

Abstract
Quinazoline derivatives are potent inhibitors of human epidermal growth factor receptor (EGFR) as anticancer agents. In this study, the cytotoxic effects of a new series of synthesized quinazoline derivatives were evaluated using MTT assay against MCF-7 and HT-29 cell lines. Using molecular docking, the binding modes of all compounds were analyzed at the binding site of EGFR. Based on the results, the compounds L1, L2, L4, L5, L6, L7, L10, L15, and L18 may be promising EGFR inhibitors based on docking score and hydrogen bonds. Consistent with the experimental data, Met769 is recognized as a key residue in the binding of potential inhibitors. According to the MTT cytotoxicity assays, Lipinski's rule of five (RO5), absorption, distribution, metabolism, excretion, and toxicity (ADMET) parameters, and docking studies, three compounds L4, L15, and L10 with IC50 values of 80, 60, and 1 μM against the MCF-7 were selected for further comparative assessments. The dynamics of free EGFR, and selected ligand-EGFR complexes were investigated using molecular dynamics (MD) simulation studies. The results indicated that the three compounds bound to EGFR active site in a stable manner during the simulation through the formation of new hydrogen bonds with Phe699, Leu694, Gly700, Lys721, Met769, Arg817, and Asp831 with the superiority of compound L15. These features can promote future drug candidate designing to produce better derivatives in the search for the anticancer agents.
Keywords: 4-Anilinoquinazoline, EGFR, Molecular docking, Molecular dynamics simulation, 4(3H)-quinazolinones
INTRODUCTION
Epidermal growth factor receptor (EGFR) is the cell-surface receptor of protein tyrosine kinase family which is over-expressed in numerous human tumors, containing ovarian, prostate, breast, bladder, lung, and colon(1,2). Several quinazoline derivatives such as erlotinib, gefitinib, and lapatinib have been synthesized as reversible inhibitors of tyrosine kinase(3). Due to developed resistance to first-generation drugs, irreversible inhibitors have been introduced with limited clinical efficacy(3). So far, numerous studies have been targeted at finding new structures based on the modification of quinazoline as potent inhibitors of EGFR in many anticancer studies(4,5,6). In addition, various compounds with substituted quinazolinone structural motif were reported as potent inhibitors of EGFR which were overexpressed in the number of cancer cell lines(7,8). Thus, quinazoline/quinazolinone-containing compounds represent an attractive scaffold for designing anticancer drugs(4,5,6,7,8,9,10).
In two previous studies, our research group have synthesized 4-anilinoquinazoline and 4(3H)-quinazolinones Schiff base derivatives reporting the antimicrobial activity(11,12). As explained above, these derivatives may be regarded as new inhibitors of human EGFR(4,5,7,8,9).
In the present study, cytotoxic effects of the 19 newly synthesized compounds were investigated against two cancer cell lines containing human breast adenocarcinoma (MCF-7) and human colon adenocarcinoma (HT-29) cell lines by MTT assay. The reason for choosing two types of cell lines was that variable levels of EGFR, human epidermal growth factor receptor 2 (HER-2), and/or HER-4 express by the growth of MCF-7 and HT-29 cell lines(1,9).
Molecular docking and molecular dynamics (MD) were used to recognize the binding sites on targets, conformational changes of biomolecules, stability, and protein folding(13). Therefore, in order to evaluate whether the synthesized compounds can potentially bind to EGFR an analysis of drug-receptor interaction was carried out by molecular modeling approaches.
To determine the suitability of the newly synthesized compounds, physicochemical properties are very important to proceed for further modifications until they achieve clinical trials(14).
Significant molecular properties for drug's pharmacokinetics in the human body were defined by the Lipinski's rule of five (RO5)(15).
Absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties which establish the pharmacokinetic outline of a drug molecule, are very crucial in evaluating its pharmacodynamics activities(16). As a result, the best compounds were introduced after evaluating its cytotoxic effects, molecular docking, in silico prediction of ADMET properties following Lipinski's RO5. Free EGFR, complexes of EGFR, and three compounds (L4, L15, and L10) were chosen for MD simulation to understand the ligand-receptor possible intermolecular interactions in details.
MATERIALS AND METHODS
Two cell lines were obtained from Pasteur Institute of Iran (Tehran, I.R. Iran). RPMI-1640 was prepared as culture media. 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), and dimethyl sulfoxide (DMSO) were purchased from Merck, Germany. Absorbance was measured using an ELISA plate reader (ELX 808, USA).
Biological studies
The cytotoxic activities of synthesized compounds against MCF-7 and HT-29 cell lines were studied by MTT as previously described(2). The cell suspension was treated with 20 μL of various concentrations (0.1-100 μM) of synthesized samples in 1% DMSO diluted with culture medium. Erlotinib and DMSO were employed as the positive control and negative control, respectively. The experiments were repeated three times. The percentage of cell viability was calculated by reported equation(2). The half maximal inhibitory concentration (IC50) values in μM against both mentioned cell lines are reported in Table 1.
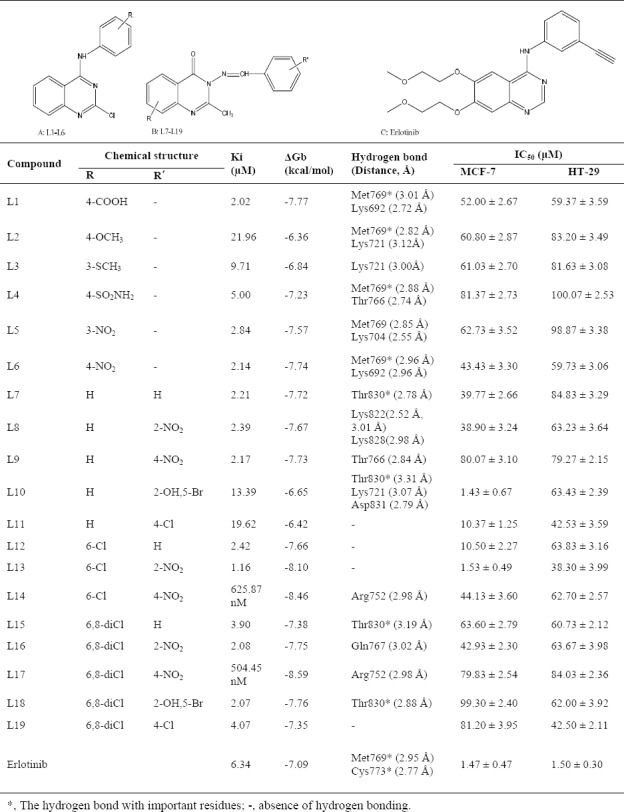
Table 1.
Estimated free energy of binding (▵Gb) and inhibition constants (Ki) for 19 novel (A) 4-anilinoquinazoline, (B) 4(3H)-quinazolinones derivatives, and (C) erlotinib as potential epidermal growth factor receptor inhibitors.
Compliance of synthesized compounds to criteria of the prospective drugs
Lipinski's RO5 is a rule of scan to evaluate drug-likeness or determine whether a chemical compound with a given biological or pharmacological activity has properties that lead to a possible oral drug in humans(15). Besides, other significant conditions and essential elements of pharmacokinetics are ADMET properties(14). Today a lot of online tools and offline software programs are available, which can help in predicting these properties of a drug candidate. The properties displayed in Table 2 are calculated using the SwissADME website(17). The ADMET profiling indications were based on statistically derived from pkCSM(16)or Tox Prediction web server(18). Here, this program was applied to calculate the essential parameters, including Ames toxicity, hepatotoxicity, P-glycoprotein substrate, and median lethal dose (LD50) (mol/kg), etc.
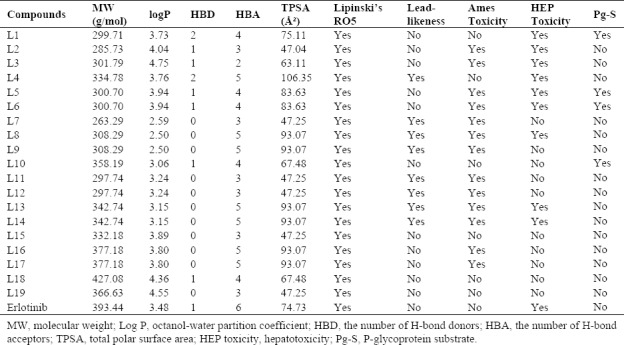
Table 2.
Drug-likeness prediction and absorption, distribution, metabolism, excretion, and toxicity prediction.
Docking studies
The theoretical binding mode of synthesized compounds to EGFR using the AutoDock 4.2.2 software(19). The target protein was derived from the RCSB protein data bank (PDB)(20). Crystal's structure of the EGFR tyrosine kinase domain with erlotinib (PDB code 1M17) at resolution 2.6 Å imported into AutoDock(21). The two-dimensional (2D) structures of the compounds were optimized using HyperChem 7.0 software as reported in previous studies(22,23).
Docking was performed with the procedure similar to the previous works(22,24). Also, to ensure the validity of docking, erlotinib as the well-known 4-anilinoquinazoline inhibitor was re-docked to the binding site with the root mean square deviations (RMSD) value relative to the crystal structure of 0.54 Å. The grid box was centered on Cα of Met769. Grid box dimensions were 50 × 50 × 50 (all in Å) with a 0.375 Å grid point spacing. A Lamarckian genetic algorithm (LGA) program was used to calculate 200 different conformers(19,24).
Molecular dynamics simulations
MD simulation was used to explore the characterization of free EGFR and interaction between EGFR and three different inhibitors. Topology file for compounds L4, L15, and L10 were generated by the PRODRG server(25). MD simulations were performed using the GROMACS package 5.3.1(26). The “pdb2gmx” program was used to generate the topology file for protein using the G43a1 united-atom force field(27). The systems inserted into dodecahedron box and was solvated with the single point charge (SPC216) water model(28). The complex 1 (EGFR-L4), complex 2 (EGFR-L15), complex 3 (EGFR-L10), and free protein were neutralized by adding two, and one negatively charged Cl counter ion, respectively. The systems were introduced to energy minimization. The Berendsen algorithm was selected for thermostat and barostat in equilibration phase(29). The lengths of hydrogen-containing bonds were constrained using linear constraint solver (LINCS) algorithm(30).
MD simulations were executed in the NVT and NPT ensemble at a weak temperature coupling (τ = 0.1 ps) and pressure coupling (τ = 1 ps) in association with position restraint procedure. The protein, ligand, solvent, and the Cl ions were separately coupled to the thermostat with a reference temperature of 300 K and at the reference pressure of 1 bar. The thermostat and barostat were the Nosé-Hoover thermostat and the isotropic Parrinello-Rahman barostat for production step(29). The long-range electrostatic interactions were preserved by the particle-mesh ewald (PME) model(31). The MD simulations times were established to 40 ns with a time step of 2 fs in the periodic boundary condition. Temperature, potential and other analyses were deliberated using GROMACS package. The flexibility and stability of receptor and ligands during MD were inspected via the residue root mean square fluctuation (RMSF) and RMSD whose square fluctuation (RMSF) and RMSD whose plots are shown in Fig. 1. Computing the number of hydrogen bonds, solvent accessible surface (SASA), the radius of gyration (Rg) and other calculations were done as mentioned previously(32) (Table 3, Figs. 1 and 2). In order to display the binding mode obtained of ligand's trajectories after MD simulation, 3D and 2D graphical tools such as the visual MD (VMD)(33)the LigPlot software(34)were employed (Fig. 2).
Fig. 1.

Analysis plots of the protein backbone and ligands structures for free epidermal growth factor receptor (EGFR), complex 1, 2, and 3 during 40 ns molecular dynamic simulation; A, the root mean square deviations (RMSD) plot; B, root mean square fluctuation (RMSF) plot; C, radius of gyration (Rg) plot; D, solvent accessible surface (SASA) plot.
Table 3.
The obtained results from structural analysis of three complexes during molecular dynamic simulation.
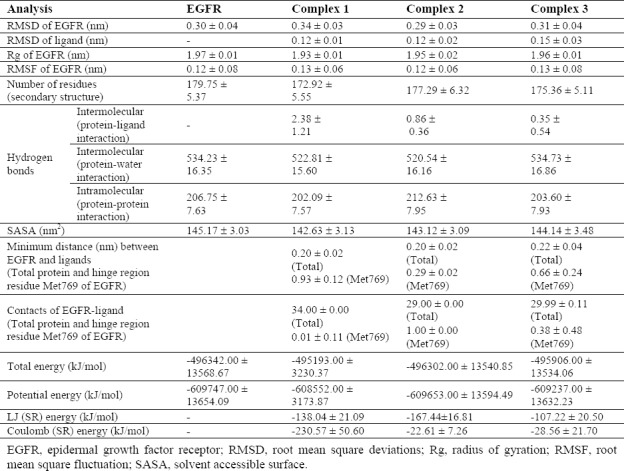
Fig. 2.

Analysis plots of the complexes 1, 2, and 3 during 40 ns molecular dynamic simulation; A, Minumum distance plot between hinge region residue Met769 and ligand; B, number of contacts plot of the hinge region residue Met769 and ligand; The H-bonding distribution plot vs time for C, L4-epidermal growth factor receptor (EGFR); D, L15-EGFR; and E, L10-EGFR.
RESULTS
MTT assay for cell viability/proliferation
All compounds were evaluated against MCF7 and HT-29 cell lines using MTT assay(2). Based on results presented in Table 1, the most of compounds were active against two cell lines. The cytotoxic activities of the compounds were comparable to erlotinib as a reference compound. Compounds L10 and L13 had potency similar to erlotinib against the MCF-7 cell line. In the other hand, replacement of H and Cl at L10 and L13 with 6,8-dichloro led to to compounds L18 and L16, respectively, which showed lower cytotoxicity than the first against one or two cell lines. Compared to erlotinib, compounds L11 and L12 were in the next priority against the MCF-7 cell line.
Drug-likeness prediction and absorption, distribution, metabolism, excretion, and toxicity prediction
All compounds have followed Lipinski's RO5 (Table 2). The ADMET predictions of some compounds have shown satisfactory results. Compound L15 violated no rule of the Lipinski's RO5 and met all ADMET parameters. In addition, compounds L1, L4, L10, L18, and L19, were not mutagenic based on the ADMET predictions and therefore may not be carcinogenic. Interestingly, all compounds were predicted to have absorption from the human intestine if administered orally based on SwissADME server. Toxic doses are often known as LD50 values in mg/kg body weight. The results of Tox prediction website showed that LD50 and predicted toxicity class of the three selected compounds L4, L15, and L10 may be 1500, 1230, and 1000 mg/kg of class IV with prediction accuracy 54.26%, 69.26%, and 67.38%, respectively. The LD50 was 125 mg/kg of class III for erlotinib. This implies that these compounds may have acceptable ADMET properties.
Molecular docking studies
The conformation with the lowest binding energy (~ -6 – -9 kcal mol-1) supports the idea that some of the compounds are well incorporated in the binding pocket. Dependent on the 4-anilinoquinazoline or 4(3H)-quinazolinone derivatives bearing Schiff base moiety, the interacting residues through hydrogen bonding with the studied compound differs. The binding patterns of different compounds are also slightly different, which may be responsible for the activity variations. It must be noted that inhibitors with 4-anilinoquinazoline scaffold have a common feature that in some cases they formed a hydrogen bond with the backbone NH of Met769 in the Hinge region(5,7). These compounds also were deeply embedded into EGFR via hydrophobic contacts that are conserved in the majority of the structures. These results were in consistent with the previously studied X-ray crystal structures, indicating the important roles of these(3,4,5,7,8,9).
Confirmation of molecular docking by molecular dynamics simulation
The trajectory stability of the free EGFR and complexes 1-3 were confirmed by the analyses (Table 3, and Figs. 1,2). As shown in the RMSD plots (Fig. 1A), the trajectories were stable during the last 25 ns simulation. It is often considered that small RMSD values of a simulation indicate a stable state of the system (Table 3). The EGFR-complex 1 showed more deviation of RMSD average with regard to the free EGFR which relatively was in agreement with RMSF (Table 3). It may be due to the more interaction of EGFR atoms in the presence of L4 that causes some conformational and structural changes. This highlights the stable binding of the L4 with EGFR leading to instability of the protein. In addition, RMSD average shows primary fluctuations in the magnitude of RMSD of ligand's atoms during MD. Therefore, three compounds obtained an equilibrium state as described by the RMSD profile (Fig. 1A). To achieve a more detailed description of flexibility of the protein residues in the absence and presence of ligands, backbone RMSF values was computed (Table 3). Very few fluctuations were seen beyond 0.5 nm during the MD simulations (Fig. 1B). Only, residues 672 and 964 exposed relatively considerable fluctuations in the vicinity of N-and C-terminus. Higher local fluctuations followed at interface between domains IIA followed at the interface between domains IIA and IIB (the regions are specified with three bars) due to the presence of loops preceding domains (Fig. 1B). In addition, there are a few regions, including residues 850-863, and 888-907 that show higher flexibility than that of the free EGFR. It was found that the complexes 1 and 3 exhibited a little more fluctuation than the free EGFR (Table 3 and Fig. 1B). In other words, complex 2 has very low fluctuations identical to free EGFR, which again emphasis on stability of EGFR after L15 binding. It can be said that the binding of ligands did not affect the proteins' overall conformational diversity significantly, as there was no major change between the RMSD and RMSF values. The plots depicted from Rg of protein displayed that EGFR in four systems had a compact structure (Fig. 1C). The Rg values are compatible upon ligands complexation with respect to free EGFR (Table 3). It can be clearly seen that the Rg value of EGFR decreases slightly upon binding of ligands implying a more compact structure after the MD simulation. Rg value was more deviated in the complex 1 and had a more compact structure with more hydrogen bonds, which confirmed more changes of complex 1 in agreement with other results.
As shown in Table 3, complex 1 exhibited relatively higher deviations (142.63 ± 3.13) of SASA with time due to making more hydrogen bonds, while native EGFR structure showed higher values. However, curve for free EGFR did not differ seriously and preserved SASA, implying that the native conformation of EGFR was mainly conserved during the production time of simulation. These results were in line with Rg analysis. Together with SASA values reduction, Rg values decreased from that for starting structure to average, which along with the interaction mode showed the effective binding of L15 to EGFR.
To further confirm of stability and the effects of ligands binding, the DSSP algorithm was obtained. The main secondary structures of the EGFR maintain rather stable during 40 ns MD simulation. There were no significant changes in structural elements (Table 3). Although complex 1 showed the largest deviation of 179.75 ± 5.37 nm2 (Table 3). Very low levels of structural changes in the protein occurred due to ligands interactions especially for compound L15 that is in agreement with other results. This further confirmed the stability of complex 2. Hence L15 is partially affecting the structural conformation of EGFR.
In addition, the average of the minimum distances between “hinge region residue” Met769 in the active site and showed the superiority of L15 of complex 2, because this was very less than complex 1, and 3 (Table 3). It results in the formation of a stable hydrogen bond through Met769 as key residue with compound L15 (Fig. 2A and Fig. 3B). However, the results of this analysis could be compared with the results of the analysis of the number of contacts for more certainty (Table 3). The number of contacts for L4 in complex 1 is very less than that in complex 2, and 3 about key residue Met769 (Fig. 2B), but it is more for other residues in the active site (data not shown for brevity). More spatial prohibition of L4 is more notable than the interaction between L15 and EGFR due to other different interactions via six new hydrogen bonds (Fig. 3A). These residues are in accordance with other results(3,4,5,7,8,9). Results of stability of hydrogen bonding in MD showed that in most conformers, one/two hydrogen bonds were found for complexes 2 and 3. This value reached to a maximum of 8 and an average of 4 for complex 1 Fig. 2C–2E).
Fig. 3.

Schematic interaction resulting from LigPlot and VMD softwares for selected three compounds L4 (A and D), L15 (B and E), and L10 (C and F) after molecular dynamic simulations. In parts A, B, and C, the compounds exposure is blue-highlighted and hydrogen bonding is in green. Hydrophobic interactions are presented by red color. The hinge region residue Met769 is in green in D-F.
Temperature and pressure extended to plateau at 300 K and 1 bar (data not shown). The more negative total energy, potential energy, LJ (SR) energy of the complex 2 indicated that this complex was more stable with more hydrophobic interactions than complex 1, but the coulomb (SR) energy was more positive than complex 1 due to less hydrogen bonds (Table 3). Negative values of LJ (SR) and coulomb (SR) energies indicate the attraction between the receptor and the mentioned compounds. The number of hydrogen bonds between receptor and water was consistent with total energy in Table 3. The small changes of hydrogen bonds in the number of hydrogen bonds between protein and water, and intramolecular hydrogen bonds of protein indicated that there are the stable and soluble structures due to the high number of hydrogen bonds (Table 3).
Comparing the binding mode of three compounds before and after simulation showed that conformational changes of the main chain in Met769 led to the formation other hydrogen bonds (Fig. 3). Some new residues such as Phe699, Leu694, Gly700, Lys721, Met769, Cys773, Arg817, and Asp831 are positioned in proximity of ligands that could participate in formation of new six hydrogen bonds with L4 and one new hydrogen bond with L15 via Met769 at distance 2.68 Å in accordance with other results(3,4,5,7,8,9). It must be noted that Lys 721 maintains a hydrogen bond with L10. Thus the end of MD simulation, new hydrogen bonds were established and previous hydrogen bonds, including Met769 and Thr830 were replaced (Fig. 3). Here the valuable application of MD simulations was indicated after docking of ligands in the binding site. Therefore, the used MD simulation was essential to specify geometry of EGFR in vicinity of water. As illustrated in (Fig. 3), compound L15 was located within this binding pocket. Thus, according to items listed above, L15 can be considered as a compound which maintains the EGFR structure stability throughout the simulation time after binding with the “hinge region residue” Met769. Overall, the analysis of MD simulation showed the superiority of complex 2 over complexes 1 and 3. The results obtained are consistent with the reports from Woods et al(35).
DISCUSSION
Cancer is one of the global health problems. Significant progress has been observed in cancer research in the last ten years(27). Considering the adverse complications and resistance to the current chemotherapeutic agents, development of more efficient anticancer agents with less harm has remained as an important subject in drug design(27). The findings of this study provide an integral part of the pre-clinical investigation of our compounds that can be further developed as anticancer agents against tumors, with excellent oral absorption. Based on The MTT results, showed that compounds L10-L13 substituted with OH, Br, Cl, and NO2 groups may play an important role in growth inhibition of the MCF-7 cell line with IC50 values of 1-10 μM compared to erlotinib. This could be attributed to the withdrawing effects on the heterocyclic ring.
The docking results showed that the residues Met769 and Thr830 play a pivotal role in making of the hydrogen bond with EGFR(5,7). The same conclusion to our docking results was reported for several other compounds(4,5,7,8,9). Compared to erlotinib, some of these compounds properly fit into the ATP binding pocket in EGFR crystal structure, suggesting that they may be potential EGFR inhibitors.
Molecular properties, Lipinski's RO5, and ADMET parameters predicted that some compounds have acceptable properties and lack the sign of mutagenicity effect. Three compounds L4, L10, and L15 violated no rule of the Lipinski's RO5 and also were not mutagenic based on the ADMET predictions and therefore may not be carcinogenic. The activity of both compounds L4 and L15 may not be correlated to multi-drug resistant tumors due to not being P-glycoprotein substrates based on Swiss ADME. Therefore, the MTT cytotoxicity assays, Lipinski's RO5, ADMET parameters, and docking studies demonstrated that the three compounds L4, L15, and L10 were selected as the best compounds for further assessments.
Clearing up of ligand binding mechanisms is the essential step to achieve more selective compounds for a given target. Complex delivered in its natural environment is required to achieve more precise ligand-receptor models(24,32). Therefore, MD simulations of complexes of the EGFR and three compounds with the weak, moderate, and strong cytotoxic effect were performed for further assessment of the effects of different ligand binding on the conformation of EGFR The three complexes immersed in water molecules stay in equilibrium during MD. We also found that the MD simulation created an improved and more relaxed structure of enzyme and ligands.
However, compound L15 exhibited nearly appropriate properties comparable to those of the standard drugs, which include good potential interaction with EGFR confirmed by MD simulation above. Compound L15, has strong interaction with ”hing region key residue“ Met769 of EGFR target, which is involved in the anticancer treatment strategies. Moreover, the overall analysis of MD simulation showed the superiority of complex 2 over complexes 1, and 3. As a result, in accordance with other results, compound L15 with quinazolinone structural motif could be a potential lead compound for identification of EGFR inhibitors(7,8).
CONCLUSION
This paper provided an approach for studying the interactions of protein with new three ligands using molecular modeling techniques. This study demonstrated the utility of other quinazoline/quinazolinone derivatives as an attractive scaffold for the development of potential EGFR inhibitors. More improvements are needed in progress to optimize new quinazoline derivatives as anticancer drug candidates. The prediction methods employed in the current study help to accelerate the analysis of the designed compounds, which were developed in our laboratory before pre-clinical, time-consuming, and high risking experimental methods. It can also be used to select the best compound among some novel proposed derivatives. Hence, it is desired that biological evaluation of the designed compounds for their inhibitory activities on EGFR is conducted using enzymatic specific tests. This research highlights the potential inhibitory activity of selected derivatives for further development.
ACKNOWLEDGMENTS
A part of this project was financially supported (Grant No. 394158) by the Vice Chancellery of Research of Isfahan University of Medical Sciences, Isfahan, I.R. Iran. We also grateful to the Yasuj University of Medical Sciences, Yasuj, I.R. Iran.
REFERENCES
- 1.Xu H, Yu Y, Marciniak D, Rishi AK, Sarkar FH, Kucuk O, et al. Epidermal growth factor receptor (EGFR)-related protein inhibits multiple members of the EGFR family in colon and breast cancer cells. Mol Cancer Ther. 2005;4(3):435–342. doi: 10.1158/1535-7163.MCT-04-0280. [DOI] [PubMed] [Google Scholar]
- 2.Nasab RR, Hassanzadeh F, Khodarahmi GA, Mirzaei M, Rostami M. Synthesis, characterization, cytotoxic screening, and density functional theory studies of new derivatives of quinazolin-4(3H)-one Schiff bases. Res Pharm Sci. 2017;12(6):444–455. doi: 10.4103/1735-5362.217425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sun J, Wang XY, Lv PC, Zhu HL. Discovery of a series of novel phenylpiperazine derivatives as EGFR TK inhibitors. Sci Rep. 2015;5:13934–13945. doi: 10.1038/srep13934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Abuelizz HA, Marzouk M, Ghabbour H, Al-Salahi R. Synthesis and anticancer activity of new quinazoline derivatives. Saudi Pharm J. 2017;25(7):1047–1054. doi: 10.1016/j.jsps.2017.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ismail RS, Ismail NS, Abuserii S, El Ella DAA. Recent advances in 4-aminoquinazoline based scaffold derivatives targeting EGFR kinases as anticancer agents. Future Journal of Pharmaceutical Sciences. 2016;2(1):9–19. [Google Scholar]
- 6.Jafari E, Khajouei MR, Hassanzadeh F, Hakimelahi GH, Khodarahmi GA. Quinazolinone and quinazoline derivatives: recent structures with potent antimicrobial and cytotoxic activities. Res Pharm Sci. 2016;11(1):1–14. [PMC free article] [PubMed] [Google Scholar]
- 7.Al-Suwaidan IA, Abdel-Aziz AA, Shawer TZ, Ayyad RR, Alanazi AM, El-Morsy AM, et al. Synthesis, antitumor activity and molecular docking study of some novel 3-benzyl-4 (3H) quinazolinone analogues. J Enzyme Inhib Med Chem. 2016;31(1):78–89. doi: 10.3109/14756366.2015.1004059. [DOI] [PubMed] [Google Scholar]
- 8.Babu YR, Bhagavanraju M, Reddy GD, Peters GJ, Prasad VV. Design and synthesis of quinazolinone tagged acridones as cytotoxic agents and their effects on EGFR tyrosine kinase. Arch Pharm (Weinheim) 2014;347(9):624–634. doi: 10.1002/ardp.201400065. [DOI] [PubMed] [Google Scholar]
- 9.Ahmed MF, Belal A, Youns M. Design, synthesis, molecular modeling and anti-breast cancer activity of novel quinazolin-4-one derivatives linked to thiazolidinone, oxadiazole or pyrazole moieties. Med Chem Res. 2015;24(7):2993–3007. [Google Scholar]
- 10.Hosseinzadeh L, Aliabadi A, Rahnama M, Sadeghi HMM, Khajouei MR. Synthesis and cytotoxic evaluation of some new 3-(2-(2-phenylthiazol-4-yl) ethyl)-quinazolin-4 (3H) one derivatives with potential anticancer effects. Res Pharm Sci. 2017;12(4):290–298. doi: 10.4103/1735-5362.212046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nasab RR, Hassanzadeh F, Khodarahmi GA, Rostami M, Mirzaei M, Jahanian-Najafabadi A, et al. Docking study, synthesis and antimicrobial evaluation of some novel 4-anilinoquinazoline derivatives. Res Pharm Sci. 2017;12(5):425–433. doi: 10.4103/1735-5362.213988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nasab RR, Mansourian M, Hassanzadeh F. Synthesis, antimicrobial evaluation and docking studies of some novel quinazolinone Schiff base derivatives. Res Pharm Sci. 2018;13(3):213–221. doi: 10.4103/1735-5362.228942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jayakanthan M, Jubendradass R, D'Cruz SC, Mathur PP. A use of homology modeling and molecular docking methods: to explore binding mechanisms of nonylphenol and bisphenol A with antioxidant enzymes. Methods Mol Biol. 2015;1268:273–289. doi: 10.1007/978-1-4939-2285-7_12. [DOI] [PubMed] [Google Scholar]
- 14.Ali AA, Lee YR, Chen TC, Chen CL, Lee CC, Shiau CY, et al. Novel anthra [1,2-c][1,2,5] thiadiazole-6,11-diones as promising anticancer lead compounds: Biological evaluation, characterization & molecular targets determination. PLoS One. 2016;11(4):e0154278. doi: 10.1371/journal.pone.0154278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lipinski CA. Lead-and drug-like compounds: the rule-of-five revolution. Drug Discov Today Technol. 2004;1(4):337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 16.Pires DE, Blundell TL, Ascher DB. pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J Med Chem. 2015;58(9):4066–4072. doi: 10.1021/acs.jmedchem.5b00104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Daina A, Michielin O, Zoete V. SwissADME: a free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci Rep. 2017;7:42717–42729. doi: 10.1038/srep42717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Drwal MN, Banerjee P, Dunkel M, Wettig M, Preissner R. ProTox: a web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014;42(Web Server issue):W53–W58. doi: 10.1093/nar/gku401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, et al. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berman HM, Battistuz T, Bhat TN, Bluhm WF, Bourne PE, Burkhardt K, et al. The protein data bank. Acta Crystallogr D Biol Crystallogr. 2002;58(1):899–907. doi: 10.1107/s0907444902003451. [DOI] [PubMed] [Google Scholar]
- 21.Stamos J, Sliwkowski MX, Eigenbrot C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J Biol Chem. 2002;277(48):46265–4672. doi: 10.1074/jbc.M207135200. [DOI] [PubMed] [Google Scholar]
- 22.Mansourian M, Fassihi A, Saghaie L, Madadkar-Sobhani A, Mahnam K, Abbasi M. QSAR and docking analysis of A2B adenosine receptor antagonists based on non-xanthine scaffold. Med Chem Res. 2015;24(1):394–407. [Google Scholar]
- 23.Coleman WF, Arumainayagam CR. HyperChem 5 (by Hypercube, Inc.) J Chem Educ. 1998;75(4):416. [Google Scholar]
- 24.Mansourian M, Madadkar-Sobhani A, Mahnam K, Fassihi A, Saghaie L. Characterization of adenosine receptor in its native environment: insights from molecular dynamics simulations of palmitoylated/glycosylated, membrane-integrated human A(2B) adenosine receptor. J Mol Model. 2012;18(9):4309–4324. doi: 10.1007/s00894-012-1427-y. [DOI] [PubMed] [Google Scholar]
- 25.SchuÈttelkopf AW, Van Aalten DM. PRODRG: a tool for high-throughput crystallography of protein-ligand complexes. Acta Crystallogr D Biol Crystallogr. 2004;60(Pt 8):1355–1363. doi: 10.1107/S0907444904011679. [DOI] [PubMed] [Google Scholar]
- 26.Hess B, Kutzner C, van der Spoel D, Lindahl E. GROMACS 4: algorithms for highly efficient, load-balanced, and scalable molecular simulation. J Chem Theory Comput. 2008;4(3):435–447. doi: 10.1021/ct700301q. [DOI] [PubMed] [Google Scholar]
- 27.Sadeghian-Rizi S, Khodarahmi GA, Sakhteman A, Jahanian-Najafabadi A, Rostami M, Mirzaei M, et al. Biological evaluation, docking and molecular dynamic simulation of some novel diaryl urea derivatives bearing quinssdynamic simulation of some novel diaryl urea derivatives bearing quinoxalindione moiety. Res Pharm Sci. 2017;12(6):500–509. doi: 10.4103/1735-5362.217430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berendsen HJC, Postma JPM, van Gunsteren WF, Hermans J. Interaction Models for Water in Relation to Protein Hydration. In: Pullman B, editor. Intermolecular Forces. Vol 14. Dordrecht: Springer; 1981. pp. 331–342. [Google Scholar]
- 29.Berendsen HJC, Postma JPM, van Gunsteren WF, DiNola A, Haak JR. Molecular dynamics with coupling to an external bath. J Chem Phys. 1984;81(8):3684–3690. [Google Scholar]
- 30.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A linear constraint solver for molecular simulations. J Comput Chem. 1998;18(12):1463–1472. [Google Scholar]
- 31.Darden TA, York DM, Pedersen LG. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98(12):10089–10092. [Google Scholar]
- 32.Mansourian M, Mahnam K, Madadkar-Sobhani A, Fassihi A, Saghaie L. Insights into the human A1 adenosine receptor from molecular dynamics simulation: structural study in the presence of lipid membrane. Med Chem Res. 2015;24(10):3645–3659. [Google Scholar]
- 33.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33–38. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 34.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: a program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8(2):127–134. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 35.Songtawee N, Gleeson MP, Choowongkomon K. Computational study of EGFR inhibition: molecular dynamics studies on the active and inactive protein conformations. J Mol Model. 2013;19(2):497–509. doi: 10.1007/s00894-012-1559-0. [DOI] [PubMed] [Google Scholar]