

A procedure for the quantitative mapping of sucrose in plant tissue with spatial resolution sufficient to measure sucrose in individual phloem bundles and across transport boundaries.
Abstract
Sucrose (Suc) is the major transport sugar in plants and plays a primary role as an energy source and signal in adaptive and stress responses. An ability to quantify Suc over time and space would serve to advance our understanding of these important processes. Current technologies used for Suc mapping are unable to quantitatively visualize its distribution within tissues. Here, we present an infrared-based microspectroscopic method that allows for the quantitative visualization of Suc at a microscopic level of resolution (∼12 µm). This method can successfully model the sugar concentration in individual vascular bundles and within a complex organ such as the stem, leaf, or seed. The sensitivity of the assay ranges from 20 to 1,000 mm. We applied this method to the cereal crop barley (Hordeum vulgare) and the model plant Arabidopsis (Arabidopsis thaliana) to highlight the potential of the procedure for resolving the spatial distribution of metabolites. We also discuss the relevance of the method for studies on carbon allocation and storage in the context of crop improvement.
Suc is the principal transport form of assimilates in the majority of plant species. Synthesized in the cytosol of mesophyll cells (source organ), it is transported to the leaf vasculature, from where it is distributed toward the various sink organs of the plant (Lalonde et al., 2004; Ruan, 2014). Suc affects almost all processes in plants due to its primary role in growth and as an energy source. In addition, Suc also is a relevant signal sensed by sophisticated signaling networks and involved in various adaptive and stress responses (Moghaddam and Van den Ende, 2013; Smeekens and Hellmann, 2014). Last but not least, Suc is the major storage form of sugars in plants. For human consumption, it is derived primarily from the sugar crops sugarcane (Saccharum spp.) and sugar beet (Beta vulgaris), in which sugar can account for up to 20% of the plant’s fresh weight.
Efforts toward an improved understanding of the metabolism, transport, and storage of Suc are central to countless research projects, including breeding and biotechnological attempts. An ability to visualize and quantify Suc over time and space (i.e. Suc mapping) would serve to advance our understanding of relevant growth- and yield-related processes. However, current technologies in use for Suc mapping are inadequate where there is a need to quantitatively visualize its distribution within a tissue. Mass spectrometry-based imaging like the matrix-assisted laser-desorption ionization technique can detect various classes of metabolites (Boughton et al., 2016; Murray et al., 2016), but its level of resolution and specificity is compromised when the detection target is, like Suc, of low Mr (issues with matrix effects). Positron emission tomography has been used to trace photoassimilate allocation, but so far it has been unable to demonstrate a sufficient level of chemical or spatial resolution when targeted to Suc (Partelová et al., 2017). Magnetic resonance imaging (MRI) is able to measure Suc in a label-free manner in vivo (Rolletschek et al., 2011), but its imaging is difficult when Suc is present at a low concentration (Borisjuk et al., 2012). The fluorescence resonance energy transfer (FRET) method, in principle, provides a high level of chemical and spatial resolution (Niittylae et al., 2009). Suc-specific FRET sensors are available and used, for example, to determine Suc transport activity (Chen et al., 2012; Bezrutczyk et al., 2018). However, in addition to some technical limitations, their practicality in the crop context is questionable, given the need to genetically transform the plant. Finally, quantitative bioluminescence has been applied for the visualization of Suc gradients within complex seed tissues (Borisjuk et al., 2002). That was actually the first visualization of Suc distribution in plants. The assay relies on complex enzymatic cocktails developed to specifically measure Suc directly in tissue sections via bioluminescence and single-photon counting. While enabling Suc imaging with a resolution close to the single-cell level, the method remained technically challenging and with limited versatility in application. It was further disadvantaged by not supporting a reasonable sample throughput.
Fourier-transform infrared (FTIR) spectroscopy has found widespread applications in medical research, bacteriology, and plant and food science (Baker et al., 2014; Salzer and Siesler, 2014). Infrared spectroscopy offers substantial analytical potential and when coupled to a microscope provides, besides chemical, also spatial resolution at the low-micrometer level. However, its exploitation for the quantitative analysis of metabolite distribution in tissue sections is so far limited by the complexity of the resulting spectral matrix, which obscures specific metabolite signatures. Therefore, it requires an algorithmic decomposition to recover a given compound-specific signal. In addition, means to standardize the analytical procedure and to reproduce the data quantitatively are lacking. To date, FTIR microscopy has not been used to image Suc distribution in plants.
The incentive to develop a novel imaging method was the need to obtain a quantitative visualization of the distribution of Suc within heterogenous tissues. The method should be versatile and easily applicable to crops (i.e. not depending on prior genetic transformation). This article describes an imaging platform based on FTIR spectroscopy and demonstrates its analytical power for the analysis of cereal crops as well as some Arabidopsis (Arabidopsis thaliana) plants. The imaging proved able to quantitatively model Suc distribution in the various plant organs at a resolution of ∼12 µm and with high mapping accuracy. Imaging data acquired for developing cereal grains were in accordance with previous assumptions, while the analysis of Arabidopsis plants uncovered some unexpected patterning of Suc in the hypocotyl of sweet mutants.
RESULTS
Experimental Design for Quantitative Imaging of Suc
The basis of the procedure is depicted in Figure 1. The sample is initially snap frozen in liquid nitrogen, then cryosectioned and placed on a membrane support. The use of a membrane (rather than the more conventional glass) to support the tissue sections allows for measurements to be made in the mid-infrared region (less than 2,500 cm−1), a part of the spectrum that is masked by glass (Fig. 1A). Along with providing a wider observation window, the membrane also is less expensive than the BaF2 platform that is generally recommended for FTIR microscopy. Most importantly, the membrane preserves a unique spectral fingerprint, thereby introducing a ubiquitous and spatially homogenous internal standard to the sample prior to imaging. This is similar to what is used routinely in chromatography and allows the spectral data to be normalized on the fingerprint of the internal standard. It ultimately enhances quality and enables comparability of the modeled data. The reliability of the internal standard could be verified experimentally (Supplemental Fig. S1): the level of variation in the signal emanating from the membrane was around 1% of the overall signal, thereby providing a highly consistent standard within each pixel of a given image. The spectral raw data represented in Figure 1B (the spectral extract of a hyperspectral data cube) show the characteristic membrane fingerprint in individual spectra but also comprise strong baseline shifts, which can be extracted readily using a baseline correction algorithm (Mie scattering extended multiplicative signal correction; see “Materials and Methods”). To achieve this, the spectral cube is subdivided into smaller pieces via K-means clustering to enhance spectral similarity in the smaller data sets. A data set derived from the spectra generated from relevant pure analytes (i.e. the external standards) is then added to each piece (Supplemental Fig. S2). After subsequent modeling, the standards are compared with their unprocessed counterparts, allowing the fit of the model to be reevaluated after each round of fitting. The procedure generates a coefficient matrix, which describes the composition of each sample spectrum from the modeled features (Fig. 1, C and D). In order to quantitatively compare these features across different samples/measurements, the matrix coefficients have to be normalized against the internal standard. The overall duration of the FTIR analysis depends on sample complexity. For example, it takes 5 h for analysis of a wheat (Triticum spp.) stem sample and up to 11 h for a cereal caryopsis sample (excluding the time needed for lyophilization).
Figure 1.

Schematic overview of the method used to acquire and analyze FTIR spectra. The left-hand images illustrate the experimental setup, while the right-hand images show average spectra derived from a glass microscope slide and a membrane slide (A), sample raw spectra from 16-µm-thick tissue sections, illustrating variation in the baseline (B), modeled baseline artifacts after four rounds of baseline correction using a partial least squares regression model (C), and baseline-corrected spectra revealing the chemical fingerprint of the sample and the internal standard after six rounds of baseline feature extraction (D). EMSC, Extended multiplicative signal correction. Abbreviations in the equation are as follows: Absn,pixel, Normalized pixel absorbance; ci, coefficient of compound I; cAbs,MMI, coefficient of membrane; Absi, spectrum derived from purified compound I; E, noise.
The ability to deliver spatially resolved information on metabolite levels is the key advantage of our method. As with any microscope, the spatial resolution (δ) is diffraction limited and, therefore, determined by the wavelength (λ) of the light in combination with physical properties (numerical aperture [NA]) of the lens. The distance between two points of a sample that can be separated is given by the formula δ(λ) = 0.61 × λ/NA. Our FTIR microscope was equipped with a 15× objective with a numerical aperture of 0.6. The resulting optical resolution within the detectable range of the detector (3,800–850 cm−1) increases according to the wavelength up to 12 µm at the far end of the detectable spectrum (Supplemental Fig. S3). The fingerprint features from 1, 800 to 1,000 cm−1 are most important for our computational approach and provide an optical resolution from 6 to 10 µm. Further increases in resolution are possible when applying different optics with a higher numerical aperture. For our instrument, two objectives are available, and the attainable spatial resolution is demonstrated in Supplemental Figure S3. It becomes clear that the resolution can be improved further to ∼2 µm when using the ATR (20×) objective. In that case, however, large-area mappings become more difficult.
Training and Validation of the Model
A validation of the model with respect to the component-specific data was performed using a three-step procedure. Initially, an in silico data set was calculated for pure Suc, subsequently used to test the effectiveness of the feature-extraction procedure (for a detailed description, see “Materials and Methods”) on a pixel-by-pixel resolution. The resulting component-specific images (Supplemental Fig. S4A) were reproduced with an R2 value greater than 0.9; there was hardly any interference with the spectra generated from the other compounds present in the matrix. The experimental data and the calculated signal intensities were clearly linearly correlated (Supplemental Fig. S4, B–D). Next, a Suc dilution series was established to estimate the correlation between known concentrations and absorbance values per imaged area: this indicated a high level of confidence, as measured by the R2 parameter (greater than 0.98) and the root mean square error of prediction (RMSEP = 0.036 pg µm−2; Fig. 2C). Finally, tissue sections imaged previously by FTIR were laser dissected, and the resulting fractions were analyzed for their Suc content by ion chromatography coupled to pulsed amperometric detection (Supplemental Fig. S5). The modeled versus experimental data sets were significantly correlated (R2 > 0.98, RMSEP = 0.016 pg µm−2; Fig. 2D). The conclusion was that the trained model accurately displayed the component-specific information and was able to reliably quantify the Suc concentration. Based on the presented data, the linear range was experimentally documented up to 3.3 pg µm−2, and the limit of detection calculated as baseline plus 3 times the RMSEP is 0.108 pg µm−2. This corresponds to a range from 19.7 to 608.9 mm Suc concentration (at a tissue section thickness of 16 µm). The theoretical signal saturation is reached at absorbance values close to 2. Therefore, an absorbance up to 1.5 still can be considered as a safe upper limit of detection and would allow for Suc concentrations up to 1,062 mm in 16-µm-thick tissue sections. Please note that the unit for Suc concentration given in the images (pg µm−2) can be translated into more conventional mass/volume data (see “Materials and Methods”).
Figure 2.

Quantitative imaging of Suc in the barley stem. A, The barley spike and a cross section through the stem enveloped by the flag leaf’s sheath. B, Color map used to visualize Suc gradients across the stem. The FTIR image was scaled to coincide with the resolution of the visual image (shown as an inset), which demonstrates the cellular structure of a vascular bundle. Phi, Inner vasculature phloem; PHo, outer vasculature phloem; XL, inner vasculature xylem. Bar = 50 µm. C, Plot showing the correlation between the relative absorbance per pixel and the Suc concentration per unit of area measured using in situ Suc standards. The in silico-generated data (shown in gray) indicate single-pixel variances (pixel size of 2.75 × 2.75 µm). D, Correlation between Suc concentrations as estimated by FTIR imaging and as measured by ion chromatography. Tissue section thickness was 16 µm.
Visualization of Local Suc Accumulation in Vascular Bundles and Surrounding Tissues in the Leaf and Stem of Barley Plants
The next priority was to test the capacity of the procedure to visualize Suc distribution in barley (Hordeum vulgare), which represents an important cereal crop model. Analysis of stem tissue samples clearly revealed that the phloem strands were associated with the highest Suc content (Fig. 2B); the levels were much lower in the cells associated with the xylem and were undetectable within the xylem itself. There was a moderate concentration of Suc in both the chlorenchyma and sclerenchyma cells lying around the stem periphery, with a negative gradient established from the outside to the center of the stem. Notably, there was some variation in the concentration of Suc in the vascular bundles at a given internode: the level was significantly lower in the outer as compared with the inner vascular ring (Supplemental Fig. S6, D and E). Such variation has been unknown before and could indicate some distinct functional properties of the respective phloem bundles. Possibly in relation to this, we observed that the diameter of inner vascular bundles was bigger than that of the outer bundles and surrounded by a thicker layer of sclerenchymal cells.
In the flag leaf, the highest levels of Suc also were associated with the phloem strands (Supplemental Fig. S6, B and C); the mean concentrations were slightly elevated compared with those estimated in the stem phloem at the same internode. The level of Suc was much lower in the leaf mesophyll cells and was slightly higher in cell layers lying closest to the epidermis.
Quantitative Imaging of Suc Distribution within the Developing Barley Grain
The Suc visualization procedure also was applied to the developing caryopsis of barley (Fig. 3). The resulting Suc maps featured a marked gradient across the caryopsis. High concentrations were associated with the ventral part of the caryopsis (Fig. 3, A and B), where phloem unloading takes place (Melkus et al., 2011). In the pericarp, high concentrations were evident in the vascular bundle and surrounding tissue, but levels decreased 2- to 5-fold toward the lateral and dorsal regions. High concentrations also were evident in the endosperm facing the vascular region of the pericarp, but the highest concentrations were found within the endosperm transfer cells (ETC). A steep Suc gradient was formed from the ETC toward the central endosperm, with minimum levels at the periphery (graph in Fig. 3C). The Suc quantification along the allocation pathway further indicates a slight drop in concentration from the vascular bundle toward the nucellar projection, while at the ETC, the level increased again (graph in Fig. 3C). Such a gradient could be indicative of transport processes occurring in the caryopsis during the main filling stage.
Figure 3.

Imaging of Suc in the barley caryopsis. A, Cross section of a caryopsis sampled at 10 DPA. The red arrows indicate the path of the Suc supply from the vascular bundle to the endosperm. B and C, Color-coded Suc map (B) and light micrograph illustrating the structure of the caryopsis in the region of the main Suc translocation pathway (C). The graph at bottom of C comprises box plots showing the Suc concentration along the pathway (target circle, median; box, interquartile range [IQR]; lines, 1.5 × IQR). VB, Vascular bundle; NP, nucellar projection; ETC, endosperm transfer cell; SE, starchy endosperm; PE, pericarp. Tissue section thickness was 16 µm. Bars = 500 µm.
Due to the limitations of technology, we could not measure Suc gradients within the caryopsis during the prestorage stage (before endosperm cellularization). During early development, the caryopsis possesses a large cavity filled with liquid endosperm that is easily lost during tissue preparation (Supplemental Fig. S7A). When such sections are imaged, measurement artifacts appear in the Suc map. In our example, the absence of Suc signal in the endosperm cavity (Supplemental Fig. S7B) could be misinterpreted as the absence of Suc in the endosperm. Suc cannot be imaged in large structureless regions. Thus, a careful and critical quality control of tissue sections always is required.
Comparison of Suc and Starch Gradients in the Same Tissue Sections
We next tested if FTIR spectral data also can be used to analyze starch distribution across the imaged sections. For this, we first identified the starch-specific FTIR signal, applied the detection scheme (as outlined in Fig. 1), applied a calibration routine, and ultimately modeled starch distribution across the same section of the caryopsis used for Suc mapping. The entire data set is presented in Figure 4. It appeared that, at this developmental stage, starch accumulation was confined mainly to the two wing regions of the endosperm. Starch imaging by FTIR was compared with an iodine-staining image of the same cross section. Both patterns show strong similarities (Fig. 4, A and C). We thus conclude that FTIR imaging data also can be used to get additional quantitative and spatially resolved information on starch distribution. The direct comparison of gradients for Suc and starch within the endosperm is shown in Figure 4D. While Suc generally declines from the ETC toward the peripheral endosperm, starch shows the opposite pattern. The decline in Suc and increase in starch were much more pronounced toward the side lobes of the endosperm. As starch is the major product of Suc conversions in the endosperm, the comparison of Suc versus starch gradients across the caryopsis adds value to the functional interpretation of data sets.
Figure 4.

Imaging of starch distribution in the barley caryopsis. A, Iodine staining illustrates the site of starch deposition. B, Calibration curve established using defined starch concentrations. C, FTIR image showing the distribution of starch. D, Endosperm gradient for Suc and starch in a barley caryopsis at 12 d after flowering starting at the endosperm transfer cell (ETC) layer toward the side lobes of the endosperm (blue arrow in A). The shaded area represents 50% of the variation within the respective data sets. PE, Pericarp; SE, starchy endosperm. Tissue section thickness was 16 µm. Bars = 500 µm.
Application of High-Resolution Suc Mapping to Arabidopsis Plants
Arabidopsis is still the most popular model plant and has been investigated thoroughly by all biological disciplines (Somerville and Koornneef, 2002). Notwithstanding, tissue-specific analysis of Suc distribution and metabolism is challenging and relies on either tissue microdissection or genetically encoded FRET sensors. In order to test the applicability of our Suc-imaging technology for Arabidopsis, we chose the genotype Columbia-0 (Col-0) and its double mutant atsweet11;12 with knockouts of the respective sugar transporters. The double mutant plant has been characterized previously and shows a characteristic reduction of growth due to altered phloem loading and sugar turnover (Chen et al., 2012; Gebauer et al., 2017).
Here, we show how the mutation affects Suc distribution in the hypocotyl/stem. We first identified two regions of interest: the inner cambial region (including the adjacent secondary growth region of the xylem) and the outer vascular region (all vascular bundles including surrounding tissues; Ragni and Greb, 2018). Representative Suc images through the hypocotyl of both genotypes are shown in Figure 5, B and C. Comparison of genotypes revealed that the cambial region of the double mutant accumulated Suc at statistically significantly higher levels compared with Col-0, while in the outer vascular region, embedding in the vascular bundles, the Suc level was lowered (Fig. 5E). The latter finding could possibly be interpreted as a consequence of lower phloem loading in the source of the mutant plant. Moreover, for Col-0, we observed Suc as more abundant in the inner cambium than in outer vascular regions (Fig. 5B). The same trend, but much more pronounced, became obvious for the double mutant plant (Fig. 5C). Such a pattern in Suc distribution was unknown before and might be related to a proliferation pattern in the hypocotyl. Sankar et al. (2014) investigated the secondary growth and uncovered the overall dynamics of cell proliferation in different regions of the Arabidopsis hypocotyl during development. Studies revealed that, during late developmental stages, phloem cell numbers are stagnating while cambial cell numbers still increase with respect to cell number. High Suc steady-state levels in the inner cambial regions could be explained by a high sink demand of the proliferating xylem tissue. A relevant aspect for the interpretation of imaging data is the consideration of protein localization and/or the spatial expression pattern of SWEET genes. These were described to some extent for the leaf/petiole (Chen et al., 2012) but remained unknown for the hypocotyl/roots.
Figure 5.

Imaging of Suc in Arabidopsis hypocotyls. A, Schematic image of Arabidopsis. B, Suc map of a Col-0 hypocotyl cross section. C, Suc map of the atsweet11;12 double mutant. D, Three-dimensional model of an Arabidopsis hypocotyl with designated shaded areas for cambium and secondary xylem (xc; red) and phloem containing ground tissue (pp; gray). E, Box plot of Suc distribution in the respective regions of Col-0 and atsweet11;12 hypocotyls (dot point, median; box, interquartile range [IQR]; lines, 1.5 × IQR; stars indicate statistical significance at P < 0.05 by the Mann-Whitney U test). Tissue section thickness was 16 µm. Bars = 500 µm.
While further investigations are needed to unravel the exact role of Suc (and sugar transporter activity) in the metabolic and developmental control of hypocotyl growth, the method is ready to use for comparative studies in various Arabidopsis models.
DISCUSSION
Plants transport assimilates largely in the form of Suc. In addition to its primary role as an energy source, Suc also is used as a signal in various adaptive and stress responses (Scialdone et al., 2013; Figueroa and Lunn, 2016). Many novel approaches to improve crop performance are targeting sugar metabolism and/or transport (Guan and Koch, 2015; Nuccio et al., 2015; Sonnewald and Fernie, 2018). Thus, an ability to quantify Suc over time and space would serve to advance our understanding of crop physiology and yield formation. This article describes a platform based on FTIR spectroscopy, allowing the quantitative visualization of the distribution of Suc within heterogenous tissues of both crops and model plants.
Advantages of FTIR Suc Mapping with Respect to Other Imaging Modalities
Our FTIR Suc mapping enables spatially and chemically resolved imaging of Suc, a feature not reached so far by mass spectrometry imaging. The latter has advantages in detecting multiple compounds, in particular lipid species (Woodfield et al., 2017), but has inherent limitations regarding Suc imaging due to unstable/unspecific small molecule ionization and matrix effects. Most recent method developments (Paek et al., 2018) seem more appropriate, but so far the method has not been successful in providing quantitative Suc maps. FTIR-based mapping can detect Suc in a label-free manner, like MRI, but it is applicable for the detection of Suc at a concentration down to 20 mm and with a spatial resolution of ∼12 µm, both values that are not reached at the current state of MRI. FTIR-based imaging is applicable to any plant and, thus, more adequate and practical in the crop context, while FRET imaging relies on prior genetic transformation. Finally, the new FTIR-based method provides substantially improved sample throughput, versatility, and convenience as compared with quantitative bioluminescence and single-photon imaging (which is the only technique providing comparable spatial and chemical resolution). Parallel sample processing and advanced mathematical algorithms/computer power are expected to further enhance throughput.
The major achievement of our approach is to permit a component-specific and standardized analysis of Suc distribution in a complex biological sample. Its chemical and spatial resolution are sufficient to quantify Suc in individual tissue types within a plant organ, such as seeds, leaf, stem, or hypocotyl (Figs. 2–4). The procedure relies on available instrumentation and materials and so can be established in any laboratory. The method could become a routine monitoring tool to track phloem Suc levels as a proxy for the loading/unloading capacities of (transgenic) crop plants. The method is of particular relevance to studies of carbon allocation in the context of crop improvement (Ruan, 2014; Jung et al., 2015). Notably, the approach can be broadened to cover a range of other metabolites, as exemplified by its use for starch imaging. Its capacity to characterize the likely heterogeneity in the metabolism of a complex organ opens novel avenues for analyzing the inner life of plants.
Specific Features, Requirements, and Limitations for FTIR-Based Suc Mapping
The Suc-imaging approach presented here outperforms the already existing approaches with respect to the ability for quantification, tissue-specific resolution, and accuracy. This was achieved by the implementation of internal standardization procedures and the combined use of various mathematical algorithms for signal decomposition and normalization (Fig. 1). The introduction of a ubiquitous and spatially homogenous internal standard prior to imaging is vital in this regard (and already common practice in chromatography/mass spectrometry), since alterations in the imaging setup (e.g. light intensity, filter usage, temperature shifts, etc.) or algorithmic computation will affect the spectral features of the internal standard in the same way the sample features would be affected. The internal standard approach enabled us to compensate for resulting alterations to spectral band intensities, increasing the quantitative reproducibility of results across samples, times, and platforms. To our knowledge, the homogenous distribution of a standard in the sample is not achievable by sample infiltration or any other means in microscopic analysis that is quantifiable on a pixel-by-pixel basis and is that restrictive in altering the sample composition. The quantification of analytes in the demonstrated cases is diffraction limited depending on the optical system used, which results in an optical resolution of ∼10 µm in the 1,000 wavenumber region. Structures close to that size cannot be resolved without interfering signals from surrounding areas, even though the digital resolution of the detector (∼3 µm) may give a different impression. Artifacts also may arise due to the preparation of sample slides in the case of large structureless regions that would be filled with solutions in vivo (Supplemental Fig. S7). Cryosections also can suffer from tissue tear where soft tissues are adjacent to hard tissues during cryocutting, as can be seen in Figure 5, B and C, in the dark blue ring between secondary xylem and cambium in the middle and outer regions of ground tissue and phloem. Lyophilization may enhance this effect due to possible tissue shrinkage during the process.
Compared with either MRI or FRET, our Suc-mapping technology has the disadvantage of being invasive. FTIR-based mapping is performed on cryosections, and its application should be performed with caution. First, mapping relies on microscopic and spectroscopic analysis. Thus, important requirements for accurate performance are the high quality of tissue cryosections and the calibration procedure as well as generating an appropriate spectral model in order to avoid overfitting and underfitting to exclude optical and numerical artifacts. Optical artifacts need to be computed along with actual chemical information in the spectral data set. This also means that any model parameter can be imaged in parallel on the same sample, whereas marker-based approaches like FRET or staining techniques often have a capacity of only one analyte per sample slide. The spectral region selected for modeling determines the selectivity on which compounds may be modeled as part of a component group or individual compound. This trend is observed generally for larger molecules, which often only can be accounted for in groups (e.g. lipids and proteins) because of broader and less individual spectral bands, whereas small molecules are easier to model due to more unique spectral characteristics. Microspectroscopy, in general, can estimate locally heterogenously distributed compounds easier than homogenously distributed ones in complex mixed spectra (e.g. biological tissue). Small hot spots of a heterogenously distributed compound would show strong spectral similarity to the pure component spectra, and noise introduced by other compounds will contribute even less in the case of strong relative differences in allocation patterns on a microscopic spatial scale.
With the presented equipment, the visualization of signal compartmentation is limited to an optical resolution of 12 µm; therefore, differences between cell compartments close to that margin remain unresolved. In order to achieve higher spatial resolution, other FTIR objectives (Supplemental Fig. S3) or more specialized approaches are required, like atomic force microscopy infrared-coupled (Dazzi et al., 2012) or synchrotron-based FTIR imaging facilities like the Diamond B22 beamline (Diamond Light Source).
Relevance of FTIR-Based Imaging for the Analysis of Suc Levels in Vascular Tissues of Plants
The introduction of new technology necessarily raises the question of whether the outcome accords with what has been measured by established methods. FTIR data allow the characterization of Suc levels in phloem (and surrounding) tissues, but why does the method provide lower concentrations as compared with published data on the phloem sap Suc concentration (Jensen et al., 2013)?
The phloem consists of several different cell types, of which the sieve element cells have received the most attention due to their solute-conducting function (Truernit, 2014). Sieve element cells are long, narrow cells that usually form a functional unit with companion cells in the architecture of the vascular bundle and are genetically and metabolically dependent on the associated companion cells (Oparka and Turgeon, 1999). While the details of phloem structure are beyond the spatial resolution of FTIR techniques (the diameter of phloem cells could be less than 12 µm), our Suc mapping informs about the concentration of Suc in phloem tissues and not in the individual phloem cells/vessels, as usually occurs by aphid/microsampling (Jensen et al., 2013). The aphid/microsampling approach estimates the concentration of Suc in sap released from individual phloem cells/vessels over time, whereas FTIR provides a snapshot of Suc distribution within the entire tissue at the moment of freezing. FTIR integrates the Suc signal across solid matter (cell walls and organelles), liquid matter (sap), and intercellular spaces (filled with fluids or air). Therefore, the lower concentration values provided by FTIR just reflect the difference of analyzed matter (i.e. sap versus tissue).
FTIR is designed to characterize the metabolic state at the tissue level, whereas both aphid-based sampling and microsampling are for investigations of individual cells. The characterization of vascular systems at different levels of structural organization (e.g. by combining different techniques) will open new perspectives for investigating the functionality of vascular systems (van Bel et al., 2002; Mullendore et al., 2010; Knoblauch and Oparka, 2012) and for understanding plant life in general.
Experiments with sap-sucking insects can be performed only under distinct growth conditions, and sap extracts often need to be collected over several hours and/or multiple aphid stylets (to acquire sufficient analytical amounts). The FTIR procedure provides substantial versatility, since it is not restricted to a specific host species or experimental (growth) condition and since it depicts the actual (sugar) status at the specific time point of sampling. The method allows access to parts of the plant that are not accessible by insects to be sampled (e.g. developing grains) and can analyze a number of tissues simultaneously (Fig. 2) as well as compare Suc levels across different phloem strands in the stem, leaves, or other organs (Supplemental Fig. S6). Based on the high spatial resolution of the FTIR method, whole-plant Suc mapping (comprising all organs/tissues like developing embryo, germinating seeds, or root) is now feasible. A related option is to systematically map all vascular bundles across the leaf or stem for the comparison of sugar levels inside the phloem. As documented here, we found distinct Suc levels in inner versus outer vascular rings of a barley stem, which was unexpected given the concept that cereal plants are rather nonsectorial (Watson and Casper, 1984). Thus, novel FTIR-based sugar mapping is a useful instrument for testing long-standing knowledge and generating new ideas on the inner life of plants.
Suc Mapping Promotes the Study of Postphloem Allocation Processes
The novel FTIR method is a reliable tool to trace Suc allocation along the postphloem pathway (Figs. 3 and 5). In the barley caryopsis, Suc intake occurs via the main vascular bundle located in the ventral pericarp, as was shown by in vivo 13C/1H-NMR imaging (Melkus et al., 2011; Radchuk and Borisjuk, 2014; Rolletschek et al., 2015). Thereby, concentration gradients are formed along the allocation pathway, which are nicely reflected in our Suc maps. The concentration of Suc in postphloem regions of the pericarp drops slightly and increases again in the ETC region. Such gradients could be indicative for passive unloading at the vein but active loading against the concentration gradient at the ETC via a set of sugar transporters (Lopato et al., 2014; Thiel, 2014). From the ETC, most of the Suc moves passively into the storage tissue, resulting in the formation of a steep negative gradient from the ETC toward the peripheral endosperm. Altogether, the Suc profiles accorded well with what has been revealed by a variety of molecular and biochemical studies. We conclude that the FTIR platform (1) visualizes the distribution of Suc at the cell-type level of resolution and (2) generates Suc maps that can reflect the postphloem allocation process.
Beside Suc, FTIR also can analyze the distribution of other metabolites like starch (Fig. 4). Our quantitative data identified a characteristic pattern of starch distribution, which was described earlier and is explained largely by distinct local conditions favoring starch biosynthesis under hypoxic conditions inside the endosperm (Rolletschek et al., 2004). The characteristic histological pattern (based on iodine staining) now has become quantitatively proven.
MATERIALS AND METHODS
Plant Material and Sampling
Barley (Hordeum vulgare ‘HOR 13170’) was grown in a greenhouse at 20°C during the day and 17°C at night. The light period lasted from 6 am for 16 h until 10 pm. The plants were grown in a substrate mixture of compost earth, white peat, and sand in 2-L (14.8-cm diameter) pots. Samples of various plant organs were harvested and immediately frozen in liquid nitrogen to be stored at −20°C. Leaf and stem samples were harvested at anthesis at the beginning of the light period (30 min of light). Leaves were cut below the ligule, and stem samples were harvested 2 cm above the third node. Developing seeds were sampled from the central spike at position 2 within the spikelet at 8 to 10 d after flowering, 8 h into the light period. Isolated seeds were husked before cryoconservation. Arabidopsis (Arabidopsis thaliana) plants were a generous gift of the Department of Integrative Plant Biology at the Institute of Plant Genetics of the Polish Academy of Sciences in Poznan. The accession Col-0 was used as a genetic background. The atsweet11;12 double mutant is a cross of SALK_073269 and SALK_031696 lines as described earlier (Chen et al., 2012). Arabidopsis plants were grown in a phytochamber at 22°C during the day and 20°C at night, with a light period of 9 h per day and 120 µmol m−2 s−1 irradiance. Hypocotyl segments were harvested 41 d after germination and immediately frozen in liquid nitrogen.
Cryosectioning and Mounting of Tissues Sections for Mapping
Cryosections were prepared from frozen plant materials. Samples were embedded in Tissue-Tek cryomolds using Tissue-Tek O.C.T. (Sakura Finetek) or 2% (w/v) carboxyl methylcellulose solution at −20°C. Embedded tissue blocks were trimmed, cross sectioned (10–20 µm thick) with a cryotome (Thermo Fisher Scientific CryoStar NX7; Microm International), and transferred onto MMI membrane slides (RNase free; product no. 50102; Molecular Machines & Industries). These slides also were used for internal standardization as described below. Tissue sections were lyophilized and stored in darkness at room temperature until analysis.
Image Acquisition
Images of tissue samples were generated using a Hyperion 3000 FTIR microscope (Bruker Optics) coupled to a Tensor 27 FTIR spectrometer (Bruker Optics) using the internal mid-infrared source. The system is equipped with a focal plane array (FPA) detector (64 × 64 pixels), which was used in transmission mode. The imaging system was purged with dry air continuously. FTIR images were recorded in the spectral range of 3,900 to 800 cm−1 at a spatial resolution of 5.5 µm for larger images (OPUS; binning 2 × 2 pixels) and 2.25 µm for smaller samples and a spectral resolution of 6 cm−1 using 15× infrared magnification objectives (Bruker Optics) with a numerical aperture of 0.6 with an optical resolution of 11.2 µm at 900 cm−1. Each spectrum comprised 64 coadded scans. A reference of a single FPA window of the empty light path was acquired prior to image acquisition and subtracted automatically from the recorded image by OPUS software (Bruker Optics).
Data Processing
Data processing was done on a Windows 7 (64-bit) desktop computer with Intel Core i7-6700K CPU (4.00 GHz) and 16 GB RAM. OPUS files were imported into Matlab (R2016; The MathWorks) as ENVI files using the multibandread function or by the irootlab toolbox (Trevisan et al., 2013). Image data were reduced to desired areas of interest and cropped to a spectral range (wave number) of 2,200 to 850 cm−1. Further data processing was done in two major steps of baseline feature extraction and metabolite feature extraction. Each of these steps followed the routine described in Figure 1, while the model for metabolite feature extraction excluded already extracted baseline features.
Iterative Processing Step 1: K-Means Clustering
The reduced data set was pooled into n data clusters according to K-means clustering, as described by Nguyen et al. (2016). The described clustering approach was performed to aliquot imaging data into smaller data sets for computation and subsequently decreasing variance within each subset. Here, clustering was computed using Euclidean distances for two to 20 clusters per iteration. The optimal classification was determined by their Pakhira-Bandyopadhyay-Maulik validity indices. Each aliquot could be divided again by K-means clustering to reduce data set sizes further if necessary.
Iterative Processing Step 2: Feature Extraction
After aliquotation of the data by K-means clustering, each aliquot was computed separately and only combined again at the end of each iteration step. The model used was different depending on whether the iterative step is part of the baseline feature extraction or the metabolite feature extraction.
For baseline feature extraction, the approach was adopted from the Mie-scatter extended multiplicative signal correction as described by various authors (Kohler et al., 2008; Bassan et al., 2010, 2012; Baker et al., 2014; Salzer and Siesler, 2014). Mie-scattering components were derived from the first five principal components of a Mie theory model based on refractive index variations between 1.1 and 1.5 at the surface of spheres with radii from 0.2 to 20 µm. The first four principal components of residual background signals (e.g. water vapor and CO2 bands extracted from air and empty membrane reference images; Supplemental Fig. S1A) were added along with a principal component matrix of a pure spectra library of proteins, lipids, carbohydrates, amino acids, and secondary metabolites common in plant tissue and the pure MMI membrane spectrum. After modeling, baseline and Mie-scattering features were subtracted from the sample spectra before the next iteration started. In general, four to six iterations were sufficient for the baseline coefficients to converge, at which point sample spectra were defined as baseline free. The final scores of the metabolite principal component matrix already could be used for statistical analysis.
Quantitative modeling of Suc, starch, and the MMI membrane was performed by further iterations. Prior to data modeling, the aliquoted data sets were extended by a set of pure spectral standards (external standards) for the parameters that were investigated (as shown in Supplemental Fig. S2). This was done to track numerical errors introduced into the data, both by finite precision limitations and truncation errors as a result approximations as a means to simplify mathematical equations. These errors would be represented similarly within the sample data and the external standard set that was processed alongside the real data. After undergoing partial least squares regression, the calculated coefficients for the remodeled external standards were compared with their original values by Pearson correlation in order to create performance validation parameters of how well the modeled standards were explained by the sample data. If the data were satisfactory, the calculated absorbance coefficients were normalized in relation to the internal standard values. This allowed the comparison of different data sets; therefore, concentration distributions could be calculated from calibration data sets. In the presented images, each metabolite was modeled by partial least squares regression against a background component matrix derived from the principal components of 42 different pure spectra from proteins, amino acids, carbohydrates, lipids, and other secondary metabolites common in plant tissue. This was sufficient to explain the variation in imaging data of the presented plant tissue samples. The calculated concentrations (cA; Eq. 1) are calculated in pg µm−2. In order to transfer these values to more conventional molar concentrations, areal concentrations are converted to a volumetric scale (tissue fresh volume) by division through cryosection thickness (Δz) and the molar mass of the compound in question (Mi; Suc, 342.2965 g mol−1), as seen in Equation 2. To give an example, the maximum value (0.244 pg Suc µm−2) in Figure 2B corresponds to 44.6 pmol Suc µm−3 tissue (given a section thickness of 16 µm). If the imaged volume contains only liquid (but no solid structures like cell walls), this value would equal to millimolar Suc concentration.
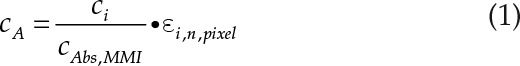 |
 |
Tests for Validation of the Reproducibility of the Internal Standard
In order to proof the reproducibility of the membrane standard, several experiments were performed to test the consistency of the membrane signal of empty slides over the field of view in one measurement, the signal of the same membrane over several measurements, the signal from different batches over several measurements, and the signal with regard to different membrane types (RNase free or not). Spatial signal variations of the membrane on a small scale (2 × 2 FPA areas) and a large scale (1 × 120 FPA diagonal across the slide) remained below 1% of the average membrane signal and were measured as quantile absolute differences. This represents a very consistent standard within each pixel of an image. Different repeated measurements of one membrane and different measurements of different membrane batches increased signal variations to 2% to 3% of the average membrane signal (Supplemental Fig. S1B), indicating that small system variations occur between measurements that can be normalized for sample features by normalization against the internal standard. A medium effect size was found only with regard to the RNase-free status of the membrane batch, which resulted in quantile absolute difference values up to 5% of the average membrane signal.
In Silico Modeling
In order to test the model accuracy, several in silico data sets were created to test feature extraction on a close to single-metabolite level. The test had to be done in silico in order to ensure the exact definition of reference data, which is either highly difficult to achieve or lacking comparable analytical methods to produce the reference data on real complex samples with the same resolution. The in silico data were designed to test the feature extraction of starch and Suc from a matrix composed of 40 individual compounds commonly found in biological plant tissue. The compound-specific images were selected from a pool of uncorrelated images (R2 < 0.3). These uncorrelated images were created by random selections of a 300 × 300-pixel window of a larger Mandelbrot simulation image. The selected images were defined by generating random coordinates for the center along with a randomized rotation angle of the 300 × 300-pixel window (Supplemental Fig. S4, D and G). Afterward, Pearson coefficients for the pairwise image correlation were determined, and only images with R2 < 0.3 were kept. From 40 of these images, a spectral cube was created as a reference for each compound and, in consequence, were merged to create a mixed spectral cube, from which the test component images were remodeled by partial least squares regression.
The data in Supplemental Figure S4 show the performance of the model in reproducing the hidden compound-specific images for the respective metabolites.
Quantification of Suc Using External Standardization
The linear correlation of signal and concentration was tested by lyophilizing frozen drops of Suc in water with a defined volume across a known area. A total of 100 mg of Suc was dissolved in 2 mL of water. Aliquots of different concentrations were prepared as a dilution series (0.001–20 g L−1). The recrystallized Suc was imaged after lyophilization (Supplemental Fig. S8). The spectral data were modeled with Suc (raw image, Supplemental Fig. S8A), the MMI membrane (raw image, Supplemental Fig. S8B), and water as parameters. The normalized Suc signal (Supplemental Fig. S8C) was averaged across the imaged area and plotted against the average areal concentration from the tested frozen drop, resulting in the linear fit presented in Figure 2C.
In order to validate Suc images after FTIR imaging, we laser dissected nine pairs of cryosections of two biological replicates of barley stems at anthesis and caryopsis at 10 d after flowering (Supplemental Fig. S5) into representable contrasting tissue areas. In this way, we could directly quantitatively compare FTIR with ion chromatography results.
Quantification of Starch Using External Standardization
Due to the low solubility of starch, the proof of linearity of the signal and concentration was demonstrated with a dilution of starch in KBr. The two powders were mixed by thorough grinding in order to achieve a homogenous concentration within a specific volume. Starch and Suc were prepared in KBr as mixed powder with ratios of 20%, 40%, 60%, 80%, and 100% (w/w). Two aliquots per concentration were measured as ATR spectra for each analyte and transformed to absorbance spectra by OPUS software. The transformed spectra for Suc were utilized to estimate an average sample volume for the ATR spectrometer. Total amounts for starch were calculated from the sample ratios and referenced against spectral coefficients as a starch calibration (Fig. 4B). Starch staining was done using iodine (Borisjuk et al., 2013).
Laser Microdissection in Cryosections
A CellCut laser microdissection (LMD) system equipped with an Olympus IX81 light microscope and CellTools 4.4 software (Molecular Machines & Industries) was used to microdissect tissues from 16- to 20-μm-thick cryosections of caryopses and internodes. Microdissection was performed under a 40× objective using 10 to 25 μm s−1 cut velocity and 1,500 to 2,000 mW laser power, each time adjusted to tissue type. The microdissected tissues were collected in transparent 0.5 mL IsolationCaps (50204; Molecular Machines & Industries).
Sugar Analysis of Microdissected Samples
Subsequently, 0.3 mL of 80% methanol was added, vortexed for 10 s, and shaken vigorously for 20 min at room temperature. To remove any particles from the fluid, Vivaclear centrifugal filters with 0.8-μm pore size (SatoriusStedim Biotech) were used (centrifugation at 2,000g for 2 min). The filtrated sample extract was used directly for sugar analysis using ion-exchange chromatography coupled to pulsed amperometric detection (ICS-3000; Thermo Fisher). Separation was carried out on a PA1 column (2 × 250 mm) and a PA1 guard column (2 × 50 mm) at 25°C by applying an isocratic run with 400 mm NaOH at a constant flow rate of 0.7 mL min−1 over 5 min. Authentic standards were used for sugar identification and external calibration. All chemicals were obtained from Sigma-Aldrich.
Statistical Image Analysis
Comparisons between digitally dissected tissue regions, as in Figures 5 and others, are based on the nonparametric analysis of signal distributions within these areas, due to nonnormal signal distribution characteristics of heterogenous tissue images. Box plots and corresponding interquartile ranges are based on pixel values of designated areas of at least three biological replicates (3 images × n pixels). Extracted features from these regions were compared by quantile absolute distance (QAD), and signal distributions for compared areas were deemed significantly different by nonparametric statistical analysis for p < 0.05 (Mann-Whitney test) and divergence effect size greater than 0.2. QAD compares the difference of quantiles of two distributions over the entire range of probabilities and is calculated from the mean difference of the quantile functions A−1 and B−1 of two populations as describes in Equation 3.
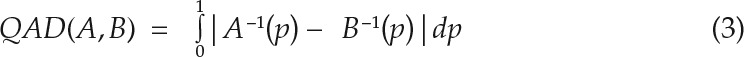 |
QAD values are always positive and will only become 0 if A and B are identical. Divergence effect size (D) is a probability distance between two cumulative distribution functions A and B regardless of their context, according to Equation 4.
Since D(A||B) ≠ D(B||A), a symmetric measure of this parameter can be achieved by Equation 5.
This effect size is a bounded parameter that can take values between 0 (identical) and 1 (no commonalities).
Supplemental Data
The following supplemental materials are available.
Supplemental Figure S1. Evaluation of the membrane as an internal standard.
Supplemental Figure S2. Algorithm employed to incorporate external standards into the sample data set.
Supplemental Figure S3. Optical resolution of IR microscopy objectives.
Supplemental Figure S4. Detection of Suc and starch in a complex in silico spectral matrix.
Supplemental Figure S5. Validation of the Suc-imaging procedure using laser dissection and ion chromatography.
Supplemental Figure S6. Imaging of Suc in the barley flag leaf and stem.
Supplemental Figure S7. Example of method limitation in samples with large structureless regions.
Supplemental Figure S8. In situ calibration used for Suc imaging.
Acknowledgments
We thank Eberhard Munz, Sabine Herrmann, Stefan Ortleb, and Karin Lipfert (Leibniz Institute of Plant Genetics and Crop Plant Research) for technical support and artwork.
Footnotes
This work was financially supported by the Deutsche Forschungsgemeinschaft (grant no. BO 1917/5-1 to L.B. and H.R.). This research was supported by Bayer Crop Science NV.
References
- Baker MJ, Trevisan J, Bassan P, Bhargava R, Butler HJ, Dorling KM, Fielden PR, Fogarty SW, Fullwood NJ, Heys KA, et al. (2014) Using Fourier transform IR spectroscopy to analyze biological materials. Nat Protoc 9: 1771–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassan P, Kohler A, Martens H, Lee J, Jackson E, Lockyer N, Dumas P, Brown M, Clarke N, Gardner P (2010) RMieS-EMSC correction for infrared spectra of biological cells: extension using full Mie theory and GPU computing. J Biophotonics 3: 609–620 [DOI] [PubMed] [Google Scholar]
- Bassan P, Sachdeva A, Kohler A, Hughes C, Henderson A, Boyle J, Shanks JH, Brown M, Clarke NW, Gardner P (2012) FTIR microscopy of biological cells and tissue: data analysis using resonant Mie scattering (RMieS) EMSC algorithm. Analyst (Lond) 137: 1370–1377 [DOI] [PubMed] [Google Scholar]
- Bezrutczyk M, Hartwig T, Horschman M, Char SN, Yang J, Yang B, Frommer WB, Sosso D (2018) Impaired phloem loading in zmsweet13a,b,c sucrose transporter triple knock-out mutants in Zea mays. New Phytol 218: 594–603 [DOI] [PubMed] [Google Scholar]
- Borisjuk L, Walenta S, Rolletschek H, Mueller-Klieser W, Wobus U, Weber H (2002) Spatial analysis of plant metabolism: sucrose imaging within Vicia faba cotyledons reveals specific developmental patterns. Plant J 29: 521–530 [DOI] [PubMed] [Google Scholar]
- Borisjuk L, Rolletschek H, Neuberger T (2012) Surveying the plant’s world by magnetic resonance imaging. Plant J 70: 129–146 [DOI] [PubMed] [Google Scholar]
- Borisjuk L, Neuberger T, Schwender J, Heinzel N, Sunderhaus S, Fuchs J, Hay JO, Tschiersch H, Braun HP, Denolf P, et al. (2013) Seed architecture shapes embryo metabolism in oilseed rape. Plant Cell 25: 1625–1640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boughton BA, Thinagaran D, Sarabia D, Bacic A, Roessner U (2016) Mass spectrometry imaging for plant biology: a review. Phytochem Rev 15: 445–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen LQ, Qu XQ, Hou BH, Sosso D, Osorio S, Fernie AR, Frommer WB (2012) Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 335: 207–211 [DOI] [PubMed] [Google Scholar]
- Dazzi A, Prater CB, Hu Q, Chase DB, Rabolt JF, Marcott C (2012) AFM-IR: combining atomic force microscopy and infrared spectroscopy for nanoscale chemical characterization. Appl Spectrosc 66: 1365–1384 [DOI] [PubMed] [Google Scholar]
- Figueroa CM, Lunn JE (2016) A tale of two sugars: trehalose 6-phosphate and sucrose. Plant Physiol 172: 7–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebauer P, Korn M, Engelsdorf T, Sonnewald U, Koch C, Voll LM (2017) Sugar accumulation in leaves of Arabidopsis sweet11/sweet12 double mutants enhances priming of the salicylic acid-mediated defense response. Front Plant Sci 8: 1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan JC, Koch KE (2015) A time and a place for sugar in your ears. Nat Biotechnol 33: 827–828 [DOI] [PubMed] [Google Scholar]
- Jensen KH, Savage JA, Holbrook NM (2013) Optimal concentration for sugar transport in plants. J R Soc Interface 10: 20130055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung B, Ludewig F, Schulz A, Meißner G, Wöstefeld N, Flügge UI, Pommerrenig B, Wirsching P, Sauer N, Koch W, et al. (2015) Identification of the transporter responsible for sucrose accumulation in sugar beet taproots. Nat Plants 1: 14001. [DOI] [PubMed] [Google Scholar]
- Knoblauch M, Oparka K (2012) The structure of the phloem: still more questions than answers. Plant J 70: 147–156 [DOI] [PubMed] [Google Scholar]
- Kohler A, Sulé-Suso J, Sockalingum GD, Tobin M, Bahrami F, Yang Y, Pijanka J, Dumas P, Cotte M, van Pittius DG, et al. (2008) Estimating and correcting Mie scattering in synchrotron-based microscopic Fourier transform infrared spectra by extended multiplicative signal correction. Appl Spectrosc 62: 259–266 [DOI] [PubMed] [Google Scholar]
- Lalonde S, Wipf D, Frommer WB (2004) Transport mechanisms for organic forms of carbon and nitrogen between source and sink. Annu Rev Plant Biol 55: 341–372 [DOI] [PubMed] [Google Scholar]
- Lopato S, Borisjuk N, Langridge P, Hrmova M (2014) Endosperm transfer cell-specific genes and proteins: structure, function and applications in biotechnology. Front Plant Sci 5: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkus G, Rolletschek H, Fuchs J, Radchuk V, Grafahrend-Belau E, Sreenivasulu N, Rutten T, Weier D, Heinzel N, Schreiber F, et al. (2011) Dynamic 13C/1H NMR imaging uncovers sugar allocation in the living seed. Plant Biotechnol J 9: 1022–1037 [DOI] [PubMed] [Google Scholar]
- Moghaddam MRB, Van den Ende W (2013) Sugars, the clock and transition to flowering. Front Plant Sci 4: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullendore DL, Windt CW, Van As H, Knoblauch M (2010) Sieve tube geometry in relation to phloem flow. Plant Cell 22: 579–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray KK, Seneviratne CA, Ghorai S (2016) High resolution laser mass spectrometry bioimaging. Methods 104: 118–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TNQ, Jeannesson P, Groh A, Piot O, Guenot D, Gobinet C (2016) Fully unsupervised inter-individual IR spectral histology of paraffinized tissue sections of normal colon. J Biophotonics 9: 521–532 [DOI] [PubMed] [Google Scholar]
- Niittylae T, Chaudhuri B, Sauer U, Frommer WB (2009) Comparison of quantitative metabolite imaging tools and carbon-13 techniques for fluxomics. Methods Mol Biol 553: 355–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuccio ML, Wu J, Mowers R, Zhou HP, Meghji M, Primavesi LF, Paul MJ, Chen X, Gao Y, Haque E, et al. (2015) Expression of trehalose-6-phosphate phosphatase in maize ears improves yield in well-watered and drought conditions. Nat Biotechnol 33: 862–869 [DOI] [PubMed] [Google Scholar]
- Oparka KJ, Turgeon R (1999) Sieve elements and companion cells: traffic control centers of the phloem. Plant Cell 11: 739–750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paek J, Kim Y, Lee D, Kim J (2018) MALDI‐MS analysis of sucrose using a charcoal matrix with different cationization agents. Bull Korean Chem Soc 39: 750–756 [Google Scholar]
- Partelová D, Kuglerová K, Konotop Y, Horník M, Lesný J, Gubišová M, Gubiš J, Kováč P, Matušíková M (2017) Imaging of photoassimilates transport in plant tissues by positron emission tomography. Nova Biotechnol Chim 16: 32–41 [Google Scholar]
- Radchuk V, Borisjuk L (2014) Physical, metabolic and developmental functions of the seed coat. Front Plant Sci 5: 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ragni L, Greb T (2018) Secondary growth as a determinant of plant shape and form. Semin Cell Dev Biol 79: 58–67 [DOI] [PubMed] [Google Scholar]
- Rolletschek H, Weschke W, Weber H, Wobus U, Borisjuk L (2004) Energy state and its control on seed development: starch accumulation is associated with high ATP and steep oxygen gradients within barley grains. J Exp Bot 55: 1351–1359 [DOI] [PubMed] [Google Scholar]
- Rolletschek H, Melkus G, Grafahrend-Belau E, Fuchs J, Heinzel N, Schreiber F, Jakob PM, Borisjuk L (2011) Combined noninvasive imaging and modeling approaches reveal metabolic compartmentation in the barley endosperm. Plant Cell 23: 3041–3054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rolletschek H, Grafahrend-Belau E, Munz E, Radchuk V, Kartäusch R, Tschiersch H, Melkus G, Schreiber F, Jakob PM, Borisjuk L (2015) Metabolic architecture of the cereal grain and its relevance to maximize carbon use efficiency. Plant Physiol 169: 1698–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan YL. (2014) Sucrose metabolism: gateway to diverse carbon use and sugar signaling. Annu Rev Plant Biol 65: 33–67 [DOI] [PubMed] [Google Scholar]
- Salzer R, Siesler HW (2014) Infrared and Raman Spectroscopic Imaging, Ed 2 Wiley-VCH Verlag, Weinheim, Germany [Google Scholar]
- Sankar M, Nieminen K, Ragni L, Xenarios I, Hardtke CS (2014) Automated quantitative histology reveals vascular morphodynamics during Arabidopsis hypocotyl secondary growth. eLife 3: e01567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scialdone A, Mugford ST, Feike D, Skeffington A, Borrill P, Graf A, Smith AM, Howard M (2013) Arabidopsis plants perform arithmetic division to prevent starvation at night. eLife 2: e00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeekens S, Hellmann HA (2014) Sugar sensing and signaling in plants. Front Plant Sci 5: 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somerville C, Koornneef M (2002) A fortunate choice: the history of Arabidopsis as a model plant. Nat Rev Genet 3: 883–889 [DOI] [PubMed] [Google Scholar]
- Sonnewald U, Fernie AR (2018) Next-generation strategies for understanding and influencing source-sink relations in crop plants. Curr Opin Plant Biol 43: 63–70 [DOI] [PubMed] [Google Scholar]
- Thiel J. (2014) Development of endosperm transfer cells in barley. Front Plant Sci 5: 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisan J, Angelov PP, Scott AD, Carmichael PL, Martin FL (2013) IRootLab: a free and open-source MATLAB toolbox for vibrational biospectroscopy data analysis. Bioinformatics 29: 1095–1097 [DOI] [PubMed] [Google Scholar]
- Truernit E. (2014) Phloem imaging. J Exp Bot 65: 1681–1688 [DOI] [PubMed] [Google Scholar]
- van Bel AJE, Ehlers K, Knoblauch M (2002) Sieve elements caught in the act. Trends Plant Sci 7: 126–132 [DOI] [PubMed] [Google Scholar]
- Watson MA, Casper BB (1984) Morphogenetic constraints on patterns of carbon distribution in plants. Annu Rev Ecol Syst 15: 233–258 [Google Scholar]
- Woodfield HK, Sturtevant D, Borisjuk L, Munz E, Guschina IA, Chapman K, Harwood JL (2017) Spatial and temporal mapping of key lipid species in Brassica napus seeds. Plant Physiol 173: 1998–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]