

Rotaviruses are causal agents of acute infantile viral diarrhea. In intestinal cells, in vitro as well as in vivo, virus assembly and exit do not imply cell lysis but rely on an active process in which the cytoskeleton plays a major role. We describe here a novel molecular mechanism by which the rotavirus spike protein VP4 drives actin remodeling. This relies on the fact that VP4 occurs in different forms. Besides its structural function within the virion, a large proportion of VP4 is expressed as free protein. Here, we show that free VP4 possesses a functional actin-binding domain. This domain, in coordination with a coiled-coil domain, promotes actin cytoskeleton remodeling, thereby providing the capacity to destabilize the cell membrane and allow efficient rotavirus exit.
KEYWORDS: VP4 protein, actin binding, actin remodeling, protein domain, rotavirus
ABSTRACT
The interactions between viruses and actin cytoskeleton have been widely studied. We showed that rotaviruses remodel microfilaments in intestinal cells and demonstrated that this was due to the VP4 spike protein. Microfilaments mainly occur in the apical domain of infected polarized enterocytes and favor the polarized apical exit of viral progeny. The present work aims at the identification of molecular determinants of actin-VP4 interactions. We used various deletion mutants of VP4 that were transfected into Cos-7 cells and analyzed interactions by immunofluorescence confocal microscopy. It has been established that the C-terminal part of VP4 is embedded within viral particles when rotavirus assembles. The use of specific monoclonal antibodies demonstrated that VP4 is expressed in different forms in infected cells: classically as spike on the outer layer of virus particles, but also as free soluble protein in the cytosol. The C terminus of free VP4 was identified as interacting with actin microfilaments. The VP4 actin binding domain is unable to promote microfilament remodeling by itself; the coiled-coil domain is also required in this process. This actin-binding domain was shown to dominate a previously identified peroxisomal targeting signal, located in the three last amino acids of VP4. The newly identified actin-binding domain is highly conserved in rotavirus strains from species A, B, and C, suggesting that actin binding and remodeling is a general strategy for rotavirus exit. This provides a novel mechanism of protein-protein interactions, not involving cell signaling pathways, to facilitate rotavirus exit.
IMPORTANCE Rotaviruses are causal agents of acute infantile viral diarrhea. In intestinal cells, in vitro as well as in vivo, virus assembly and exit do not imply cell lysis but rely on an active process in which the cytoskeleton plays a major role. We describe here a novel molecular mechanism by which the rotavirus spike protein VP4 drives actin remodeling. This relies on the fact that VP4 occurs in different forms. Besides its structural function within the virion, a large proportion of VP4 is expressed as free protein. Here, we show that free VP4 possesses a functional actin-binding domain. This domain, in coordination with a coiled-coil domain, promotes actin cytoskeleton remodeling, thereby providing the capacity to destabilize the cell membrane and allow efficient rotavirus exit.
INTRODUCTION
Rotaviruses (RV) belong to the Reoviridae family and are the most widespread causal agents of acute infantile viral diarrhea (1, 2). Mature virions contain 11 double-stranded RNA segments surrounded by three concentric protein layers, the most external being composed of assemblies of the VP7 coat and VP4 spike proteins (3–5). Rotaviruses infect enterocytes from the luminal side. After replication, new virions are released at the apical pole by an atypical polarized trafficking pathway that has been partially described (6). The VP4 spike protein is reported to play a major role in both rotavirus entry and exit thanks to several molecular interactions. VP4 is indeed thought to mediate primary interactions with cellular components through its N-terminal subunit, VP8*, which can interact with cell surface receptors (7, 8) and/or coreceptors, which contain specific glycan moieties (9). VP5*, the C-terminal subunit of VP4, has been shown to interact with several putative partners, including integrins (10), membranes via a fusion domain which allows virus entry (11), other VP4 molecules to form oligomers (12), and peroxisomes (13). Previous studies have shown that VP4 also specifically interacts with the apical actin cytoskeleton and induces its remodeling, leading to a disorganization of the brush border (14). We showed that VP4 interacts with actin microfilaments by perturbing their treadmilling-based renewal, a specific mechanism involved in actin dynamics (15), and we have proposed that this favors the access of virions to the apical pole of enterocytes for cellular exit (14), since the actin network is essential for exocytosis (16). Finally, we demonstrated that blocking the treadmilling by using jasplakinolide leads to a fully depolarized release of new virions (15). A similar process has been recently demonstrated by another group in MA104 cells (17), a nonpolarized cell line, indicating that our initial observation is not restricted to polarized enterocytes.
Actin microfilaments represent a common tool in the wide spectrum of strategies developed by viruses to take control of their target cells (18). Some viruses, such as vaccinia virus, baculoviruses, Epstein-Barr virus, herpes simplex virus, frog virus 3, and murine mammary tumor virus, promote the polymerization of these cytoskeletal filaments to assist their intracellular movements, assembly, and budding (19). In some instances, F-actin projections are induced to promote moving of viral particles and budding, as seen with measles virus, vaccinia virus, or p815 retrovirus (20). Human immunodeficiency virus type 1 (HIV-1) induces cell shape changes and the generation of intercellular synapses, involving actin microfilaments, to facilitate virus budding and spreading (21). Baculoviruses also induce the localization of actin toward an ectopic compartment, the nucleus (22). Other viruses, such as influenza virus, only show colocalization of some of their proteins with actin cytoskeleton (23). Actin or actin-related proteins have also been considered a structural component of the viral particle for rabies virus and HIV-1 (24, 25). At least some viruses use actin as cofactors for their replication, e.g., human parainfluenza 3, where polymerized actin interaction with the transcribing ribonucleoprotein is required for mRNA synthesis (26). Interestingly, only a few viruses, such as adenovirus, human cytomegalovirus, vesicular stomatitis virus, and rubella virus, have been shown to disrupt actin microfilaments, by inducing their cleavage, to facilitate virion release (27–30).
Our previous work underlined the crucial role of the spike rotavirus protein interaction with actin (14, 15). However, mechanisms of actin binding and remodeling by VP4 remained unknown. Analysis of the primary amino acid sequence of VP4 did not reveal any already identified actin-binding domain or any homology with known actin-binding protein. In the present work, a detailed molecular dissection of the VP4 sequence was performed in order to define how VP4 interacts with actin microfilaments and how it promotes remodeling. A new actin-binding domain was discovered in the C-terminal part of VP4, which we named rotavirus actin-binding domain (RV-ABD). When expressed alone, this motif does not induce obvious reorganization of the actin cytoskeleton. Microfilament remodeling requires the cooperation of RV-ABD and the coiled-coil domain of VP4. Interestingly, these domains are buried within the viral particles (5), but we were able to shown here that a pool of free, nonassembled VP4 is responsible for its actin binding and remodeling activities. Bioinformatics studies indicate that this RV-ABD was present in several strains of species A, B, and C rotaviruses, suggesting that actin binding and remodeling is a general strategy for rotavirus replication. Our results describe a novel and original pathogen-driven microfilament reorganization strategy based on simple protein-protein interactions, without the involvement of signaling pathways to facilitate virus exit from the cells.
RESULTS AND DISCUSSION
VP4, the rotavirus spike protein, is expressed both within virions and as a free protein.
Previous observations have shown that the spike rotavirus protein VP4 is able to bind and remodel actin during rotavirus infection of MA104 and Caco-2 cells (14, 15). To understand the molecular mechanisms of this remodeling process, we first analyzed the subcellular localizations of VP4 during infection by using two distinct monoclonal antibodies (MAb): 7.7, a neutralizing antibody described to mainly recognize VP5* in both free and assembled VP4 (31, 32), and 2G4, an antibody that recognizes VP8* when assembled within the viral particles (33). Confocal microscopy and double immunolabeling of RV-infected MA104 cells showed that these two antibodies did not display identical patterns (Fig. 1). A large part of VP4, as revealed by 7.7 MAb, was distributed along filament-like structures, reminiscent of actin microfilaments, as previously observed (14), as well as in dots that did not colocalize with 2G4 staining. VP4 was also present in dots that were labeled by 2G4 MAb but only partially by 7.7 MAb. This pattern corresponds to the final steps of rotavirus assembly in these cells (15). The results indicate that VP4 is always expressed in three forms: (i) assembled with virion progeny, (ii) along intact actin microfilaments, and (iii) within remodeled microfilaments. These results also confirmed that 2G4 MAb only recognizes VP4 when assembled in rotavirus progeny particles and that rotavirus-infected cells always express VP4 both as assembled virions and as a free protein.
FIG 1.

VP4 is expressed both within virions and as a free protein. MA104 cells were infected by rotavirus RF stain and analyzed by confocal microscopy and immunofluorescence at 6 h postinfection (h.p.i.) using two different monoclonal antibodies. MAb 7.7 (A and D) mainly recognizes VP8* either on assembled or unassembled VP4, and 2G4 (B and E) recognizes assembled VP5*. Panels C and F represent merged images of panels A and B and panels D and E, respectively. MAb 7.7 staining was revealed using FITC-labeled secondary antibody (green), and 2G4 staining was revealed using Alexa-547 dye (red) directly coupled to the antibody. Arrows point to some 2G4-labeled dots that colocalize faintly with 7.7 staining. Arrowheads point to some 7.7-labeled dots that do not colocalize with 2G4 staining. Scale bar, 5 µm. Images are representative of three independent experiments.
The C-terminal part of VP4 contains signals for actin binding and remodeling.
Since VP4 is involved in actin reorganization and most of the VP5* subunit is buried within the viral shell in assembled rotavirus (5), we hypothesized that the effects of VP4 on actin microfilament reorganization are either due to the VP8*-containing domains on the outer and middle layers of virions or to VP5* from the nonassembled VP4. However, results from Fig. 1 indicated that the antibody recognizing VP8*, 2G4, never stained filament-like structures, indicating that VP8* is unlikely to be involved in actin reorganization. Therefore, we decided to focus subsequent experiments on the VP5* subunit of VP4. To delineate the molecular bases of actin binding and reorganization, we designed a strategy in which we expressed VP4 or fragments thereof outside the context of infection. To this end, Cos-7 cells were transiently transfected by a series of VP4 constructs (Fig. 2) that will be detailed below. Control experiments (Fig. 3) demonstrated that the addition of green fluorescent protein (GFP) does not modify VP4 expression and localization, as shown by colocalization between VP4-GFP and MAb 7.7 staining. Figure 3 also shows that 2G4 is unable to recognize VP4-GFP, confirming that 2G4 only recognizes the virion-assembled form of VP4. As expected from previous results, VP4-GFP displayed both a filament-like and a punctate structure. VP8*-GFP is expressed in the soluble fraction of the cell, again making its role in actin remodeling highly unlikely. Interestingly, VP8*-GFP was also recognized by 7.7, suggesting that this antibody can also interact with a portion of VP8*.
FIG 2.

Summary of the VP4-GFP constructs used in the present study. Each construct was obtained using the backbone plasmid pEGFP-C1, and all truncated-VP4 constructs encoding amino acid stretches 481 to 776, 481 to 773, 481 to 757, 562 to 776, 641 to 776, 641 to 773, 641 to 757, 702 to 776, 713 to 776, 713 to 773, and 718 to 776, as well as the constructs containing deletions (481-776 del 574-670, 481-776 del 574-713, and 481-776 del 574-718), were generated by PCR and inserted within pEGFP-C1 vector via EcoRI and BamHI sites. Sp100 coiled-coil fused to VP4 (562-776) was made by insertion of PCR-amplified Sp100 coiled-coil 29-152 in VP4 (562-776). Note that GFP-truncated VP4 encoding amino acid stretches 713 to 776, 718 to 776, 718 to 773, and 733 to 776 were made by insertion of amplified segments from the SA11 strain in pEGFP-C1 via XhoI and BamHI sites.
FIG 3.

2G4 antibody only recognizes assembled VP4. Cos-7 cells were transiently transfected using either a VP4-GFP construct or a VP8*-GFP construct. Three days later, cells were fixed, permeabilized, and analyzed by confocal microscopy and immunofluorescence using 7.7 or 2G4 MAbs. Transfected GFP constructs appear in green, and Alexa-547-labeled secondary antibodies (red) were used to reveal MAb 7.7 or 2G4. Scale bars, 5 µm. Images are representative of two independent experiments.
Various VP4 deletion constructs were produced (Fig. 2) and then tested by transfection in Cos-7 cells, followed by confocal microscopy analysis. The first three constructs consisted of the removing of either the 480 amino acids (aa) of the N-terminal part, keeping the coiled-coil domain, the first 561 aa, removing the coiled-coil domain (aa 481 to 574), or keeping only the C-terminal part (aa 718 to 776) of VP4. These three constructs provided patterns very distinct (Fig. 4) from those of the full-length VP4 construct, which displayed both filament-like structures and puncta that colocalized with phalloidin staining (Fig. 4A to C). Transfection of the 481-776 construct mainly displayed large puncta and a few filament-like structures that also colocalized with phalloidin (Fig. 4D to F). Removal of the first 561 amino acids resulted in a pattern where the VP4 fragment colocalized with nonremodeled actin microfilaments (Fig. 4G to I). Finally, the C-terminal VP4 construct displayed a distinct pattern with several small dots that did not colocalize with actin microfilaments (Fig. 4J to L). These results suggested that (i) a domain in the C-terminal part of VP4 is required to promote interactions with actin microfilaments; (ii) binding to actin microfilaments and microfilament remodeling should be independent, since it was possible to observe actin-VP4 colocalization without any puncta (Fig. 4G to I); and (iii) the most C-terminal part of VP4 contained another signal promoting interactions with another subcellular compartment that will be characterized in the following paragraph.
FIG 4.
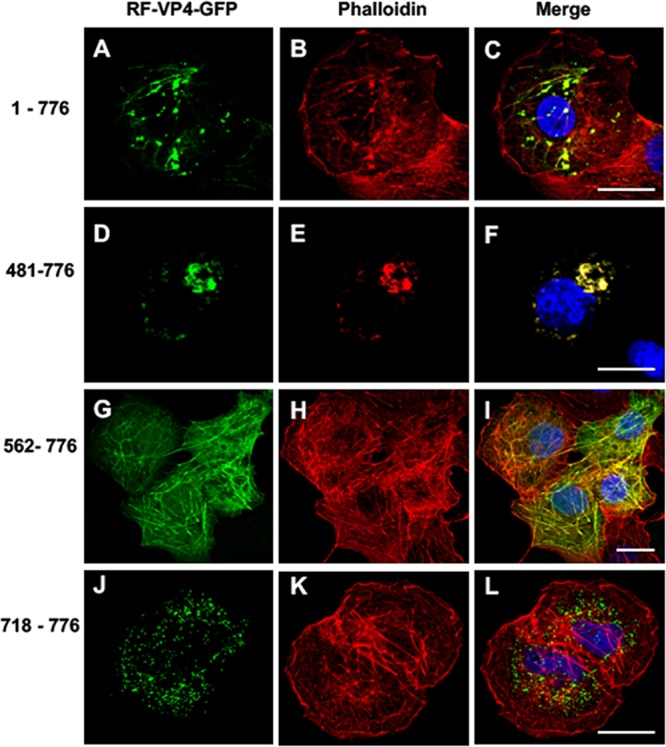
Effects of successive N-terminal deletions on the colocalization of VP4-GFP constructs with actin microfilaments. Cos-7 cells were transiently transfected using four RF VP4-GFP constructs (green) (A, D, G, and J) containing either the full-length VP4 (construct 1-776) (A to C), an N-terminal deletion keeping the coiled-coil domain (construct 481-776) (D to F), an N-terminal deletion removing the main part of the coiled-coil domain (construct 562-776) (G to I), or a construct containing only the last 59 C-terminal amino acids of VP4 (construct 718-776) (J to L). Three days after transfection, cells were fixed, permeabilized, labeled with Alexa-547-phalloidin (red) (B, E, H, and K), and analyzed by confocal fluorescence microscopy. Panels C, F, I, and L represent the corresponding merged images. Scale bar, 5 µm. Images are representative of four independent experiments.
A novel mechanism for virus-microfilament interactions.
The above-described results indicated that (i) the full-length form of free VP4 both promotes colocalization with actin microfilaments and induces microfilaments remodeling, (ii) the N-terminal part of VP4 is not involved in interactions with actin microfilaments, and (iii) the C-terminal part of VP4 contains a signal(s) necessary for colocalization with microfilaments (Fig. 4G to I). The next step was to define more precisely the VP4 domain involved in actin binding and to explain the molecular mechanism of microfilament remodeling. Several other constructs, also diagrammatically shown in Fig. 2, were tested using transient transfection of Cos-7 cells to delineate a minimal putative rotavirus-actin-binding domain. As shown in Table 1, colocalization of VP4 constructs with actin microfilaments was always observed, except for 2 constructs, namely, 713-758 and 718-776, indicating that RV-ABD should be located between aa 713 (Thr) and aa 773 (Gln) of the RV strain. Interestingly, Table 1 also indicated that microfilament remodeling was only observed when the expressed fragment contained the entire coiled-coil domain together with RV-ABD. The coiled-coil domain by itself was unable to promote microfilament colocalization and consequently their remodeling. Transfection of VP4 constructs containing the coiled-coil domain and RV-ABD (aa 481 to 776) (Fig. 4A to C and 5A to C) resulted in actin remodeling, whereas transfection of VP4 constructs in which the coiled-coil domain has been removed while the RV-ABD has been conserved resulted in actin colocalization without remodeling (Fig. 4G to I and 5D to F). Transfection of VP4 constructs with internal deletions allowed us to better define the precise limits of RV-ABD. As shown in Table 1 and Fig. 5, the VP4 constructs 481-776 del 574-670 (Fig. 5G to I) and 481-776 del 574-713 (Fig. 5J to L), in which both the coiled-coil and the RV-ABD domains are conserved, were still able to remodel actin microfilaments. In contrast, removal of both the coiled-coil and the RV-ABD domains (construct 481-776 del 574-718) suppressed both microfilament colocalization and actin remodeling (Table 1). Together, these results strongly suggested that VP4 is able to bind actin microfilaments via its RV-ABD (aa 713 to 773) and to remodel actin microfilaments through the joined interaction of both the coiled-coil domain and the RV-ABD. To demonstrate this, we used two distinct approaches. First, a construct containing the rotavirus RV-ABD fused to an irrelevant coiled-coil domain from the nuclear antigen Sp100, a component of PML nuclear bodies (34), was transfected in Cos-7 cells and resulted in both microfilament colocalization and actin remodeling, as shown on Table 1 and Fig. 5M to O. Second, Cos-7 cells were transfected with two VP4 481-776 constructs inserted either in GFP- or mCherry-containing plasmids. As expected, these two constructs displayed perfect colocalization (Fig. 6A to C). This model also allowed us to perform fluorescent resonance energy transfer (FRET) experiments in which cells were illuminated only with a blue light (488 nm) to excite GFP, and fluorescence was collected either in the green range (500 to 530 nm) or in the red range (570 to 650 nm). As shown in Fig. 6D and E, in cells where both VP4-GFP and VP4-mCherry constructs were expressed (Fig. 6D, upper row), a large proportion of the fluorescence was recovered in the red range, indicating that the two constructs were in close interactions, allowing energy transfer (FRET) from GFP to mCherry. This strongly suggested that they oligomerize even when expressed outside the context of viral infection. In contrast, in control cells, where GFP- and mCherry-containing constructs were transfected without VP4 (Fig. 6D, lower line), a small proportion of FRET could be detected. Quantification of fluorescence in each channel (Fig. 6E) confirmed these results.
TABLE 1.
Actin microfilament binding, remodeling capacities, and peroxisomal targeting of different VP4 constructs used in this studya
| Construct | Microfilament remodeling |
Microfilament binding |
Peroxisomal localization |
|---|---|---|---|
| 1-776 | + | + | − |
| 481-776 | + | + | − |
| 562-776 | − | + | − |
| 574-776 | − | + | ND |
| 670-776 | − | + | ND |
| 702-776 | − | + | ND |
| 702-773 | − | + | − |
| 713-776 | − | + | − |
| 713-773 | − | + | − |
| 713-758 | − | − | − |
| 718-776 | − | − | + |
| 773-776 | − | − | + |
| 481-776 del 574-670 | + | + | ND |
| 481-776 del 574-713 | + | + | ND |
| 481-776 del 574-718 | − | − | ND |
| Sp100 CC-VP4 574-776 | + | + | ND |
Microfilament binding and remodeling capacities were evaluated by confocal microscopy. ND, not determined.
FIG 5.

Role of RV-ABD and coiled-coil domain in actin binding and remodeling. Cos-7 cells were transiently transfected using five of the RF VP4-GFP constructs described in the legend to Fig. 3, namely, constructs 481-776 (A to C), 670-776 (D to F), 481-776 del-574-670 (G to I), 481-776 del-574-713 (J to L), and the construct containing the exogenous coiled-coil domain Sp100 CC VP4 574-776 (M to O). Three days after transfection, cells were fixed, permeabilized, labeled with Alexa-547-phalloidin, and analyzed by confocal fluorescence microscopy. Panels A, D, G, J, and M represent GFP fluorescence (green); panels B, E, H, K, and N represent phalloidin fluorescence (red); panels C, F, I, L, and O correspond to the respective merged images. Scale bar, 5 µm. Images are representative of three to five independent experiments.
FIG 6.

Homo-oligomerization of free VP4. Cos-7 cells were transiently transfected using plasmids containing the construct VP4 481-776, in line with either GFP or mCherry fluorescent proteins. Three days after transfection, Cos-7 cells were observed by confocal microscopy. (A) Typical image obtained with the GFP-containing construct; (B) typical image obtained with the mCherry-containing construct; (C) merged image. Scale bar represents 5 µm. (D) FRET experiment was performed using Cos-7 cells expressing both VP4-GFP and VP4-mCherry (upper) or GFP and mCherry alone (lower). Cells were only illuminated with a 488-nm laser beam, and fluorescence was recovered in the green range (500 to 530 nm) and in the red range (red filter, 570 to 650 nm). Eight different fields were analyzed. Blue color represents the background signal, green color the signal recorded for the GFP channel, and red color the signal for the mCherry channel. In the control lower line, the main signal detected was in the green range, whereas in the upper line a strong red fluorescence was detected in the eight fields analyzed, indicating a FRET signal. Scale bar represents 500 µm. (E) Images from panel D were quantified using ImageJ software, and the ratio of red fluorescence to green fluorescence was calculated for each field. Means and SD were calculated for each experiment and are reported as the percentage of red fluorescence over total fluorescence.
The present results indicate that actin remodeling results from the concerted action of two distinct domains, i.e., RV-ABD and coiled-coil. The most likely molecular mechanism by which VP4 remodels actin microfilaments is that at least two molecules of VP4 bind to actin microfilaments through RV-ABD domains and oligomerize through coiled-coil domains, allowing microfilament bridging. The simultaneous occurrence of the RV-ABD and coiled-coil domains within a single protein chain should promote VP4 oligomerization and in the meantime will induce the remodeling of preassembled F-actin bundles. Such an interaction results in the formation of what we previously called actin bodies (14), which are focal clusters of F-actin fully colocalized with phalloidin. This mechanism explains how VP4 may induce a destabilization of the subcortical microfilaments, favoring the crossing of viral particles through the apical cell membrane and their release in the extracellular space without significant cell death (6, 17). This mode of action of VP4 is in agreement with the modifications of the actin cytoskeleton previously observed during rotavirus infection of MA104, Cos-7, and Caco-2 cells, where microfilaments are progressively gathered in actin bodies (14, 15). These cytoskeleton modifications observed during rotavirus infection are also observed during cellular transfection of the pEGFP-C1-VP4 expression plasmid (14), meaning that this process does not depend on viral particle assembly but requires the expression of free VP4.
Viruses have developed various strategies to hijack cellular machineries, especially those of the actin cytoskeleton (18). The present study reveals a new way by which a virus protein may interact with microfilaments, as VP4 acts through a simple mechanism based on protein-protein interactions and bridging properties. Furthermore, VP4 microfilament remodeling activities do not seem to require any signaling events, contrary to most of the virus-driven actin rearrangements involving Rho GTPase-mediated signaling pathways (35). This conclusion, which is in line with our previously published data (15), reinforces the observation that microfilament remodeling is a progressive process that only depends on the amount of VP4 produced in the target cells and not on the VP4 molecules incorporated in viral particles.
The VP4 peroxisomal targeting signal is functional but masked by an actin-binding domain.
As mentioned above, the construct RF-VP4-GFP 718-776 was localized in another intracellular compartment that did not colocalize with phalloidin (Fig. 4J to L). Such a type of staining has been already described for VP4 by Mohan et al. (13) and reported to be due to a canonical peroxisome targeting signal (PTS1) in the last four C-terminal amino acids of the SA11 rotavirus strain. To explore this further, the reactivity of four VP4 constructs that contained various lengths of its C-terminal part was studied (Fig. 7). For this experiment, the SA11 strain-specific sequence of VP4 was used in order to compare our data with those of Mohan et al. (13). The transfection of the VP4 last four C-terminal amino acids (773 to 776) (Fig. 7A to C and Table 1) coupled with GFP resulted in a pattern that fully colocalized with a peroxisome marker, confirming the functionality of this short domain. The addition of 65 extra C-terminal amino acids (718 to 776) resulted in a pattern where peroxisome colocalization was maintained. However, a filament-like distribution was also clearly visible (Fig. 7D to F). Addition of five more C-terminal amino acids (713 to 776) totally suppressed the peroxisome localization of this VP4 construct, which appeared to be exclusively made of large filament-like structures (Fig. 7G to I). Finally, removal of the four last C-terminal amino acids (718 to 773) abolished the peroxisome colocalization (Fig. 7J to L and Table 1). This demonstrated that VP4 contains an authentic PTS1 that is in competition with another signal that favors interactions with actin microfilaments and suggests that the actin-binding domain dominates over the PTS1 signal. This may explain why peroxisome colocalization has never been observed during rotavirus infection, even if one considers that VP4 is also expressed as a free protein during the infection process.
FIG 7.
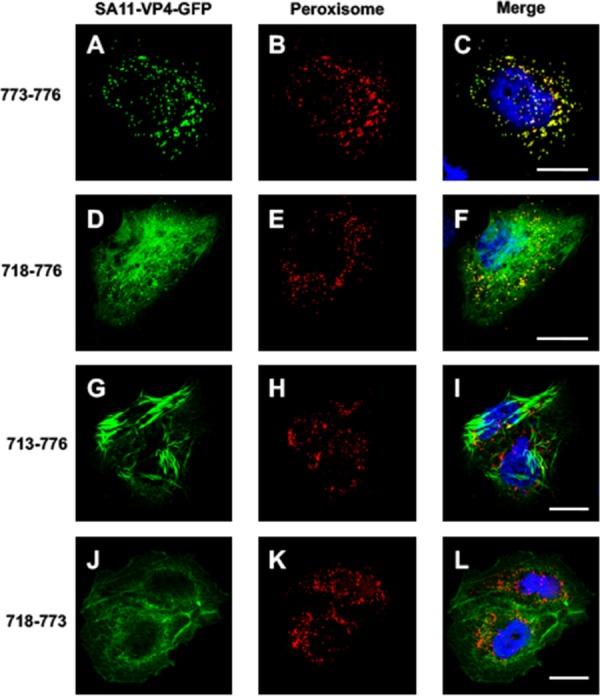
Functional peroxisome-targeting signal is present on VP4 but masked by RV-ABD. Cos-7 cells were transiently transfected using four VP4-GFP (A, D, G, and J) (green) constructs containing (i) the last four amino acids of SA11 strain VP4, namely, QCRL (a typical PTS-1 sequence), termed construct 773-776 (A to C), (ii) the last 59 amino acids of VP4 (construct 718-776) (D to F), (iii) the last 64 amino acids (construct 713-776) (G to I), or (iv) the same construct as construct ii but lacking the PTS signal (construct 718 to 773) (J to L). Three days after transfection, cells were fixed, permeabilized, labeled with Alexa-547-labeled anti-peroxisome antibody (D, E, H, K) (red), and analyzed by confocal fluorescence microscopy. (C, F, I, and L) Respective merged images. Scale bar, 5 µm. Images are representative of three independent experiments.
Actin binding and remodeling: a common strategy among rotaviruses.
The experiments described in Fig. 3, 4, 5, and 7 indicated that VP4 constructs from either the RV or SA11 strains behave similarly regarding interaction with actin microfilaments. Further bioinformatics comparisons of VP4 sequences from several animal strains of species A rotavirus (RRV, RF, SA11, OSU, and chicken), including a human Wa strain, show high conservation, especially within the RV-ABD domain (Fig. 8). Interestingly, even the quite divergent chicken rotavirus, which is characterized by major sequence variations compared to mammalian strains in species A rotaviruses (36), reveals high conservation in the RV-ABD domain compared to full-length VP4 sequence (Fig. 8). Interestingly, this domain was also found in the available sequences of species B and C rotavirus (Fig. 8). Bioinformatics studies indicated that all of the strains studied displayed a high degree of conservation in the coiled-coil domain. In addition, analysis of the C-terminal sequence of several VP4 proteins from the same strains also showed that the four last amino acids were either identical or similar (Fig. 8), consisting of an authentic PTS1 signal, indicating that rotavirus integrates peroxisome interactions in its strategy, as suggested by Mohan et al. (13). However, as mentioned above, the RV-ABD appears dominant over the PTS1 domain.
FIG 8.

Sequence alignment of RV-ABD domains from several rotavirus strains. Sequences of VP4 proteins from different strains of species A, B, or C rotavirus were aligned using BLAST. An asterisk indicates amino acid identity, a colon indicates amino acid similarity, a period indicates amino acid divergence (also indicated by black color), and no sign indicates unrelated amino acids. Note that the line below species B and C rotavirus sequences summarizes the comparison of the eight strains studied here. Blue letter codes correspond to the PTS-1 signal. Amino acid numbering was aligned with the numbering of the RF strain.
This set of data indicates that the ability to bind and remodel actin microfilaments was present in a rotavirus ancestor and was selected a long time ago for an efficient strategy of the virus. Since all these rotavirus strains contained both an actin-binding domain and a coiled-coil domain, it is tempting to hypothesize that VP4 actin binding and remodeling are a common theme for the rotavirus’ replication cycle. However, this conclusion will be difficult to demonstrate as RVC are difficult, and RVB almost impossible, to grow in cell culture.
As mentioned above, RV-ABD is located in the C-terminal part of VP4 and is buried when VP4 is assembled within viral particles. To explain how VP4 may still display its capacity to bind and remodel actin microfilaments, we analyzed the only available three-dimensional (3D) structure of VP4 assembled in viral particles (5, 37). Using data from the Protein Data Bank (PDB) (accession number 4V7Q), we analyzed the location of RV-ABD in a rotavirus particle. As shown in Fig. 9A and B, RV-ABD was indeed located deep inside the particle, under the shell formed by VP7. This observation confirmed that only the pool of free VP4 is involved in actin binding (Fig. 1). Surprisingly, sequence comparison with several known actin-binding domains, such as those of myosin 1a (UniProt accession number Q9UBC5), alpha-actinin (UniProt accession number P12814), ezrin (UniProt accession number P15311), and villin (UniProt accession number P09327), did not provide any significant similarities, suggesting that this actin-binding domain belongs to a new family. In addition, it should be pointed out that the coiled-coil domain, also involved in the actin-remodeling process, was engaged within the stalk of trimeric VP4 when assembled within viral particles (5, 38).
FIG 9.

Structural analysis of RV-ABD. (A and B) The 3D structure of a unit of assembled rotavirus particle was retrieved from the PDB (accession number 4V7Q) and is displayed in panel A. The sequence corresponding to RV-ABD was searched, and a zoomed image of this region is displayed in panel B, corresponding to the square in panel A. Mark 1 corresponds to aa 701 (Ile) and mark 2 to aa 776 (Leu) according to RF numbering. Only one of the three VP4 chains (the one annotated BX in PDB) is underlined in black between marks 1 and 2.
Conclusions.
Several studies have already defined putative functional domains from the analysis of the primary structure of VP4. The galectin-like domain has been crystalized (32) and functionally studied (39). An integrin-binding domain (40), a fusion domain (11), a coiled-coil domain (40, 41), a dynein-binding domain (42), and a peroxisomal targeting signal (13) have been functionally studied. None of these domains accounts for actin-binding and remodeling activity. The present discovery of an original RV-ABD domain and of its likely mechanism of action, together with the demonstration of VP4 expression as a free protein during infection, strongly support the idea that free VP4 plays an important role in rotavirus cellular exit from the apical domain of polarized cells, as shown in our previous studies (6, 14, 15). Interestingly, a recent study (17) proposes that actin is part of rotavirus assembly and exit even in MA104 nonpolarized cells, indicating that cell lysis is not required. These data, together with our previous findings, point again to the general importance of rotavirus-actin interactions in virus morphogenesis and trafficking.
Rotavirus displays a very striking property that consists of the apically polarized release of viral progeny without immediate cell death in vitro (6) as well as in vivo (43). Since this release process does not involve any conventional vesicular traffic step, one can hypothesize that the apical membrane of intestinal cells has to be transiently destabilized to permit virus exit. Two main factors ensure the high stability of the intestinal brush border membrane, namely its particular “superraft”-based lipid composition (44) and the highly packed actin microfilament bundles (45). The present results provide a molecular mechanism for VP4-driven brush border membrane destabilization through its actin microfilament remodeling activity. Interestingly, it has been shown that another viral protein, namely, NSP4, may also disturb the actin cytoskeleton and, although not colocalizing with F-actin, causes F-actin reorganization (46). These authors have shown that, in NSP4-transfected cells, actin reorganization occurs in a calcium-dependent manner, causing an increase in F-actin content and a decrease in phosphocofilin, the inactive form of cofilin. These data are in line with the present work and point to a coordinated action of viral NSP4 and VP4 proteins. By increasing intracellular calcium levels, NSP4 activates cofilin, resulting in the generation of additional barbed ends, which are the targets of VP4 (15). In the cells thus prepared for VP4 action, the spike protein would apply its microfilament bridging activity. The most striking feature of this viral strategy is that neither the action of NSP4 (46, 47) nor that of VP4 (15) is dependent on the Rho GTPases involved in the physiological regulation of F-actin network dynamics and represents a novel mode of action for a virus.
MATERIALS AND METHODS
Cells infection and transfections.
MA104 cells were maintained in Dulbecco's modified Eagle medium supplemented with 10% fetal bovine serum (Invitrogen, UK), 100 U/ml penicillin, and 100 µg/ml of streptomycin (all reagents were from Life Technologies, Saint-Aubin, France) at 37°C with 5% CO2. Production and titration of the bovine rotavirus RF strain were carried out as previously described (14, 15). Infections of cells were performed at a multiplicity of infection of 10 PFU per cell, as previously described (14, 15). Cos-7 monkey kidney cell lines were maintained in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum, 100 U penicillin, and 0.1 mg streptomycin/ml (Invitrogen, Cergy-Pontoise, France). Transfections were realized using Exgen500 (Euromedex, Souffelweyersheim, France) or Effectene (Qiagen, Courtaboeuf, France). Briefly, 24 h after transfection, cells were trypsinized and seeded on glass. Experiments were carried out 24 to 48 h posttransfection.
Plasmid construction.
The full-length VP4 (of bovine rotavirus RF strain) cDNA (GenBank accession number U65924) was a gift of Jean Cohen (CNRS, Gif-sur-Yvette, France). Unless stated otherwise, VP4 was from the RF strain. VP4 (SA11 strain) cDNA was kindly provided by Satoshi Komoto and Koki Taniguchi (Department of Virology and Parasitology, School of Medicine, Fujita Health University, Japan). Plasmid bearing Sp100 cDNA was kindly provided by Hugues de Thé (CNRS U944/INSERM UMR7212, Paris, France). All constructs encoding GFP-truncated VP4 were made using the backbone plasmid pEGFP-C1 (Clontech, Ozyme, Montigny le Bretonneux, France). Truncated VP4 constructs encoding stretches of aa 481 to 776, 481 to 773, 481 to 757, 562 to 776, 641 to 776, 641 to 773, 641 to 757, 702 to 776, 713 to 776, 713 to 773, and 718 to 776 were generated by PCR and inserted within the pEGFP-C1 vector via EcoRI and BamHI sites. Arbitrarily, each GFP-truncated VP4 construct was designated by the limits of amino acid sequences preceding VP4; e.g., VP4 with full-length GFP (aa 1 to 776) is designated VP4 (1-776). Insertion of VP4 coiled-coil segment VP4 (481-574) in VP4 (713-776) was made in the EcoRI site. Sp100 coiled-coil segment fused to VP4 (562-776) was made by insertion of PCR-amplified Sp100 coiled-coil aa 129 to 182 in VP4 (562-776). GFP-truncated SA11 VP4, encoding amino acid stretches 713 to 776, 718 to 776, 718 to 773, and 733 to 776, were made by insertion of amplified segments in pEGFP-C1 via XhoI and BamHI sites. For PCR experiments, the following primers were used: 481RF forward, 5ʹ-CTTCGAATTCTACACCGAAACTGTATGGAGTG-3ʹ; 562RF forward, 5ʹ-CTTCGAATTCTTCAACGCTCACTGATTCATTGTCTG-3ʹ; 641RF forward, 5ʹ-CTTCGAATTCTTCAACGCTCACTGATTCATTGTCTG-3ʹ; 702RF forward, 5ʹ-CTTCGAATTCTCCATTTGATGTACAAAAATTCGCTG-3ʹ; 713RF forward, 5ʹ-CTTCGAATTCTACTGACTCACCAGTTATATCGGCAA-3ʹ; 718RF forward, 5ʹ-CTTCGAATTCTATATCGGCAATAATTGACTTTAAAAC-3ʹ; 757RF reverse, 5ʹ-GAGGTGGATCCTTAATTAATAAATTCACGTAATAG-3ʹ; 773RF reverse, 5ʹ-GAGGTGGATCCTTATTGCATTATCAAACTTTC-3ʹ; 776RF reverse, 5ʹ-GAGGTGGATCCTTACAAGCGACATTGCATTATC-3ʹ.
The coiled-coil fragment of Sp100 was PCR amplified using the following primers: Sp100CC forward, 5ʹ-CTTCGGAATCCTGTCAACATGCAGGAATACCCCGA-3ʹ; Sp100CC reverse, 5ʹ-AACTACTTAAGAGCTTTTCAAAAGTGGTC-3ʹ. For FRET experiments, an additional 481-776 construct was inserted in an mCherry plasmid via EcoRI and BamHI sites.
Immunofluorescence and FRET experiments.
Cells were cultured on 12-mm-diameter circular coverslips. For immunofluorescence experiments, cells were washed three times in phosphate-buffered saline (PBS; GIBCO) and fixed with 4% paraformaldehyde for 20 min at room temperature. Permeabilization was performed in PBS–0.2% Triton X-100 for 10 min. MAbs used (7.7 and 2G4) were generous gifts of Jean Cohen and Mary Estes. Secondary antibodies were anti-mouse Alexa Fluor 488 antibody (Molecular Probes, USA) and anti-mouse Cy3-conjugated antibody (Jackson Immunoresearch lab.). For actin staining, samples were incubated with Alexa-547-phalloidin (FluoProbes, Montluçon, France) in PBS–0.1% Tween 20 for 2 h at room temperature. Peroxisomes were localized using a PTS1-dsRed probe (Clontech, Saint-Germain-en-Laye, France). After extensive washes, cells stained with 4ʹ,6-diamidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich, Lyon, France) for 3 min were treated for 10 min with 100-mg/ml 1,4-diazabicyclo-[2.2.2]-octane antifading reagent (Sigma-Aldrich, Lyon, France) and mounted with Fluoprep (bioMérieux, France). Observations were made by a confocal microscopy LSM 510 META with a Zeiss 63× (numeric aperture, 1.4) oil immersion lens, and data acquisition was performed sequentially for each channel. Confocal microscopy images were deconvoluted with AutoDeblur Software (AutoQuant Imaging Inc., Troy, NY). Images were mounted using Photoshop in .tiff format.
For FRET experiments, Cos-7 cells were transiently transfected with either VP4 (481-776)-GFP together with VP4 (481-776)-mCherry or with control plasmids containing only GFP and mCherry. Measurements were performed on the same microscope, using the 488-nm laser line. Emitted fluorescence was recovered in eight different fields by adjusting the emission windows at 500 to 530 nm to collect green fluorescence or at 570 to 650 nm to collect the red fluorescence. The two channels were collected sequentially to avoid any interference. Fluorescence intensity in each channel was measured using ImageJ software. The red/green fluorescence ratio was then calculated for each field. Means and standard deviations (SD) were calculated for each experiment.
Protein sequence analysis.
Sequences of VP4 proteins from different strains of species A rotavirus (Wa, GenBank accession number KT694942.1; OSU, GenBank accession number KJ450845.1; RF, GenBank accession number KF729690.1; RRV, GenBank accession number AY033150.1; SA11, GenBank accession number LC178567.1; chicken, GenBank accession number ACN22281.1), species B rotavirus (IDIR, GenBank accession number X16949), or species C rotavirus (Shintoku, GenBank accession number AB738415.1) were aligned using BLAST software (NCBI). Coiled-coil domain prediction was made using the tool available online from PRABI (Pole Rhone-Alpes de Bioinformatique; https://npsa-prabi.ibcp.fr/cgi-bin/npsa_automat.pl?page=npsa_lupas.html).
Structural prediction.
Structural models were constructed using different prediction tools and web servers that are freely available (SAM-T08 [48], Robetta [49], I-Tasser [50], HHPred [51], Phyre [52], PCons [53], and LocPred [54]). Models were further ranked using cutting-edge evaluation scores, Qmean (55), ModFold (56), or Pcons (53). Final selected models result from a compromise between the evaluation scores and the structural agreement of the models obtained for rotavirus from species A (RF and SA11) and species C (Shintoku) sequences. Models for the 3D structure of VP4 embedded within viral particles were retrieved from PDB (accession number 4V7Q). The sequence of RV-ABD was directly identified on the 3D model. Only one of the three VP4 chains (the one denoted BX in PDB 4V7Q) present on the model was underlined.
ACKNOWLEDGMENTS
This work was supported by institutional funding of INSERM, University Pierre et Marie Curie (UPMC). W.C. is a recipient of a postdoc grant from ANR (ANR ProbDOM). We thank Philippe Fontanges and Romain Morichon for image production at the Imaging Platform of Centre de Recherches Saint-Antoine (INSERM-UPMC, Paris, France).
REFERENCES
- 1.Lundgren O, Svensson L. 2001. Pathogenesis of rotavirus diarrhea. Microbes Infect 3:1145–1156. doi: 10.1016/S1286-4579(01)01475-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parashar UD, Gibson CJ, Bresee JS, Glass RI. 2006. Rotavirus and severe childhood diarrhea. Emerg Infect Dis 12:304–306. doi: 10.3201/eid1202.050006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Prasad BV, Wang GJ, Clerx JP, Chiu W. 1988. Three-dimensional structure of rotavirus. J Mol Biol 199:269–275. doi: 10.1016/0022-2836(88)90313-0. [DOI] [PubMed] [Google Scholar]
- 4.Pesavento JB, Crawford SE, Estes MK, Prasad BVV. 2006. Rotavirus proteins: structure and assembly. Curr Top Microbiol Immunol 309:189–219. [DOI] [PubMed] [Google Scholar]
- 5.Settembre EC, Chen JZ, Dormitzer PR, Grigorieff N, Harrison SC. 2011. Atomic model of an infectious rotavirus particle. EMBO J 30:408–416. doi: 10.1038/emboj.2010.322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jourdan N, Maurice M, Delautier D, Quero AM, Servin AL, Trugnan G. 1997. Rotavirus is released from the apical surface of cultured human intestinal cells through nonconventional vesicular transport that bypasses the Golgi apparatus. J Virol 71:8268–8278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez S, Arias CF. 2004. Multistep entry of rotavirus into cells: a Versaillesque dance. Trends Microbiol 12:271–278. doi: 10.1016/j.tim.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Estes MK, Greenberg HB. 2013. Rotaviruses, p 1347–1401. In Knipe DM, Howley PM, Cohen JI, Griffin DE, Lamb RA, Martin MA, Racaniello VR, Roizman B (ed), Fields virology, 6th ed, Wolters Kluwer Health/Lippincott Williams & Wilkins, Philadelphia, PA. [Google Scholar]
- 9.Yu X, Coulson BS, Fleming FE, Dyason JC, Itzstein von M, Blanchard H. 2011. Novel structural insights into rotavirus recognition of ganglioside glycan receptors. J Mol Biol 413:929–939. doi: 10.1016/j.jmb.2011.09.005. [DOI] [PubMed] [Google Scholar]
- 10.Graham KL, Fleming FE, Halasz P, Hewish MJ, Nagesha HS, Holmes IH, Takada Y, Coulson BS. 2005. Rotaviruses interact with alpha4beta7 and alpha4beta1 integrins by binding the same integrin domains as natural ligands. J Gen Virol 86:3397–3408. doi: 10.1099/vir.0.81102-0. [DOI] [PubMed] [Google Scholar]
- 11.Gilbert JM, Greenberg HB. 1998. Cleavage of rhesus rotavirus VP4 after arginine 247 is essential for rotavirus-like particle-induced fusion from without. J Virol 72:5323–5327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yoder JD, Dormitzer PR. 2006. Alternative intermolecular contacts underlie the rotavirus VP5* two- to three-fold rearrangement. EMBO J 25:1559–1568. doi: 10.1038/sj.emboj.7601034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohan KVK, Som I, Atreya CD. 2002. Identification of a type 1 peroxisomal targeting signal in a viral protein and demonstration of its targeting to the organelle. J Virol 76:2543–2547. doi: 10.1128/jvi.76.5.2543-2547.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gardet A, Breton M, Fontanges P, Trugnan G, Chwetzoff S. 2006. Rotavirus spike protein VP4 binds to and remodels actin bundles of the epithelial brush border into actin bodies. J Virol 80:3947–3956. doi: 10.1128/JVI.80.8.3947-3956.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gardet A, Breton M, Trugnan G, Chwetzoff S. 2007. Role for actin in the polarized release of rotavirus. J Virol 81:4892–4894. doi: 10.1128/JVI.02698-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Muallem S, Kwiatkowska K, Xu X, Yin HL. 1995. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. J Cell Physiol 128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trejo-Cerro Ó, Eichwald C, Schraner EM, Silva-Ayala D, Lopez S, Arias CF. 2018. Actin-dependent nonlytic rotavirus exit and infectious virus morphogenetic pathway in nonpolarized cells. J Virol 92:e02076-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang I-H, Burckhardt CJ, Yakimovich A, Greber UF. 2018. Imaging, tracking and computational analyses of virus entry and egress with the cytoskeleton. Viruses 10:166. doi: 10.3390/v10040166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Radtke K, Dohner K, Sodeik B. 2006. Viral interactions with the cytoskeleton: a hitchhiker's guide to the cell. Cell Microbiol 8:387–400. doi: 10.1111/j.1462-5822.2005.00679.x. [DOI] [PubMed] [Google Scholar]
- 20.Cudmore S, Reckmann I, Way M. 1997. Viral manipulations of the actin cytoskeleton. Trends Microbiol 5:142–148. doi: 10.1016/S0966-842X(97)01011-1. [DOI] [PubMed] [Google Scholar]
- 21.Nobile C, Rudnicka D, Hasan M, Aulner N, Porrot F, Machu C, Renaud O, Prevost MC, Hivroz C, Schwartz O, Sol-Foulon N. 2010. HIV-1 Nef inhibits ruffles, induces Filopodia, and modulates migration of infected lymphocytes. J Virol 84:2282–2293. doi: 10.1128/JVI.02230-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Charlton CA, Volkman LE. 1991. Sequential rearrangement and nuclear polymerization of actin in baculovirus-infected Spodoptera frugiperda cells. J Virol 65:1219–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bucher D, Popple S, Baer M, Mikhail A, Gong YF, Whitaker C, Paoletti E, Judd A. 1989. M protein (M1) of influenza virus: antigenic analysis and intracellular localization with monoclonal antibodies. J Virol 63:3622–3633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Naito S, Matsumoto S. 1978. Identification of cellular actin within the rabies virus. Virology 91:151–163. doi: 10.1016/0042-6822(78)90363-X. [DOI] [PubMed] [Google Scholar]
- 25.Ott DE, Coren LV, Johnson DG, Kane BP, Sowder RCII, Kim YD, Fisher RJ, Zhou XZ, Lu KP, Henderson LE. 2000. Actin-binding cellular proteins inside human immunodeficiency virus type 1. Virology 266:42–51. doi: 10.1006/viro.1999.0075. [DOI] [PubMed] [Google Scholar]
- 26.De BP, Burdsall AL, Banerjee AK. 1993. Role of cellular actin in human parainfluenza virus type 3 genome transcription. J Biol Chem 268:5703–5710. [PubMed] [Google Scholar]
- 27.Jackson P, Bellett AJ. 1985. Reduced microfilament organization in adenovirus type 5-infected rat embryo cells: a function of early region 1a. J Virol 55:644–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jones NL, Lewis JC, Kilpatrick BA. 1986. Cytoskeletal disruption during human cytomegalovirus infection of human lung fibroblasts. Eur J Cell Physiol 41:304–312. [PubMed] [Google Scholar]
- 29.Simon KO, Whitaker-Dowling PA, Youngner JS, Widnell CC. 1990. Sequential disassembly of the cytoskeleton in BHK21 cells infected with vesicular stomatitis virus. Virology 177:289–297. doi: 10.1016/0042-6822(90)90482-7. [DOI] [PubMed] [Google Scholar]
- 30.Bowden DS, Pedersen JS, Toh BH, Westaway EG. 1987. Distribution by immunofluorescence of viral products and actin-containing cytoskeletal filaments in Rubella virus-infected cells. Arch Virol 92:211–219. doi: 10.1007/BF01317478. [DOI] [PubMed] [Google Scholar]
- 31.Burns JW, Greenberg HB, Shaw RD, Estes MK. 1988. Functional and topographical analyses of epitopes on the hemagglutinin (VP4) of the simian rotavirus SA11. J Virol 62:2164–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Monnier N, Higo-Moriguchi K, Sun ZYJ, Prasad BVV, Taniguchi K, Dormitzer PR. 2006. High-resolution molecular and antigen structure of the VP8* core of a sialic acid-independent human rotavirus strain. J Virol 80:1513–1523. doi: 10.1128/JVI.80.3.1513-1523.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen DY, Estes MK, Ramig RF. 1992. Specific interactions between rotavirus outer capsid proteins VP4 and VP7 determine expression of a cross-reactive, neutralizing VP4-specific epitope. J Virol 66:432–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Szostecki C, Guldner HH, Netter HJ, Will H. 1990. Isolation and characterization of cDNA encoding a human nuclear antigen predominantly recognized by autoantibodies from patients with primary biliary cirrhosis. J Immunol 145:4338–4347. [PubMed] [Google Scholar]
- 35.Munter S, Way M, Frischknecht F. 2006. Signaling during pathogen infection. Sci STKE 2006:re5. doi: 10.1126/stke.3352006re5. [DOI] [PubMed] [Google Scholar]
- 36.Trojnar E, Otto P, Johne R. 2009. The first complete genome sequence of a chicken group A rotavirus indicates independent evolution of mammalian and avian strains. Virology 386:325–333. doi: 10.1016/j.virol.2009.01.034. [DOI] [PubMed] [Google Scholar]
- 37.Trask SD, Dormitzer PR. 2006. Assembly of highly infectious rotavirus particles recoated with recombinant outer capsid proteins. J Virol 80:11293–11304. doi: 10.1128/JVI.01346-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dormitzer PR, Nason EB, Prasad BV, Harrison SC. 2004. Structural rearrangements in the membrane penetration protein of a non-enveloped virus. Nature 430:1053–1058. doi: 10.1038/nature02836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hu L, Crawford SE, Czako R, Cortes-Penfield NW, Smith DF, Le Pendu J, Estes MK, Prasad BVV. 2012. Cell attachment protein VP8* of a human rotavirus specifically interacts with A-type histo-blood group antigen. Nature 485:256–259. doi: 10.1038/nature10996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham KL, Halasz P, Tan Y, Hewish MJ, Takada Y, Mackow ER, Robinson MK, Coulson BS. 2003. Integrin-using rotaviruses bind alpha2beta1 integrin alpha2 I domain via VP4 DGE sequence and recognize alphaXbeta2 and alphaVbeta3 by using VP7 during cell entry. J Virol 77:9969–9978. doi: 10.1128/JVI.77.18.9969-9978.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Enouf V, Chwetzoff S, Trugnan G, Cohen J. 2003. Interactions of rotavirus VP4 spike protein with the endosomal protein Rab5 and the prenylated Rab acceptor PRA1. J Virol 77:7041–7047. doi: 10.1128/JVI.77.12.7041-7047.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Martínez-Moreno M, Navarro-Lérida I, Roncal F, Albar JP, Alonso C, Gavilanes F, Rodríguez-Crespo I. 2003. Recognition of novel viral sequences that associate with the dynein light chain LC8 identified through a pepscan technique. FEBS Lett 544:262–267. doi: 10.1016/S0014-5793(03)00516-7. [DOI] [PubMed] [Google Scholar]
- 43.Davidson GP, Gall DG, Petric M, Butler DG, Hamilton JR. 1977. Human rotavirus enteritis induced in conventional piglets. Intestinal structure and transport. J Clin Investig 60:1402–1409. doi: 10.1172/JCI108901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Braccia A, Villani M, Immerdal L, Niels-Christiansen L-L, Nystrøm BT, Hansen GH, Danielsen EM. 2003. Microvillar membrane microdomains exist at physiological temperature. Role of galectin-4 as lipid raft stabilizer revealed by “superrafts.” J Biol Chem 278:15679–15684. doi: 10.1074/jbc.M211228200. [DOI] [PubMed] [Google Scholar]
- 45.Tyska MJ, Mooseker MS. 2004. A role for myosin-1A in the localization of a brush border disaccharidase. J Cell Physiol 165:395–405. doi: 10.1083/jcb.200310031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berkova Z, Crawford SE, Blutt SE, Morris AP, Estes MK. 2007. Expression of rotavirus NSP4 alters the actin network organization through the actin remodeling protein cofilin. J Virol 81:3545–3553. doi: 10.1128/JVI.01080-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Berkova Z, Crawford SE, Trugnan G, Yoshimori T, Morris AP, Estes MK. 2006. Rotavirus NSP4 induces a novel vesicular compartment regulated by calcium and associated with viroplasms. J Virol 80:6061–6071. doi: 10.1128/JVI.02167-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karplus K. 2009. SAM-T08, HMM-based protein structure prediction. Nucleic Acids Res 37:W492–W497. doi: 10.1093/nar/gkp403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim DE, Chivian D, Baker D. 2004. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res 32:W526–W531. doi: 10.1093/nar/gkh468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang J, Zhang Y. 2015. I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43:W174–W181. doi: 10.1093/nar/gkv342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Söding J, Biegert A, Lupas AN. 2005. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res 33:W244–W248. doi: 10.1093/nar/gki408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJE. 2015. The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc 10:845–858. Published online 2015 May 7. doi: 10.1038/nprot.2015.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wallner B, Larsson P, Elofsson A. 2007. Pcons.net: protein structure prediction meta server. Nucleic Acids Res 35:W369–W374. doi: 10.1093/nar/gkm319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.De Brevern AG, Benros C, Gautier R, Valadié H, Hazout S, Etchebest C. 2004. Local backbone structure prediction of proteins. In Silico Biol 4:381–386. [PMC free article] [PubMed] [Google Scholar]
- 55.Benkert P, Künzli M, Schwede T. 2009. QMEAN server for protein model quality estimation. Nucleic Acids Res 37:W510–W514. doi: 10.1093/nar/gkp322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maghrabi AHA, McGuffin LJ. 2017. ModFOLD6: an accurate web server for the global and local quality estimation of 3D protein models. Nucleic Acids Res 45:W416–W421. doi: 10.1093/nar/gkx332. [DOI] [PMC free article] [PubMed] [Google Scholar]