subtypes A1 and D because only two individuals in the latter group expressed B*57:02. Our ability to determine why Gag T242N is selected by B*57:02 but not by the more common HLA-B*57:03 in subtype A1 was similarly limited, though it is worth noting that the absence of selection by the latter is not attributable to a lack of statistical power: whereas 4 of 6 subtype A1-infected individuals expressing B*57:02 harbored T242N, leading to a statistically significant association after phylogenetic correction, T242N was observed in only 1 of 15 B*57:03-expressing subtype A1-infected individuals, who also expressed B*57:02. Even more intriguingly, all 8 instances of T242N in subtype A1, of which 4 occurred in B*57:02-expressing individuals, never occurred with the subtype consensus proline at the adjacent codon 243, suggesting that B*57:02 was pressuring HIV to escape via P243X in subtype A1. Indeed, B*57:02 carriage was negatively associated with Gag P243 in subtype A1 with an lnOR of −∞ (P = 0.007), but this association did not survive our stringent multiple-comparison correction and, thus, is not featured on our immune escape map. Nevertheless, this suggests that differences in TW10 epitope presentation between B*57:02 and B*57:03 in subtype A1 may alter immune selection pressures on this epitope. This notion is supported by descriptions of B*57-mediated selection of P243T in subtype A1 (41), as well as the fact that B*57:03 and B*57:02 differ by a single residue in their peptide-binding groove that likely interacts with this region of TW10 (76). Indeed, selection of distinct escape mutations by closely related HLA alleles presenting the same viral epitope has previously been described (77–79).
The well-matched clinical profiles of our cohort subsets are also worth mentioning, given the accumulating evidence that HIV subtype D is associated with more rapid progression than subtype A1 (30–34). We did not deliberately select cohort subsets with comparable clinical profiles. Rather, the comparable clinical profiles likely reflect the fact that we studied the baseline (pretherapy) cross section of two HIV treatment cohorts (see Materials and Methods), and treatment eligibility is guided in part by immunological (CD4+ T-cell count) criteria. Though it is possible that cohort subsets differ in terms of infection duration (this information was not known for the study participants), the majority of HLA-driven escape and reversion events occur well before CD4+ T-cell counts reach the levels observed in the present study (51). In fact, HLA-associated polymorphisms in other HIV subtypes have been determined using the baseline (treatment-naive) cross section of a HIV treatment cohort(s) with CD4 and pVL profiles comparable to those of the present cohort (7–10, 12, 13, 45, 80).
The value of integrated bioinformatic and molecular studies like the present one is worth stating directly. While techniques such as gamma interferon enzyme-linked immunosorbent spot (ELISpot) assays can identify HIV epitopes recognized in vitro in one or more viral subtypes, these may not necessarily be relevant in vivo. In contrast, HLA-associated polymorphisms identified at the population level identify specific HIV regions under strong and reproducible selection (that is, immunogenic viral regions) in vivo. Integrated bioinformatic and molecular studies thus offer a complementary approach to rational immunogen selection that begins with an in vivo-relevant observation (i.e., the identification of viral sites under differential HLA-mediated selective pressure) and subsequently proceeds to mechanistic characterization to inform immunogen selection. While only two examples of differential escape were investigated in the present study, in many cases a putative mechanism could be proposed for future validation. For example, epitope binding predictions and subtype-specific sequence conservation data suggest that the lack of selection of both RT 514D and RT 179I by HLA-B*49:01 in subtype D may be attributable to subtype-specific fitness costs, while the lack of B*42:01-mediated selection of Nef 20M in subtype A1 is likely attributable to suboptimal epitope binding.
In conclusion, our study represents the first characterization of HLA-driven adaptation pathways in HIV subtypes A1 and D and the first statistically rigorous characterization of differential HLA-driven escape across viral subtypes. The results support a considerable impact of viral genetic context on HIV adaptation to host HLA, where differences are attributable to subtype-specific sequence variations that influence epitope-HLA binding or determine the mutational barrier to immune escape. A framework to identify viral regions that differ in terms of epitope binding and the functional costs of immune escape across HIV subtypes could aid in vaccine design. The present study highlights the power of combined bioinformatic and mechanistic studies, paired with knowledge of epitope immunogenicity, toward this goal.
MATERIALS AND METHODS
Participants and ethics statement.
Participants comprised a subset of the 513 individuals originally recruited to two established Ugandan HIV treatment cohorts, the Adherence Monitoring Uganda (AMU) cohort and the Uganda AIDS Rural Treatment Outcomes (UARTO) cohort (46–48). All participants were antiretroviral naive at the baseline (cohort entry), which is when HIV genotyping was performed for the present analysis. While the duration of infection was unknown, the CD4 count and viral load distributions indicate that the majority of participants were likely to be in chronic infection (Table 1), which is appropriate for identifying HLA-associated polymorphisms statistically, as by this stage a majority of HLA-driven escape (and reversion) will have occurred, thereby increasing the statistical power to identify such associations (51). All participants were enrolled under Research Ethics Board (REB)-approved protocols and provided written informed consent. Ethics approval to conduct this study was obtained from the institutional review boards of Providence Health Care/University of British Columbia, Simon Fraser University, and the Mbarara University of Science and Technology.
Host and viral genotyping.
Total nucleic acids were extracted from a 500-µl blood plasma sample using standard methods. HIV gag, protease-reverse transcriptase (amplified together), integrase, and nef were amplified via reverse transcriptase (RT)-PCR (using a SuperScript III one-step RT-PCR system with Platinum Taq high-fidelity DNA polymerase; Invitrogen), followed by a nested round of PCR (using an Expand high-fidelity PCR system; Roche) using gene-specific primers optimized for pan-HIV group M binding. Amplicons were bulk (directly) sequenced using a 3730xl or 3130xl automated DNA sequencer (Applied Biosystems). Chromatograms were analyzed using the Sequencher (version 5.0.1) (Gene Codes) or RECall (81) program, with nucleotide mixtures being called if the height of the secondary peak exceeded 25% of the dominant peak height (Sequencher) or 20% of the dominant peak area (RECall). Analysis was restricted to gag, protease-RT, integrase, and nef sequences identified as pure subtype A1 or D sequences, determined by first screening sequences with the Recombinant Identification Program (RIP; version 3.0) at a window size of 100 (https://www.hiv.lanl.gov/content/sequence/RIP/RIP.html) and then inferring and visualizing gene-specific maximum likelihood phylogenies as follows. HIV gene-specific sequence data sets were aligned using the HIValign program (options, MAFFT; codon alignment) (82–84) and inspected using the AliView program (85). Maximum likelihood phylogenies were constructed using the PhyML program (https://www.hiv.lanl.gov/content/sequence/PHYML/interface.html) and visualized using the FigTree program (version 1.3.1).
High-level resolution HLA class I typing was performed using sequence-based methods (86). Intercohort differences in HLA allele frequency were assessed using the Los Alamos HIV Immunology Database’s HLA comparison tool (http://www.hiv.lanl.gov/content/immunology/hla/hla_compare.html). Multiple comparisons were addressed using q values, the P value analogue of the false discovery rate (FDR). The FDR is the expected proportion of false-positive results among results deemed significant at a given P value threshold (e.g., at a q value of ≤0.2, we expect 20% of identified associations to be false positive).
Identification of HLA-associated polymorphisms.
Established phylogenetically corrected statistical methods were used to identify HLA-associated polymorphisms in subtypes A1 and D separately (9, 49). To preserve universal HIV codon numbering conventions, the HIV subtype B reference strain HXB2 sequence was added to each HIV data set and aligned as described above, and all columns where HXB2 was gapped were stripped out. Evidence for selection by HLA class I and/or covarying amino acids in HIV Gag, protease-RT, integrase, and Nef was assessed using a weighted logistic regression approach. In this model, the amino acid is assumed to evolve independently down the tree until it reaches the tips, which represent the studied HIV sequences. On each tree tip, the selection pressure arising from HLA alleles expressed by the participant is directly modeled using a stochastic additive process that treats each HLA allele as a binary variable (i.e., the presence or absence of the allele in question). Covarying HIV amino acids are also included as model variables. To identify the factors that contribute to the observed HIV sequence at the tree tips, a forward selection approach is employed. By this approach, the most significant HLA-HIV amino acid (or HIV amino acid-HIV amino acid) association is iteratively added. Here, the null hypothesis that the observed HIV amino acid at the tree tips can be explained by neutral evolution along the phylogeny is tested against the alternative hypothesis that its presence is better explained by the presence of an HLA class I allele(s) and/or a covarying HIV amino acid(s). P values were computed using the likelihood ratio test, and a q value of <0.2 was used to define statistical significance, as in previous studies (7–9, 12, 13, 45). HLA-associated polymorphisms were grouped into two categories: adapted (inferred escaped) forms, which represent HIV amino acids significantly overrepresented in the presence of the HLA allele, and nonadapted (inferred susceptible) forms, which represent HIV amino acids significantly underrepresented in the presence of the allele in question.
A novel modification to an existing weighted logistic regression approach was used to identify instances of differential selection of HLA-associated polymorphisms across HIV subtypes (9, 10, 49, 51). This test was applied to all polymorphisms identified in subtype A1 and/or D where at least three individuals in both cohorts harbored the restricting HLA allele. Briefly, the null model treats each HIV polymorphism as the outcome variable and the single restricting HLA allele as the predictor. This model is then compared to a more expressive one that includes an interaction term for subtype using a likelihood ratio test. To increase the stringency of the test, polymorphisms differentially selected at the more conservative threshold of a P value of <0.05 and a q value of <0.1 were considered significant.
Peptide conformational modeling.
In silico-predicted peptide structures were generated using the PEP-FOLD (version 2.0) program (87–89) and visualized using the PyMOL molecular graphics system (version 2.0.7; Schrödinger, LLC). The root mean square deviation (RMSD), calculated as the square root of the mean of the square of the distances between the matched atoms, between structures was determined using the Visual Molecular Dynamics program (version 1.9.4) (54) to assess the similarity of the structures.
Recombinant virus generation and replicative assessment.
The Gag T242N mutation was engineered into one arbitrarily selected subtype A1 and one subtype D participant-derived Gag-protease sequence using overlap extension PCR, where the resulting amplicons were designed to have an approximately 20-bp overlap with a pNL4.3 (subtype B) reference strain plasmid lacking Gag-protease (ΔGag-protease), which has been described previously (90). The consensus (Los Alamos National Laboratory HIV sequence database) and T242N mutant HIV subtype A1 Gag-protease sequences were synthesized as three roughly equally sized gBlocks (Integrated DNA Technologies) with approximately 20 bp of overlap between fragments as well as with the ΔGag-protease pNL4.3 reference strain plasmid. Inserts were fused into the ΔGag-protease pNL4.3 plasmid using the Gibson enzymatic assembly (91) (NEBuilder; New England BioLabs) at a 3:1 insert/backbone molar ratio. The entire HIV coding region of the resulting recombinant plasmids was sequenced to confirm carriage of only the mutations of interest. Viral stocks pseudotyped with the vesicular stomatitis virus G (VSVg) envelope (58) were obtained by cotransfecting 13 µg of recombinant plasmid into 2.8 million HEK 293T cells along with 1.3 µg of a VSVg plasmid (which originated from the study described in reference 92) (Lipofectamine LTX; Thermo Fisher Scientific). VSVg pseudotyping was done in order to ensure that all recombinant viruses were capable of at least a first round of infection. Culture supernatants containing recombinant virions were harvested at 48 h posttransfection and frozen prior to use. The titers of the recombinant viral stocks were determined by infecting a CEM-derived reporter CD4+ T-cell line harboring an HIV LTR-driven green fluorescent protein (GFP) expression cassette (the CEM-GXR cell line) (59) with serially diluted viral inocula and measuring the percentage of GFP-expressing (i.e., infected) cells at 48 h postinfection (day 2). The volume of viral stock required to achieve 0.3% GFP-positive (GFP+) cells on day 2 was then determined by linear extrapolation.
Viral replication was assessed using a published multicycle reporter assay (59). Briefly, the CEM-GXR GFP reporter T-cell line was infected at a multiplicity of infection of 0.003, that is, to achieve 0.3% GFP-positive (i.e., infected) cells on day 2. To quantify subsequent viral spread, the percentage of infected cells was measured via flow cytometry (Guava 8HT; Millipore) daily for up to 8 days. The replication capacity was then calculated as the natural log of the slope of viral spread during the exponential phase. The replication capacities of viruses harboring T242N were compared to those of their subtype parental or consensus control. The replication of each participant-derived recombinant virus was assessed in a minimum of three independent experiments; consensus-derived recombinant viruses were assessed in duplicate.
Nef-mediated CD4, HLA, and SERINC5 downregulation.
K105R was engineered into subtype A1 or D consensus Nef (Los Alamos National Laboratory HIV sequence database) gBlocks (Integrated DNA Technologies) via overlap extension PCR. Amplicons were cloned into pSELECT-GFPzeo, which contains dual promoters driving the expression of GFP as well as the gene of interest, and the sequences were confirmed. The ability of each Nef clone to downregulate cell surface CD4 and HLA class I was determined as previously described (93). Briefly, a CEM CD4+ T-cell line engineered to express high levels of HLA-A*02 (CEM-A*02) was transfected with consensus or mutant Nef clones by electroporation. Cells were stained at 20 to 24 h posttransfection with allophycocyanin (APC)-conjugated mouse anti-human CD4 antibody and phycoerythrin-conjugated mouse anti-human HLA-A*02 antibody (BD Biosciences). The median fluorescence intensity (MFI) of GFP+ (Nef-expressing) cells was determined by flow cytometry (Guava 8HT; Millipore) and analyzed using FlowJo (version 9.9.5) software. The CD4 or HLA downregulation function of each Nef mutant was normalized to that of its subtype consensus control using the following equation: (MFInegative control − MFImutant)/(MFInegative control − MFIsubtype consensus). The function of each mutant and consensus Nef sequence was assessed in a minimum of three independent experiments.
To assess SERINC5 downregulation function, CEM-A*02 cells were cotransfected with 1 µg of the Nef plasmid and 5 µg of a plasmid expressing an internally hemagglutinin (HA)-tagged SERINC5 by electroporation (94). Cells were stained at 20 to 22 h posttransfection with APC-conjugated mouse anti-HA antibody (BioLegend). The MFI of SERINC5+ GFP+ cells was determined by flow cytometry (Guava 8HT; Millipore) and analyzed using FlowJo (version 9.9.5) software. The Nef-mediated SERINC5 downregulation function of each mutant was normalized to that of its corresponding subtype consensus control using the same equation presented above. The function of each Nef mutant and consensus sequence was assessed in a minimum of three independent experiments.
Epitope competitive binding assay.
Subtype-specific epitope binding to HLA-B*15:03 was assessed using a competition-based cellular binding assay (66–68). Candidate subtype A1 and D epitopes spanning Nef residue 105 that displayed the highest affinity for HLA-B*15:03 were identified using netMHC (version 4.0) software (65). To generate a B*15:03-specific reference peptide, the B*15:03-restricted epitope IQQEFGIPY (HIV integrase codons 135 to 143 [69]) sequence was modified to contain a cysteine at position 4 to yield IQQCGFIPY (IY9) and synthesized (GenScript), and the fluorophore 5-(iodoacetamido)fluorescein (5-IAF; Thermo Fisher) was conjugated to the cysteine. Fluorescently labeled IY9 was titrated on acid-stripped (pH 2.9) parental HLA-null K562-derived cells (95) and K562-derived cells retrovirally transduced to stably express HLA-B*15:03 to confirm HLA specificity and to determine the optimal concentration of the reference peptide for use in competitive binding assays. In the competitive binding assay, a fixed concentration of fluorescently labeled IY9 was mixed with a titration of the candidate or control epitope (0 to 104 µM) and then incubated with acid-stripped B*15:03-positive K562-derived cells at 4°C for 24 h in the dark. The median fluorescence intensity was then determined by flow cytometry (Guava 8HT; Millipore) and analyzed using FlowJo (version 9.9.5) software. Three independent experiments were performed.
Accession number(s).
Subtype A1 and D HIV gag, integrase, and nef sequences have been deposited into GenBank under accession numbers MH971615 to MH971951, MH925338 to MH925677, and MH971287 to MH971614, respectively. Partial protease-reverse transcriptase sequences for a subset of participants were previously submitted as part of another study (96) and have been updated to include the entire protease and reverse transcriptase sequences under accession numbers KJ906625, KJ906627, KJ906631, KJ906635 to KJ906637, KJ906641, KJ906642, KJ906646, KJ906649, KJ906650, KJ906653 to KJ906655, KJ906658, KJ906660, KJ906661, KJ906665, KJ906666, KJ906670 to KJ906672, KJ906676, KJ906678, KJ906679, KJ906681, KJ906682, KJ906685 to KJ906692, KJ906694 to KJ906698, KJ906701, KJ906706, KJ906708, KJ906711 to KJ906714, KJ906716, KJ906717, KJ906720, KJ906721, KJ906724, KJ906725, KJ906728, KJ906730, KJ906731, KJ906733, KJ906735 to KJ906738, KJ906740 to KJ906746, KJ906749, KJ906751 to KJ906755, KJ906758, KJ906759, KJ906761, KJ906763, KJ906766, KJ906767, KJ906769 to KJ906771, KJ906774, KJ906775, KJ906777, KJ906778, KJ906780, KJ906781 to KJ906785, KJ906787, KJ906788, KJ906790 to KJ906792, KJ906796 to KJ906803, KJ906805 to KJ906808, KJ906810, KJ906813 to KJ906819, KJ906823, to KJ906832, KJ906834 to KJ906838, KJ906840 to KJ906844, KJ906846 to KJ906850, KJ906852 to KJ906854, KJ906856 to KJ906860, KJ906862, KJ906864 to KJ906873, KJ906875, KJ906876, KJ906878, KJ906880, KJ906881, KJ906884, KJ906891 to KJ906896, KJ906899, KJ906900, KJ906903, KJ906907, KJ906908, KJ906910 to KJ906917, KJ906919 to KJ906928, KJ906930 to KJ906942, KJ906944 to KJ906950, KJ906952 to KJ906954, KJ906956 to KJ906958, KJ906960 to KJ906965, KJ906967 to KJ906976, KJ906978 to KJ906983, KJ906985, KJ906987, KJ906989, KJ906990, KJ906992 to KJ906999, KJ907001 to KJ907006, KJ907008, KJ907010, KJ907011, KJ907013, KJ907015, KJ907018, KJ907021 to KJ907024, KJ907026 to KJ907028, KJ907030, KJ907032 to KJ907035, KJ907037 to KJ907039, KJ907041 to KJ907044, KJ907048 to KJ907052, KJ907055 to KJ907057, KJ907060 to KJ907065, KJ907067, KJ907070, KJ907071, KJ907073, KJ907075, KJ907077, KJ907079, KJ907080, KJ907083, KJ907086, KJ907088, KJ907089, KJ907091, KJ907093, KJ907094, KJ907096, KJ907097, KJ907099, KJ907101 to KJ907107, KJ907112, KJ907116, KJ907117, KJ907119, KJ907121, KJ907122, KJ907124 to KJ907127, KJ907129, KJ907131 to KJ907133, KJ907135, KJ907136, KJ907138, KJ907140 to KJ907142, KJ907144, KJ907146, KJ907149, KJ907150, KJ907152 to KJ907156, KJ907158, to KJ907163, KJ907165, KJ907167, KJ907168, KJ907171 to KJ907173, KJ907175 to KJ907177, KJ907179, KJ907180, KJ907182. The remaining protease-reverse transcriptase sequences have been deposited under the accession numbers MH925335 to MH925337.
Supplementary Material
ACKNOWLEDGMENTS
We thank Gursev Anmole, Philip Mwimanzi, Tallie Kuang, Kevin Rey, and Theresa Mo for technical assistance and Rupert Kaul, Lyle McKinnon, Christian Brander, Clayton Moore, and Hassan Rasouli for helpful discussions. We gratefully acknowledge the contribution of the original participants of the UARTO and AMU cohorts, without whom this study would not be possible.
The following reagent was obtained through the AIDS Reagent Program, Division of AIDS, NIAID, NIH: pHEF-VSVG from Lung-Ji Chang (catalog number 4693).
This work was supported by a Canadian Institutes for Health Research (CIHR) project grant (PJT-148621) to G.Q.L., D.R.B., P.R.H., M.A.B., and Z.L.B. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research under contract no. HHSN261200800001E. This research was supported in part by the Intramural Research Program of the NIH, Frederick National Laboratory Center for Cancer Research. N.N.K. and S.W.J. were supported by Frederick Banting and Charles Best CIHR MSc awards. M.A.B. holds a Canada Research Chair, Tier 2, in viral pathogenesis and immunity. Z.L.B. is supported by a scholar award from the Michael Smith Foundation for Health Research.
The content of this publication does not necessarily reflect the views or policies of the U.S. Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JVI.01502-18.
REFERENCES
- 1.Phillips RE, Rowland-Jones S, Nixon DF, Gotch FM, Edwards JP, Ogunlesi AO, Elvin JG, Rothbard JA, Bangham CR, Rizza CR. 1991. Human immunodeficiency virus genetic variation that can escape cytotoxic T cell recognition. Nature 354:453–459. doi: 10.1038/354453a0. [DOI] [PubMed] [Google Scholar]
- 2.Borrow P, Lewicki H, Wei X, Horwitz MS, Peffer N, Meyers H, Nelson JA, Gairin JE, Hahn BH, Oldstone MB, Shaw GM. 1997. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med 3:205–211. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 3.Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat Med 3:212–217. doi: 10.1038/nm0297-212. [DOI] [PubMed] [Google Scholar]
- 4.Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. 2001. Clustered mutations in HIV-1 gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J Exp Med 193:375–386. doi: 10.1084/jem.193.3.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moore CB, John M, James IR, Christiansen FT, Witt CS, Mallal SA. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439–1443. doi: 10.1126/science.1069660. [DOI] [PubMed] [Google Scholar]
- 6.Bhattacharya T, Daniels M, Heckerman D, Foley B, Frahm N, Kadie C, Carlson J, Yusim K, McMahon B, Gaschen B, Mallal S, Mullins JI, Nickle DC, Herbeck J, Rousseau C, Learn GH, Miura T, Brander C, Walker B, Korber B. 2007. Founder effects in the assessment of HIV polymorphisms and HLA allele associations. Science 315:1583–1586. doi: 10.1126/science.1131528. [DOI] [PubMed] [Google Scholar]
- 7.Brumme ZL, Brumme CJ, Heckerman D, Korber BT, Daniels M, Carlson J, Kadie C, Bhattacharya T, Chui C, Szinger J, Mo T, Hogg RS, Montaner JS, Frahm N, Brander C, Walker BD, Harrigan PR. 2007. Evidence of differential HLA class I-mediated viral evolution in functional and accessory/regulatory genes of HIV-1. PLoS Pathog 3:e94. doi: 10.1371/journal.ppat.0030094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Avila-Rios S, Ormsby CE, Carlson JM, Valenzuela-Ponce H, Blanco-Heredia J, Garrido-Rodriguez D, Garcia-Morales C, Heckerman D, Brumme ZL, Mallal S, John M, Espinosa E, Reyes-Teran G. 2009. Unique features of HLA-mediated HIV evolution in a Mexican cohort: a comparative study. Retrovirology 6:72. doi: 10.1186/1742-4690-6-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Carlson JM, Brumme CJ, Martin E, Listgarten J, Brockman MA, Le AQ, Chui CK, Cotton LA, Knapp DJ, Riddler SA, Haubrich R, Nelson G, Pfeifer N, Deziel CE, Heckerman D, Apps R, Carrington M, Mallal S, Harrigan PR, John M, Brumme ZL. 2012. Correlates of protective cellular immunity revealed by analysis of population-level immune escape pathways in HIV-1. J Virol 86:13202–13216. doi: 10.1128/JVI.01998-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chikata T, Carlson JM, Tamura Y, Borghan MA, Naruto T, Hashimoto M, Murakoshi H, Le AQ, Mallal S, John M, Gatanaga H, Oka S, Brumme ZL, Takiguchi M. 2014. Host-specific adaptation of HIV-1 subtype B in the Japanese population. J Virol 88:4764–4775. doi: 10.1128/JVI.00147-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rousseau CM, Daniels MG, Carlson JM, Kadie C, Crawford H, Prendergast A, Matthews P, Payne R, Rolland M, Raugi DN, Maust BS, Learn GH, Nickle DC, Coovadia H, Ndung'u T, Frahm N, Brander C, Walker BD, Goulder PJ, Bhattacharya T, Heckerman DE, Korber BT, Mullins JI. 2008. HLA class I-driven evolution of human immunodeficiency virus type 1 subtype C proteome: immune escape and viral load. J Virol 82:6434–6446. doi: 10.1128/JVI.02455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carlson JM, Schaefer M, Monaco DC, Batorsky R, Claiborne DT, Prince J, Deymier MJ, Ende ZS, Klatt NR, DeZiel CE, Lin TH, Peng J, Seese AM, Shapiro R, Frater J, Ndung'u T, Tang J, Goepfert P, Gilmour J, Price MA, Kilembe W, Heckerman D, Goulder PJ, Allen TM, Allen S, Hunter E. 2014. HIV transmission. Selection bias at the heterosexual HIV-1 transmission bottleneck. Science 345:1254031. doi: 10.1126/science.1254031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Van Tran G, Chikata T, Carlson JM, Murakoshi H, Nguyen DH, Tamura Y, Akahoshi T, Kuse N, Sakai K, Sakai S, Cobarrubias K, Oka S, Brumme ZL, Van Nguyen K, Takiguchi M, NHTD Treatment-Naive Cohort Study Group. 2016. A strong association of human leukocyte antigen-associated Pol and Gag mutations with clinical parameters in HIV-1 subtype A/E infection. AIDS 30:681–689. doi: 10.1097/QAD.0000000000000969. [DOI] [PubMed] [Google Scholar]
- 14.Allen TM, Altfeld M, Yu XG, O’Sullivan KM, Lichterfeld M, Le Gall S, John M, Mothe BR, Lee PK, Kalife ET, Cohen DE, Freedberg KA, Strick DA, Johnston MN, Sette A, Rosenberg ES, Mallal SA, Goulder PJ, Brander C, Walker BD. 2004. Selection, transmission, and reversion of an antigen-processing cytotoxic T-lymphocyte escape mutation in human immunodeficiency virus type 1 infection. J Virol 78:7069–7078. doi: 10.1128/JVI.78.13.7069-7078.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat Med 10:282–289. doi: 10.1038/nm992. [DOI] [PubMed] [Google Scholar]
- 16.Friedrich TC, Dodds EJ, Yant LJ, Vojnov L, Rudersdorf R, Cullen C, Evans DT, Desrosiers RC, Mothe BR, Sidney J, Sette A, Kunstman K, Wolinsky S, Piatak M, Lifson J, Hughes AL, Wilson N, O’Connor DH, Watkins DI. 2004. Reversion of CTL escape-variant immunodeficiency viruses in vivo. Nat Med 10:275–281. doi: 10.1038/nm998. [DOI] [PubMed] [Google Scholar]
- 17.Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J Virol 80:3617–3623. doi: 10.1128/JVI.80.7.3617-3623.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li B, Gladden AD, Altfeld M, Kaldor JM, Cooper DA, Kelleher AD, Allen TM. 2007. Rapid reversion of sequence polymorphisms dominates early human immunodeficiency virus type 1 evolution. J Virol 81:193–201. doi: 10.1128/JVI.01231-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J Virol 81:12608–12618. doi: 10.1128/JVI.01369-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothe BR, Kuntzen T, Oniangue-Ndza C, Trocha A, Yu XG, Brander C, Sette A, Walker BD, Allen TM. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J Virol 82:5594–5605. doi: 10.1128/JVI.02356-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J Virol 81:12382–12393. doi: 10.1128/JVI.01543-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delport W, Scheffler K, Seoighe C. 2008. Frequent toggling between alternative amino acids is driven by selection in HIV-1. PLoS Pathog 4:e1000242. doi: 10.1371/journal.ppat.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shahid A, Olvera A, Anmole G, Kuang XT, Cotton LA, Plana M, Brander C, Brockman MA, Brumme ZL. 2015. Consequences of HLA-B*13-associated escape mutations on HIV-1 replication and Nef function. J Virol 89:11557–11571. doi: 10.1128/JVI.01955-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carlson JM, Le AQ, Shahid A, Brumme ZL. 2015. HIV-1 adaptation to HLA: a window into virus-host immune interactions. Trends Microbiol 23:212–224. doi: 10.1016/j.tim.2014.12.008. [DOI] [PubMed] [Google Scholar]
- 25.Hemelaar J. 2013. Implications of HIV diversity for the HIV-1 pandemic. J Infect 66:391–400. doi: 10.1016/j.jinf.2012.10.026. [DOI] [PubMed] [Google Scholar]
- 26.Coutsinos D, Invernizzi CF, Xu H, Moisi D, Oliveira M, Brenner BG, Wainberg MA. 2009. Template usage is responsible for the preferential acquisition of the K65R reverse transcriptase mutation in subtype C variants of human immunodeficiency virus type 1. J Virol 83:2029–2033. doi: 10.1128/JVI.01349-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bar-Magen T, Donahue DA, McDonough EI, Kuhl BD, Faltenbacher VH, Xu H, Michaud V, Sloan RD, Wainberg MA. 2010. HIV-1 subtype B and C integrase enzymes exhibit differential patterns of resistance to integrase inhibitors in biochemical assays. AIDS 24:2171–2179. doi: 10.1097/QAD.0b013e32833cf265. [DOI] [PubMed] [Google Scholar]
- 28.Koning FA, Castro H, Dunn D, Tilston P, Cane PA, Mbisa JL. 2013. Subtype-specific differences in the development of accessory mutations associated with high-level resistance to HIV-1 nucleoside reverse transcriptase inhibitors. J Antimicrob Chemother 68:1220–1236. doi: 10.1093/jac/dkt012. [DOI] [PubMed] [Google Scholar]
- 29.Quashie PK, Oliviera M, Veres T, Osman N, Han YS, Hassounah S, Lie Y, Huang W, Mesplede T, Wainberg MA. 2015. Differential effects of the G118R, H51Y, and E138K resistance substitutions in different subtypes of HIV integrase. J Virol 89:3163–3175. doi: 10.1128/JVI.03353-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaleebu P, French N, Mahe C, Yirrell D, Watera C, Lyagoba F, Nakiyingi J, Rutebemberwa A, Morgan D, Weber J, Gilks C, Whitworth J. 2002. Effect of human immunodeficiency virus (HIV) type 1 envelope subtypes A and D on disease progression in a large cohort of HIV-1-positive persons in Uganda. J Infect Dis 185:1244–1250. doi: 10.1086/340130. [DOI] [PubMed] [Google Scholar]
- 31.Vasan A, Renjifo B, Hertzmark E, Chaplin B, Msamanga G, Essex M, Fawzi W, Hunter D. 2006. Different rates of disease progression of HIV type 1 infection in Tanzania based on infecting subtype. Clin Infect Dis 42:843–852. doi: 10.1086/499952. [DOI] [PubMed] [Google Scholar]
- 32.Baeten JM, Chohan B, Lavreys L, Chohan V, McClelland RS, Certain L, Mandaliya K, Jaoko W, Overbaugh J. 2007. HIV-1 subtype D infection is associated with faster disease progression than subtype A in spite of similar plasma HIV-1 loads. J Infect Dis 195:1177–1180. doi: 10.1086/512682. [DOI] [PubMed] [Google Scholar]
- 33.Kiwanuka N, Laeyendecker O, Robb M, Kigozi G, Arroyo M, McCutchan F, Eller LA, Eller M, Makumbi F, Birx D, Wabwire-Mangen F, Serwadda D, Sewankambo NK, Quinn TC, Wawer M, Gray R. 2008. Effect of human immunodeficiency virus type 1 (HIV-1) subtype on disease progression in persons from Rakai, Uganda, with incident HIV-1 infection. J Infect Dis 197:707–713. doi: 10.1086/527416. [DOI] [PubMed] [Google Scholar]
- 34.Venner CM, Nankya I, Kyeyune F, Demers K, Kwok C, Chen PL, Rwambuya S, Munjoma M, Chipato T, Byamugisha J, Van Der Pol B, Mugyenyi P, Salata RA, Morrison CS, Arts EJ. 2016. Infecting HIV-1 subtype predicts disease progression in women of sub-Saharan Africa. EBioMedicine 13:305–314. doi: 10.1016/j.ebiom.2016.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Frahm N, Kiepiela P, Adams S, Linde CH, Hewitt HS, Sango K, Feeney ME, Addo MM, Lichterfeld M, Lahaie MP, Pae E, Wurcel AG, Roach T, St John MA, Altfeld M, Marincola FM, Moore C, Mallal S, Carrington M, Heckerman D, Allen TM, Mullins JI, Korber BT, Goulder PJ, Walker BD, Brander C. 2006. Control of human immunodeficiency virus replication by cytotoxic T lymphocytes targeting subdominant epitopes. Nat Immunol 7:173–178. doi: 10.1038/ni1281. [DOI] [PubMed] [Google Scholar]
- 36.Kloverpris HN, Adland E, Koyanagi M, Stryhn A, Harndahl M, Matthews PC, Shapiro R, Walker BD, Ndung’u T, Brander C, Takiguchi M, Buus S, Goulder P. 2014. HIV subtype influences HLA-B*07:02-associated HIV disease outcome. AIDS Res Hum Retroviruses 30:468–475. doi: 10.1089/aid.2013.0197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matthews PC, Koyanagi M, Kloverpris HN, Harndahl M, Stryhn A, Akahoshi T, Gatanaga H, Oka S, Juarez Molina C, Valenzuela Ponce H, Avila Rios S, Cole D, Carlson J, Payne RP, Ogwu A, Bere A, Ndung’u T, Gounder K, Chen F, Riddell L, Luzzi G, Shapiro R, Brander C, Walker B, Sewell AK, Reyes Teran G, Heckerman D, Hunter E, Buus S, Takiguchi M, Goulder PJ. 2012. Differential clade-specific HLA-B*3501 association with HIV-1 disease outcome is linked to immunogenicity of a single Gag epitope. J Virol 86:12643–12654. doi: 10.1128/JVI.01381-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat Med 13:46–53. doi: 10.1038/nm1520. [DOI] [PubMed] [Google Scholar]
- 39.Payne RP, Branch S, Kloverpris H, Matthews PC, Koofhethile CK, Strong T, Adland E, Leitman E, Frater J, Ndung'u T, Hunter E, Haubrich R, Mothe B, Edwards A, Riddell L, Chen F, Harrigan PR, Brumme ZL, Mallal S, John M, Jooste JP, Shapiro R, Deeks SG, Walker BD, Brander C, Landis C, Carlson JM, Prado JG, Goulder PJ. 2014. Differential escape patterns within the dominant HLA-B*57:03-restricted HIV Gag epitope reflect distinct clade-specific functional constraints. J Virol 88:4668–4678. doi: 10.1128/JVI.03303-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chopera DR, Cotton LA, Zawaira A, Mann JK, Ngandu NK, Ntale R, Carlson JM, Mlisana K, Woodman Z, de Assis Rosa D, Martin E, Miura T, Pereyra F, Walker BD, Gray CM, Martin DP, Ndung’u T, Brockman MA, Karim SA, Brumme ZL, Williamson C. 2012. Intersubtype differences in the effect of a rare p24 gag mutation on HIV-1 replicative fitness. J Virol 86:13423–13433. doi: 10.1128/JVI.02171-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McKinnon LR, Capina R, Peters H, Mendoza M, Kimani J, Wachihi C, Kariri A, Kimani M, Richmond M, Kiazyk SK, Fowke KR, Jaoko W, Luo M, Ball TB, Plummer FA. 2009. Clade-specific evolution mediated by HLA-B*57/5801 in human immunodeficiency virus type 1 clade A1 p24. J Virol 83:12636–12642. doi: 10.1128/JVI.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Serwanga J, Nakiboneka R, Mugaba S, Magambo B, Ndembi N, Gotch F, Kaleebu P. 2015. Frequencies of Gag-restricted T-cell escape “footprints” differ across HIV-1 clades A1 and D chronically infected Ugandans irrespective of host HLA B alleles. Vaccine 33:1664–1672. doi: 10.1016/j.vaccine.2015.02.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kawashima Y, Pfafferott K, Frater J, Matthews P, Payne R, Addo M, Gatanaga H, Fujiwara M, Hachiya A, Koizumi H, Kuse N, Oka S, Duda A, Prendergast A, Crawford H, Leslie A, Brumme Z, Brumme C, Allen T, Brander C, Kaslow R, Tang J, Hunter E, Allen S, Mulenga J, Branch S, Roach T, John M, Mallal S, Ogwu A, Shapiro R, Prado JG, Fidler S, Weber J, Pybus OG, Klenerman P, Ndung’u T, Phillips R, Heckerman D, Harrigan PR, Walker BD, Takiguchi M, Goulder P. 2009. Adaptation of HIV-1 to human leukocyte antigen class I. Nature 458:641–645. doi: 10.1038/nature07746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Karakas A, Brumme ZL, Poon AF. 2015. Global database-driven assessment of HIV-1 adaptation to the immune repertoires of human populations. J Virol 89:10693–10695. doi: 10.1128/JVI.01355-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soto-Nava M, Avila-Rios S, Valenzuela-Ponce H, Garcia-Morales C, Carlson JM, Tapia-Trejo D, Garrido-Rodriguez D, Alva-Hernandez SN, Garcia-Tellez TA, Murakami-Ogasawara A, Mallal SA, John M, Brockman MA, Brumme CJ, Brumme ZL, Reyes-Teran G. 2018. Weaker HLA footprints on HIV in the unique and highly genetically admixed host population of Mexico. J Virol 92:e01128-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hunt PW, Cao HL, Muzoora C, Ssewanyana I, Bennett J, Emenyonu N, Kembabazi A, Neilands TB, Bangsberg DR, Deeks SG, Martin JN. 2011. Impact of CD8+ T-cell activation on CD4+ T-cell recovery and mortality in HIV-infected Ugandans initiating antiretroviral therapy. AIDS 25:2123–2131. doi: 10.1097/QAD.0b013e32834c4ac1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bebell LM, Siedner MJ, Musinguzi N, Boum Y, Bwana BM, Muyindike W, Hunt PW, Martin JN, Bangsberg DR. 2017. Trends in one-year cumulative incidence of death between 2005 and 2013 among patients initiating antiretroviral therapy in Uganda. Int J STD AIDS 28:800–807. doi: 10.1177/0956462416671431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Musinguzi N, Mocello RA, Boum Y, Hunt PW, Martin JN, Haberer JE, Bangsberg DR, Siedner MJ. 2017. Duration of viral suppression and risk of rebound viremia with first-line antiretroviral therapy in rural Uganda. AIDS Behav 21:1735–1740. doi: 10.1007/s10461-016-1447-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Carlson JM, Brumme ZL, Rousseau CM, Brumme CJ, Matthews P, Kadie C, Mullins JI, Walker BD, Harrigan PR, Goulder PJ, Heckerman D. 2008. Phylogenetic dependency networks: inferring patterns of CTL escape and codon covariation in HIV-1 Gag. PLoS Comput Biol 4:e1000225. doi: 10.1371/journal.pcbi.1000225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, Yu XG, Meier A, Rockstroh JK, Allen TM, Jessen H, Rosenberg ES, Carrington M, Walker BD. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med 3:e403. doi: 10.1371/journal.pmed.0030403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martin E, Carlson JM, Le AQ, Chopera DR, McGovern R, Rahman MA, Ng C, Jessen H, Kelleher AD, Markowitz M, Allen TM, Milloy MJ, Carrington M, Wainberg MA, Brumme ZL. 2014. Early immune adaptation in HIV-1 revealed by population-level approaches. Retrovirology 11:64. doi: 10.1186/s12977-014-0064-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, Vogel M, Yu XG, Draenert R, Johnston MN, Strick D, Allen TM, Feeney ME, Kahn JO, Sekaly RP, Levy JA, Rockstroh JK, Goulder PJ, Walker BD. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581–2591. doi: 10.1097/00002030-200312050-00005. [DOI] [PubMed] [Google Scholar]
- 53.Serwanga J, Mugaba S, Pimego E, Nanteza B, Lyagoba F, Nakubulwa S, Heath L, Nsubuga RN, Ndembi N, Gotch F, Kaleebu P. 2012. Profile of T cell recognition of HIV type 1 consensus group M Gag and Nef peptides in a clade A1- and D-infected Ugandan population. AIDS Res Hum Retroviruses 28:384–392. doi: 10.1089/aid.2011.0175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Humphrey W, Dalke A, Schulten K. 1996. VMD: visual molecular dynamics. J Mol Graph 14:33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 55.Holm L, Sander C. 1993. Protein structure comparison by alignment of distance matrices. J Mol Biol 233:123–138. doi: 10.1006/jmbi.1993.1489. [DOI] [PubMed] [Google Scholar]
- 56.Tsai HH, Tsai CJ, Ma B, Nussinov R. 2004. In silico protein design by combinatorial assembly of protein building blocks. Protein Sci 13:2753–2765. doi: 10.1110/ps.04774004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bordogna A, Pandini A, Bonati L. 2011. Predicting the accuracy of protein-ligand docking on homology models. J Comput Chem 32:81–98. doi: 10.1002/jcc.21601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bartz SR, Vodicka MA. 1997. Production of high-titer human immunodeficiency virus type 1 pseudotyped with vesicular stomatitis virus glycoprotein. Methods 12:337–342. doi: 10.1006/meth.1997.0487. [DOI] [PubMed] [Google Scholar]
- 59.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Virol Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 60.Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J Virol 83:2460–2468. doi: 10.1128/JVI.01970-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guy B, Kieny MP, Riviere Y, Le Peuch C, Dott K, Girard M, Montagnier L, Lecocq JP. 1987. HIV F/3′ orf encodes a phosphorylated GTP-binding protein resembling an oncogene product. Nature 330:266–269. doi: 10.1038/330266a0. [DOI] [PubMed] [Google Scholar]
- 62.Marsh JW. 1999. The numerous effector functions of Nef. Arch Biochem Biophys 365:192–198. doi: 10.1006/abbi.1999.1208. [DOI] [PubMed] [Google Scholar]
- 63.Usami Y, Wu Y, Gottlinger HG. 2015. SERINC3 and SERINC5 restrict HIV-1 infectivity and are counteracted by Nef. Nature 526:218–223. doi: 10.1038/nature15400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rosa A, Chande A, Ziglio S, De Sanctis V, Bertorelli R, Goh SL, McCauley SM, Nowosielska A, Antonarakis SE, Luban J, Santoni FA, Pizzato M. 2015. HIV-1 Nef promotes infection by excluding SERINC5 from virion incorporation. Nature 526:212–217. doi: 10.1038/nature15399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Andreatta M, Nielsen M. 2016. Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics 32:511–517. doi: 10.1093/bioinformatics/btv639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kessler JH, Benckhuijsen WE, Mutis T, Melief CJ, van der Burg SH, Drijfhout JW. 2004. Competition-based cellular peptide binding assay for HLA class I. Curr Protoc Immunol Chapter 18:Unit 18.12. [DOI] [PubMed] [Google Scholar]
- 67.Kessler JH, Mommaas B, Mutis T, Huijbers I, Vissers D, Benckhuijsen WE, Schreuder GM, Offringa R, Goulmy E, Melief CJ, van der Burg SH, Drijfhout JW. 2003. Competition-based cellular peptide binding assays for 13 prevalent HLA class I alleles using fluorescein-labeled synthetic peptides. Hum Immunol 64:245–255. doi: 10.1016/S0198-8859(02)00787-5. [DOI] [PubMed] [Google Scholar]
- 68.van der Burg SH, Ras E, Drijfhout JW, Benckhuijsen WE, Bremers AJ, Melief CJ, Kast WM. 1995. An HLA class I peptide-binding assay based on competition for binding to class I molecules on intact human B cells. Identification of conserved HIV-1 polymerase peptides binding to HLA-A*0301. Hum Immunol 44:189–198. doi: 10.1016/0198-8859(95)00105-0. [DOI] [PubMed] [Google Scholar]
- 69.Pereyra F, Heckerman D, Carlson JM, Kadie C, Soghoian DZ, Karel D, Goldenthal A, Davis OB, DeZiel CE, Lin T, Peng J, Piechocka A, Carrington M, Walker BD. 2014. HIV control is mediated in part by CD8+ T-cell targeting of specific epitopes. J Virol 88:12937–12948. doi: 10.1128/JVI.01004-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kloverpris H, Karlsson I, Bonde J, Thorn M, Vinner L, Pedersen AE, Hentze JL, Andresen BS, Svane IM, Gerstoft J, Kronborg G, Fomsgaard A. 2009. Induction of novel CD8+ T-cell responses during chronic untreated HIV-1 infection by immunization with subdominant cytotoxic T-lymphocyte epitopes. AIDS 23:1329–1340. doi: 10.1097/QAD.0b013e32832d9b00. [DOI] [PubMed] [Google Scholar]
- 71.Apps R, Qi Y, Carlson JM, Chen H, Gao X, Thomas R, Yuki Y, Del Prete GQ, Goulder P, Brumme ZL, Brumme CJ, John M, Mallal S, Nelson G, Bosch R, Heckerman D, Stein JL, Soderberg KA, Moody MA, Denny TN, Zeng X, Fang J, Moffett A, Lifson JD, Goedert JJ, Buchbinder S, Kirk GD, Fellay J, McLaren P, Deeks SG, Pereyra F, Walker B, Michael NL, Weintrob A, Wolinsky S, Liao W, Carrington M. 2013. Influence of HLA-C expression level on HIV control. Science 340:87–91. doi: 10.1126/science.1232685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung’u T, Lakhi S, Gilmour J, Goepfert P, Walker BD, Kaslow R, Mulenga J, Allen S, Goulder PJ, Hunter E. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J Exp Med 206:909–921. doi: 10.1084/jem.20081984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Granier C, Battivelli E, Lecuroux C, Venet A, Lambotte O, Schmitt-Boulanger M, Delaugerre C, Molina JM, Chakrabarti LA, Clavel F, Hance AJ. 2013. Pressure from TRIM5alpha contributes to control of HIV-1 replication by individuals expressing protective HLA-B alleles. J Virol 87:10368–10380. doi: 10.1128/JVI.01313-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ackerson B, Rey O, Canon J, Krogstad P. 1998. Cells with high cyclophilin A content support replication of human immunodeficiency virus type 1 Gag mutants with decreased ability to incorporate cyclophilin A. J Virol 72:303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sokolskaja E, Berthoux L, Luban J. 2006. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J Virol 80:2855–2862. doi: 10.1128/JVI.80.6.2855-2862.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Robinson J, Halliwell JA, McWilliam H, Lopez R, Marsh SG. 2012. IPD—the Immuno Polymorphism Database. Nucleic Acids Res 41:D1234–D1240. doi: 10.1093/nar/gks1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Carlson JM, Listgarten J, Pfeifer N, Tan V, Kadie C, Walker BD, Ndung’u T, Shapiro R, Frater J, Brumme ZL, Goulder PJ, Heckerman D. 2012. Widespread impact of HLA restriction on immune control and escape pathways of HIV-1. J Virol 86:5230–5243. doi: 10.1128/JVI.06728-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yagita Y, Kuse N, Kuroki K, Gatanaga H, Carlson JM, Chikata T, Brumme ZL, Murakoshi H, Akahoshi T, Pfeifer N, Mallal S, John M, Ose T, Matsubara H, Kanda R, Fukunaga Y, Honda K, Kawashima Y, Ariumi Y, Oka S, Maenaka K, Takiguchi M. 2013. Distinct HIV-1 escape patterns selected by cytotoxic T cells with identical epitope specificity. J Virol 87:2253–2263. doi: 10.1128/JVI.02572-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Leslie A, Price DA, Mkhize P, Bishop K, Rathod A, Day C, Crawford H, Honeyborne I, Asher TE, Luzzi G, Edwards A, Rousseau CM, Rosseau CM, Mullins JI, Tudor-Williams G, Novelli V, Brander C, Douek DC, Kiepiela P, Walker BD, Goulder PJR. 2006. Differential selection pressure exerted on HIV by CTL targeting identical epitopes but restricted by distinct HLA alleles from the same HLA supertype. J Immunol 177:4699–4708. doi: 10.4049/jimmunol.177.7.4699. [DOI] [PubMed] [Google Scholar]
- 80.Brumme ZL, John M, Carlson JM, Brumme CJ, Chan D, Brockman MA, Swenson LC, Tao I, Szeto S, Rosato P, Sela J, Kadie CM, Frahm N, Brander C, Haas DW, Riddler SA, Haubrich R, Walker BD, Harrigan PR, Heckerman D, Mallal S. 2009. HLA-associated immune escape pathways in HIV-1 subtype B Gag, Pol and Nef proteins. PLoS One 4:e6687. doi: 10.1371/journal.pone.0006687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Woods CK, Brumme CJ, Liu TF, Chui CK, Chu AL, Wynhoven B, Hall TA, Trevino C, Shafer RW, Harrigan PR. 2012. Automating HIV drug resistance genotyping with RECall, a freely accessible sequence analysis tool. J Clin Microbiol 50:1936–1942. doi: 10.1128/JCM.06689-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gaschen B, Kuiken C, Korber B, Foley B. 2001. Retrieval and on-the-fly alignment of sequence fragments from the HIV database. Bioinformatics 17:415–418. doi: 10.1093/bioinformatics/17.5.415. [DOI] [PubMed] [Google Scholar]
- 83.Katoh K, Standley DM. 2013. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30:772–780. doi: 10.1093/molbev/mst010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Eddy SR. 1998. Profile hidden Markov models. Bioinformatics 14:755–763. doi: 10.1093/bioinformatics/14.9.755. [DOI] [PubMed] [Google Scholar]
- 85.Larsson A. 2014. AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30:3276–3278. doi: 10.1093/bioinformatics/btu531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Moonsamy PV, Williams T, Bonella P, Holcomb CL, Hoglund BN, Hillman G, Goodridge D, Turenchalk GS, Blake LA, Daigle DA, Simen BB, Hamilton A, May AP, Erlich HA. 2013. High throughput HLA genotyping using 454 sequencing and the Fluidigm access array system for simplified amplicon library preparation. Tissue Antigens 81:141–149. doi: 10.1111/tan.12071. [DOI] [PubMed] [Google Scholar]
- 87.Maupetit J, Derreumaux P, Tuffery P. 2009. PEP-FOLD: an online resource for de novo peptide structure prediction. Nucleic Acids Res 37:W498–W503. doi: 10.1093/nar/gkp323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thevenet P, Shen Y, Maupetit J, Guyon F, Derreumaux P, Tuffery P. 2012. PEP-FOLD: an updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res 40:W288–W293. doi: 10.1093/nar/gks419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Shen Y, Maupetit J, Derreumaux P, Tuffery P. 2014. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J Chem Theory Comput 10:4745–4758. doi: 10.1021/ct500592m. [DOI] [PubMed] [Google Scholar]
- 90.Miura T, Brockman MA, Brumme ZL, Brumme CJ, Pereyra F, Trocha A, Block BL, Schneidewind A, Allen TM, Heckerman D, Walker BD. 2009. HLA-associated alterations in replication capacity of chimeric NL4-3 viruses carrying gag-protease from elite controllers of human immunodeficiency virus type 1. J Virol 83:140–149. doi: 10.1128/JVI.01471-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gibson DG, Young L, Chuang R-Y, Venter JC, Hutchison CA, Smith HO. 2009. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6:343–345. doi: 10.1038/nmeth.1318. [DOI] [PubMed] [Google Scholar]
- 92.Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther 6:715–728. doi: 10.1038/sj.gt.3300895. [DOI] [PubMed] [Google Scholar]
- 93.Mann JK, Byakwaga H, Kuang XT, Le AQ, Brumme CJ, Mwimanzi P, Omarjee S, Martin E, Lee GQ, Baraki B, Danroth R, McCloskey R, Muzoora C, Bangsberg DR, Hunt PW, Goulder PJR, Walker BD, Harrigan P, Martin JN, Ndung’u T, Brockman MA, Brumme ZL. 2013. Ability of HIV-1 Nef to downregulate CD4 and HLA class I differs among viral subtypes. Retrovirology 10:100. doi: 10.1186/1742-4690-10-100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jin SW, Alsahafi N, Kuang X, Mwimanzi P, Gottlinger H, Brumme Z, FInzi A, Brockman M. 2018. Impact of natural HIV-1 Nef polymorphisms on SERINC5 antagonism, abstr 202 2018 Conference on Retroviruses and Opportunistic Infections (CROI), Boston, MA, 4 to 7 March 2018. [Google Scholar]
- 95.Suhoski MM, Golovina TN, Aqui NA, Tai VC, Varela-Rohena A, Milone MC, Carroll RG, Riley JL, June CH. 2007. Engineering artificial antigen-presenting cells to express a diverse array of co-stimulatory molecules. Mol Ther 15:981–988. doi: 10.1038/mt.sj.6300134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lee GQ, Bangsberg DR, Muzoora C, Boum Y, Oyugi JH, Emenyonu N, Bennett J, Hunt PW, Knapp D, Brumme CJ, Harrigan PR, Martin JN. 2014. Prevalence and virologic consequences of transmitted HIV-1 drug resistance in Uganda. AIDS Res Hum Retroviruses 30:896–906. doi: 10.1089/aid.2014.0043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.