

Abstract
AIM
To elucidate the underlying mechanism that microRNA-22 (miR-22) promotes the apoptosis of rat pancreatic acinar cells (AR42J) and the elements that regulate the expression of miR-22.
METHODS
One hundred nanomoles per liter of caerulein (Cae) was administrated to induce the apoptosis of AR42J cells and the apoptosis rate was detected by flow cytometry analysis. An amylase assay kit was used to measure the amylase expression level in the supernatant. Quantitative real-time PCR (qRT-PCR) was adopted to measure miR-22 expression. We used online tools to predict the potential transcription promoter of miR-22 and the binding sites, which was further identified by using luciferase reporter analysis, chromatin immunoprecipitation (ChIP) and ChIP-qPCR assays. Then, a mimic of miR-22, Nr3c1 plasmid encoding the glucocorticoid receptor (GR), and si-Nr3c1 were used to transfect AR42J cells, respectively. The mRNA expression of miR-22, Nr3c1, and Erb-b2 receptor tyrosine kinase 3 (ErbB3) was confirmed by qRT-PCR and the apoptosis rate of AR42J cells was detected by flow cytometry analysis. Western blot was used to detect the expression of ErbB3, GR, PI3k, PI3k-p85α, Akt, p-Akt, Bad, Bax, Bcl-xl, Bcl-2, and cleaved caspase3.
RESULTS
After inducing apoptosis of AR42J cells in vitro, the expression of miR-22 was significantly increased by 2.20 ± 0.26 and 4.19 ± 0.54 times, respectively, at 3 h and 6 h in comparison with the control group. As revealed by qRT-PCR assay, the expression of miR-22 was 78.25 ± 6.61 times higher in the miR-22 mimic group relative to the miRNA control group, accompanied with an obviously increased acinar cell apoptosis rate (32.53 ± 1.15 vs 18.07 ± 0.89, P = 0.0006). The upregulation of miR-22 could suppress its target gene, ErbB3, and the phosphorylation of PI3k and Akt. Furthermore, we predicted the potential transcription promoter of miR-22 and the binding sites using online tools. Luciferase reporter analysis and site-directed mutagenesis indicated that the binding site (GACAGCCATGTACA) of the GR, which is encoded by the Nr3c1 gene. Downregulation of the expression of GR could upregulate the expression of miR-22, which further promoted the apoptosis of AR42J cells.
CONCLUSION
GR transcriptionally represses the expression of miR-22, which further promotes the apoptosis of pancreatic acinar cells by downregulating the downstream signaling pathway.
Keywords: MicroRNA-22, Apoptosis, Pancreatic acinar cells, Erb-b2 receptor tyrosine kinase 3, Glucocorticoid receptor
Core tip: The severity of acute pancreatitis (AP) is inversely related to the rate of apoptosis of pancreatic acinar cells. MicroRNA-22 (miR-22) might promote caerulein-induced apoptosis of pancreatic acinar cells (AR42J) via down-regulating the expression of its target gene, Erb-b2 receptor tyrosine kinase 3 (ErbB3) and the PI3k/Akt signaling pathway. Glucocorticoid receptor transcriptionally repressed the expression of miR-22 by binding to the miR-22 promoter transcription start site. The upregulation of miR-22 expression resulting from silencing Nr3c1 contributed to the apoptosis of AR42J cells.
INTRODUCTION
Acute pancreatitis (AP), which has had high morbidity and mortality rates in recent years, is characterized by acute inflammatory changes in the pancreas and destruction of the acinar cells[1]. Until now, the pathogenesis of AP has remained unclear. Two patterns of pancreatic acinar cell death (apoptosis and necrosis) are involved in AP[2]. Apoptosis is a physiological and programmed form of cell death, and it is thought to be the best method of cell death[3]. The relationship between apoptosis and AP has been extensively investigated, and it has been demonstrated that the severity of AP is inversely related to the rate of apoptosis[4].
MicroRNAs (miRNAs), noncoding small RNAs that are 18 to 24 nucleotides in length, play essential roles in various physiological and pathological processes in animals and plants[5]. By binding to the 3’ untranslated region (UTR) of their target mRNA molecules, miRNAs can downregulate target gene expression and block the translation of mRNA at the posttranscriptional level[6,7]. Recently, many studies have shown that miRNAs are essential to different cellular processes, regulating almost 80% of genes in processes such as development, proliferation, apoptosis, metabolism, and morphogenesis in multiple cell types under physiological and pathological conditions[8,9].
Our previous study showed that microRNA-22 (miR-22) is important in the process of pancreatic acinar cell apoptosis. The upregulation of miR-22 promotes the apoptosis of pancreatic acinar cells induced by tumor necrosis factor alpha (TNF-α). We demonstrated the role of miR-22 in promoting cell apoptosis by repressing its target gene, Erb-b2 receptor tyrosine kinase 3 (ErbB3). However, the underlying mechanism has not been fully elucidated[10]. Currently, most miRNA studies have focused on the regulation of downstream target gene expression, and there have been few studies on upstream miRNA transcription factors[11]. An intergenic miRNA has its own independent transcription start site (TSS), while an intragenic miRNA is generally transcribed with its cohost gene[12]. MiR-22, an exonic miRNA, has its own host gene promoter[13].
In this study, we elucidated the downstream signaling pathways that miR-22 regulates in pancreatic acinar cell apoptosis. Furthermore, we identified the transcriptional promoter of miR-22 and verified its function in pancreatic acinar cell apoptosis.
MATERIALS AND METHODS
MiR-22 mimic, Nr3c1 plasmid encoding the glucocorticoid receptor and si-Nr3c1 construct
The mimic of miR-22, Nr3c1 plasmid encoding the glucocorticoid receptor (GR), and si-Nr3c1 were designed and chemically synthesized by RiboBio (Guangzhou, China).
Cell culture and transfection
The pancreatic acinar cell line AR42J (American Type Culture Collection, United States) was cultured in Dulbecco’s modified Eagle’s medium (DMEM)-F12 (Gibco, United States) containing 20% fetal bovine serum (Gibco, United States) in a humidified incubator. AR42J cells (1 × 106/well) were seeded in 6-well plates 12 h before transfection. The cells were transfected with the Nr3c1 plasmid encoding the GR (100 nmol/L) with Lipofectamine™ 2000 (Invitrogen, United States). The cells were transfected with the miR-22 mimic (100 nmol/L) and si-Nr3c1 (50 nmol/L) using transfection reagents (RiboBio, Guangzhou, China). After transfection for 48 h, the AR42J cells were collected for the next experiment.
Amylase detection
AR42J cells at 5 × 105/well were seeded into 6-well plates and incubated with DMEM-F12 containing 100 nmol/L caerulein (Cae) for 24 h. The supernatant was collected. An amylase assay kit (Jiancheng Bio, Nanjing, China) was used to measure the amylase expression level in the supernatant, following the manufacturer’s instructions.
Flow cytometry analysis of apoptosis
AR42J cells were harvested after treatment with 100 nmol/L Cae for 24 h. Then, the apoptosis rate of AR42J cells was dectected using Annexin V-APC apoptosis kit (KeyGEN Bio, China) according to the manufacturer’s instructions.
Quantitative reverse-transcription polymerase chain reaction
Total RNA was extracted and reverse-transcribed into cDNA using PrimeScript RT Master Mix (Takara, Japan). qRT-PCR was performed using the SYBR Premix Ex TaqTM kit (Takara, Japan). The expression levels of miR-22, ErbB3, and Nr3c1 relative to the expression level of GAPDH were determined using the 2-ΔΔCT method. The specific primer sequences are as follows: ErbB3 (forward), 5’-TCTGCATTAAAGTCATCGAGGAC-3’ and ErbB3 (reverse), 5’-CAGCCGTACAATGTGGGCAT-3’; Nr3c1 (forward), 5’-CTTGAGAAACTTACACCTCGATGACC-3’ and Nr3c1 (reverse), 5’-AGCAGTAGGTAAGGAGATTCTCAACC-3’; and GAPDH (forward), 5’-TCTCTGCTCCTCCCTGTTCT-3’ and GAPDH (reverse), 5’-TACGGCCAAATCCGTTCACA-3’. The miR-22 primer was designed and synthesized by RiboBio (Guangzhou, China).
Western blot analysis
Total protein from cultured AR42J cells was extracted according to the manufacturer’s instructions (Beyotime Bio, Wuhan, China). Forty micrograms of protein of each sample was loaded and separated on a 12% SDS polyacrylamide gel and electrophoretically transferred onto PVDF membranes (Millipore, United States). Then the membranes were incubated with anti-rat monoclonal Bad, Bax, Bcl-xl, Cleaved-Caspase3, PI3k, Akt, p-Akt, GR, (CST, United States), Bcl-2, PI3k-p85α, and ErbB3 antibodies (Santa Cruz Bio, United States) or the anti-β-actin antibody (CST, United States) at 4 °C overnight, and subsequently HRP-labeled secondary antibodies (1:5000) at 37 °C for 2 h; then the signals were visualized with an electrochemiluminescence kit (Pierce, Rockford, IL, United States).
Transcription factor search
The potential miR-22 transcriptional promoter and binding sites were predicted using the online tools (http://www.genomatix.de/; http://jaspar.binf.ku.dk/cgi-bin/jaspar_db.pl; and http://www.gene-regulation.com). The transcriptional promoter with the highest score was chosen for further analysis.
Luciferase reporter assay
AR42J cells were seeded in 24-well plates and, after 24 h, transfected with 100 ng Nr3c1 control plasmid (Nr3c1-NC) or the Nr3c1 plasmid encoding the GR (Nr3C1-OE), together with 50 nmol/L miR-22 promoter plasmid (miR-22 promoter) contained by psiCHECKTM-2 vector (RiboBio, China) with Lipofectamine™ 2000 (Invitrogen, United States); cells transfected with only the miR-22 promoter control plasmid (miR-22 promoter NC) served as the control group. Binding site mutations were generated with mutagenic primers using a MutanBEST Kit (Takara). The mutant miR-22 promoter plasmids (miR-22 promoter mut1 and miR-22 promoter mut2) were cotransfected with Nr3c1-OE. The luciferase activity was measured 48 h after transfection.
Chromatin immunoprecipitation (ChIP) and ChIP-qPCR assays
ChIP assays were performed following the instructions provided with the ChIP assay kit (Beyotime, Wuhan, China). First, the chromatin in AR42J cells was cross-linked with 1% formaldehyde for 10 min at 37 °C, and then the cells were washed three times with cold PBS. The cells were collected, lysed, and sonicated. The nuclear lysates were sonicated, and an equal amount of chromatin was immunoprecipitated at 4 °C overnight with 3 μg of GR and IgG anti-rat monoclonal antibody (CST, United States). The immunoprecipitated products were collected after incubation on protein A + G-coated magnetic beads; then, the beads were washed, and the bound chromatin was eluted in ChIP elution buffer. The protein was digested with proteinase K for 4 h at 45 °C. The DNA was purified using a DNA purification kit (Beyotime). The DNA fragments of the GR binding sites in the miR-22 promoter were designed and synthesized by RiboBio (Guangzhou, China). After immunoprecipitation, the GR binding site was evaluated using qPCR and normalized to the total chromatin. Total chromatin was used as the input. IgG and a non-specific GR binding site (Nbs) were used as controls. The primers used to amplify the DNA fragments of the GR binding sites in the miR-22 promoter were also designed and synthesized by RiboBio (Guangzhou, China). The ChIP-qPCR conditions were based on a three-step method.
Statistical analysis
The results are expressed as the mean ± SD from at least three separate experiments. Statistical analyses were performed using SPSS 22.0 software, and comparisons were made using Student’s t-tests and one-way ANOVA. aP < 0.05, bP < 0.01, and cP < 0.001 was considered statistically significant.
RESULTS
Cae induced apoptosis of AR42J cells
Flow cytometry was used to detect apoptosis of AR42J cells. As shown in Figure 1A, the apoptosis rate of AR42J cells increased significantly after treatment with 100 nmol/L Cae for 24 h. The amylase level in the medium was higher compared with that in the control group (Figure 1B). Compared with the control cells, the Cae-treated cells had increased protein expression levels of Bad, Bax, and cleaved caspase-3 and significantly decreased protein expression levels of Bcl-2 and Bcl-xl (Figure 1C). The expression of miR-22 was confirmed by qRT-PCR. As shown in Figure 1D, the expression level of miR-22 was higher in AR42J cells exposed to Cae for 3 h and 6 h than in the control cells.
Figure 1.
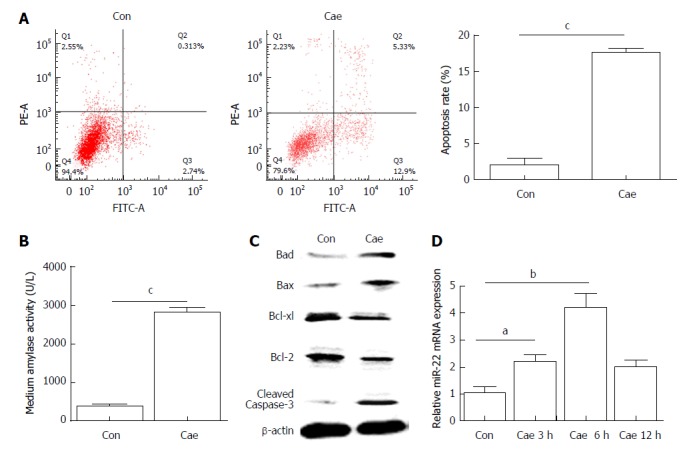
The apoptosis rate and levels of amylase, apoptosis-associated proteins, and microRNA-22 in caerulein-induced AR42J cells. A: The apoptosis rate of AR42J cells after incubation with caerulein for 24 h; B: Amylase levels in the medium; C: Western blot analysis of the levels of apoptosis-associated proteins in AR42J cells; D: MicroRNA-22 levels in AR42J cells. Data were obtained from three independent experiments performed in triplicate and are shown as the mean ± SD. aP < 0.05, bP < 0.01, cP < 0.001 vs control group. Cae: Caerulein; miR-22: MicroRNA-22.
Upregulation of miR-22 promotes the apoptosis of AR42J cells by suppressing the PI3k/Akt signaling pathway
AR42J cells were transfected with the miR-22 mimic as described. The expression of miR-22 was significantly elevated in the cells transfected with the mimic compared with the miRNA NC cells (Figure 2A). As shown in Figure 2B and C, the mRNA and protein expression levels of ErbB3, the target gene of miR-22, were significantly lower in cells overexpressing miR-22 than in the control cells. After AR42J cells were induced with Cae, the apoptosis rate, amylase level, and the protein expression levels of PI3k, p-PI3k, Akt, p-Akt, and apoptosis-associated protein were detected. The apoptosis rate in the miR-22 mimic + Cae group was significantly higher than that in the miRNA NC + Cae group (Figure 2D), while the amylase level did not differ significantly between the two groups (Figure 2E). Compared with the miRNA NC cells, cells with upregulated miR-22 had higher expression levels of Bad, Bax, and activated caspase-3. The expression levels of the proteins that promoted cellular proliferation were clearly reduced in the miR-22 mimic + Cae group. In addition, the upregulation of miR-22 significantly reduced the phosphorylation of PI3k and Akt induced by Cae.
Figure 2.
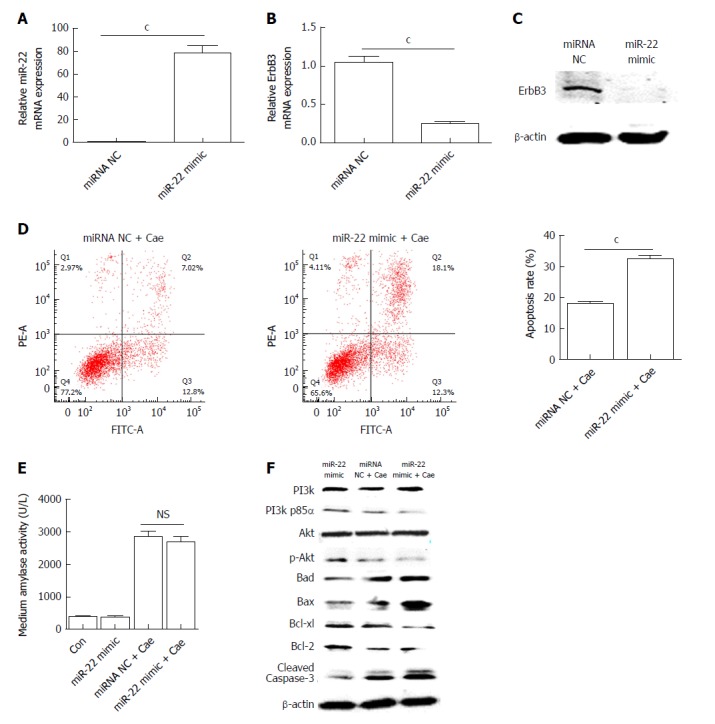
Upregulation of microRNA-22 promotes the apoptosis of AR42J cells by suppressing the PI3k/Akt signaling pathway. A: MicroRNA-22 expression level; B: Erb-b2 receptor tyrosine kinase 3 (ErbB3) mRNA expression level; C: ErbB3 protein expression level; D: The apoptosis rate of AR42J cells induced with caerulein (Cae) after transfection; E: Amylase levels in the medium; F: Western blot analysis of the levels of PI3k, p-PI3k, Akt, p-Akt, and apoptosis-associated proteins in AR42J cells. Data were obtained from three independent experiments performed in triplicate and are shown as the mean ± SD. NSP > 0.05, cP < 0.001 vs miRNA NC or miRNA NC + Cae groups. ErbB3: Erb-b2 receptor tyrosine kinase 3; Cae: Caerulein; miRNA: MicroRNA; miR-22: MicroRNA-22.
Prediction and verification of the transcription factor of miR-22
Using online programs, we predicted the transcription factor and binding sites of the miR-22 promoter. The possible transcription factors are shown in Figure 3A, of which Nr3c1 had the highest score. The 5’-flanking region of miR-22 was cloned into the Xba1-site of the pGL3-luciferase reporter vector, and the empty pGL3-luciferase reporter was used as a control group. The results showed that overexpression of Nr3c1 led to a significant decrease in luciferase activity compared with that of the Nr3c1 NC + miR-22 promoter group (Figure 3B). We further identified the binding sites of Nr3c1 using TF search software. To determine the functional importance of Nr3c1 binding sites in the miR-22 promoter, site-directed mutagenesis was performed. The base sequences of the predicted binding sites and mutated sequences are shown in Figure 3C. The luciferase reporter results demonstrated that compared with the wild-type group, the group with the mutated first binding site had significantly higher luciferase activity, while the group with the mutated second binding site had activity that was significantly higher than that of the wild-type but not as high as that of the group with the mutated first binding site (Figure 3D), indicating that Nr3c1 might bind to the first site (GACAGCCATGTACA) to regulate miR-22 promoter activity. Furthermore, ChIP analysis was performed in AR42J cells to determine whether GR bound to the miR-22 promoter. As shown in Figure 3E, the ChIP and ChIP-qPCR assays showed that GR interacted with the miR-22 promoter within the GACAGCCATGTACA site.
Figure 3.
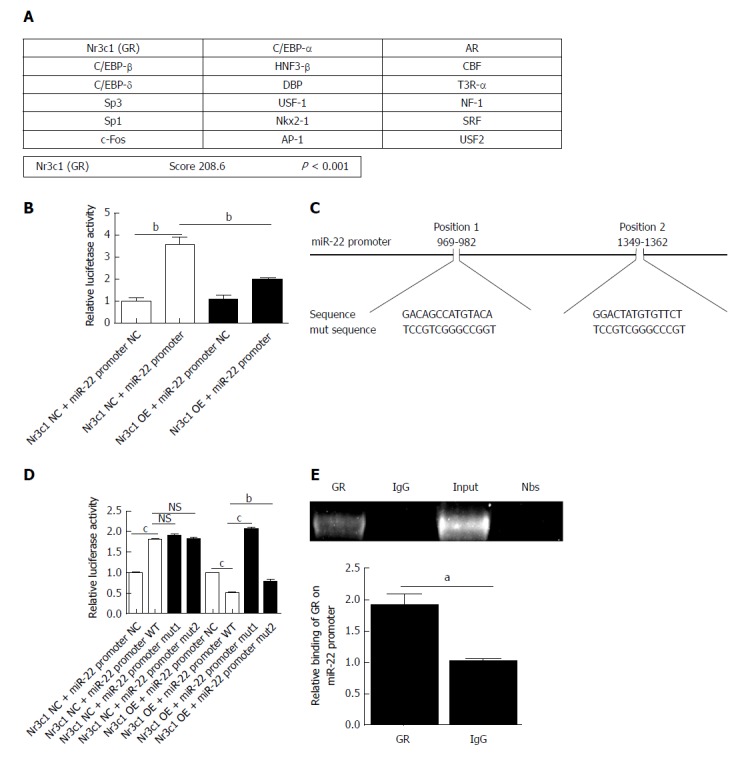
Prediction of the transcription factors of microRNA-22 and the luciferase reporter. A: The possible transcription factors of microRNA-22 (miR-22) were predicted, of which Nr3c1 had the highest score; B: The luciferase reporter expression after the overexpression of Nr3c1; C: The predicted glucocorticoid receptor binding sites within the miR-22 promoter and the mutant versions generated by site mutagenesis are shown; D: The luciferase reporter expression after mutagenesis. MiR-22 promoter NC, mut 1, or mut 2 plasmid was co-transfected with Nr3c1 NC or Nr3c1 OE plasmid into AR42J cells, respectively. Dual luciferase reporter assays were performed 48 h after transfection; E: Results of the chromatin immunoprecipitation (ChIP) assay and ChIP-qPCR. Data were obtained from three independent experiments performed in triplicate and are shown as the mean ± SD. NSP > 0.05, aP < 0.05, bP < 0.01, cP < 0.001 vs Nr3c1 NC + miR-22 promoter NC, Nr3c1 NC + miR-22 promoter, Nr3c1 NC + miR-22 promoter WT, Nr3c1 OE + miR-22 promoter NC, Nr3c1 OE + miR-22 promoter WT or IgG groups. ChIP: Chromatin immunoprecipitation; GR: Glucocorticoid receptor; mut: Mutagenesis; OE: Overexpression; miR-22: MicroRNA-22.
Nr3c1 regulates the expression of miR-22
To investigate the influence of Nr3c1 on the expression of miR-22, the Nr3c1 plasmid encoding the GR and si-Nr3c1 were used to regulate the expression of Nr3c1. As shown in Figure 4A, overexpression of Nr3c1 significantly reduced miR-22 expression compared with that of the control. In contrast, si-Nr3c1 downregulated the expression levels of Nr3c1 mRNA and the GR protein, which promote the expression of miR-22 (Figure 4B).
Figure 4.
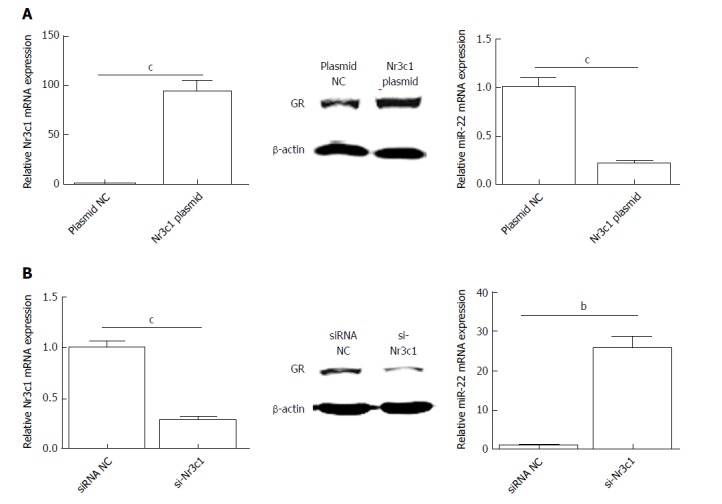
Expression of Nr3c1 mRNA, glucocorticoid receptor protein, and microRNA-22 level after transfection. A: The levels of Nr3c1 mRNA, GR protein, and miR-22 after transfection with Nr3c1 plasmid; B: The levels of Nr3c1 mRNA, glucocorticoid receptor protein, and miR-22 after transfection with si-Nr3c1. Data were obtained from three independent experiments in triplicate and are shown as the mean ± SD. bP < 0.01, cP < 0.001 vs plasmid NC or siRNA NC groups. GR: Glucocorticoid receptor; miR-22: MicroRNA-22.
Si-Nr3c1 promotes the apoptosis of AR42J cells
Downregulation of Nr3c1 promoted the expression of miR-22. The genes involved in the pathway downstream of miR-22 that promoted apoptosis were detected. As shown in Figure 5A and B, the mRNA and protein expression levels of ErbB3 were significantly lower when miR-22 was upregulated than in the control. After AR42J cells were induced with Cae, the apoptosis rate increased significantly (Figure 5C). However, the amylase level was not significantly different between the two groups (Figure 5D). As shown in Figure 5E, the expression levels of p-PI3k and p-Akt were clearly lower in the cells transfected with si-Nr3c1 than in the siRNA NC group. Downregulation of Nr3c1 increased the expression of Bad, Bax, and activated caspase-3, while Bcl-2 and Bcl-xl protein expression levels were obviously reduced. Together, these data suggest that downregulation of Nr3c1 promoted the expression of miR-22 and the apoptosis of AR42J cells.
Figure 5.
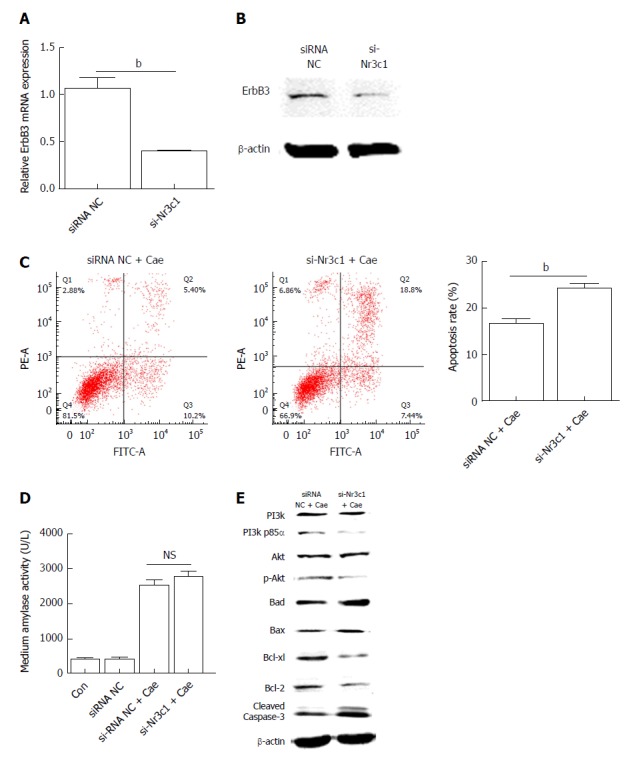
Down-regulation of Nr3c1 by using si-Nr3c1 promotes the apoptosis of AR42J cells by suppressing the PI3k/Akt signaling pathway. A: Erb-b2 receptor tyrosine kinase 3 (ErbB3) mRNA expression level; B: ErbB3 protein level; C: The apoptosis rate of AR42J cells after the effect of caerulein for 24 h; D: Amylase level in medium; E: Western blot analysis for p-PI3k, p-Akt, and apoptosis associated proteins in AR42J cells. Data were obtained from three independent experiments in triplicate and are shown as the mean ± SD. bP < 0.01 vs siRNA NC groups. ErbB3: Erb-b2 receptor tyrosine kinase 3; Cae: Caerulein; miR-22: MicroRNA-22.
DISCUSSION
Some studies have demonstrated that miR-22 plays important roles in regulating the expression levels of its target genes and that it is associated with various diseases, such as autoimmune diseases, cardiovascular diseases, emphysema, and cancer[14-18]. Our previous results showed that the expression of miR-22 is clearly higher in acute edema pancreatitis (AEP) in vivo. Elevating miR-22 expression using a miR-22 mimic could promote the activity of activated caspase-3 and the rate of apoptosis of pancreatic acinar cells (AR42J) induced with TNF-α in vitro. Moreover, the target genes of miR-22 were predicted by bioinformatics, and the luciferase reporter gene confirmed that ErbB3 was the target gene of miR-22[10]. Furthermore, we identified the signaling pathway by which miR-22 regulates the apoptosis of AR42J cells. The results demonstrated that miR-22 represses the expression of its target gene ErbB3. ErbB3, which belongs to the ErbB family, can be transactivated by forming heterodimers with other ErbB family members, especially ErbB2. ErbB3 lacks intrinsic kinase activity and cannot autophosphorylate due to the evolutionary acquisition of several changes within the kinase domain[19,20].
Among the members of the ErbB family, ErbB3 has the highest affinity for PI3k because of its six YXXM motifs that can directly bind to the p85 regulatory subunit of PI3k after tyrosine phosphorylation of ErbB3[21,22]. Therefore, the activation of ErbB3 results in a strong activation of the PI3k/Akt signaling pathway[23]. The activation of the PI3k/Akt signaling pathway can lead to apoptosis resistance in cancers such as ovarian, thyroid, breast, hepatic, cervical, prostate, lung, pancreatic, and colon cancers. Many studies have demonstrated that inhibiting the activation of PI3k/Akt may lead to cell apoptosis[24-31]. Our results showed that miR-22 could upregulate the expression of genes that promote apoptosis and reduce the expression of genes that promote proliferation by suppressing the phosphorylation of PI3k and Akt in Cae-induced apoptosis, which results in promoting the activity of caspase 3. Caspases are a family of cysteine proteases that are present in the cytosol as inactive proenzymes, and they become activated when apoptosis is initiated, playing essential roles in various stages of apoptosis[32]. MiR-22 might promote the apoptosis of AR42J cells by repressing the PI3k/Akt pathway via inhibition of its target gene, ErbB3.
Transcription factors are a group of proteins that bind to a specific sequence at the 5’-end of a gene to ensure that the target gene is expressed at a specific time, in a specific location, and with a specific intensity. In fact, transcriptional regulators play pivotal roles during developmental and pathophysiological processes[33,34]. MiR-22, which belongs to the category of intergenic miRNAs, has its own independent transcription factors. Recent studies have shown that the transcription factors of miR-22 include Jak3, STAT3, STAT5, and FosB. FosB promotes the expression of miR-22, while Jak3, STAT3, and STAT5 are transcriptional repressors[35,36]. In this study, to identify the miR-22 transcription factor involved in regulating the apoptosis of pancreatic acinar cells, we first predicted the transcription factor and TSS. The prediction results showed that Nr3c1 might be a transcription factor regulating miR-22. Nr3c1 is a vital GR gene; it can receive stimulation from glucocorticoids and then influence downstream transcription factors by changing the protein configuration, thereby playing an indispensable role in gene regulation[37]. We cloned the 5’ flanking regions of miR-22 and analyzed the promoter regions. The first site (GACAGCCATGTACA) demonstrated the highest promoter activity, as measured by the luciferase reporter assay. The results of the site-directed mutagenesis and ChIP-qPCR confirmed that Nr3c1 binds to the miR-22 core promoter. The upregulation of miR-22 expression resulting from silencing Nr3c1 contributed to the apoptosis of AR42J cells. Apoptosis is a physiological and programmed form of cell death, which is considered the best method of cell death. It is characterized by cell shrinkage, nuclear chromatin condensation, and the retention of organelles[38]. The severity of AP is inversely related to the degree of apoptosis, suggesting that apoptosis may be a teleologically beneficial response to acinar cell injury in general and especially in AP[39].
In conclusion, our results indicated that GR transcriptionally repressed the expression of miR-22 by binding to the miR-22 promoter TSS. Downregulating the expression of GR could promote the expression of miR-22. The upregulation of miR-22 promoted Cae-induced apoptosis of AR42J cells by targeting ErbB3 and further suppressed the PI3k/Akt signaling pathway. The upregulation of miR-22 might have therapeutic potential for AP.
ARTICLE HIGHLIGHTS
Research background
The severity of acute pancreatitis (AP) is inversely related to the rate of apoptosis of pancreatic acinar cells. Our previous study showed that microRNA-22 (miR-22) promotes the apoptosis of pancreatic acinar cells by targeting Erb-b2 receptor tyrosine kinase 3 (ErbB3). However, the underlying mechanism has not been fully elucidated, and the elements that regulate the expression of miR-22 remain unclear.
Research motivation
The downstream signaling pathways that miR-22 regulates pancreatic acinar cell apoptosis have not been fully elucidated. Besides, miR-22 is an exonic microRNA and has its own host gene promoter. In this study, we identified the transcriptional promoter of miR-22 and verified their functions in pancreatic acinar cell apoptosis.
Research objectives
This research aimed to elucidate the underlying mechanism that miR-22 promotes the apoptosis of rat pancreatic acinar cells (AR42J) and identify the transcriptional promoter of miR-22.
Research methods
MiR-22 promoted the apoptosis of AR42J cells by targeting the ErbB3 gene, and the downstream signaling pathway (PI3k/Akt signaling pathway) was identified using caerulein (Cae)-induced apoptosis of AR42J cells. Furthermore, we predicted the potential transcription promoter of miR-22 and the binding sites using online tools. Luciferase reporter analysis and site-directed mutagenesis indicated the binding site of the glucocorticoid receptor (GR). The binding of GR to the miR-22 promoter in cell culture was identified by a chromatin immunoprecipitation assay.
Research results
The results of this study indicated that GR transcriptionally repressed the expression of miR-22 by binding to the miR-22 promoter transcription start site. Downregulation of the expression of GR could upregulate the expression of miR-22. The upregulation of miR-22 promoted the Cae-induced apoptosis of AR42J by targeting ErbB3 and further suppressing the PI3k/Akt signaling pathway.
Research conclusions
GR transcriptionally repressed the expression of miR-22 and downregulation of the expression of GR could upregulate the expression of miR-22, which further promoted the Cae-induced apoptosis of AR42J cells.
Research perspectives
This study found that GR transcriptionally repressed the expression of miR-22, which might be a target to regulate the expression of miR-22. The further research is to explore the treatment measures for AP by using drugs targeting GR in in vitro cell models and in vivo AP models.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B, B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Institutional review board statement: This study was reviewed and approved by the Ethics Committee of People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital).
Conflict-of-interest statement: We declare that we have no financial and personal relationships with other people or organizations that can inappropriately influence our work, and there is no professional or other personal interest of any nature or kind in any products, service and/or company that could be construed as influencing the position presented in, or the review of, the manuscript.
Data sharing statement: No additional data are available.
ARRIVE guidelines statement: The manuscript has been revised according to the ARRIVE guidelines.
Peer-review started: September 5, 2018
First decision: October 24, 2018
Article in press: November 13, 2018
P- Reviewer: Demonacos C, Gonzalez A, Mendez I S- Editor: Ma RY L- Editor: Wang TQ E- Editor: Bian YN
Contributor Information
Qiang Fu, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Chuan-Jiang Liu, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Xu Zhang, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Zhen-Sheng Zhai, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Yu-Zhu Wang, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Ming-Xing Hu, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Xian-Ling Xu, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Hong-Wei Zhang, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China.
Tao Qin, Department of Hepatobiliary and Pancreatic Surgery, People’s Hospital of Zhengzhou University (Henan Provincial People’s Hospital), School of Medicine, Zhengzhou University, Zhengzhou 450003, Henan Province, China. m18937638396@163.com.
References
- 1.Pan LL, Li J, Shamoon M, Bhatia M, Sun J. Recent Advances on Nutrition in Treatment of Acute Pancreatitis. Front Immunol. 2017;8:762. doi: 10.3389/fimmu.2017.00762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mareninova OA, Sung KF, Hong P, Lugea A, Pandol SJ, Gukovsky I, Gukovskaya AS. Cell death in pancreatitis: caspases protect from necrotizing pancreatitis. J Biol Chem. 2006;281:3370–3381. doi: 10.1074/jbc.M511276200. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs Y, Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat Rev Mol Cell Biol. 2015;16:329–344. doi: 10.1038/nrm3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bhatia M. Apoptosis versus necrosis in acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2004;286:G189–G196. doi: 10.1152/ajpgi.00304.2003. [DOI] [PubMed] [Google Scholar]
- 5.Khan S, Ansarullah, Kumar D, Jaggi M, Chauhan SC. Targeting microRNAs in pancreatic cancer: microplayers in the big game. Cancer Res. 2013;73:6541–6547. doi: 10.1158/0008-5472.CAN-13-1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–379. doi: 10.1146/annurev-biochem-060308-103103. [DOI] [PubMed] [Google Scholar]
- 7.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 8.Mahmoudi E, Cairns MJ. MiR-137: an important player in neural development and neoplastic transformation. Mol Psychiatry. 2017;22:44–55. doi: 10.1038/mp.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Qin T, Fu Q, Pan YF, Liu CJ, Wang YZ, Hu MX, Tang Q, Zhang HW. Expressions of miR-22 and miR-135a in acute pancreatitis. J Huazhong Univ Sci Technolog Med Sci. 2014;34:225–233. doi: 10.1007/s11596-014-1263-7. [DOI] [PubMed] [Google Scholar]
- 11.Berindan-Neagoe I, Monroig Pdel C, Pasculli B, Calin GA. MicroRNAome genome: a treasure for cancer diagnosis and therapy. CA Cancer J Clin. 2014;64:311–336. doi: 10.3322/caac.21244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He C, Li Z, Chen P, Huang H, Hurst LD, Chen J. Young intragenic miRNAs are less coexpressed with host genes than old ones: implications of miRNA-host gene coevolution. Nucleic Acids Res. 2012;40:4002–4012. doi: 10.1093/nar/gkr1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A. Identification of mammalian microRNA host genes and transcription units. Genome Res. 2004;14:1902–1910. doi: 10.1101/gr.2722704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang B, Yao Q, Xu D, Zhang JA. MicroRNA-22-3p as a novel regulator and therapeutic target for autoimmune diseases. Int Rev Immunol. 2017;36:176–181. doi: 10.1080/08830185.2017.1281272. [DOI] [PubMed] [Google Scholar]
- 15.Huang ZP, Wang DZ. miR-22 in Smooth Muscle Cells: A Potential Therapy for Cardiovascular Disease. Circulation. 2018;137:1842–1845. doi: 10.1161/CIRCULATIONAHA.118.033042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brusselle GG, Bracke KR. MicroRNA miR-22 drives T(H)17 responses in emphysema. Nat Immunol. 2015;16:1109–1110. doi: 10.1038/ni.3295. [DOI] [PubMed] [Google Scholar]
- 17.Mansini AP, Lorenzo Pisarello MJ, Thelen KM, Cruz-Reyes M, Peixoto E, Jin S, Howard BN, Trussoni CE, Gajdos GB, LaRusso NF, et al. MicroRNA (miR)-433 and miR-22 dysregulations induce histone-deacetylase-6 overexpression and ciliary loss in cholangiocarcinoma. Hepatology. 2018;68:561–573. doi: 10.1002/hep.29832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang X, Hu C, Arnovitz S, Bugno J, Yu M, Zuo Z, Chen P, Huang H, Ulrich B, Gurbuxani S, et al. miR-22 has a potent anti-tumour role with therapeutic potential in acute myeloid leukaemia. Nat Commun. 2016;7:11452. doi: 10.1038/ncomms11452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pellat A, Vaquero J, Fouassier L. Role of ErbB/HER family of receptor tyrosine kinases in cholangiocyte biology. Hepatology. 2017 doi: 10.1002/hep.29350. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 20.Yarden Y, Sliwkowski MX. Untangling the ErbB signalling network. Nat Rev Mol Cell Biol. 2001;2:127–137. doi: 10.1038/35052073. [DOI] [PubMed] [Google Scholar]
- 21.Soltoff SP, Carraway KL 3rd, Prigent SA, Gullick WG, Cantley LC. ErbB3 is involved in activation of phosphatidylinositol 3-kinase by epidermal growth factor. Mol Cell Biol. 1994;14:3550–3558. doi: 10.1128/mcb.14.6.3550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HH, Sierke SL, Koland JG. Epidermal growth factor-dependent association of phosphatidylinositol 3-kinase with the erbB3 gene product. J Biol Chem. 1994;269:24747–24755. [PubMed] [Google Scholar]
- 23.Hennessy BT, Smith DL, Ram PT, Lu Y, Mills GB. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]
- 24.Chen C, Chang YC, Lan MS, Breslin M. [Corrigendum] Leptin stimulates ovarian cancer cell growth and inhibits apoptosis by increasing cyclin D1 and Mcl-1 expression via the activation of the MEK/ERK1/2 and PI3K/Akt signaling pathways. Int J Oncol. 2016;49:847. doi: 10.3892/ijo.2016.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazumdar M, Adhikary A, Chakraborty S, Mukherjee S, Manna A, Saha S, Mohanty S, Dutta A, Bhattacharjee P, Ray P, et al. Targeting RET to induce medullary thyroid cancer cell apoptosis: an antagonistic interplay between PI3K/Akt and p38MAPK/caspase-8 pathways. Apoptosis. 2013;18:589–604. doi: 10.1007/s10495-013-0803-0. [DOI] [PubMed] [Google Scholar]
- 26.Yuan L, Wang J, Xiao H, Xiao C, Wang Y, Liu X. Isoorientin induces apoptosis through mitochondrial dysfunction and inhibition of PI3K/Akt signaling pathway in HepG2 cancer cells. Toxicol Appl Pharmacol. 2012;265:83–92. doi: 10.1016/j.taap.2012.09.022. [DOI] [PubMed] [Google Scholar]
- 27.Kim MS, Kim JH, Bak Y, Park YS, Lee DH, Kang JW, Shim JH, Jeong HS, Hong JT, Yoon DY. 2,4-bis (p-hydroxyphenyl)-2-butenal (HPB242) induces apoptosis via modulating E7 expression and inhibition of PI3K/Akt pathway in SiHa human cervical cancer cells. Nutr Cancer. 2012;64:1236–1244. doi: 10.1080/01635581.2012.718405. [DOI] [PubMed] [Google Scholar]
- 28.Harashima N, Inao T, Imamura R, Okano S, Suda T, Harada M. Roles of the PI3K/Akt pathway and autophagy in TLR3 signaling-induced apoptosis and growth arrest of human prostate cancer cells. Cancer Immunol Immunother. 2012;61:667–676. doi: 10.1007/s00262-011-1132-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou L, Luan H, Liu Q, Jiang T, Liang H, Dong X, Shang H. Activation of PI3K/Akt and ERK signaling pathways antagonized sinomenine-induced lung cancer cell apoptosis. Mol Med Rep. 2012;5:1256–1260. doi: 10.3892/mmr.2012.798. [DOI] [PubMed] [Google Scholar]
- 30.Hu W, Shen T, Wang MH. Cell cycle arrest and apoptosis induced by methyl 3,5-dicaffeoyl quinate in human colon cancer cells: Involvement of the PI3K/Akt and MAP kinase pathways. Chem Biol Interact. 2011;194:48–57. doi: 10.1016/j.cbi.2011.08.006. [DOI] [PubMed] [Google Scholar]
- 31.Roy SK, Srivastava RK, Shankar S. Inhibition of PI3K/AKT and MAPK/ERK pathways causes activation of FOXO transcription factor, leading to cell cycle arrest and apoptosis in pancreatic cancer. J Mol Signal. 2010;5:10. doi: 10.1186/1750-2187-5-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Budihardjo I, Oliver H, Lutter M, Luo X, Wang X. Biochemical pathways of caspase activation during apoptosis. Annu Rev Cell Dev Biol. 1999;15:269–290. doi: 10.1146/annurev.cellbio.15.1.269. [DOI] [PubMed] [Google Scholar]
- 33.Niwa H. The principles that govern transcription factor network functions in stem cells. Development. 2018:145. doi: 10.1242/dev.157420. [DOI] [PubMed] [Google Scholar]
- 34.Omatsu Y, Nagasawa T. The critical and specific transcriptional regulator of the microenvironmental niche for hematopoietic stem and progenitor cells. Curr Opin Hematol. 2015;22:330–336. doi: 10.1097/MOH.0000000000000153. [DOI] [PubMed] [Google Scholar]
- 35.Sibbesen NA, Kopp KL, Litvinov IV, Jønson L, Willerslev-Olsen A, Fredholm S, Petersen DL, Nastasi C, Krejsgaard T, Lindahl LM, et al. Jak3, STAT3, and STAT5 inhibit expression of miR-22, a novel tumor suppressor microRNA, in cutaneous T-Cell lymphoma. Oncotarget. 2015;6:20555–20569. doi: 10.18632/oncotarget.4111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahmad HM, Muiwo P, Muthuswami R, Bhattacharya A. FosB regulates expression of miR-22 during PMA induced differentiation of K562 cells to megakaryocytes. Biochimie. 2017;133:1–6. doi: 10.1016/j.biochi.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 37.Palma-Gudiel H, Córdova-Palomera A, Leza JC, Fañanás L. Glucocorticoid receptor gene (NR3C1) methylation processes as mediators of early adversity in stress-related disorders causality: A critical review. Neurosci Biobehav Rev. 2015;55:520–535. doi: 10.1016/j.neubiorev.2015.05.016. [DOI] [PubMed] [Google Scholar]
- 38.Kiraly G, Simonyi AS, Turani M, Juhasz I, Nagy G, Banfalvi G. Micronucleus formation during chromatin condensation and under apoptotic conditions. Apoptosis. 2017;22:207–219. doi: 10.1007/s10495-016-1316-4. [DOI] [PubMed] [Google Scholar]
- 39.Kaiser AM, Saluja AK, Sengupta A, Saluja M, Steer ML. Relationship between severity, necrosis, and apoptosis in five models of experimental acute pancreatitis. Am J Physiol. 1995;269:C1295–C1304. doi: 10.1152/ajpcell.1995.269.5.C1295. [DOI] [PubMed] [Google Scholar]