

Influenza A virus (IAV) causes significant morbidity and mortality annually worldwide, despite the availability of new formulations of the vaccine each season. There is a critical need to develop more-efficacious vaccines. However, testing novel vaccines in the human population in controlled studies is difficult due to the limited availability and efficacy of intervention strategies should the vaccine fail. There are also significant safety concerns for work with highly pathogenic IAV strains in the laboratory. Therefore, novel strategies are needed to improve the safety of vaccine studies and of research on highly pathogenic IAV. In this study, we developed an IAV strain engineered to contain a small-molecule-mediated safety switch. This tag, when attached to an essential viral protein, allows for the regulation of IAV replication in vitro and in vivo. This strategy provides a platform for the regulation of virus replication without targeting viral proteins directly.
KEYWORDS: biotechnology, influenza vaccines, virus engineering
ABSTRACT
Influenza A virus (IAV) remains a global health concern despite the availability of a seasonal vaccine. It is difficult to predict which strains will circulate during influenza season, and therefore, it is extremely challenging to test novel vaccines in the human population. To overcome this obstacle, new vaccines must be tested in challenge studies. This approach poses significant safety problems, since current pharmacological interventions for IAV are poorly efficacious. New methods are needed to enhance the safety of these challenge studies. In this study, we have generated a virus expressing a small-molecule-assisted shutoff (SMASh) tag as a safety switch for IAV replication. The addition of the SMASh tag to an essential IAV protein allows for small-molecule-mediated inhibition of replication. Treatment with this drug controls the replication of a SMASh-tagged virus in vitro and in vivo. This model for restriction of viral replication has potential for broad applications in vaccine studies, virotherapy, and basic virus research.
IMPORTANCE Influenza A virus (IAV) causes significant morbidity and mortality annually worldwide, despite the availability of new formulations of the vaccine each season. There is a critical need to develop more-efficacious vaccines. However, testing novel vaccines in the human population in controlled studies is difficult due to the limited availability and efficacy of intervention strategies should the vaccine fail. There are also significant safety concerns for work with highly pathogenic IAV strains in the laboratory. Therefore, novel strategies are needed to improve the safety of vaccine studies and of research on highly pathogenic IAV. In this study, we developed an IAV strain engineered to contain a small-molecule-mediated safety switch. This tag, when attached to an essential viral protein, allows for the regulation of IAV replication in vitro and in vivo. This strategy provides a platform for the regulation of virus replication without targeting viral proteins directly.
INTRODUCTION
Influenza A viruses (IAVs) are respiratory viruses that cause significant morbidity and mortality annually worldwide. The virus has a segmented negative-sense single-stranded RNA genome. The virus polymerase lacks a proofreading function, contributing to the high mutation rates that have been observed for IAV genes (1). This is cause for concern, since antigenic evolution can greatly reduce the effectiveness of IAV vaccines from season to season. Additionally, because the genome is segmented, coinfection of a single cell with two distinct strains of IAV can result in the reassortment of segments and the generation of novel strains (2, 3). The high rate of mutation also increases the resistance of IAV to antiviral drugs. Amantadine, which targets the M2 ion channel, is no longer prescribed to treat IAV due to the high incidence of preexisting resistance in circulating strains. Neuraminidase inhibitors are the current standard of care for IAV infection. These antivirals only mildly reduce symptoms, and there is also growing resistance to widely prescribed drugs such as oseltamivir (OST) (4). A new drug that targets the IAV polymerase has shown promise; however, escape mutants readily emerged after only a single nucleotide change (5). The increasing antiviral resistance of IAV highlights the need for the development of novel strategies to combat IAV infection.
Vaccination is the best method for preventing IAV infection and can also reduce symptoms if infection does occur (6). Unfortunately, even when properly matched to circulating strains, seasonal vaccines can be poorly efficacious (7). This issue underscores the critical need for new vaccine strategies and approaches. However, testing experimental vaccines in humans is challenging, because it is difficult to predict which strains will be prevalent in any given IAV season. One way to overcome this difficulty is to conduct large-scale population studies, which involve assessment of the humoral immune response following vaccination and/or tracking of IAV infections in vaccinated individuals (8, 9). These studies typically take place over many years and involve hundreds to thousands of individuals, which makes them costly and time-consuming. To overcome these hurdles, vaccine challenge studies can be carried out in which individuals are given a vaccine and are subsequently infected with a strain-matched live virus (10–12). In the event of vaccine failure and uncontrolled virus replication, intervention methods are required to prevent the onset of disease. Unfortunately, intervention strategies are limited. Ineffective intervention could have dire consequences, including severe lower-respiratory-tract infection, which occurs in approximately 20% of participants (13), and potentially death. Despite quarantine, the transmission of challenge strains is still a concern, putting health care providers and the greater population at risk (14). The development of novel intervention methods that could be applied in addition to current strategies is necessary to improve the safety of these challenge studies.
Research involving highly pathogenic IAV strains for both vaccine design and basic science is limited due to safety concerns. Previously, species-specific restriction using endogenous host microRNAs (miRNAs) expressed in humans but absent in ferrets allowed for the restriction of laboratory strains to this model host (15). However, this strategy cannot be used in human cell lines or in differentiated primary human lung epithelium. Alternative safety strategies that can be employed in the event of containment failure are needed to mitigate biosafety concerns when one is working with human pathogens within relevant model systems.
The expression of viral proteins, and consequently virus replication, can be regulated using the recently developed small-molecule-assisted shutoff (SMASh) tag (16). This tag consists of the hepatitis C virus (HCV) NS3 protease (NS3p) followed by the NS4a helical domain, which functionally serves as a degron domain, triggering polyubiquitination and proteasomal degradation. The tag is fused to the N or C terminus of the protein of interest via the NS3 cleavage site. Under normal conditions, the protease cleaves the tag from the protein. The tag is degraded, while the liberated protein is left undisturbed. Following the administration of an NS3p-specific protease inhibitor, cleavage does not occur and the tagged fusion protein is degraded. Asunaprevir (ASV) is one such inhibitor that binds noncovalently to the active site of NS3p (17) and is currently in stage III clinical trials as a treatment for HCV infection (18–20). The SMASh tag has been applied previously to regulate the expression of proteins encoded by paramyxovirus (16) and pneumovirus (21) in the context of recombinant virus strains, demonstrating that this tag can be stably expressed as part of a viral genome. As demonstrated for measles virus (16), it can be used as a mechanism to shut down virus replication if attached to an essential viral protein.
In this study, we explored the use of the SMASh tag to generate a drug-controllable IAV, applying the technology for the first time to a segmented RNA virus. By adding the SMASh tag to the C terminus of the IAV polymerase acidic (PA) protein, we demonstrate drug-mediated control over IAV replication both in vitro and in vivo. Importantly, the tag remains sensitive to treatment over multiple virus replication cycles, demonstrating the stability of the SMASh system in IAV. This study provides proof of principle for the use of exogenous sequences for the restriction of IAV replication. The model established here could be applied to other viruses and refined for broad applications in vaccine safety and design, virotherapy, and basic virology research.
RESULTS
The replication of IAV_SMASh is restricted by asunaprevir in vitro.
Control of IAV replication using antiviral drugs is currently limited to the inhibition of virus neuraminidase. However, resistance to antiviral drugs is rapidly emerging, and treatment with these drugs can be poorly efficacious even in sensitive strains (22). While there are major efforts to develop antivirals that target other IAV proteins, we sought to develop a system where replication could be experimentally controlled via a tag rather than direct targeting of IAV proteins. We generated a small-molecule-controllable IAV via addition of the SMASh tag to the C terminus of the polymerase acidic (PA) protein of IAV PR8 (IAV_SMASh) (Fig. 1A). To determine if treatment with ASV can lead to a shutoff of IAV replication, cells were first infected with IAV_SMASh and then exposed to ASV or an equivalent volume of dimethyl sulfoxide (DMSO) as a vehicle control. ASV treatment reduced IAV_SMASh replication in both MDCK and A549 cells as measured by reduced IAV NP expression (Fig. 1B and C), although higher doses of ASV were required in MDCK cells. These data demonstrate that ASV is able to regulate IAV_SMASh replication in both model host and human cells. Additionally, IAV_SMASh was responsive to ASV treatment in a dose-dependent manner. As little as 0.5 µM ASV was able to noticeably reduce IAV NP levels in A549 cells (Fig. 1C). Together, these data demonstrate that in vitro replication of IAV_SMASh can be regulated by treatment with ASV.
FIG 1.
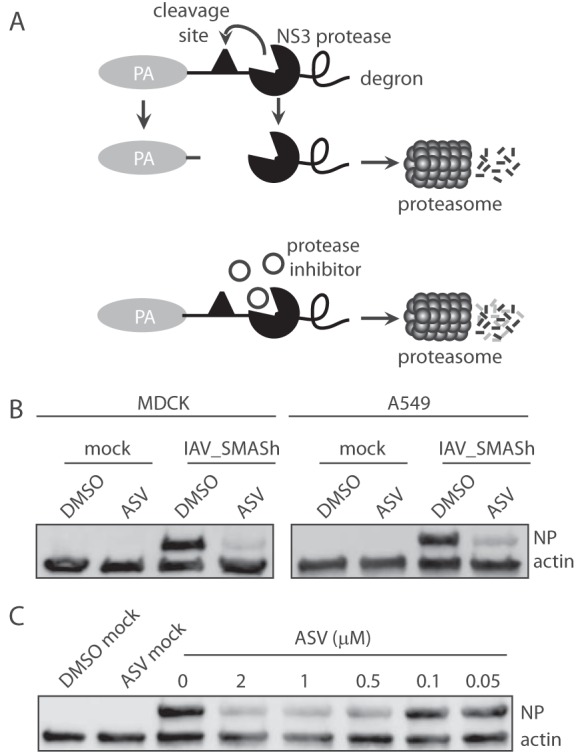
IAV_SMASh replication is restricted by asunaprevir treatment in vitro. (A) Model depicting small-molecule-mediated degradation of the IAV PA protein. (B) (Left) MDCK cells infected with IAV_SMASh at an MOI of 0.3 were analyzed for NP and actin in the presence of 20 µM ASV or DMSO at 24 hpi. (Right) A549 cells infected with IAV_SMASh at an MOI of 0.2 were analyzed for NP and actin in the presence of 2 µM ASV or DMSO at 24 hpi. (C) A549 cells infected with IAV_SMASh at an MOI of 0.2 were analyzed for NP and actin in the presence of DMSO or various concentrations of ASV at 24 hpi. “Mock” refers to uninfected samples treated as indicated. Data are representative of results from 2 to 4 independent experiments.
IAV_SMASh is responsive to asunaprevir through multiple replication cycles in vitro.
To determine if ASV is able to restrict IAV_SMASh replication and spread, we performed in vitro multicycle growth analysis in MDCK cells in the presence or absence of ASV. Treatment with ASV reduced IAV_SMASh titers by 1 order of magnitude at 48 h postinfection (hpi) from those with DMSO treatment (Fig. 2A). Importantly, ASV treatment did not affect the titer of control IAV (IAV_ctrl). These results were replicated in A549 cells (Fig. 2B). In agreement with the results shown in Fig. 1C, IAV_SMASh is responsive to ASV in a dose-dependent manner over multiple replication cycles (Fig. 2C). Treatment with 0.5 µM ASV resulted in a 2-fold reduction, while 10 µM ASV yielded a >10-fold reduction, in viral titers at 72 hpi. Based on these data, 50% effective concentrations (EC50) were calculated, and these values were similar at 24 and 48 hpi, indicating that ASV treatment was equally efficacious at these two time points (Fig. 2D). Together, these data demonstrate that IAV_SMASh replication can be controlled by treatment with ASV over multiple rounds of replication.
FIG 2.

IAV_SMASh is responsive to asunaprevir through multiple replication cycles in vitro. (A and B) MDCK (A) or A549 (B) cells infected with IAV_SMASh or IAV_ctrl at an MOI of 0.05 were grown in the presence of 2 µM ASV or DMSO. Supernatants were harvested at 0, 12, 24, 48, and 72 hpi, and titers were determined on MDCK cells. Error bars represent standard deviations. (C) A549 cells infected with IAV_SMASh at an MOI of 0.05 were grown in the presence of DMSO or various concentrations of ASV. Supernatants were harvested at 0, 12, 24, 48, and 72 hpi, and titers were determined on MDCK cells. The experiments for which results are shown in panels A through C were performed once or twice, with 3 replicates per group. (D) Four-parameter variable slope regression modeling was applied to the data from panel C in order to calculate active concentrations at 24 and 48 hpi. EC50 and 95% confidence intervals (in parentheses) are shown on the right.
Asunaprevir and oseltamivir cooperate to control virus replication in vitro.
Oseltamivir (OST) is the current standard of care for IAV infections and for interventions during vaccine challenge studies. Because ASV and OST target different stages of the virus life cycle, they may be combined to better control IAV replication. Dose titrations of ASV and OST individually were performed at 48 hpi to determine an appropriate dose for combination treatments (Fig. 3A and B). Treatment with ASV, OST, or both significantly reduced IAV_SMASh titers at 48 hpi from those with the control treatments (Fig. 3C). However, treatment with both ASV and OST was more effective than either drug alone (Fig. 3D), indicating that treatments that target two different stages of the viral life cycle can cooperate to control virus replication. Importantly, the combined treatment did not increase cytotoxicity over that with individual or control treatments (Fig. 3E).
FIG 3.

ASV and OST cooperate to control IAV_SMASh replication in vitro. (A and B) A549 cells infected with IAV_SMASh were treated with the indicated concentrations of ASV (A) or OST (B). Supernatants were harvested at 48 hpi, and titers were determined on MDCK cells. Error bars represent standard deviations. (C) A549 cells were infected with IAV_SMASh and were treated with either 2 μM ASV, 0.1 μM OST, both, or equivalent volumes of DMSO and phosphate-buffered saline as a control. Supernatants were harvested at 48 hpi, and titers were determined on MDCK cells. (D) Titers in drug-treated cells relative to those in control-treated cells (from data in panel C). P values were calculated using Tukey’s multiple-comparison test. (E) A549 cells were treated as described in the legend to panel C, and viability was measured. Experiments were performed either twice with three replicates per group (A, C, and D), once with three replicates per group (B), or once with five replicates per group (E).
IAV_SMASh replication is restricted by asunaprevir in vivo.
To determine the virulence of IAV_SMASh and its suitability for use in vaccine challenge studies and other in vivo applications, we assessed the level of attenuation of IAV_SMASh in C57BL/6 mice. We have shown previously that higher doses of recombinant IAV can cause a level of disease similar to that with lower doses of wild-type (wt) IAV (23). Therefore, mice were infected with 40 PFU of wt IAV, 100 PFU of IAV_ctrl, or 100 PFU of IAV_SMASh. Importantly, the 2.5× dose of IAV_SMASh drove the same degree of disease as wt IAV, as measured by weight loss (Fig. 4A). These data demonstrate that despite the insertion of an exogenous sequence into the PA segment, IAV_SMASh causes physiological disease in vivo similar to that with the wt virus. Given that ASV is a drug designed to target a liver-tropic virus, it is important to determine the capacity of this drug to function within the lung, the primary site of IAV replication. To test the responsiveness of IAV_SMASh in vivo to small-molecule-mediated attenuation, ASV or control treatments were given intranasally (i.n.) at the time of IAV_SMASh infection and following infection (Fig. 4B). ASV treatment reduced IAV_SMASh titers in the lung at 72 hpi by approximately 3-fold from those for control-treated mice (Fig. 4C). Similar results were obtained when higher doses of ASV were administered intraperitoneally (i.p.) (data not shown). To ensure that the SMASh tag remains susceptible to ASV-mediated attenuation over multiple replication cycles under selective pressure, we conducted an ex vivo escape assay in which cells were infected with IAV_SMASh recovered from mouse lungs and were treated with ASV or DMSO (Fig. 4D). Virus from ASV-treated mice was still sensitive to ASV treatment in vitro (Fig. 4E), indicating that the SMASh tag is stable over multiple rounds of replication under selective pressure. Critically, ASV treatment protected mice from a lethal dose of IAV_SMASh (Fig. 4F). Together, these data indicate that IAV_SMASh is virulent in vivo but that its replication can be controlled by administration of ASV.
FIG 4.

IAV_SMASh replication is restricted by asunaprevir treatment in vivo. (A) Mice were infected with 40 PFU of wt IAV or 100 PFU of IAV_ctrl or IAV_SMASh and were monitored for weight loss. Error bars represent standard deviations. dpi, days postinfection. (B) Infection and treatment schedule. (C) Animals were infected and treated as diagramed in panel B with 374 µg of ASV per treatment. Lungs were harvested at 72 hpi, and titers were determined on MDCK cells. Data are representative of the results of two independent experiments with 3 to 4 mice per group. (D) A549 cells were infected with virus recovered from the lungs of mice for which results are shown in panel C and were treated with ASV or DMSO. (E) ASV sensitivity was analyzed by probing for NP by Western blotting (left), and bands were quantified (right). (F) Survival of mice infected with 50 PFU of IAV_SMASh and treated as diagramed in panel B with DMSO (n = 10) or 500 µg of ASV (n = 5) per treatment.
SMASh tag expression is stable through multiple rounds of replication.
ASV inhibits NS3p activity by binding to the protease active site (17). Mutations in the active site that reduce ASV sensitivity may also reduce protease activity and lead to increased degradation of PA. Additionally, mutations in the cleavage site that affect NS3p activity would lead to increased degradation of PA. It is therefore unlikely for the SMASh tag to mutate in such a way that ASV treatment no longer controls IAV_SMASh replication. However, mutations in the degron that prevent the destruction of the targeted protein could occur. Additionally, although a complete packaging sequence is placed downstream of the tag, truncations could eliminate the entire tag and cause IAV_SMASh to revert to a wt-like state with no sensitivity to ASV. Sequencing of virus recovered from the lungs of infected mice (Fig. 4C) revealed a mutant virus with a deletion resulting in loss of degron expression. Given this result, we plaque-purified IAV_SMASh to perform an in vitro escape assay where we serially passaged IAV_SMASh in MDCK cells in the presence or absence of ASV. IAV_SMASh was still sensitive to ASV treatments after two passages (Fig. 5A). Additionally, the viruses recovered after the first and second passages retained the full-length SMASh tag (Fig. 5B) and contained few mutations (Fig. 5C). Importantly, there was no increase in mutation frequency in the ASV-treated group, suggesting that the SMASh tag can remain stable under selective pressure.
FIG 5.
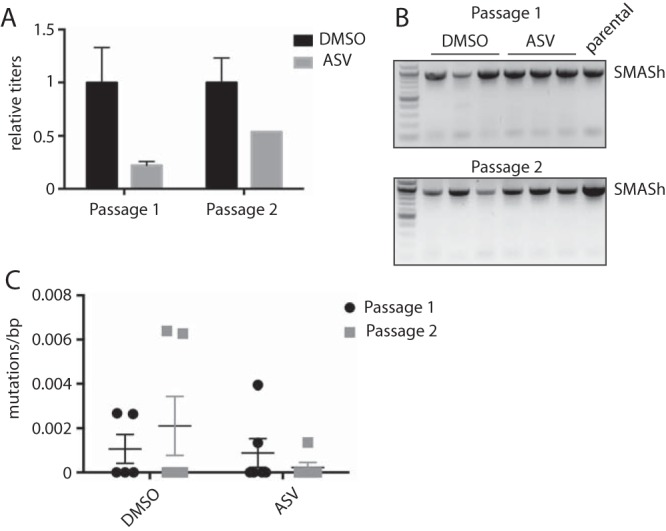
SMASh tag expression is stable through multiple rounds of replication in vitro. (A) ASV sensitivity was analyzed by plaque assays after the first and second passages in MDCK cells. Error bars represent standard deviations. (B) The SMASh tag was amplified from purified virus by reverse transcription-PCR after the first and second passages. Results for the parental virus, IAV_SMASh, are also shown. (C) The number of mutations was calculated following Sanger sequencing. Data are representative of results from 2 (A and B) or 1 (C) independent experiment with three replicates per group.
DISCUSSION
Research involving highly pathogenic IAV strains raises biosafety concerns, and steps must be taken to mitigate potential safety issues for gain-of-function studies. Safety is also a concern during vaccine challenge studies, which are an important step in the development and testing of novel vaccine and therapeutic strategies against IAV. To reduce the potential for severe disease, challenge strains are administered intranasally, which is less physiologically relevant than the aerosol route (14). Nevertheless, vaccine failure can lead to severe infection in the absence of effective intervention strategies (13). In this study, we used the HCV NS2b/NS3 protease-based SMASh tag to allow for small-molecule-mediated restriction of IAV replication. IAV_SMASh is specifically controlled by treatment with an NS2b/NS3 protease inhibitor, and the virus can express the tag over multiple replication cycles under selective pressure. This proof-of-principle model demonstrates the successful use of exogenous sequences for enhancing safety in working with pathogenic IAV, including during vaccine challenge studies. Importantly, this approach could also be applied to other viruses, including those for which human vaccine studies are urgently needed. This system could also be used as a novel rheostat vaccine that would behave as both a live attenuated vaccine and a natural infection. In the absence of ASV treatment, the SMASh tag is cleaved from PA, and the virus can replicate similarly to a wild-type virus. ASV could be administered after the initiation of antiviral immunity but before virus-driven pathogenesis. This strategy could be applied to viruses for which vaccines are poorly immunogenic.
We demonstrated that the addition of the SMASh tag to IAV PA allows for ASV-mediated regulation of IAV replication. PA is a component of the IAV polymerase heterotrimer. In addition to its direct role in replication and in the transcription of the IAV genome, PA has endonuclease activity to facilitate cap-snatching from host mRNAs (24). These essential functions of PA make it an ideal target for small-molecule-mediated degradation. We have demonstrated previously that targeting of NP by endogenous microRNAs results in significant attenuation of virus replication in vitro and in vivo (25). Furthermore, miRNA targeting of other segments resulted in various levels of virus attenuation (15), with NP conferring the highest degree of viral suppression (26). These data suggest that addition of the SMASh tag to different segments may allow for customizable attenuation (26, 27). This shutoff system could be used as a tool with which to study the roles of specific proteins at different stages of infection by treating with the protease inhibitor at different time points.
Alternative protease-inhibitor pairs could be used to further optimize the application of this method to IAV. ASV has been designed to target HCV in the liver. When delivered systemically, ASV is undetectable in serum and in organs outside the liver by 24 h postadministration (17). Since the inhibition of IAV_SMASh using ASV is dose dependent (Fig. 2C), effective delivery of the drug to the lung would increase the efficiency of drug-mediated inhibition of virus replication. Redesigning the SMASh tag using a protease with an inhibitor that is more effectively delivered to the lung could overcome the limitations of ASV. An additional concern with using the NS3 protease in an IAV system is that NS3p is known to cleave MAVS (28), which could hinder the innate immune response to IAV. However, we did not see an increase in IAV_SMASh titers over IAV_ctrl titers in the absence of ASV in human cells. This may be due to the rapid degradation of the SMASh tag (16), preventing the targeting of MAVS. Redesigning the SMASh tag could optimize the system for use with IAV and mitigate the potential cleavage of host proteins.
The data shown in Fig. 5 indicate that IAV_SMASh grown under the selective pressure of ASV treatment remains responsive to ASV treatment in vitro. However, sequencing of virus from the experiments for which results are shown in Fig. 4 revealed that some viruses contain deletion mutations in the SMASh tag, showing that escape can occur. One concern would be the emergence of ASV-resistant mutants, compromising the efficacy of treatment in vivo. However, treatment with ASV led to 80% survival in mice infected with IAV_SMASh, indicating that even if ASV-insensitive mutants arise, ASV treatment reduces the overall viral load and remains protective in vivo. Fukuyama and colleagues built an IAV containing a reporter in NS1, and after 6 passages, they were able to select a virus with mutations in HA and PB2 that conferred increased stability (29). A similar strategy could be employed to select SMASh-containing virus clones with enhanced stability. We also demonstrate that treatment of IAV_SMASh with both ASV and OST results in increased viral repression. These data suggest that combinatorial treatment can increase intervention-mediated attenuation and may decrease the chance of virus escape to virulence.
Overall, our results demonstrate the use of the SMASh tag to allow for small-molecule-mediated regulation of IAV replication. ASV treatment specifically inhibits the replication of IAV containing the SMASh tag, and ASV sensitivity is stable over multiple rounds of replication. This method of viral restriction could be used for other viruses, with broad applications in vaccine design, oncolytic virotherapy, and fundamental virology.
MATERIALS AND METHODS
Cell culture.
Madin-Darby canine kidney (MDCK) cells, human embryonic kidney (HEK) 293T cells, and human lung adenocarcinoma A549 cells (all from ATCC) were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin.
Plasmid design and virus rescue.
The influenza A/Puerto Rico/8/1934 (PR8) PA coding sequence was mutated to disrupt the packaging signal upstream of the stop codon and to eliminate the stop codon. This segment, the SMASh tag, and a complete 184-nucleotide (nt) 3′ viral RNA (vRNA) packaging signal were amplified and recombined into the pDZ IAV rescue vector via In-Fusion HD cloning (TaKaRa Bio, Inc.). As a control, Cre recombinase (Cre) was cloned following a PTV-1 2A site after the stop codon of PA. Packaging signals upstream were mutated, and a complete packaging signal was added to the end of Cre as described above. This virus, termed IAV_ctrl, serves as a control for the insertion of an exogenous coding sequence on the 5′ end of the PA vRNA. Viruses were rescued via HEK293T transfection and were amplified in embryonated chicken eggs as described previously (30). Rescued viruses were plaque purified, their sequences confirmed, and their titers determined on MDCK cells. Virus titers were calculated as PFU per milliliter.
In vitro multicycle growth analysis.
Confluent MDCK or A549 cells were infected with wt IAV, IAV_ctrl, or IAV_SMASh at a multiplicity of infection (MOI) of 0.05 for 1 h and were incubated at 37°C in viral growth medium (DMEM with 2.5% bovine serum albumin [BSA] fraction V, 2.5% HEPES buffer, and 1% penicillin-streptomycin) supplemented with 1 to 2 µg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK) trypsin. Asunaprevir (ASV; Adooq/MCE) dissolved in DMSO or an equivalent volume of DMSO as a vehicle control was added at 0 hpi. The medium was supplemented with additional ASV or DMSO at 24 and 48 hpi. At 0, 12, 24, 48, and 72 hpi, culture supernatants were collected and titers were determined on MDCK cells. Virus titers were calculated as PFU per milliliter.
Western blot analysis.
MDCK or A549 cells were infected with IAV_SMASh at an MOI of 0.2 for 1 h and were incubated at 37°C in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and DMSO or ASV at the concentrations indicated in the figures or figure legends. At 24 hpi, cells were lysed, and lysates were separated by SDS-PAGE (4% to 15% gel). Proteins were transferred to a nitrocellulose membrane and were blocked with 5% milk. The membrane was incubated in a mouse anti-IAV NP monoclonal antibody (1:1,000; clone IC5-1B7; BEI Resources) and a mouse pan-actin primary antibody (1:1,000; clone ACTN05; Thermo Fisher) followed by horseradish peroxidase (HRP)-conjugated sheep anti-mouse IgG (1:1,000; catalog no. 45-001-275; GE Healthcare). Images were obtained using a Li-Cor Odyssey Fc imaging system.
ASV-OST cooperation assay.
A549 cells in infection medium were infected with plaque-purified IAV_SMASh at an MOI of 0.05 for 1 h and were incubated at 37°C in viral growth medium supplemented with TPCK and either 2 μM ASV, 0.1 μM oseltamivir carboxylate (OST; MCE), or both. Supernatants were harvested at 48 hpi and were titrated on MDCK cells. Viability assays were performed on A549 cells using the CellTiter-Glo luminescent cell viability assay (Promega). Luminescence was calculated using a Synergy H1 microplate reader (BioTek).
Mouse experiments.
C57BL/6 mice (Jackson Laboratory) were anesthetized with ketamine-xylazine and were infected i.n. with the doses of wt IAV, IAV_ctrl, or IAV_SMASh indicated in the figures or figure legends. Treatment with ASV or the vehicle control DMSO was administered either i.n. prior to infection, at the time of infection, or at various times postinfection (374 or 500 µg ASV per treatment) or i.p. postinfection (820 µg ASV per treatment). Animals were sacrificed at various times postinfection, and lungs were harvested. All experiments involving mice were performed as dictated by the University of Minnesota Institutional Animal Care and Use Committee.
Ex vivo escape assay.
The titers of IAV_SMASh recovered from the lungs of mice treated with ASV or the DMSO vehicle control were determined on MDCK cells. A549 cells were infected with IAV_SMASh from either condition at an MOI of 0.02 for 2 h and were incubated at 37°C in DMEM supplemented with 10% FBS, 1% penicillin-streptomycin, and 1 µM ASV or an equivalent volume of DMSO. At 24 hpi, cells were lysed and were analyzed by Western blotting as described above using a mouse anti-IAV NP monoclonal antibody (1:500) followed by HRP-conjugated sheep anti-mouse IgG (1:1,000).
In vitro escape assay.
MDCK cells in infection medium were infected with plaque-purified IAV_SMASh at an MOI of 0.05 for 1 h at 37°C. Cells were grown in viral growth medium supplemented with TPCK and 3 μM ASV or DMSO. Supernatants were harvested at 48 hpi and were titrated on MDCK cells. MDCK cells were infected as described above with the round 1 supernatant at an MOI of 0.001 and were grown in viral growth medium supplemented with 300 nM ASV or DMSO. At 48 hpi, the supernatant was harvested and was titrated on MDCK cells. Virus RNAs from both rounds of infection were extracted, and the sequence of the SMASh tag was confirmed by Sanger sequencing.
Statistical analysis.
Statistical analysis was performed using GraphPad Prism 7 software. Comparisons between two groups were performed using a two-tailed Student t test, and a P value of < 0.05 was considered statistically significant. Comparisons between three or more groups were performed using one-way analysis of variance (ANOVA) and the multiple-comparison test indicated in the relevant figure legend. To determine 50% effective concentrations (EC50), four-parameter variable slope regression modeling was employed using the Prism 7 software package.
ACKNOWLEDGMENTS
We thank Stephen Rice for critical reading of the manuscript and Courtney Aldrich for helpful discussions.
This work was supported by startup funds from the University of Minnesota Department of Microbiology and Immunology, NIH NIAID grant K22 AI110581, NIH NIAID grant R01 AI132962 to R.A.L., and Public Health Service grant AI071002 from the NIH NIAID to R.K.P. E.J.F. was supported by NIH NIAID award T32 AI007313.
REFERENCES
- 1.Kash JC, Taubenberger JK. 2015. The role of viral, host, and secondary bacterial factors in influenza pathogenesis. Am J Pathol 185:1528–1536. doi: 10.1016/j.ajpath.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao H, Li L, White MC, Steel J, Lowen AC. 2015. Influenza A virus coinfection through transmission can support high levels of reassortment. J Virol 89:8453–8461. doi: 10.1128/JVI.01162-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schrauwen EJ, de Graaf M, Herfst S, Rimmelzwaan GF, Osterhaus AD, Fouchier RA. 2014. Determinants of virulence of influenza A virus. Eur J Clin Microbiol Infect Dis 33:479–490. doi: 10.1007/s10096-013-1984-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marjuki H, Mishin VP, Chesnokov AP, De La Cruz JA, Davis CT, Villanueva JM, Fry AM, Gubareva LV. 2015. Neuraminidase mutations conferring resistance to oseltamivir in influenza A(H7N9) viruses. J Virol 89:5419–5426. doi: 10.1128/JVI.03513-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hayden FG, Sugaya N, Hirotsu N, Lee N, de Jong MD, Hurt AC, Ishida T, Sekino H, Yamada K, Portsmouth S, Kawaguchi K, Shishido T, Arai M, Tsuchiya K, Uehara T, Watanabe A, Baloxavir Marboxil Investigators Group . 2018. Baloxavir marboxil for uncomplicated influenza in adults and adolescents. N Engl J Med 379:913–923. doi: 10.1056/NEJMoa1716197. [DOI] [PubMed] [Google Scholar]
- 6.Grijalva CG, Zhu Y, Williams DJ, Self WH, Ampofo K, Pavia AT, Stockmann CR, McCullers J, Arnold SR, Wunderink RG, Anderson EJ, Lindstrom S, Fry AM, Foppa IM, Finelli L, Bramley AM, Jain S, Griffin MR, Edwards KM. 2015. Association between hospitalization with community-acquired laboratory-confirmed influenza pneumonia and prior receipt of influenza vaccination. JAMA 314:1488–1497. doi: 10.1001/jama.2015.12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tricco AC, Chit A, Soobiah C, Hallett D, Meier G, Chen MH, Tashkandi M, Bauch CT, Loeb M. 2013. Comparing influenza vaccine efficacy against mismatched and matched strains: a systematic review and meta-analysis. BMC Med 11:153. doi: 10.1186/1741-7015-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ohmit SE, Petrie JG, Cross RT, Johnson E, Monto AS. 2011. Influenza hemagglutination-inhibition antibody titer as a correlate of vaccine-induced protection. J Infect Dis 204:1879–1885. doi: 10.1093/infdis/jir661. [DOI] [PubMed] [Google Scholar]
- 9.Black S, Nicolay U, Vesikari T, Knuf M, Del Giudice G, Della Cioppa G, Tsai T, Clemens R, Rappuoli R. 2011. Hemagglutination inhibition antibody titers as a correlate of protection for inactivated influenza vaccines in children. Pediatr Infect Dis J 30:1081–1085. doi: 10.1097/INF.0b013e3182367662. [DOI] [PubMed] [Google Scholar]
- 10.Sobhanie M, Matsuoka Y, Jegaskanda S, Fitzgerald T, Mallory R, Chen Z, Luke C, Treanor J, Subbarao K. 2016. Evaluation of the safety and immunogenicity of a candidate pandemic live attenuated influenza vaccine (pLAIV) against influenza A(H7N9). J Infect Dis 213:922–929. doi: 10.1093/infdis/jiv526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jegaskanda S, Luke C, Hickman HD, Sangster MY, Wieland-Alter WF, McBride JM, Yewdell JW, Wright PF, Treanor J, Rosenberger CM, Subbarao K. 2016. Generation and protective ability of influenza virus-specific antibody-dependent cellular cytotoxicity in humans elicited by vaccination, natural infection, and experimental challenge. J Infect Dis 214:945–952. doi: 10.1093/infdis/jiw262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Memoli MJ, Shaw PA, Han A, Czajkowski L, Reed S, Athota R, Bristol T, Fargis S, Risos K, Powers JH, Davey RT Jr, Taubenberger JK. 2016. Evaluation of antihemagglutinin and antineuraminidase antibodies as correlates of protection in an influenza A/H1N1 virus healthy human challenge model. mBio 7:e00417-16. doi: 10.1128/mBio.00417-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrat F, Vergu E, Ferguson NM, Lemaitre M, Cauchemez S, Leach S, Valleron AJ. 2008. Time lines of infection and disease in human influenza: a review of volunteer challenge studies. Am J Epidemiol 167:775–785. doi: 10.1093/aje/kwm375. [DOI] [PubMed] [Google Scholar]
- 14.Killingley B, Enstone J, Booy R, Hayward A, Oxford J, Ferguson N, Nguyen Van-Tam J, Influenza Transmission Strategy Development Group . 2011. Potential role of human challenge studies for investigation of influenza transmission. Lancet Infect Dis 11:879–886. doi: 10.1016/S1473-3099(11)70142-6. [DOI] [PubMed] [Google Scholar]
- 15.Langlois RA, Albrecht RA, Kimble B, Sutton T, Shapiro JS, Finch C, Angel M, Chua MA, Gonzalez-Reiche AS, Xu K, Perez D, Garcia-Sastre A, tenOever BR. 2013. microRNA-based strategy to mitigate the risk of gain-of-function influenza studies. Nat Biotechnol 31:844–847. doi: 10.1038/nbt.2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chung HK, Jacobs CL, Huo Y, Yang J, Krumm SA, Plemper RK, Tsien RY, Lin MZ. 2015. Tunable and reversible drug control of protein production via a self-excising degron. Nat Chem Biol 11:713–720. doi: 10.1038/nchembio.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McPhee F, Sheaffer AK, Friborg J, Hernandez D, Falk P, Zhai G, Levine S, Chaniewski S, Yu F, Barry D, Chen C, Lee MS, Mosure K, Sun LQ, Sinz M, Meanwell NA, Colonno RJ, Knipe J, Scola P. 2012. Preclinical profile and characterization of the hepatitis C virus NS3 protease inhibitor asunaprevir (BMS-650032). Antimicrob Agents Chemother 56:5387–5396. doi: 10.1128/AAC.01186-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muir AJ, Poordad F, Lalezari J, Everson G, Dore GJ, Herring R, Sheikh A, Kwo P, Hezode C, Pockros PJ, Tran A, Yozviak J, Reau N, Ramji A, Stuart K, Thompson AJ, Vierling J, Freilich B, Cooper J, Ghesquiere W, Yang R, McPhee F, Hughes EA, Swenson ES, Yin PD. 2015. Daclatasvir in combination with asunaprevir and beclabuvir for hepatitis C virus genotype 1 infection with compensated cirrhosis. JAMA 313:1736–1744. doi: 10.1001/jama.2015.3868. [DOI] [PubMed] [Google Scholar]
- 19.Poordad F, Sievert W, Mollison L, Bennett M, Tse E, Brau N, Levin J, Sepe T, Lee SS, Angus P, Conway B, Pol S, Boyer N, Bronowicki JP, Jacobson I, Muir AJ, Reddy KR, Tam E, Ortiz-Lasanta G, de Ledinghen V, Sulkowski M, Boparai N, McPhee F, Hughes E, Swenson ES, Yin PD, UNITY-1 Study Group . 2015. Fixed-dose combination therapy with daclatasvir, asunaprevir, and beclabuvir for noncirrhotic patients with HCV genotype 1 infection. JAMA 313:1728–1735. doi: 10.1001/jama.2015.3860. [DOI] [PubMed] [Google Scholar]
- 20.Toyoda H, Kumada T, Tada T, Takaguchi K, Ishikawa T, Tsuji K, Zeniya M, Iio E, Tanaka Y. 2016. Safety and efficacy of dual direct-acting antiviral therapy (daclatasvir and asunaprevir) for chronic hepatitis C virus genotype 1 infection in patients on hemodialysis. J Gastroenterol 51:741–747. doi: 10.1007/s00535-016-1174-4. [DOI] [PubMed] [Google Scholar]
- 21.Yan D, Weisshaar M, Lamb K, Chung HK, Lin MZ, Plemper RK. 2015. Replication-competent influenza virus and respiratory syncytial virus luciferase reporter strains engineered for co-infections identify antiviral compounds in combination screens. Biochemistry 54:5589–5604. doi: 10.1021/acs.biochem.5b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jefferson T, Jones M, Doshi P, Spencer EA, Onakpoya I, Heneghan CJ. 2014. Oseltamivir for influenza in adults and children: systematic review of clinical study reports and summary of regulatory comments. BMJ 348:g2545. doi: 10.1136/bmj.g2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heaton NS, Langlois RA, Sachs D, Lim JK, Palese P, tenOever BR. 2014. Long-term survival of influenza virus infected club cells drives immunopathology. J Exp Med 211:1707–1714. doi: 10.1084/jem.20140488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dias A, Bouvier D, Crepin T, McCarthy AA, Hart DJ, Baudin F, Cusack S, Ruigrok RW. 2009. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 458:914–918. doi: 10.1038/nature07745. [DOI] [PubMed] [Google Scholar]
- 25.Langlois RA, Varble A, Chua MA, Garcia-Sastre A, tenOever BR. 2012. Hematopoietic-specific targeting of influenza A virus reveals replication requirements for induction of antiviral immune responses. Proc Natl Acad Sci U S A 109:12117–12122. doi: 10.1073/pnas.1206039109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Waring BM, Sjaastad LE, Fiege JK, Fay EJ, Reyes I, Moriarity B, Langlois RA. 2018. microRNA-based attenuation of influenza virus across susceptible hosts. J Virol 92:e01741-17. doi: 10.1128/JVI.01741-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiege JK, Langlois RA. 2015. Investigating influenza A virus infection: tools to track infection and limit tropism. J Virol 89:6167–6170. doi: 10.1128/JVI.00462-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ferreira AR, Magalhaes AC, Camoes F, Gouveia A, Vieira M, Kagan JC, Ribeiro D. 2016. Hepatitis C virus NS3-4A inhibits the peroxisomal MAVS-dependent antiviral signalling response. J Cell Mol Med 20:750–757. doi: 10.1111/jcmm.12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fukuyama S, Katsura H, Zhao D, Ozawa M, Ando T, Shoemaker JE, Ishikawa I, Yamada S, Neumann G, Watanabe S, Kitano H, Kawaoka Y. 2015. Multi-spectral fluorescent reporter influenza viruses (Color-flu) as powerful tools for in vivo studies. Nat Commun 6:6600. doi: 10.1038/ncomms7600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hai R, Martinez-Sobrido L, Fraser KA, Ayllon J, Garcia-Sastre A, Palese P. 2008. Influenza B virus NS1-truncated mutants: live-attenuated vaccine approach. J Virol 82:10580–10590. doi: 10.1128/JVI.01213-08. [DOI] [PMC free article] [PubMed] [Google Scholar]