

Viruses of the Flaviviridae family cause major illness and death around the world and thus pose a great threat to human health. Here we show that IFN-inducible MxB restricts several members of the Flaviviridae, including HCV, Japanese encephalitis virus, and dengue virus. This finding not only suggests an active role of MxB in combating these major pathogenic human viruses but also significantly expands the antiviral spectrum of MxB. Our study further strengthens the link between virus dependence on CypA and susceptibility to MxB restriction and also suggests that MxB may employ a common mechanism to inhibit different viruses. Elucidating the antiviral functions of MxB advances our understanding of IFN-mediated host antiviral defense and may open new avenues to the development of novel antiviral therapeutics.
KEYWORDS: CypA, Flaviviridae, MxB, NS5A, hepatitis C virus
ABSTRACT
Type I interferon (IFN) inhibits viruses by inducing the expression of antiviral proteins. The IFN-induced myxovirus resistance B (MxB) protein has been reported to inhibit a limited number of viruses, including HIV-1 and herpesviruses, but its antiviral coverage remains to be explored further. Here we show that MxB interferes with RNA replication of hepatitis C virus (HCV) and significantly inhibits viral replication in a cyclophilin A (CypA)-dependent manner. Our data further show that MxB interacts with the HCV protein NS5A, thereby impairing NS5A interaction with CypA and NS5A localization to the endoplasmic reticulum, two events essential for HCV RNA replication. Interestingly, we found that MxB significantly inhibits two additional CypA-dependent viruses of the Flaviviridae family, namely, Japanese encephalitis virus and dengue virus, suggesting a potential link between virus dependence on CypA and virus susceptibility to MxB inhibition. Collectively, these data have identified MxB as a key factor behind IFN-mediated suppression of HCV infection, and they suggest that other CypA-dependent viruses may also be subjected to MxB restriction.
IMPORTANCE Viruses of the Flaviviridae family cause major illness and death around the world and thus pose a great threat to human health. Here we show that IFN-inducible MxB restricts several members of the Flaviviridae, including HCV, Japanese encephalitis virus, and dengue virus. This finding not only suggests an active role of MxB in combating these major pathogenic human viruses but also significantly expands the antiviral spectrum of MxB. Our study further strengthens the link between virus dependence on CypA and susceptibility to MxB restriction and also suggests that MxB may employ a common mechanism to inhibit different viruses. Elucidating the antiviral functions of MxB advances our understanding of IFN-mediated host antiviral defense and may open new avenues to the development of novel antiviral therapeutics.
INTRODUCTION
Myxovirus resistance (Mx) proteins, a small family of interferon (IFN)-induced proteins, are known for their protection of mice from infection by influenza A virus (1, 2). Mx genes are evolutionarily conserved among vertebrates ranging from fish to primates (3). The mouse genome contains two Mx genes, named Mx1 and Mx2, both of which exert broad antiviral activity against DNA and RNA viruses. The majority of inbred mice carry an inactive Mx1 gene, rendering them more susceptible to influenza virus infection (4, 5). Humans also have two Mx genes: MxA and MxB. It is interesting that, during evolution, rodents lost the ortholog of the human MxB gene and carry two MxA orthologs (Mx1 and Mx2) (6). Transgenic mice bearing the human MxA gene are resistant to influenza virus infection (7, 8). In contrast to MxA, the human MxB protein was long considered nonantiviral (9, 10), until human immunodeficiency virus type 1 (HIV-1) was reported to be inhibited by MxB in 2013 (11–13).
Mx proteins are dynamin-like large GTPases (14). Their expression is stimulated by type I interferon and, to a lesser extent, type III interferon (15, 16). The crystal structures of human MxA and MxB proteins show that their GTPase domains fold into independent globular structures which are connected via the bundle signaling element (BSE) hinge to the helical stalk domain (14). The stalk domain mediates the dimerization and oligomerization of the MxA and MxB proteins, which are required for the antiviral function of both proteins (17, 18). Current data indicate that MxA is more dependent on its GTPase activity than MxB for inhibition of viruses (19, 20). Further, MxA uses its loop 4 to recognize the nucleoprotein (NP) of influenza A virus (21, 22), whereas MxB targets the HIV-1 capsid core structure with its N-terminal sequence, which is absent in MxA (13, 23).
Our understanding of the antiviral function of MxB and appreciation of its importance in host antiviral defense will greatly benefit from defining the antiviral breadth of MxB and further characterizing the underlying antiviral mechanisms. In our quest for new target viruses of MxB, we found that hepatitis C virus (HCV) is significantly inhibited by MxB. We further observed that MxB inhibition of HCV is correlated with HCV dependence on cyclophilin A (CypA), a peptidyl prolyl isomerase that binds to the HCV protein NS5A and promotes HCV replication (24). Interestingly, our results show that two other Cyp-dependent viruses, dengue virus (DENV) (CypA dependent) and Japanese encephalitis virus (JEV) (CypB dependent), are also inhibited by MxB, which suggests that MxB may have a relatively broad antiviral spectrum given that many viruses depend on Cyp for efficient replication (25).
RESULTS
MxB inhibits HCV infection.
With the aim of determining whether MxB inhibits viruses other than lentiviruses, we tested the effect of MxB on HCV infection. We first generated a Huh7.5.1 cell line that was stably transduced with a tetracycline-inducible retroviral vector carrying the MxB cDNA. We produced the Jc1-Luc HCVcc virus, which expresses Gaussia luciferase (Gluc) as a reporter, and used this virus to infect the MxB Huh7.5.1 cells in the presence of doxycycline to induce MxB expression (Fig. 1A). The results of Western blotting showed that MxB expression reduced the level of HCV core protein in the infected cells approximately 3-fold (Fig. 1A), which was corroborated by a 2-fold decrease in HCV RNA (Fig. 1B). The levels of infectious HCV virions in the culture supernatants were determined by infecting regular Huh7.5.1 cells and then measuring the Gluc activity. The results showed that MxB diminished the production of infectious HCV 2-fold (Fig. 1C). We further examined the inhibitory effect of MxB on HCV infection by immunostaining HCV core protein in the infected cells. The results of confocal microscopy showed a profound suppression of HCV core expression when MxB was induced by doxycycline (Fig. 1D). Approximately 70% of the cells expressed MxB with doxycycline induction, and HCV infection did not change this MxB-positive percentage (data not shown). Pearson and Manders coefficient analysis revealed a very small amount of overlap between the red fluorescent signals for MxB and the green fluorescent signals for HCV core protein, with coefficient values of 0.27 and 0.56, respectively (Fig. 1E), which indicates a very potent inhibition of HCV core expression by MxB and suggests a possible underestimation of the anti-HCV potency of MxB shown by the results in Fig. 1A to C, which also incorporate the values from the MxB-negative cells.
FIG 1.

MxB inhibits HCV infection. (A) Jc1 HCVcc was used to infect Huh7.5.1 cells, which were treated with doxycycline (Dox) to express MxB. Levels of MxB protein and HCV core proteins were determined by Western blotting at 72 hpi. The band densities for core and actin were quantified by use of NIH ImageJ software. (B) Levels of HCV RNA in infected cells were quantified by qRT-PCR. The GAPDH mRNA level was also determined, and the data were used as an internal control to normalize the level of viral RNA. (C) HCV infection was determined by measuring Gluc activity in the supernatant. (D) Immunofluorescence staining of HCV core protein (green) and MxB (red). Nuclei were visualized by DAPI staining (blue). (E) Colocalization coefficients (Pearson and Manders coefficient values) of the HCV core protein (green) and MxB (red) shown in panel D were determined for randomly selected cells (>30) by use of Image-Pro Plus 7.0C software. Data are representative of at least three independent experiments, and values are expressed as means ± SD. For panels A to C, data are normalized to the control group, with the control value arbitrarily set to 1.
MxB expression was highly induced by IFN-α-2b in HEK293T, Huh7, and Huh7.5.1 cells (Fig. 2A). To determine the role of endogenous MxB in IFN-mediated inhibition of HCV, we depleted MxB in IFN-α-2b-treated Huh7 cells by use of small interfering RNA (siRNA) and then infected these cells with Jc1-Luc HCVcc. The results of Western blotting showed that all three siRNAs significantly reduced the level of endogenous MxB compared to that with the control siRNA (Fig. 2B). IFN-α-2b (50 IU/ml) diminished the production of infectious HCV by 50%, which was reversed to the level of control infection without IFN-α-2b when MxB was knocked down (Fig. 2B). These data demonstrate the important role of MxB in IFN-mediated inhibition of HCV infection.
FIG 2.

MxB plays an important role in IFN-mediated inhibition of HCV infection. (A) Expression levels of MxB in different cell lines. HEK293T, Huh7, and Huh7.5.1 cells were treated with IFN-α-2b (500 IU/ml) for 48 h. Levels of MxB and SamHD1 were determined by Western blotting. (B and D) Jc1 HCVcc was used to infect Huh7 cells that were transfected with either siRNA targeting MxB or control scrambled siRNA (siNT), followed by treatment with 50 IU/ml IFN-α-2b (B) or 1,000 IU/ml IFN-α-2b (D). Levels of MxB were determined by Western blotting. HCV infection was assessed by measuring Gluc activity at 72 hpi. (C) Huh7 cells were treated with IFN-α-2b (0, 50, and 500 IU/ml) for 48 h. The mRNA copies of MxB, MxA, Viperin, and ISG56 were quantified by qRT-PCR. (E) Huh7 cells were transfected with either siRNA targeting MxB or control scrambled siRNA (siNT) and treated with 50 IU/ml IFN-α-2b for 48 h, and mRNA levels of MxB, MxA, Viperin, and ISG56 were detected by qRT-PCR. (F) Huh7 cells were transfected with a plasmid expressing MxB-Flag for 48 h, and mRNA levels of MxB, MxA, Viperin, and ISG56 were detected by qRT-PCR. Data are representative of at least three independent experiments, and values are expressed as means ± SD. For panels B and D to F, data are normalized to the control group, with the control value arbitrarily set to 1.
It is notable that multiple interferon-stimulated genes (ISGs), including MxA, Viperin, and ISG56, have been reported to restrict HCV replication (50). To understand why knockdown of endogenous MxB alone almost completely eliminated the inhibitory effect of IFN on HCV replication, we determined the expression levels of several ISGs in Huh7.5.1 cells that were treated with different doses of IFN. We found that at 50 IU/ml IFN-α-2b, the mRNA level of MxB was much higher than that of MxA, Viperin, or ISG56 (Fig. 2C), which suggests that MxB may play a dominant role in inhibiting HCV replication. However, when cells were treated with a higher dose of IFN (500 IU/ml), the expression level of MxA surpassed that of MxB 6-fold (Fig. 2C). Accordingly, with treatment at 1,000 IU/ml IFN-α-2b, which diminished the HCV RNA level almost 100-fold, knockdown of MxB still increased HCV RNA expression 3-fold but was unable to reverse the inhibitory effect of IFN-α-2b, likely because the high levels of MxA and other restrictive ISGs still profoundly suppressed HCV infection (Fig. 2D).
In addition, we also assessed the effect of MxB on the expression levels of other ISGs, and we found no significant change in mRNA levels of MxA, Viperin, and ISG56 in either MxB knockdown cells treated with IFN (Fig. 2E) or MxB-overexpressing cells (Fig. 2F) compared to those in the control group. This result demonstrates that MxB does not regulate the transcription of the ISGs tested, suggesting that MxB plays no role in regulating their anti-HCV activity by inducing their expression.
The first 25 amino acids of MxB have been shown to be essential for inhibiting HIV-1 (13, 23). In a similar fashion, deletion of this 25-amino-acid fragment ablated the anti-HCV activity of MxB (Fig. 3). However, the anti-HCV function of MxB was restored when the first 25 amino acids, which contain a nuclear localization signal (NLS) motif, were replaced with an NLS (PKKKRKV) sequence derived from the simian virus 40 (SV40) large T antigen (Fig. 3). These results further confirm the role of the N-terminal sequence of MxB in its antiviral function against HIV-1 and HCV and suggest that MxB may employ similar mechanisms to inhibit these two viruses.
FIG 3.

The NLS of MxB is required for its anti-HCV activity. (A) Schematic representation of wild-type (WT) and mutant MxB proteins. The position of each domain is illustrated by amino acid numbers. (B) The anti-HCV activities of MxB mutants were examined by transfecting Huh7.5.1 cells with plasmids expressing Flag-tagged wild-type and mutated MxB, followed by infection with Jc1 HCVcc. Data are representative of at least three independent experiments, and values are expressed as means ± SD. Data are normalized to the control group, with the control value arbitrarily set to 1.
MxB suppresses HCV RNA replication.
Next, we investigated which step of HCV infection is inhibited by MxB. To ensure the physiological relevance of MxB overexpression in our work, we first compared the levels of MxB expression in Huh7.5.1 cells either transfected with an MxB-expressing vector or treated with IFN. For better quantification of Western blots, we used an Odyssey infrared imaging system (Li-Cor) to obtain digital readings of Western blots. The result showed that transfection of 500 ng of pMxB-Flag DNA, which was used in our experiments, generated a level of MxB similar to that induced with 500 IU/ml IFN-α-2b, demonstrating the physiological relevance of MxB overexpression in our study (Fig. 4A). Huh7.5.1 cells transiently expressing Flag-tagged MxB were infected with the JFH1 virus strain. We then quantified viral protein and viral RNA in the infected cells and determined virus production and viral infectivity in an equal volume of supernatant at 72 h postinfection (hpi). The results showed that the overexpressed MxB caused 2-fold reductions in viral protein (Fig. 4B), viral RNA (Fig. 4C), virus production (Fig. 4D), and the infectivity of progeny viruses (Fig. 4E). This suggests that MxB mainly affects the early stage of HCV replication. We did not observe any effect of MxB on HCV attachment to target cells (Fig. 4F) or on HCV entry (Fig. 4G). We further found that overexpression of MxB led to 50% reductions of the HCV protein NS3 (Fig. 4H) and of viral RNA (Fig. 4I) in a Huh7 cell line containing the JFH1-derived subgenomic replicon (JFH1; HCV subtype 2a), which suggests that MxB inhibits viral gene expression at the transcription and/or translation level. To further determine which step of HCV infection is inhibited by MxB, we measured the effect of MxB on viral protein expression from mutated HCV JFH1/GND, which contains a mutation in the HCV RNA polymerase NS5B and is unable to replicate viral RNA. Unlike that of wild-type HCV, the viral protein expression of the HCV mutant was insensitive to MxB, suggesting that MxB does not affect the translation of viral mRNA (Fig. 4J). Therefore, we conclude that MxB mainly inhibits viral RNA replication.
FIG 4.

MxB inhibits RNA replication of HCV. (A) Huh7.5.1 cells were either treated with IFN-α-2b (0, 50, or 500 IU/ml) or transfected with MxB-Flag DNA (0, 500, or 1,000 ng) for 48 h, and MxB levels were determined by Western blotting and quantified by use of a Li-Cor Odyssey system. (B) Huh7.5.1 cells were transfected with a plasmid expressing MxB-Flag or with empty vector DNA (EV) as the control and then infected with JFH1 HCVcc (MOI = 0.5). Levels of MxB and HCV core were determined by Western blotting. (C) Levels of HCV RNA in infected cells were quantified by qRT-PCR. The level of GAPDH mRNA was used as the internal control to normalize the level of viral RNA. (D) HCV production was determined by measuring the HCV core level in the supernatant by enzyme-linked immunosorbent assay (ELISA). (E) Levels of infectious progeny viruses in supernatants were determined by infecting Huh7.5.1 cells, followed by qRT-PCR quantification of viral RNA in the newly infected cells at 72 hpi. (F) Effect of MxB on HCV binding to target cells. Huh7.5.1 cells were incubated with JFH1 HCVcc (MOI = 0.5) at 4°C for 1 h. Anti-CD81 antibody was used to prevent HCV binding to its receptor, CD81. The attached HCV particles on the cell surface were quantified by determining the amount of viral genomic RNA by qRT-PCR. (G) Effect of MxB on HCV entry. After incubating Huh7.5.1 cells with JFH1 HCVcc, HCV that had entered Huh7.5.1 cells was quantified by determining the level of viral genomic RNA by qRT-PCR. IFN-α-2b was used as a control treatment of cells for 24 h before HCV infection. (H) Huh7 cells harboring the JFH1-derived subgenomic replicon were transfected with MxB-Flag plasmid DNA, followed by Western blotting to measure levels of MxB and HCV NS3. (I) Levels of HCV RNA were determined by qRT-PCR. (J) Effect of MxB on translation of HCV RNA. Huh7.5.1 cells were cotransfected with MxB-Flag plasmid DNA together with in vitro-synthesized JFH1 mRNA or JFH1(GND) mRNA. Levels of MxB and HCV core proteins were determined by Western blotting. Data are representative of at least three independent experiments, and values are expressed as means ± SD. For panels B to J, data are normalized to the control group, with the control value arbitrarily set to 1.
Knockdown of CypA abrogates MxB inhibition of HCV.
We previously found that depletion of CypA impairs MxB inhibition of HIV-1 (13). We therefore asked whether CypA also affects the anti-HCV activity of MxB. To answer this question, we knocked down CypA by using short hairpin RNA (shRNA), and we observed that HCV production decreased by 50% and that MxB did not further diminish HCV production (Fig. 5A). In support of this observation, the CypA inhibitor cyclosporine (CsA) neutralized the anti-HCV effect of MxB (Fig. 5B). In contrast, MxB was still able to reduce HCV infection by 50% in the presence of the NS5B (viral RNA-dependent RNA polymerase) inhibitor sofsbuvir (Fig. 5B). In this work, a dose close to the 50% inhibitory concentration (IC50) was chosen for both sofsbuvir and CsA. Moreover, a CypA-independent HCV mutant, DEYN (26), which harbors the D316E and Y317N mutations in the NS5A protein and is refractory to CsA (Fig. 5C), also resisted MxB inhibition (Fig. 5D). Together these data suggest that MxB targets a CypA-dependent step during HCV replication.
FIG 5.

Inhibition of HCV by MxB is CypA dependent. (A) Effect of CypA knockdown on MxB inhibition of HCV in Huh7.5.1 cells. Endogenous CypA in Huh7.5.1 cells was knocked down by use of shRNA, followed by transfection with MxB-Flag plasmid DNA and infection with Jc1 HCVcc for 72 h. Levels of MxB and CypA were determined by Western blotting. Levels of HCV replication were determined by measuring Gluc activity. (B) Effects of cyclosporine (CsA) and sofsbuvir (SOF) on MxB inhibition of HCV infection. Huh7.5.1 cells were transiently transfected with MxB-Flag DNA and then infected with Jc1 HCVcc in the presence of CsA or SOF. Expression of MxB was examined by Western blotting. HCV infection was monitored by measuring Gluc activity. (C) Effect of CsA on infection by the JFH1 mutant DEYN. Huh7.5.1 cells were infected with JFH1 or the DEYN mutant at an MOI of 0.5, followed by treatment with CsA at the indicated concentrations for 72 h. HCV replication was determined by quantification of viral genomic RNA. (D) Effect of MxB on infection by JFH1 or the JFH1 mutant DEYN. The level of HCV replication was determined by qRT-PCR quantification of viral RNA in infected cells. Data are representative of at least three independent experiments, and values are expressed as means ± SD. For all panels, data are normalized to the control group, with the control value arbitrarily set to 1 or 100%.
MxB disrupts interaction of CypA with the HCV protein NS5A.
Since CypA contributes to HCV RNA replication through interaction with the viral protein NS5A, we examined whether MxB affects the formation of CypA-NS5A complexes. We first transfected cells with plasmid DNAs expressing MxB-Myc, CypA-HA, and NS5A-Flag and then immunoprecipitated CypA-HA by using an anti-HA antibody. The results in Fig. 6A and further quantification in Fig. 6B show that NS5A-Flag was coprecipitated with CypA-HA and that wild-type MxB-Myc or the MxB(26-715+SV40-NLS)-Myc mutant significantly reduced the level of NS5A-Flag that was coprecipitated with CypA-HA. In contrast, the MxB(26-715)-Myc mutant exerted no effect in this regard, consistent with its inability to inhibit HCV. These results are in agreement with the data from immunofluorescence staining experiments (Fig. 6C), which showed that the strong colocalization of CypA-HA and NS5A-Flag was disrupted by either MxB(WT)-Myc or MxB(26-715+SV40-NLS)-Myc but not by MxB(26-715)-Myc. This is further illustrated by the results of quantification analysis, which showed an approximately 10-fold reduction in the percentage of NS5A colocalization with CypA in the presence of MxB or MxB(26-715+SV40-NLS) compared to that for the control group or in the presence of the MxB(26-715)-Myc mutant (Fig. 6D). Together these data demonstrate that MxB disrupts the interaction between NS5A and CypA, which may have caused the decrease of HCV infection.
FIG 6.

MxB disrupts interaction of CypA with HCV protein NS5A. (A) Effects of MxB and its mutants on interaction of NS5A with CypA. MxB-Myc, NS5A-Flag, and CypA-HA were coexpressed. Immunoprecipitation was performed to pull down CypA-HA by use of anti-HA antibody. The level of coprecipitated NS5A-Flag was determined by Western blotting (IB). (B) Protein band intensities in the Western blots in panel A were determined by use of NIH ImageJ software. Data are normalized to the control group, with the control value arbitrarily set to 1. (C) Colocalization of NS5A and CypA is disrupted by MxB. Huh7.5.1 cells were cotransfected with NS5A-Flag, CypA-HA, and MxB-Myc or its mutants. Cells were fixed and stained with anti-Flag (red), anti-HA (green), and anti-Myc (blue) antibodies. Representative images are shown. Bars, 5 μm. (D) Colocalization of CypA (green) and NS5A (red) was assessed for randomly selected cells (n > 30) by use of Image-Pro Plus 7.0C software. Data are representative of at least three independent experiments, and values are expressed as means ± SD.
MxB associates with HCV NS5A.
We next investigated how MxB interferes with the formation of NS5A-CypA complexes. One possibility is that MxB binds to NS5A and prevents the binding of CypA to NS5A. To test this, we performed coimmunoprecipitation (co-IP) experiments and observed that NS5A was coprecipitated with MxB as well as with the MxB(26-715+SV40-NLS) mutant, but not with MxB(26-715) (Fig. 7B). The MxB association with NS5A is specific, as we did not detect any association of MxB with other HCV proteins, including NS3 and NS4A (Fig. 7A). We further determined which sequence of NS5A associates with MxB. NS5A comprises an N-terminal AH motif and domains I, II, and III (Fig. 7C). The AH motif anchors NS5A to the endoplasmic reticulum (ER), and domains II and III are involved in the interaction with CypA (27). We constructed NS5A mutants with each of these four regions deleted individually and examined their association with MxB. The results showed that NS5A lost interaction with MxB only when domain I was deleted (Fig. 7D), suggesting that domain I is primarily responsible for the interaction of NS5A with MxB. Furthermore, removal of domain I did not affect the NS5A association with CypA (Fig. 7E), in agreement with previous reports showing that CypA interacts with domains II and III of NS5A (27). As a result, MxB did not disrupt the association of domain I-deleted NS5A with CypA (Fig. 7E). These data suggest that MxB binds to NS5A through domain I, which prevents CypA from accessing domain II of NS5A, likely by creating a steric hindrance.
FIG 7.

Domain I of NS5A is required for MxB and NS5A interaction. (A) MxB interacts with NS5A protein. MxB-Myc was coexpressed with Flag-tagged NS3, NS4A, or NS5A. Immunoprecipitation was performed to pull down Flag-tagged proteins by use of anti-Flag antibody. The level of coprecipitated MxB-Myc was determined by Western blotting. (B) MxB-Myc interacts with NS5A-Flag. HEK293T cells were transfected with NS5A-Flag and MxB-Myc wild-type or mutant DNA. After 48 h, immunoprecipitation was performed with anti-Myc antibodies to pull down MxB. The presence of NS5A-Flag in the precipitated materials was detected by use of anti-Flag antibodies for Western blotting. (C) Schematic representation of the key domains of NS5A. The position of each domain is illustrated by amino acid numbers. (D) Deletion of domain I in NS5A eliminates its interaction with MxB. NS5A-Flag and its deletion mutants were coexpressed with MxB-Myc in HEK293T cells. NS5A-Flag and its mutant proteins were precipitated with anti-Flag antibody, and the presence of MxB-Myc in the precipitated materials was detected by use of anti-Myc antibodies for Western blotting. (E and F) MxB disrupts the interaction of CypA and either NS5A(DI) (E) or NS5A(DEYN) (F). Immunoprecipitation was performed with anti-HA antibodies to pull down CypA-HA. Levels of coprecipitated NS5A-Flag and its mutants were determined by Western blotting using anti-Flag antibody. Data are representative of at least three independent experiments.
Next, we examined the interaction between the DEYN NS5A protein and CypA by co-IP, and we observed an association of the mutated NS5A protein with CypA (Fig. 7F). Interestingly, as opposed to the disruption of wild-type NS5A binding to CypA by MxB, binding of the DEYN NS5A mutant to CypA was not affected by MxB (Fig. 7F). These results suggest that MxB does not affect the interaction of DEYN NS5A and CypA, which may contribute to the resistance of the DEYN HCV strain to MxB.
MxB prevents NS5A localization to the ER.
NS5A is localized to the ER and is an essential component of the HCV replication complex (28). MxB was minimally colocalized with the ER marker protein disulfide isomerase (PDI) (Fig. 8A), which does not explain the interaction of these two proteins which we observed in the coimmunoprecipitation experiments. One possibility is that MxB might have altered the subcellular localization of NS5A. Indeed, as opposed to strong colocalization of NS5A with PDI when it was expressed alone, MxB or MxB(26-715+SV40-NLS), but not MxB(26-715), profoundly diminished this colocalization (Fig. 8B). This is further supported by the results of quantification analysis, which showed that the percentage of NS5A colocalization with PDI decreased from 23% in the control group to 3% and 2% in the presence of MxB and MxB(26-715+SV40-NLS), respectively (Fig. 8C). Therefore, in addition to disrupting NS5A association with CypA, MxB also prevents NS5A localization to the ER. Both defects together dampen HCV infection.
FIG 8.

MxB disrupts NS5A localization to the ER. (A) Minimal colocalization of MxB with the ER marker PDI. Huh7.5.1 cells were transfected with MxB-Flag plasmid DNA for 48 h, and MxB-Flag and PDI were stained with anti-Flag (red) and anti-PDI (green) antibodies. The nuclei were visualized by staining with DAPI. The last panel is a magnified image of the selected area. (B) Colocalization of PDI and NS5A is disrupted by MxB. NS5A-Flag, MxB-Myc, and MxB-Myc mutants were transiently expressed in Huh7.5.1 cells and detected by staining with anti-Flag (red) and anti-Myc (blue) antibodies. The ER marker PDI is stained green. Representative images are shown. Bars, 5 μm. (C) The ratio of colocalization between NS5A (red) and PDI (green) was determined for randomly selected cells (>30) and plotted as a histogram. Data are representative of at least three independent experiments, and values are expressed as means ± SD.
There is currently no evidence supporting the role of domain I in the association of NS5A with the ER. Nonetheless, our data suggest that MxB binding to domain I alters the localization of NS5A from the ER, although the detailed mechanism awaits further investigation. It is possible that MxB binding to domain I prevents the AH domain from interacting with the ER, likely because these two domains are adjacent to each other. In addition, a recent study showed that mutations in domain I which impair NS5A-CypA interaction alter the subcellular localization of NS5A (29), which suggests the involvement of CypA in regulating NS5A transportation. Therefore, disassociation of CypA from NS5A via MxB probably also affects localization of NS5A to the ER.
MxB inhibits dengue virus and Japanese encephalitis virus.
In addition to HCV, we further tested whether MxB inhibits other members of the Flaviviridae, including JEV, Zika virus (ZIKV), and DENV. The pcDNA4-MxB plasmid DNA was transfected into Vero cells to transiently express MxB, and the cells were challenged by JEV, ZIKV, or DENV. Levels of JEV and ZIKV replication were determined by quantifying viral RNA in the infected cells by quantitative reverse transcription-PCR (qRT-PCR). The results showed that JEV infection was reduced 2-fold (Fig. 9A), whereas ZIKV infection was not affected by MxB (Fig. 9B). Levels of infectious DENV in the culture supernatants were determined by performing viral plaque assays, and the results showed a 10-fold decrease of DENV production from MxB-expressing cells compared to that from cells that were transfected with empty vector (Fig. 9C). Therefore, in addition to HIV-1 and HCV, MxB also inhibits specific flaviviruses. It is worth noting that in agreement with previous work, both HCV and JEV were inhibited by CsA, whereas Zika virus resisted both MxB and CsA (Fig. 9D). Together with an early study showing that JEV replication is dependent on CypB (30), these data show that there is a potential link between virus dependence on CypA/B and viral susceptibility to MxB inhibition.
FIG 9.

MxB inhibits dengue virus and Japanese encephalitis virus. (A) Vero cells were transiently transfected with MxB-Flag DNA and then infected with JEV (SA14-14-2) at an MOI of 0.01. After 48 h, levels of JEV RNA in the infected cells were determined by qRT-PCR. Expression of MxB-Flag was examined by Western blotting. (B) Zika virus (PLCal_ZV) (MOI = 0.5) was used to infect Vero cells transiently expressing MxB-Flag. At 48 hpi, the level of Zika virus RNA in the infected cells was determined by qRT-PCR. (C) Vero cells were transiently transfected with MxB-Flag DNA and then infected with DENV2 (Tr1751) (MOI = 0.5). After 48 h, the supernatants were collected for plaque assay to measure the titers of DENV2. (D) Effects of CsA on replication of HCV, JEV, and ZIKV. HCV (MOI = 0.5), JEV (MOI = 0.01), and ZIKV (MOI = 0.5) were used to infect Huh7.5.1 cells in the presence of different doses of CsA (0.25 μM, 0.5 μM, 1 μM, and 2 μM). After 48 h, levels of viral RNA was determined by qRT-PCR. (E and F) Effects of MxB on expression of NS5. Either JEV NS5-Flag (E) or DENV NS5-Flag (F) was coexpressed with various amounts of MxB-Myc for 24 h, followed by treatment with MG132. The expression of NS5 and MxB was determined by Western blotting. DMSO, dimethyl sulfoxide. (G) MxB interacts with NS5A(DEYN), JEV NS5, and DENV NS5. Immunoprecipitation was performed with anti-Flag antibody to pull down Flag-tagged NS5A(DEYN), JEV NS5, and DENV NS5. Levels of coprecipitated MxB-Myc were determined by Western blotting.
Flaviviruses do not encode NS5A. However, a previous study reported the interaction of West Nile virus (WNV) NS5 with CypA in an in vitro glutathione S-transferase (GST) pulldown assay, suggesting that the NS5 protein may represent a target of CypA (31). We thus decided to test whether MxB interacts with the NS5 proteins of JEV and DENV. When we coexpressed MxB and the NS5 protein of JEV or DENV, we found a significant reduction of NS5 expression (Fig. 9E and F), suggesting that MxB inhibits the expression of the NS5 proteins of both JEV and DENV. Addition of the proteasome inhibitor MG132 restored NS5 expression, which demonstrates that MxB induces NS5 degradation through the proteasomal pathway (Fig. 9E and F). The results of co-IP analysis further showed that MxB was associated with the NS5 proteins of both JEV and DENV (Fig. 9G). We therefore propose that MxB inhibits JEV and DENV infection by interacting with NS5 and causing NS5 degradation via proteasomes.
DISCUSSION
Upon IFN stimulation, hundreds of ISGs are upregulated. These ISGs exert a broad range of antiviral activity by hampering different stages of the virus life cycle. The antiviral function of MxB has been much less well known than the well-characterized antiviral role of MxA. Shortly after the cloning of human MxB cDNA in 1989 (32), its antiviral activity against several viruses, including influenza A virus, mengovirus, encephalomyocarditis virus, Semliki forest virus, herpes simplex virus type 1, and vesicular stomatitis virus, was tested in murine 3T3 cells, and no antiviral effects against these viruses were observed (33). MxB was considered nonantiviral for quite a long time, until in 2011 the results of a screening study for the antiviral function of ISGs presented a weak inhibition of HIV-1 by MxB (28). In 2013, three groups convincingly showed that MxB indeed strongly inhibits HIV-1 and that HIV-1 is able to escape from MxB inhibition by changing the sequence of its viral capsid protein (11–13). Certain simian immunodeficiency virus (SIV) strains, including SIVMND (from mandrill monkeys) (12), and herpesviruses (34, 35) were subsequently shown to be inhibited by MxB. The potentially broad antiviral function of MxB is supported by an evolutionary analysis of MxB proteins in primates showing that MxB has been undergoing positive selection yet this positive selection has not been driven by lentiviruses (36). In support of this conclusion, the results of the present study show that members of the Flaviviridae, including HCV, JEV, and DENV, are inhibited by MxB. This suggests that MxB has evolved to control a range of different pathogenic viruses, thus playing an important role in host antiviral defense.
We noted that MxB inhibited HCV and JEV at a moderate level (2-fold). One possibility is that these two viruses might have evolved partial resistance to MxB after a sufficiently long period of transmission in the human population. Alternatively, different genotypes of HCV may exhibit different levels of susceptibility to MxB inhibition. In support of this speculation, it has been reported that sequence heterogeneity of different HCV genotypes accounts for different levels of sensitivity to HCV NS5A inhibitors (37). As a result, a given HCV NS5A inhibitor is often used to treat infections by certain HCV genotypes. Since domain I of NS5A serves as the binding site for its inhibitors, which MxB also likely targets, it is possible that the sequence polymorphism in domain I of NS5A also modulates the sensitivity of HCV to MxB inhibition, which warrants further investigation.
Both HIV-1 and HCV recruit CypA as a cofactor to promote viral infection (38–40). CypA binds to a loop structure in the N-terminal domain of the HIV-1 capsid protein (41), regulates the uncoating of the viral core after viral entry, and helps to shield viral RNA and DNA from recognition by innate immune sensors (39, 42). CypA promotes the function of HCV NS5A by interacting with domains II and III (24, 27). HCV infection depends on CypA to such a great extent that a CypA inhibitor, alisporivir, has been developed and tested in clinical trials to treat HCV-infected patients (43). Interestingly, depletion of CypA or treatment with CsA alleviates MxB inhibition of HIV-1 or HCV (13). Although we cannot formally rule out the possibility that CypA acts as a cofactor of MxB, the results of this study support a mechanism by which MxB inhibits CypA-dependent viruses by deterring viral access to CypA. In the case of HCV, our data show that MxB associates with the viral protein NS5A, disrupts NS5A binding to CypA, and prevents NS5A localization to the ER, as illustrated in Fig. 10. This antiviral mechanism is further supported by the resistance of the CypA-independent HCV mutant DEYN to MxB, although it is still able to interact with MxB (Fig. 9G). Similarly, mutations in the CypA-binding loop in the HIV-1 capsid protein generate resistance to MxB (11–13, 44), even though the viral core structure built with these mutated capsid proteins still associates with MxB (44, 45), suggesting that MxB loses its inhibition when the virus becomes independent of CypA. It will be interesting to test whether such a mechanism operates in MxB inhibition of other viruses that also depend on CypA.
FIG 10.
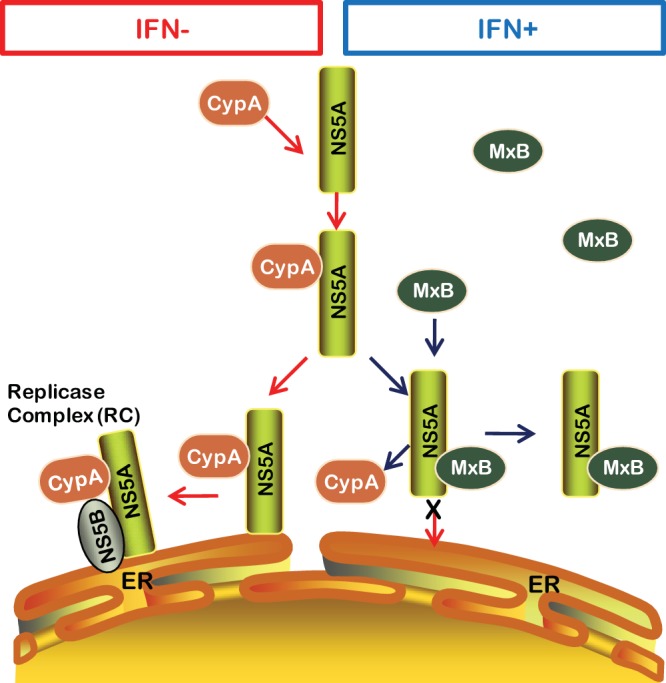
Schematic presentation of MxB inhibition of HCV replication through disruption of the interaction of NS5A and CypA. In the absence of IFN, MxB is not expressed. CypA binds to NS5A and promotes HCV replication at the ER. When MxB is expressed upon IFN stimulation, the interaction of CypA with NS5A is disrupted by MxB binding to NS5A. As a result, NS5A cannot localize to the ER, and HCV infection is restricted.
MxB may use a similar mechanism to recognize the HIV-1 capsid core and the HCV NS5A protein. Results of mutagenesis studies have shown that the N-terminal sequence of MxB, including the 11-RRR-14 motif, determines its specific recognition of the HIV-1 core structure (46, 47). Our study showed that deletion of the first 25 amino acids of MxB abrogates its inhibition of HCV and impairs MxB association with NS5A. Although the residues within this 25-amino-acid region that mediate the latter recognition event remain to be determined, it is clear that the N-terminal region of MxB is involved in specific targeting of both HIV-1 and HCV.
MxB appears to interact with a tertiary structure that is only available in the HIV-1 capsid core, since neither the monomer, dimer, nor hexamer of capsid protein associates with the MxB protein (18). However, this putative tertiary structure has not yet been identified. In any event, MxB is readily coprecipitated with NS5A, and this association depends on domain I of NS5A (Fig. 7). Further, MxB disrupts the binding of CypA to wild-type NS5A but not to an NS5A mutant with domain I deleted (Fig. 7). One possible scenario is that after binding of domain I, MxB dimerization and oligomerization interfere with domain II of NS5A interacting with CypA. In addition, an early study showed that mutation of NS5A domain I impairs the NS5A-CypA interaction (29), which suggests a role of domain I in facilitating the formation of the NS5A-CypA complex. Therefore, it is possible that binding of MxB to domain I abrogates the latter role of domain I in the NS5A-CypA interaction. In any event, the detailed mechanisms warrant further study.
Our study suggests a potential link between virus dependence on CypA/B and viral susceptibility to MxB inhibition. For example, not only are HIV-1 and HCV sensitive to CsA inhibition (25), but JEV and DENV were also reported to be inhibited by CsA (30, 31). In contrast to these four viruses that are subjected to MxB restriction, ZIKV resists MxB as well as CsA inhibition (Fig. 6). This plausible correlation remains to be investigated further and, if proven, may help to illuminate the possible common mechanism behind the antiviral action of MxB against different viruses.
In summary, we have discovered that MxB inhibits members of the Flaviviridae, which has expanded the antiviral coverage of MxB beyond restricting lentiviruses and herpesviruses. The mechanistic study of MxB inhibition of HCV suggests that MxB may target and suppress infection by CypA-dependent viruses by impairing viral association with CypA. Collectively, our results further demonstrate the important role of MxB in IFN-mediated antiviral defense against different virus families.
MATERIALS AND METHODS
Plasmid DNA and reagents.
Construction of Retro-X-MxB and Retro-X-CypA plasmid DNAs was described previously (13). MxB and CypA cDNA sequences were inserted between the BamHI and NotI restriction sites in the pcDNA4/TO expression vector, with Flag, Myc, and hemagglutinin (HA) tag sequences attached to the C termini of both proteins. Flag-tagged NS3, NS4A, and NS5A sequences of HCV and NS5 sequences of JEV and DENV were cloned into pcDNA4/TO. The infectious viral DNA HCV clones [JFH1 and JFH1(GND)] were kindly provided by Takaji Wakita. NS5A point mutations (D316E and Y317N) were generated in the context of JFH1 viral DNA by use of a site-directed mutagenesis kit (SBS). The antibodies used for Western blotting included mouse anti-core (ab2740; Abcam), anti-NS3 (ab65407; Abcam), anti-Flag M2 (F3165; Sigma-Aldrich), anti-PDI (ab2792; Abcam), anti-HA (sc-7392; Santa Cruz), and anti-beta-actin (ab8224; Abcam) monoclonal antibodies as well as rabbit anti-Flag (B1020; Biodragon), anti-MxB (NBP1-81018; Novus), and anti-PPIA (AV51352; Sigma-Aldrich) polyclonal antibodies and goat anti-Myc antibody (ab9132; Abcam). Alexa Fluor-conjugated secondary antibodies were purchased from Thermo Fisher.
Cell culture and transfection.
Huh7 cells (Rongtuan Lin, McGill University), Huh7.5.1 cells (Rongtuan Lin, McGill University), MxB-expressing Huh7.5.1 cells (Fei Guo, CAMS), HEK293T cells (CRL-11268; ATCC), and Vero cells (CCL-81; ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) supplemented with 10% fetal bovine serum (FBS) at 37°C with 5% CO2. The MxB-expressing Huh7.5.1 cell line was generated by stable transduction with the doxycycline-inducible Retro-X-MxB retroviral vector under puromycin selection. HEK293T and Huh7.5.1 cells were transfected by use of Lipofectamine 2000 (Invitrogen) and Vigofect (Vigorous), respectively, in accordance with the manufacturers’ instructions.
In vitro RNA transcription and production of infectious HCV.
In vitro synthesis of JFH1 RNA or replication-incompetent JFH1/GND RNA was performed as described by Takaji Wakita (48), using an in vitro RNA transcription kit (Ambion). HCV RNA was then transfected into Huh7.5.1 cells by use of Lipofectamine RNAi Max (Invitrogen) according to the manufacturer’s protocol. Immunofluorescence staining for core protein was performed 72 h after transfection and showed that 60 to 80% of the cells were positive for HCV core protein, indicating a high level of transfection and HCV replication in Huh7.5.1 cells. The transfected cells were passaged to allow sufficient HCV particles to accumulate in the culture supernatants, and viruses were harvested by centrifugation and filtration through 0.45-µm filters to remove cell debris and then stored at −80°C. The 50% tissue culture infective dose (TCID50) of the HCV stock was determined by limiting dilution assay. The reporter Jc1 HCVcc is an intragenotypic recombinant infectious virus between the JFH1 and J6-CF strains which expresses Gaussia luciferase (Gluc) as a reporter (49). Huh7.5.1 cells were infected with Jc1 HCVcc at a multiplicity of infection (MOI) of 0.1, and the infected cells were passaged for 7 days to amplify infectious viral particles.
Virus infections.
Huh7.5.1 cells were seeded into 6-well plates (5 × 105 per well) 1 day prior to transfection with pMxB and pcDNA4. At 48 h posttransfection, cells were infected with JFH1 or Jc1 HCVcc at an MOI of 1. After 72 h, cells were harvested. Cell lysates were used for Western blotting to measure the level of HCV core protein. Total RNA was extracted from the infected cells, and the level of HCV RNA was determined by qRT-PCR. Amounts of infectious Jc1 HCVcc in the supernatant were measured by infecting Huh7.5.1 cells and then determining the activity of Gluc that was secreted into the culture supernatants by the infected Huh7.5.1 cells, using a Centro XS3 LB 960 luminometer. Vero cells overexpressing MxB were infected with JEV (SA14-14-2), ZIKV (PLCal_ZV), or DENV2 (Tr1751) for 48 h, viral RNA was quantified by qRT-PCR, and the viral titer was measured by plaque assay in order to determine viral infectivity.
Western blotting and immunoprecipitation.
Cells were lysed in buffer containing 25 mM Tris, pH 7.4, 150 mM NaCl, 1% NP-40, 1 mM EDTA, and 5% glycerol (Pierce) on ice for 1 h and then centrifuged at 12,000 rpm for 10 min to remove cell debris. Cellular extracts were subjected to SDS-PAGE. Proteins were transferred onto polyvinylidene difluoride (PVDF) membranes and probed with the indicated antibodies at predetermined concentrations. For immunoprecipitation, 8% of the whole-cell lysates (8% WCL) were directly examined by Western blotting, and the remaining lysates were incubated with antibodies for 4 h and then further incubated with protein A+G agarose gel beads overnight with gentle agitation at 4°C. The antibody beads were washed with lysis buffer, and the bound proteins were examined by Western blotting. The protein band intensities on the Western blots were determined using NIH ImageJ software.
Gene silencing.
To knock down MxB, Huh7.5.1 cells were transfected with control siRNA or siRNA targeting MxB (Ribobio) (siMxB-1, GCACGATTGAAGACATAAA; siMxB-2, GGGACGCCTTCACAGAATA; and siMxB-3, GGAGAATGAGACCCGTTTA) by use of Lipofectamine RNAi Max (Invitrogen). The expression of CypA was silenced by use of shCypA (TRCN0000049228; Sigma).
qRT-PCR.
Total RNA was extracted from the infected cells or supernatant by use of an RNA extraction kit (Tiandz). The level of viral RNA was determined by performing qRT-PCR analysis by use of a one-step SYBR PrimeScript RT-PCR kit. The primer pair (5′-GCGTTAGTATGAGTGTCGTG-3′ and 5′-TCGCAAGCACCCTATCAG-3′) amplifies the 5′ untranslated region (UTR) of HCV. Other primers were designed to have the following sequences: for HCV, 5′-GCGTTAGTATGAGTGTCGTG-3′ and 5′-TCGCAAGCACCCTATCAG-3′; for JEV, 5′-ACAATCATGGCAAACGACAA-3′ and 5′-CTTCTCGTTGTGGGCTTCTC-3′; for ZIKV, 5′-CCACGCACTGATAACAT-3′ and 5′-AAGTAGCAAGGCCTGCTCT-3′; for MxA, 5′-GTGCATTGCAGAAGGTCAGA-3′ and 5′-TTCAGGAGCCAGCTGTAGGT-3′; for MxB, 5′-AAGCAGTATCGAGGCAAGGA-3′ and 5′-TCGTGCTCTGAACAGTTTGG-3′; for Viperin, 5′-CTGAGAGGGCCAGATGAGAC-3′ and 5′-GAAATGGCTCTCCACCTGAA-3′; and for ISG56, 5′-ACAAGCTGGCTGGAAAGAAA-3′ and 5′-GTACACGAAGGTGCTGCTCA-3′. Levels of cellular glyceraldehyde-3-phosphate dehydrogenase (GAPDH) RNA were amplified with primers 5′-ATCATCCCTGCCTCTACTGG-3′ and 5′-GTCAGGTCCACCACTGACAC-3′; the results served as an internal control to normalize the level of HCV RNA.
Immunofluorescence staining.
Cells were fixed in 4% paraformaldehyde (in 1× phosphate-buffered saline [PBS]) and permeabilized with 0.2% Triton X-100–PBS prior to incubation with primary antibodies for 1 h at room temperature with gentle shaking. After washing with 1× PBS three times, Alexa Fluor-conjugated secondary antibodies (Alexa Fluor 488–donkey anti-mouse, Alexa Fluor 555–donkey anti-rabbit, and Alexa Fluor 640–donkey anti-goat) were added for a further incubation of 30 min. Nuclei were stained with DAPI (4′,6-diamidino-2-phenylindole). Images were recorded with a PerkinElmer Ultra View VoX confocal imaging system. Colocalization coefficients (Pearson and Manders coefficient values) were determined for randomly selected cells (>30) by use of Image-Pro Plus 7.0C software.
Treatment with CsA and sofsbuvir.
Huh7.5.1 cells were seeded into 24-well plates 1 day prior to transfection with pMxB or pcDNA4 for 48 h and then infected with Jc1-Luc HCVcc at an MOI of 0.5 in the presence of cyclosporine (CsA) (0.1 μg/ml) or sofsbuvir (0.02 μg/ml) for 4 h. The virus inoculum was washed off, and cells were cultured for an additional 72 h. Cells were harvested for Western blotting, and the Gaussia luciferase activity in the supernatants was measured.
Binding and entry tests.
For binding tests, 4 × 105 Huh7.5.1 cells were seeded into 6-well plates overnight and then transfected with pMxB and pcDNA4 by use of Vigofect transfection reagent for 48 h. We incubated CD81 antibodies (final concentration, 1.5 μg/ml) with pcDNA4-transfected cells for 1 h as a positive control before JFH1 HCVcc infection (MOI = 0.5), and then virus-infected cells were incubated at 4°C for 1 h, followed by washing with PBS, and the HCV absorption on the cell surface was quantified by qPCR. To test for virus entry, cells incubated with virus particles at 4°C for 1 h were shifted to 37°C for 4 h and then treated with 50 μg/ml proteinase K to remove remnant viral particles from the cell surface, and the HCV RNA in cells was used to evaluate the efficiency of virus entry. For the entry test, pcDNA4-transfected cells were treated with 1,000 IU/ml IFN-α-2b for 24 h as a positive control.
Statistical analysis.
All data are presented as means ± standard deviations (SD). Statistical analyses were performed with two-tailed, unpaired Student’s t test, available in GraphPad Prism software. The significance of differences is indicated in the figures (*, P < 0.05; **, P < 0.01; ***, P < 0.001; and n.s., not significant).
ACKNOWLEDGMENTS
This work was supported by The National Key Research and Development Program of China (grant 2016YFD0500307 to S.C.), The National Mega-Project for Infectious Disease (grant 2018ZX10301408 to S.C.), CIHR (grant HOP-143171 to C.L.), the Peking Union Medical College Youth Fund (grant 3332018096 to D.-R.Y.), The National Natural Science Foundation of China (grant 81772205 to S.C.), the NSFC and FRSQ International Exchange Program (grant 31711520716 to S.C. and a 2017-2018 grant to C.L.), the CAMS Innovation Fund for Medical Sciences (grant 2016-I2M-2-002 to X.-Y.L. and grant 2017-I2M-1-012 to Y.-X.Z.), and a Xiehe scholarship (to S.C.).
We thank the National Infrastructure of Microbial Resources (NIMR-2014-3) for providing valuable reagents.
REFERENCES
- 1.Horisberger MA, Staeheli P, Haller O. 1983. Interferon induces a unique protein in mouse cells bearing a gene for resistance to influenza virus. Proc Natl Acad Sci U S A 80:1910–1914. doi: 10.1073/pnas.80.7.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Staeheli P, Haller O, Boll W, Lindenmann J, Weissmann C. 1986. Mx protein: constitutive expression in 3T3 cells transformed with cloned Mx cDNA confers selective resistance to influenza virus. Cell 44:147–158. doi: 10.1016/0092-8674(86)90493-9. [DOI] [PubMed] [Google Scholar]
- 3.Haller O, Staeheli P, Kochs G. 2007. Interferon-induced Mx proteins in antiviral host defense. Biochimie 89:812–818. doi: 10.1016/j.biochi.2007.04.015. [DOI] [PubMed] [Google Scholar]
- 4.Staeheli P, Grob R, Meier E, Sutcliffe JG, Haller O. 1988. Influenza virus-susceptible mice carry Mx genes with a large deletion or a nonsense mutation. Mol Cell Biol 8:4518–4523. doi: 10.1128/MCB.8.10.4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Haller O, Acklin M, Staeheli P. 1987. Influenza virus resistance of wild mice: wild-type and mutant Mx alleles occur at comparable frequencies. J Interferon Res 7:647–656. doi: 10.1089/jir.1987.7.647. [DOI] [PubMed] [Google Scholar]
- 6.Haller O, Staeheli P, Schwemmle M, Kochs G. 2015. Mx GTPases: dynamin-like antiviral machines of innate immunity. Trends Microbiol 23:154–163. doi: 10.1016/j.tim.2014.12.003. [DOI] [PubMed] [Google Scholar]
- 7.Deeg CM, Hassan E, Mutz P, Rheinemann L, Gotz V, Magar L, Schilling M, Kallfass C, Nurnberger C, Soubies S, Kochs G, Haller O, Schwemmle M, Staeheli P. 2017. In vivo evasion of MxA by avian influenza viruses requires human signature in the viral nucleoprotein. J Exp Med 214:1239–1248. doi: 10.1084/jem.20161033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Manz B, Dornfeld D, Gotz V, Zell R, Zimmermann P, Haller O, Kochs G, Schwemmle M. 2013. Pandemic influenza A viruses escape from restriction by human MxA through adaptive mutations in the nucleoprotein. PLoS Pathog 9:e1003279. doi: 10.1371/journal.ppat.1003279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King MC, Raposo G, Lemmon MA. 2004. Inhibition of nuclear import and cell-cycle progression by mutated forms of the dynamin-like GTPase MxB. Proc Natl Acad Sci U S A 101:8957–8962. doi: 10.1073/pnas.0403167101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Melen K, Keskinen P, Ronni T, Sareneva T, Lounatmaa K, Julkunen I. 1996. Human MxB protein, an interferon-alpha-inducible GTPase, contains a nuclear targeting signal and is localized in the heterochromatin region beneath the nuclear envelope. J Biol Chem 271:23478–23486. doi: 10.1074/jbc.271.38.23478. [DOI] [PubMed] [Google Scholar]
- 11.Kane M, Yadav SS, Bitzegeio J, Kutluay SB, Zang T, Wilson SJ, Schoggins JW, Rice CM, Yamashita M, Hatziioannou T, Bieniasz PD. 2013. MX2 is an interferon-induced inhibitor of HIV-1 infection. Nature 502:563–566. doi: 10.1038/nature12653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goujon C, Moncorge O, Bauby H, Doyle T, Ward CC, Schaller T, Hue S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu Z, Pan Q, Ding S, Qian J, Xu F, Zhou J, Cen S, Guo F, Liang C. 2013. The interferon-inducible MxB protein inhibits HIV-1 infection. Cell Host Microbe 14:398–410. doi: 10.1016/j.chom.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 14.Haller O, Gao S, von der Malsburg A, Daumke O, Kochs G. 2010. Dynamin-like MxA GTPase: structural insights into oligomerization and implications for antiviral activity. J Biol Chem 285:28419–28424. doi: 10.1074/jbc.R110.145839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Holzinger D, Jorns C, Stertz S, Boisson-Dupuis S, Thimme R, Weidmann M, Casanova JL, Haller O, Kochs G. 2007. Induction of MxA gene expression by influenza A virus requires type I or type III interferon signaling. J Virol 81:7776–7785. doi: 10.1128/JVI.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mordstein M, Kochs G, Dumoutier L, Renauld JC, Paludan SR, Klucher K, Staeheli P. 2008. Interferon-lambda contributes to innate immunity of mice against influenza A virus but not against hepatotropic viruses. PLoS Pathog 4:e1000151. doi: 10.1371/journal.ppat.1000151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gao S, von der Malsburg A, Dick A, Faelber K, Schroder GF, Haller O, Kochs G, Daumke O. 2011. Structure of myxovirus resistance protein A reveals intra- and intermolecular domain interactions required for the antiviral function. Immunity 35:514–525. doi: 10.1016/j.immuni.2011.07.012. [DOI] [PubMed] [Google Scholar]
- 18.Fribourgh JL, Nguyen HC, Matreyek KA, Alvarez FJD, Summers BJ, Dewdney TG, Aiken C, Zhang P, Engelman A, Xiong Y. 2014. Structural insight into HIV-1 restriction by MxB. Cell Host Microbe 16:627–638. doi: 10.1016/j.chom.2014.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pitossi F, Blank A, Schroder A, Schwarz A, Hussi P, Schwemmle M, Pavlovic J, Staeheli P. 1993. A functional GTP-binding motif is necessary for antiviral activity of Mx proteins. J Virol 67:6726–6732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ponten A, Sick C, Weeber M, Haller O, Kochs G. 1997. Dominant-negative mutants of human MxA protein: domains in the carboxy-terminal moiety are important for oligomerization and antiviral activity. J Virol 71:2591–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mitchell PS, Patzina C, Emerman M, Haller O, Malik HS, Kochs G. 2012. Evolution-guided identification of antiviral specificity determinants in the broadly acting interferon-induced innate immunity factor MxA. Cell Host Microbe 12:598–604. doi: 10.1016/j.chom.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Patzina C, Haller O, Kochs G. 2014. Structural requirements for the antiviral activity of the human MxA protein against Thogoto and influenza A virus. J Biol Chem 289:6020–6027. doi: 10.1074/jbc.M113.543892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goujon C, Moncorge O, Bauby H, Doyle T, Barclay WS, Malim MH. 2014. Transfer of the amino-terminal nuclear envelope targeting domain of human MX2 converts MX1 into an HIV-1 resistance factor. J Virol 88:9017–9026. doi: 10.1128/JVI.01269-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Verdegem D, Badillo A, Wieruszeski JM, Landrieu I, Leroy A, Bartenschlager R, Penin F, Lippens G, Hanoulle X. 2011. Domain 3 of NS5A protein from the hepatitis C virus has intrinsic alpha-helical propensity and is a substrate of cyclophilin A. J Biol Chem 286:20441–20454. doi: 10.1074/jbc.M110.182436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dawar FU, Tu J, Khattak MN, Mei J, Lin L. 2017. Cyclophilin A: a key factor in virus replication and potential target for anti-viral therapy. Curr Issues Mol Biol 21:1–20. doi: 10.21775/cimb.021.001. [DOI] [PubMed] [Google Scholar]
- 26.Yang F, Robotham JM, Grise H, Frausto S, Madan V, Zayas M, Bartenschlager R, Robinson M, Greenstein AE, Nag A, Logan TM, Bienkiewicz E, Tang H. 2010. A major determinant of cyclophilin dependence and cyclosporine susceptibility of hepatitis C virus identified by a genetic approach. PLoS Pathog 6:e1001118. doi: 10.1371/journal.ppat.1001118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Badillo A, Receveur-Brechot V, Sarrazin S, Cantrelle FX, Delolme F, Fogeron ML, Molle J, Montserret R, Bockmann A, Bartenschlager R, Lohmann V, Lippens G, Ricard-Blum S, Hanoulle X, Penin F. 2017. Overall structural model of NS5A protein from hepatitis C virus and modulation by mutations confering resistance of virus replication to cyclosporin A. Biochemistry 56:3029–3048. doi: 10.1021/acs.biochem.7b00212. [DOI] [PubMed] [Google Scholar]
- 28.Moradpour D, Penin F, Rice CM. 2007. Replication of hepatitis C virus. Nat Rev Microbiol 5:453–463. doi: 10.1038/nrmicro1645. [DOI] [PubMed] [Google Scholar]
- 29.Shanmugam S, Nichols AK, Saravanabalaji D, Welsch C, Yi M. 2018. HCV NS5A dimer interface residues regulate HCV replication by controlling its self-interaction, hyperphosphorylation, subcellular localization and interaction with cyclophilin A. PLoS Pathog 14:e1007177. doi: 10.1371/journal.ppat.1007177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kambara H, Tani H, Mori Y, Abe T, Katoh H, Fukuhara T, Taguwa S, Moriishi K, Matsuura Y. 2011. Involvement of cyclophilin B in the replication of Japanese encephalitis virus. Virology 412:211–219. doi: 10.1016/j.virol.2011.01.011. [DOI] [PubMed] [Google Scholar]
- 31.Qing M, Yang F, Zhang B, Zou G, Robida JM, Yuan Z, Tang H, Shi PY. 2009. Cyclosporine inhibits flavivirus replication through blocking the interaction between host cyclophilins and viral NS5 protein. Antimicrob Agents Chemother 53:3226–3235. doi: 10.1128/AAC.00189-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Aebi M, Fah J, Hurt N, Samuel CE, Thomis D, Bazzigher L, Pavlovic J, Haller O, Staeheli P. 1989. cDNA structures and regulation of two interferon-induced human Mx proteins. Mol Cell Biol 9:5062–5072. doi: 10.1128/MCB.9.11.5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pavlovic J, Zurcher T, Haller O, Staeheli P. 1990. Resistance to influenza virus and vesicular stomatitis virus conferred by expression of human MxA protein. J Virol 64:3370–3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Crameri M, Bauer M, Caduff N, Walker R, Steiner F, Franzoso FD, Gujer C, Boucke K, Kucera T, Zbinden A, Munz C, Fraefel C, Greber UF, Pavlovic J. 2018. MxB is an interferon-induced restriction factor of human herpesviruses. Nat Commun 9:1980. doi: 10.1038/s41467-018-04379-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schilling M, Bulli L, Weigang S, Graf L, Naumann S, Patzina C, Wagner V, Bauersfeld L, Goujon C, Hengel H, Halenius A, Ruzsics Z, Schaller T, Kochs G. 2018. Human MxB protein is a pan-herpesvirus restriction factor. J Virol 92:e01056-18. doi: 10.1128/JVI.01056-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mitchell PS, Young JM, Emerman M, Malik HS. 2015. Evolutionary analyses suggest a function of MxB immunity proteins beyond lentivirus restriction. PLoS Pathog 11:e1005304. doi: 10.1371/journal.ppat.1005304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gamal N, Gitto S, Andreone P. 2016. Efficacy and safety of daclatasvir in hepatitis C: an overview. J Clin Transl Hepatol 4:336–344. doi: 10.14218/JCTH.2016.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Luban J, Bossolt KL, Franke EK, Kalpana GV, Goff SP. 1993. Human immunodeficiency virus type 1 Gag protein binds to cyclophilins A and B. Cell 73:1067–1078. doi: 10.1016/0092-8674(93)90637-6. [DOI] [PubMed] [Google Scholar]
- 39.Shah VB, Shi J, Hout DR, Oztop I, Krishnan L, Ahn J, Shotwell MS, Engelman A, Aiken C. 2013. The host proteins transportin SR2/TNPO3 and cyclophilin A exert opposing effects on HIV-1 uncoating. J Virol 87:422–432. doi: 10.1128/JVI.07177-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chatterji U, Bobardt M, Selvarajah S, Yang F, Tang H, Sakamoto N, Vuagniaux G, Parkinson T, Gallay P. 2009. The isomerase active site of cyclophilin A is critical for hepatitis C virus replication. J Biol Chem 284:16998–17005. doi: 10.1074/jbc.M109.007625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gamble TR, Vajdos FF, Yoo S, Worthylake DK, Houseweart M, Sundquist WI, Hill CP. 1996. Crystal structure of human cyclophilin A bound to the amino-terminal domain of HIV-1 capsid. Cell 87:1285–1294. doi: 10.1016/S0092-8674(00)81823-1. [DOI] [PubMed] [Google Scholar]
- 42.Rasaiyaah J, Tan CP, Fletcher AJ, Price AJ, Blondeau C, Hilditch L, Jacques DA, Selwood DL, James LC, Noursadeghi M, Towers GJ. 2013. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature 503:402–405. doi: 10.1038/nature12769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chatterji U, Lim P, Bobardt MD, Wieland S, Cordek DG, Vuagniaux G, Chisari F, Cameron CE, Targett-Adams P, Parkinson T, Gallay PA. 2010. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J Hepatol 53:50–56. doi: 10.1016/j.jhep.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu Z, Pan Q, Liang Z, Qiao W, Cen S, Liang C. 2015. The highly polymorphic cyclophilin A-binding loop in HIV-1 capsid modulates viral resistance to MxB. Retrovirology 12:1. doi: 10.1186/s12977-014-0129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fricke T, White TE, Schulte B, de Souza Aranha Vieira DA, Dharan A, Campbell EM, Brandariz-Nuñez A, Diaz-Griffero F. 2014. MxB binds to the HIV-1 core and prevents the uncoating process of HIV-1. Retrovirology 11:68. doi: 10.1186/s12977-014-0068-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goujon C, Greenbury RA, Papaioannou S, Doyle T, Malim MH. 2015. A triple-arginine motif in the amino-terminal domain and oligomerization are required for HIV-1 inhibition by human MX2. J Virol 89:4676–4680. doi: 10.1128/JVI.00169-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Busnadiego I, Kane M, Rihn SJ, Preugschas HF, Hughes J, Blanco-Melo D, Strouvelle VP, Zang TM, Willett BJ, Boutell C, Bieniasz PD, Wilson SJ. 2014. Host and viral determinants of Mx2 antiretroviral activity. J Virol 88:7738–7752. doi: 10.1128/JVI.00214-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marukian S, Jones CT, Andrus L, Evans MJ, Ritola KD, Charles ED, Rice CM, Dustin LB. 2008. Cell culture-produced hepatitis C virus does not infect peripheral blood mononuclear cells. Hepatology 48:1843–1850. doi: 10.1002/hep.22550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485. doi: 10.1038/nature09907. [DOI] [PMC free article] [PubMed] [Google Scholar]