

Abstract
Background Despite advances in the diagnosis of pancreatic ductal adenocarcinoma (PDA), histological evaluation of small and poorly defined masses in the pancreas is uncomfortable and unsafe.
Methods We herein report a case of early stage PDA, in which multiple KRAS mutations were detected in the pancreatic juice preoperatively. A small hypoechoic area adjacent to the portal vein was detected through endoscopic ultrasound in the pancreatic body. KRAS mutations were evaluated using plasma, and the pancreatic juice by digital PCR.
Results Pancreatic duct biopsy and pancreatic juice cytology were performed with no evidence of malignancy; however, KRAS mutations, KRAS G12V and G12D, were detected in the pancreatic juice. Histological assessment of the resected specimen demonstrated a solid tumor with desmoplastic reaction accompanied by carcinoma in situ in the main pancreatic duct where KRAS G12V mutation was identified. More detailed analysis demonstrated KRAS G12D mutation in the cluster of low grade pancreatic intraepithelial neoplasia, implying that the lesion developed independently.
Conclusions Our study indicates the potential of “endoscopic liquid biopsy” to capture the driver gene for PDA diagnosis.
Introduction
Pancreatic ductal adenocarcinoma (PDA) is one of the most dismal types of cancer, and novel methods for early detection are highly warranted 1 . The standard diagnostics for PDA include tumor tissue collection during endoscopy and, in particular, the sensitivity and specificity values of endoscopic ultrasound-guided fine needle aspiration/biopsy (EUS-FNA/B) are over 90 % 2 . However, tissue sampling sometimes results in inadequate examination due to insufficient material. It is also challenging to fully uncover tumor heterogeneity using a small piece of specimen. Another issue of the modalities is severe complications, reported in up to 1 % – 2 % and 12.0 % of EUS-FNA/B and endoscopic retrograde cholangiopancreatography (ERCP)-related procedures, respectively 3 .
In patients with cancer, the presence of cell-free DNA (cfDNA) in the blood and its potential utilities have been recognized, and their clinical relevance has attracted widespread attention 4 . The primary limitation of such “liquid biopsy” is the modest availability of circulating tumor-derived DNA. Given the extremely low tumor cellularity, detecting tumor-derived DNAs in the blood of patients is challenging 5 . Here, we describe a case of early pancreatic cancer with successful detection of mutant KRAS variants in the pancreatic juice collected through the Vater-papilla during endoscopic cytology. The latest analytical tools for tumor genotyping are powerful enough to identify the tumor-derived DNA, even from early lesions in the pancreatic juice.
Case report
A 53-year-old woman was referred to our hospital due to a complaint of epigastric and dorsal pain. Before the genetic study, written informed consent was obtained from the patient for publication of the case. The laboratory data showed a slight elevation of the pancreatic enzymes (110 U/L) and normal levels of carbohydrate antigen 19 – 9 (CA19 – 9) and IgG4. Images of the pancreas obtained using ultrasound, CT, and MRI showed dilation of the main pancreatic duct (MPD; 6 mm) with no sign of the solid tumor ( Fig. 1a ). EUS revealed an 8-mm hypoechoic area in the body of the pancreas connected to the dilated MPD ( Fig. 1b ).
Fig. 1.
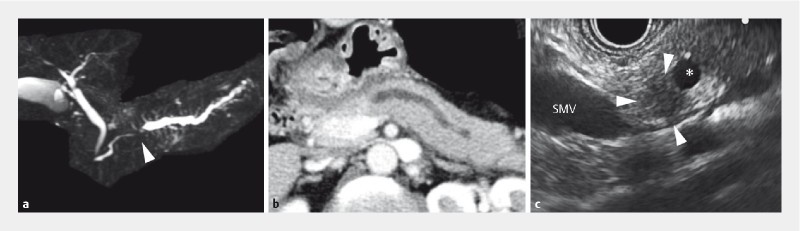
Imaging diagnosis of the primary pancreatic tumor. a Magnetic resonance cholangiopancreatography (MRCP) demonstrates obstruction of the main pancreatic duct (MPD; arrowhead) accompanied by distal dilatation. b CT shows dilation of the MPD with no sign of the solid tumor. c Hypoechoic area is visualized by endoscopic ultrasound in the body of the pancreas (surrounded by arrowheads; 8 mm in diameter) connected to the dilated MPD (asterisk). Note that there are no other detectable tumors or cystic lesions. MPD; main pancreatic duct, SMV; superior mesenteric vein.
The patient was informed about the possible malignant tumor closing the portal vein; however, she refused to undergo FNA/B testing for histological assessment. Instead, a biopsy from the stricture of the MPD and pancreatic juice cytology were performed to diagnose the tumor, resulting in no evidence of malignancy. Pancreaticoduodenectomy was selected as the treatment option for this tumor, which was highly suspected to be PDA.
Before the surgical intervention, we performed a genetic test in a droplet digital PCR platform to capture mutant KRAS and GNAS using the pancreatic juice (500 µL). The fresh pancreatic juice collected via the ERCP catheter was frozen immediately after the procedure (within 10 min). In the purified DNA, two major variants, KRAS G12V (13.3 %) and G12D (3.0 %), were detected ( Fig. 2 ), whereas mutant GNAS was not detected (see the Supplementary Method for details). We also used the pancreatic juice drained through the endoscopic nasopancreatic drainage tube, a liquid left behind out of the organ for several hours; however, the yield of DNA was much lower than the fresh sample and too heavily fragmented for assay.
Fig. 2.
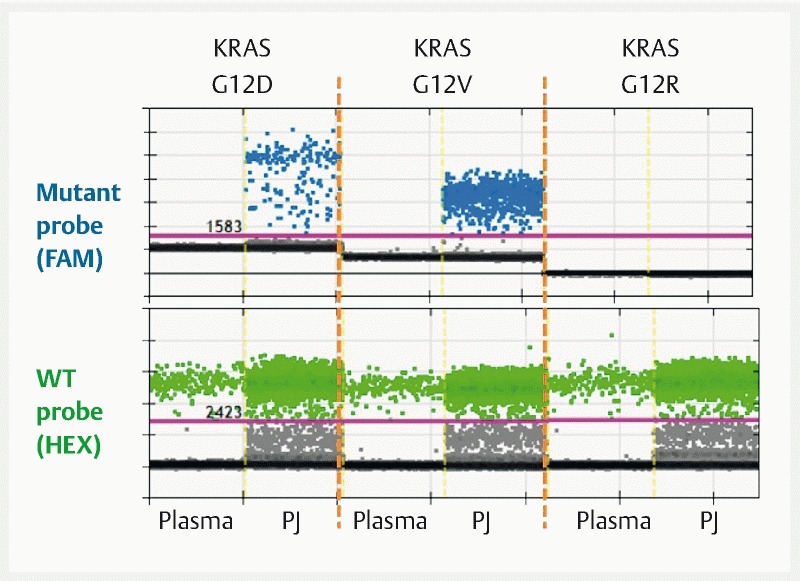
Droplet digital PCR confirming KRAS mutation in the pancreatic juice. KRAS (exon 2)-specific PCR fragments in cell-free DNA from the plasma and pancreatic juice are analyzed by droplet digital PCR. Mutation in KRAS is selectively detected by mutant-specific probe against mutant KRAS at codon 12/13 (upper panel). A larger p.G12V mutant frequency is demonstrated in the pancreatic juice. Additionally, the presence of a smaller proportion of p.G12D mutant alleles is also shown, whereas no signals for other KRAS variants are detected (result using p.G12R-specific probe is shown). PJ, pancreatic juice; FAM, 6-carboxyfluorescein; HEX, hexachloro-6-carboxyfluorescein.
In the resected specimen, focal fibrosis was found proximal to the MPD stricture, and the size of the fibrotic area corresponded well with the hypoechoic lesion visualized by EUS ( Fig. 3a ). Pathologically, well-differentiated adenocarcinoma was evident with no sign of metastasis (stage IA; UICC 8 th edition).
Fig. 3.
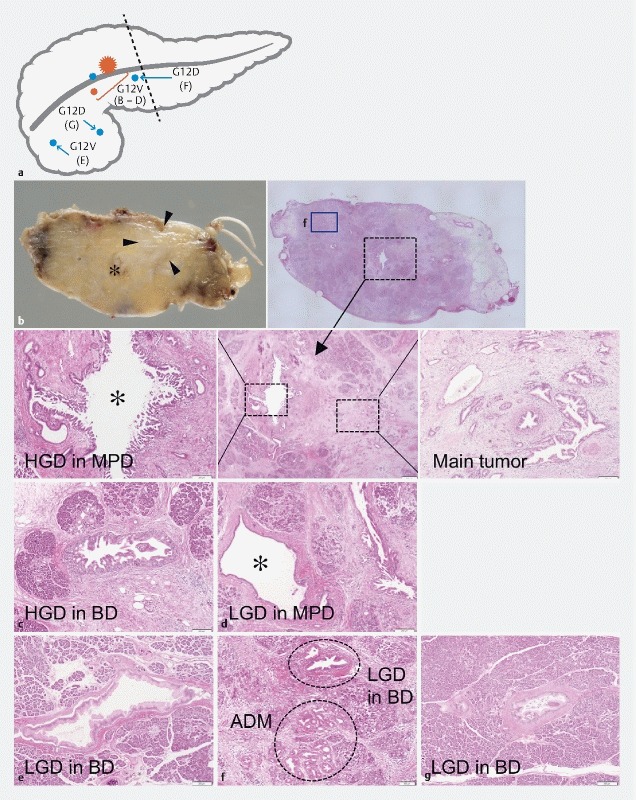
Gross appearance of the resected pancreas and pathological mapping of the main tumor and microscopic lesions. a The whole resected specimen is used to define the distribution of the lesions. The primary ductal adenocarcinoma (PDA; red star) is surrounded by multiple low grade (LGD; blue circles) and high grade dysplasias (HGD; red circles) as illustrated. Dashed line indicates resection margin. b Macroscopic findings for the main tumor. c – g Microscopic lesions associated with the main tumor. The main PDA and closely located HGD in the MPD harbor KRAS G12V ( b ). In the surrounding normal-looking pancreas, LGD and HGDs with KRAS G12V are identified ( c – e ), whereas LGDs, one of which is accompanied by acinar-ductal metaplasia, are marked by KRAS G12D ( f , g ). MPD; main pancreatic duct, BD; branch duct. Asterisk indicates MPD.
We next performed additional genetic analysis using a resected specimen; in the main tumor with dense desmoplasia, KRAS G12V mutation was demonstrated by targeted amplicon sequencing ( Fig. 3b, c and Supplementary Table 1 ; see the Supplementary Methods for details). The series of lesions continuing to the MPD possessed an identical type of KRAS mutation, suggesting a cancerization from the primary PDA ( Fig. 3d, e ). Other abnormal ductal structures corresponding to acinar-to-ductal metaplasia (ADM) to low grade pancreatic intraepithelial neoplasias were noted (PanINs; Fig. 3f, g ). These lesions were micro-dissected and sequenced. Interestingly, some lesions harbored KRAS G12D, a distinct type of variant from the main tumor, indicating an independent development of additional precursor(s). All tumors had no other mutations in the common PDA-related tumor suppressor genes, such as TP53, CDNK2A/p16, SMAD4, or RNF43. Mutations in GNAS were also not evident in the tumors including abnormal ductal structures in the normal looking pancreas.
Supplementary Table 1. Result of multiregion sequencing (targeted amplicon sequencing).
| Main tumor (B) | HGD in MPD (B) | HGD (C) | LGD in MPD (D) | LGD (E) | LGD (F) | ADM (F) | LGD (G) | |
| KRAS | G12V (10.3 %) 1 | G12V (11.5 %) 1 | G12V (19.2 %) 1 | G12V (1 %) 1 | G12V (2 %) 1 | G12D (5.2 %) 1 | G12D (2 %) 1 | G12D (1 %) 1 |
| TP53 | WT | WT | WT | WT | WT | WT | WT | WT |
| CDKN2A | WT | WT | WT | WT | WT | WT | WT | WT |
| SMAD4 | WT | WT | WT | WT | WT | WT | WT | WT |
| GNAS | WT | WT | WT | WT | WT | WT | WT | WT |
| RNF43 | WT | WT | WT | WT | WT | WT | WT | WT |
| BRAF | WT | WT | WT | WT | WT | WT | WT | WT |
| PIK3CA | WT | WT | WT | WT | WT | WT | WT | WT |
| STK11 | WT | WT | WT | WT | WT | WT | WT | WT |
| IDH1 | WT | WT | WT | WT | WT | WT | WT | WT |
| CTNNB1 | WT | WT | WT | WT | WT | WT | WT | WT |
| MAP2K4 | WT | WT | WT | WT | WT | WT | WT | WT |
| TGFBR1 | WT | WT | WT | WT | WT | WT | WT | WT |
| TGFBR2 | WT | WT | WT | WT | WT | WT | WT | WT |
| ARID1A | WT | WT | WT | WT | WT | WT | WT | WT |
| SF3B1 | WT | WT | WT | WT | WT | WT | WT | WT |
| RBM10 | WT | WT | WT | WT | WT | WT | WT | WT |
| KDM6A | WT | WT | WT | WT | WT | WT | WT | WT |
LGD, low grade dysplasia; HGD, high grade dysplasia; ADM, acinar-to-ductal metaplasia; WT, wild-type.
Values in parentheses indicate the multiregions shown in Fig. 3 .
Discussion
Reliable and feasible detection of early stage pancreatic cancer and premalignant lesions, such as PanIN, will greatly aid in patient survival. However, obtaining trustworthy evidence for the malignancy is not always easy, especially when a small lesion is targeted. Recently, cell-free DNA in plasma has attracted attention as a diagnostic tool for various types of cancer 4 . In human PDAs, mutations in KRAS are found in over 90 % of patients 1 , and significant effort has been made to detect the driver gene in patient specimens, such as pancreatic juice or plasma 5 . In the current report, KRAS genotyping of pancreatic juice indicated the presence of “neoplasia” in the pancreas, although pancreatic duct biopsy and pancreatic juice cytology did not provide morphological evidence of malignancy. Insufficient material is a common reason for failure of cytology/biopsy, and diagnostic accuracy is hindered by the invasiveness of tissue collection. In contrast, a genetic approach can detect the tumor-associated alterations irrespective of the tumor size, providing very sensitive diagnostics relative to conventional methods.
It should be noted that, in the current case, two types of KRAS variants were detected in the pancreatic juice. Among the precursor lesions of invasive pancreatic cancer, PanIN may progress to PDA sequentially from a monoclonal precursor, whereas intraductal papillary mucinous neoplasms (IPMNs) are frequently accompanied by multi-centric clones 6 . Interestingly, in our case, targeted amplicon sequencing of the resected specimen demonstrated that the primary PDA and cancerization observed in the main duct were marked by KRAS G12V, and other small areas composed of low grade PanIN (LG-PanIN) associated with ADM on the caudal side of the tumor harbored KRAS G12D. Imaging studies before the surgical intervention did not detect either IPMNs or other satellite lesions to the hypoechoic area; however, genetic tests using pancreatic juice and resected specimens provide evidence of multi-centric clones within the pancreas. Additionally, the KRAS G12V mutation was found in other LG-PanINs located in branch ducts distant from the primary PDA. Such lesions may also develop independently from the primary PDA, and the “shared” mutation in KRAS may be incidental, although the possibility of ductal spread (early dissemination via the pancreatic duct) cannot be entirely excluded 7 .
Detection of a KRAS mutation alone is not sufficient for a definitive diagnosis of PDA, since it is the earliest genetic event during tumorigenesis and most precursor lesions harbor the abnormality 8 . Additional mutations and aberrant protein expression of the tumor suppressor genes, such as TP53 and SMAD 4, can accumulate 1 , and significant time, perhaps over a decade, may be required for progression from the emergence of the initiating mutation to the acquisition of malignant potential 9 . On the other hand, a small subset of PDAs lack the typical stepwise mutations and aberrant protein expressions 10 . Indeed, a smaller number of mutations in the major PDA-associated driver genes, KRAS, CDKN2A, TP53, and SMAD4, is an independent predictor of better overall survival 11 . Therefore, positive KRAS mutations with no other genetic abnormalities observed by liquid biopsy indicate a wide range of conditions from an occurrence of the earliest precursor to PDA with metastatic potential.
Endoscopy is an indispensable tool for the detection of small PDAs, and advanced options, such as genetic and molecular tests, may further enhance diagnostic accuracy. Although tissue collection by EUS-FNA/B is a standard procedure for providing histological evidence of the tumor, the possibility of procedure-related dissemination has to be borne in mind, specifically for candidates with a high probability of a cure 12 . A genetic test may compensate for the invasiveness with variable sensitivity and specificity depending on the source of the samples (i. e. plasma, duodenal and pancreatic juice, and other body fluids). It is necessary to integrate advanced imaging and feasible molecular profiling for precise diagnosis of the tumor.
We report a case of an early stage PDA with genetic evidence of neoplasia (i. e. KRAS mutation) in pancreatic juice before surgical intervention. Sequencing study of the resected specimens indicated the utility of “liquid biopsy” for diagnosis when the findings from pathological examination are not informative. Early detection of PDA is still challenging, and more accurate and less invasive options that can uncover both tumor grade and heterogeneity may bring out the full potential of endoscopic diagnostics without hampering the possibility of a cure.
Supplementary methods
Pancreatic juice collection and cfDNA extraction
Pancreatic juice (< 1 mL) was used, immediately frozen (– 80 °C) after collection via the transpapillary route using an ERCP catheter and drained through an endoscopic nasopancreatic drainage tube. DNA was isolated using the QIAamp Circulating Nucleic Acids Kit (Qiagen; Hilden, Germany) according to the manufacturer’s instructions. The sample was quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific; Waltham, Massachusetts, United States) and a Qubit2.0 fluorometer (Thermo Fisher Scientific).
Mutation detection by digital PCR
Mutant KRAS variants (codons 12 and 13) or mutant GNAS variants (codon 201) were analyzed using a QX200 Droplet Digital PCR System (Bio-Rad; Hercules, California, United States) as described previously 1 . Custom probes and primers were designed for eight major mutations in KRAS codons 12 and 13 or GNAS codon 201 1 . The reaction mixture was prepared as described in Supplementary Table 2 . Purified DNA was partitioned into ~22 000 droplets per sample by mixing with 70 µL of Droplet Generation Oil in a QX200 droplet generator (Bio-Rad). Droplets were then subjected to thermal cycling, as detailed in Supplementary Table 3 . Samples were transferred to a QX200 droplet reader (Bio-Rad) for fluorescence measurement of 6-fluorescein amidite and hexachlorofluorescein probes. Droplets were scored as positive or negative based on their fluorescence intensity, which was determined by the gating threshold defined using positive and negative controls. Finally, absolute copy number input in the reaction and the ratio of mutated fragments were calculated by QuantaSoft (ver 1.7; Bio-Rad) software based on the Poisson distribution.
Supplementary Table 2. Preparation of ddPCR reaction mixture.
| Component | Final concentration |
| ddPCR Supermix for Probes (no dUTP) | 1 × |
| Template DNA 1 | – |
| Additional dNTP mixture | 0.91 mM |
| Primers (forward and reverse primer) | 0.45 µM each |
| Probes (input as a pair of WT and each mutant probe) | |
|
0.45 µM |
|
0.77 µM |
|
0.45 µM |
|
0.45 µM |
|
0.05 µM |
|
0.68 µM |
|
0.07 µM |
|
0.68 µM |
|
0.68 µM |
|
0.45 µM |
|
0.34 µM |
|
0.45 µM |
Total, 22 µL reaction volume.
1 – 4 µL of purified DNA were utilized.
Supplementary Table 3. ddPCR thermal cycling conditions.
| Step | No. of cycles | Temperature, °C | Time, min |
| 1 | 1 | 95 | 10 |
| 2 | 40 | 94 | 0.5 |
| 58 (KRAS)/60 (GNAS) | 1 | ||
| 3 | 1 | 98 | 10 |
Mutation profiling of tumors and abnormal lesions
Each specimen was prepared as formalin-fixed paraffin embedded (FFPE) blocks and slides. Genomic (g) DNA was then purified and isolated using the GeneRead DNA FFPE Kit (Qiagen; Hilden, Germany) according to the manufacturer’s instructions, and finally eluted with 30 µL of elution buffer.
Mutation profiles were determined by target amplicon sequencing using a next generation sequencer as described previously 2 . A 20- to 60-ng portion of gDNA was amplified by PCR using an IPMN/PDA-related gene panel which we designed, which consisted of 18 genes, and 220 amplicons (Ion AmpliSeq Custom DNA Panel) ( Supplementary Table 4 ). Sequencing was performed using an Ion Personal Genome Machine System and the Ion PGM 200 Sequencing Kit (both from Thermo Fisher Scientific) according to the manufacturer’s instructions. Sequence reads were demultiplexed, quality-filtered, and aligned to the human reference genome (GRCh37) using Torrent Suite software (ver. 5.0.4; Thermo Fisher Scientific). Variants were identified with the Variant Caller software (ver. 5.0.4.0; Thermo Fisher Scientific). To identify somatic mutations, independent genotyping of each lesion and normal sample (duodenum) was subtracted; variants found in the normal sample were excluded from the molecular profiling. The variant calling analysis was operated using the somatic variant calling mode optimized to detect low-frequency variants, which was set with the following parameters: minimum allele frequency of 0.02 and minimum coverage of 100. We also excluded putative false-negative variants by evaluating the Phred-scaled variation call quality calculated by this plugin, and by manually confirming the alignment with IGV software (version 2.3.59).
Supplementary Table 4. Targeted regions of the 18 genes explored by the AmpliSeq custom panel.
| Gene | No. of amplicons | Total amplicon length, bp |
| KRAS | 4 | 309 |
| TP53 | 14 | 1317 |
| CDKN2A | 3 | 307 |
| SMAD4 | 11 | 912 |
| GNAS | 2 | 170 |
| RNF43 | 36 | 3349 |
| BRAF | 4 | 342 |
| PIK3CA | 4 | 311 |
| STK11 | 6 | 553 |
| IDH1 | 2 | 153 |
| CTNNB1 | 2 | 152 |
| MAP2K4 | 12 | 978 |
| TGFBR1 | 21 | 1712 |
| TGFBR2 | 12 | 1071 |
| ARID1A | 47 | 2934 |
| SF3B1 | 8 | 628 |
| RBM10 | 15 | 1371 |
| KDM6A | 17 | 1394 |
Acknowledgments
We would like to thank Munehiko Ogata (Sapporo Higashi Tokushukai Hospital) for tissue sample preparation and Atsuko Sumi (Sapporo Higashi Tokushukai Hospital) for technical support on the genetic analyses. We would like to thank Editage ( www.editage.jp ) for English language editing.
Competing interests None
These authors have contributed equally.
References
- 1.Patra K C, Bardeesy N, Mizukami Y. Diversity of precursor lesions for pancreatic cancer: the genetics and biology of intraductal papillary mucinous neoplasm. Clin Transl Gastroenterol. 2017;8:e86. doi: 10.1038/ctg.2017.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Eloubeidi M A, Chen V K, Eltoum I A et al. Endoscopic ultrasound-guided fine needle aspiration biopsy of patients with suspected pancreatic cancer: diagnostic accuracy and acute and 30-day complications. Am J Gastroenterol. 2003;98:2663–2668. doi: 10.1111/j.1572-0241.2003.08666.x. [DOI] [PubMed] [Google Scholar]
- 3.Katsinelos P, Lazaraki G, Chatzimavroudis G et al. Risk factors for therapeutic ERCP-related complications: an analysis of 2,715 cases performed by a single endoscopist. Ann Gastroenterol. 2014;27:65–72. [PMC free article] [PubMed] [Google Scholar]
- 4.Wan J CM, Massie C, Garcia-Corbacho J et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–238. doi: 10.1038/nrc.2017.7. [DOI] [PubMed] [Google Scholar]
- 5.Takai E, Totoki Y, Nakamura H et al. Clinical utility of circulating tumor DNA for molecular assessment in pancreatic cancer. Sci Rep. 2015;5:18425. doi: 10.1038/srep18425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Izawa T, Obara T, Tanno S et al. Clonality and field cancerization in intraductal papillary-mucinous tumors of the pancreas. Cancer. 2001;92:1807–1817. doi: 10.1002/1097-0142(20011001)92:7<1807::aid-cncr1697>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Rhim A D, Thege F I, Santana S M et al. Detection of circulating pancreas epithelial cells in patients with pancreatic cystic lesions. Gastroenterology. 2014;146:647–651. doi: 10.1053/j.gastro.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanda M, Matthaei H, Wu J et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142:730–733 e739. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yachida S, Jones S, Bozic I et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114–1117. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Witkiewicz A K, McMillan E A, Balaji U et al. Whole-exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun. 2015;6:6744. doi: 10.1038/ncomms7744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi H, Kohno T, Ueno H et al. Utility of assessing the number of mutated KRAS, CDKN2A, TP53, and SMAD4 genes using a targeted deep sequencing assay as a prognostic biomarker for pancreatic cancer. Pancreas. 2017;46:335–340. doi: 10.1097/MPA.0000000000000760. [DOI] [PubMed] [Google Scholar]
- 12.Katanuma A, Maguchi H, Hashigo S et al. Tumor seeding after endoscopic ultrasound-guided fine-needle aspiration of cancer in the body of the pancreas. Endoscopy. 2012;44 02:E160–161. doi: 10.1055/s-0031-1291716. [DOI] [PubMed] [Google Scholar]
References (Supplementary methods)
- 1.Ono Y, Sugitani A, Karasaki H et al. An improved digital polymerase chain reaction protocol to capture low-copy KRAS mutations in plasma cell-free DNA by resolving subsampling issues. Mol Oncol. 2017;11:1448–1458. doi: 10.1002/1878-0261.12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Imai K, Karasaki H, Ono Y et al. Metachronous pancreatic cancer originating from disseminated founder pancreatic intraductal neoplasias (PanINs) J Pathol Clin Res. 2015;1:76–82. doi: 10.1002/cjp2.8. [DOI] [PMC free article] [PubMed] [Google Scholar]