

Abstract
Osteoporosis is a common skeletal disease characterized by bone loss and subsequent increased risk of fragility fractures. Recent advances in our mechanistic understanding of molecular communications among osteoblasts, osteoclasts, and osteocytes give insight into the important roles of the canonical Wnt/β-catenin pathway and the RANK/RANKL/OPG pathway in the process of bone remodeling. Due to the translation of the canonical Wnt/β-catenin pathway and the RANK/RANKL/OPG pathway in the regulation of osteoblasts and osteoclasts, new targets have been studied in recent years, such as sclerostin and receptor activator of NF-κB ligand (RANKL). In this review, we first introduce the signaling pathways involved in interactions among osteoblasts, osteoclasts, and osteocytes. Next, we describe clinical trials of denosumab and romosozumab, which are monoclonal antibodies that target RANKL and sclerostin, respectively. We analyze the efficacy of these drugs and provide a profile for the management of osteoporosis.
MeSH Keywords: Osteoporosis, Therapeutic Uses, Wnt Signaling Pathway
Background
Osteoporosis is a skeletal disease that occurs worldwide and is characterized by bone loss, microarchitectural deterioration, and compromised bone strength. Osteoporosis leads to increasing bone fragility and propensity for fracture, particularly in postmenopausal women [1–3]. Low bone mineral density (BMD), evaluated by the current criterion standard, dual-energy x-ray absorptiometry (DXA), is a predictor of fracture risk [1,3]. Osteoporosis is defined by either a fragility fracture or by a BMD T score of −2.5 or lower in the femoral neck or hip bones [3,4]. Among fragility fractures, vertebral and hip fractures pose the greatest risk of morbidity and mortality [5].
Several drugs are available in for the management of osteoporosis. These drugs are divided into antiresorptive agents and anabolic agents, such as bisphosphonates estrogens and parathyroid hormones [2–4,6,7]. Despite their efficacy in the treatment of osteoporosis, adverse effects and low adherence may impede long-term use. Nowadays, long-acting bisphosphonates may be the best choice for a patient with poor adherence because of their long half-life in the skeleton. In recent years, receptor activator of NF-κB ligand (RANKL) and sclerostin have been promising therapeutic targets. Denosumab, a monoclonal antibody that binds to RANKL, mimics osteoprotegerin (OPG), thereby inhibiting osteoclastogenesis [8–10]. Romosozumab, a monoclonal antibody that binds to and inhibits sclerostin, has the dual effects of promoting bone formation and inhibiting bone resorption [11,12].
In this review, we give a brief introduction to the canonical Wnt/β-catenin pathway and the RANK/RANKL/OPG pathway, which includes the functional mechanisms of sclerostin and RANKL. Then, we review the clinical trials of denosumab and romosozumab in detail, including efficacy and safety profiles. We pay special attention to BMD gains at the lumbar spine, total hip, and femoral neck. We compare concurrent therapy and sequential therapy among bisphosphonates, parathyroid hormone, denosumab, and romosozumab, with the goal of identifying the best therapies for management of osteoporosis.
Signaling Pathways Involved in Bone Remodeling
Bone remodeling is a dynamic process in which bone formation and bone resorption interact with each other [6]. To maintain bone homeostasis, bone formation and bone resorption must be dynamically balanced. Osteoblasts conduct bone formation, whereas osteoclasts conduct bone resorption. Osteocytes interact with osteoblasts and osteoclasts, influencing bone formation and bone resorption by adjusting the secretion of important proteins in response to changes in mechanical loading (Figure 1) [13–17].
Figure 1.
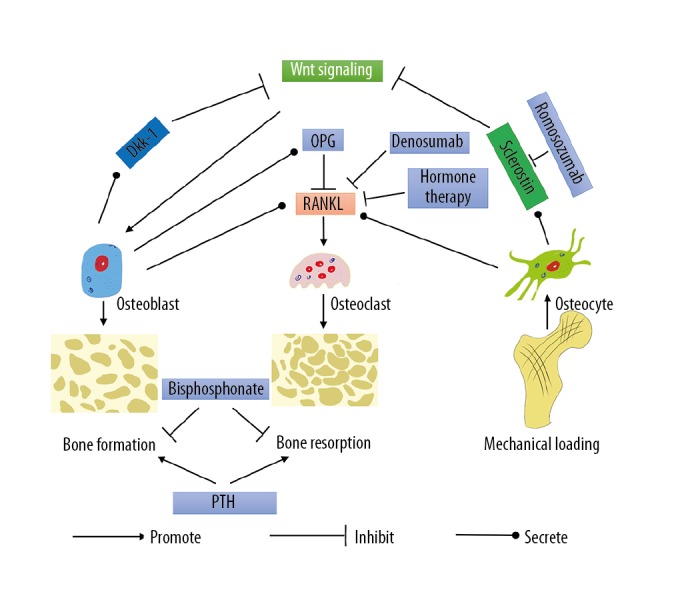
The signaling pathways involved in bone remodeling. Dkk-1 – Dickkopf-related protein 1; OPG – osteoprotegerin; PTH – parathyroid hormone; RANK – receptor activator of NF-κB; RANKL – receptor activator of NF-κB ligand.
Bone remodeling is a complicated process involving many cells and molecules. Bone formation is conducted by osteoblasts and bone resorption is conducted by osteoclasts. Osteoblast is a source of OPG, RANKL, and Dkk-1. Osteocyte is the only source of sclerostin and the main source of RANKL. RANK/RANKL interactions play a key role in osteoclastogenesis. OPG, the decoy receptor of RANKL, can block the interactions between RANK and RANKL. The canonical Wnt/β-catenin pathway plays a pivotal role in osteoblastogenesis. Sclerostin and Dkk-1 can inhibit the canonical Wnt/β-catenin pathway through binding to Wnt co-receptors. Importantly, mechanical loading can regulate the expression of sclerostin. As a result, activation of the canonical Wnt/β-catenin pathway is changed, followed by adjustment in bone formation and bone resorption. Osteocytes may play a more important role than previously recognized.
Osteoblasts are derived from the mesenchymal stem cell lineage [6,18]. The canonical Wnt/β-catenin pathway plays a pivotal role in the differentiation of osteoblast progenitors to mature osteoblasts and the process of bone formation [17,19]. The canonical Wnt/β-catenin pathway can be antagonized by Dickkopf-related protein 1 (Dkk-1) and sclerostin through binding to the Wnt low-density lipoprotein (LDL) receptor-related protein co-receptor 5 (LRP5) and LDL receptor-related protein co-receptor 6 (LRP6) [17,20,21]. Recently, the study of sclerostin has been of great interest. Sclerostin, encoded by SOST, is secreted by osteocytes [13,14]. The secretion of sclerostin is influenced by mechanical loading [17,22–24]. Mutations in SOST can cause sclerosteosis [25]. In SOST knockout mice, elevations in bone formation and bone strength have been observed [26]. Bone loss of ovariectomized rats was completely reversed after administration of anti-sclerostin antibody, and greater increases in bone mass and bone strength were observed than in normal rats [27]. In female cynomolgus monkeys, treatment with anti-sclerostin antibody led to increased bone formation, bone mass, and bone strength [28]. In conclusion, blockade of sclerostin has significant anabolic effects and could be beneficial for bone gain.
The canonical Wnt/β-catenin pathway also plays a role in regulating osteoclastogenesis and subsequent bone resorption. Experiments causing gain and loss of function of β-catenin showed that β-catenin promoted osteoblasts expressing OPG, thereby blocking the differentiation of osteoclasts [29]. Additionally, deletion of β-catenin resulted in accelerated differentiation from osteoclast progenitors to mature osteoclasts, demonstrating that β-catenin can inhibit the rate of osteoclastogenesis [30]. The results of one study [17] suggested that Wnt signaling directly inhibits osteoclast progenitors, independent of OPG; however, OPG was not measured directly [17].
Osteoclasts express receptor activator of NF-κB (RANK) on the cell membrane, and are derived from hematopoietic stem cells of the monocyte and macrophage lineage [6,18,31]. The differentiation from osteoclast progenitors to mature osteoclasts is dependent on the presence of the RANK Ligand (RANKL) and macrophage colony-stimulating factor (M-CSF) [6]. RANKL is mainly expressed by osteocytes and osteoblasts, and plays a key role in osteoclast differentiation and activation through binding to RANK [15,31–34]. Mutant mice lacking RANKL exhibit severe osteopetrosis and complete defects of osteoclastogenesis, verifying the importance of RANKL for osteoclastogenesis [32]. However, the effect of RANKL can be blocked by OPG, a soluble decoy receptor, both in vitro and in vivo [33–35]. OPG-deficient mice exhibit a decrease in bone density [36]. Administration of recombinant murine OPG caused increased bone density in normal mice and blocked bone loss in ovariectomized rats [35]. In male Sprague-Dawley rats, significant increases in bone volume and density were observed when recombinant human OPG was administered [37]. In ovariectomized mice, transgenic overexpression of human OPG caused a significant bone gain effect [38]. Importantly, estrogen deficiency induced an increase in RANKL in bone marrow cells, demonstrating a role for RANKL in the increased bone resorption in postmenopausal women [39], and a clinical trial showed that subcutaneous injection of OPG effectively reduced bone resorption in postmenopausal women [40]. In conclusion, these protective effects of OPG show that RANKL may be an effective therapeutic target for osteoporosis.
Therapeutic Agents
Recently, understanding of the canonical Wnt/β-catenin pathway and the RANKL/RANK/OPG pathway has been translated to the clinical level. Antagonists that block sclerostin or RANKL have been tested in clinical trials. In this section of the review, we begin with a brief introduction to bisphosphonates and parathyroid hormone, because these are usually used as control treatments in clinical trials. Then, we describe clinical trials of denosumab and romosozumab (Table 1). Calcium and vitamin D supplements are provided as a basic treatment in these studies.
Table 1.
Efficacies of denosumab and romosozumab.
| Agent | Duration of treatment (years) | Improvement of BMD | Reference |
|---|---|---|---|
| Denosumab | 1 | LS 5.0%, TH 2.9%, FN 1.3% | [12] |
| 2 | LS 7.7%, TH 4.0%, FN 3.3% | [80] | |
| 3 | LS 9.2%, TH 6.0% | [81] | |
| 3 | LS 9.4%, TH 4.8%, FN 4.0% | [82] | |
| 5 | LS 13.7%, TH 7.0%, FN 6.1% | [80] | |
| 5 | LS 13.1%, TH 6.2%, FN 5.7% | [83] | |
| 6 | LS 15.2%, TH 7.5%, FN 6.7% | [82] | |
| 7 | LS 16.5%, TH 7.4%, FN 7.1% | [10] | |
| 8 | LS 18.4%, TH 8.3%, FN 7.8% | [83] | |
| 10 | LS 21.7%, TH 9.2%, FN 9.0% | [10] | |
| Romosozumab | 1 | LS 11.3%, TH 4.1%, FN 3.7% | [11] |
| 1 | LS 13.7%, TH 6.2% | [85] | |
| 1 | LS 13.3%, TH 6.8%, FN 5.2% | [12] |
LS – lumbar spine; TH – total hip; FN – femoral neck; BMD – bone mineral density.
Bisphosphonates
Alendronate, zoledronic acid, risedronate, and ibandronate are the first-line bisphosphonates used in clinical applications. They work by inhibiting farnesyl diphosphate synthase in the mevalonate pathway, thereby blocking the formation and function of osteoclasts [41,42]. As a result, they can reduce both bone formation markers and bone resorption markers; increase the BMD at the lumbar spine, femoral neck, and the total hip; and reduce the risk of vertebral fractures, nonvertebral fractures, and hip fractures [43–50]. Notably, the therapeutic effect is sustained for up to several years [47,51–53]. Although some adverse events have been reported, the incidence is low [53,54]. Overall, the use of bisphosphonates is safe [43–45,47,55–58].
Parathyroid hormone (1–84) and teriparatide
Parathyroid hormone (1–84) and teriparatide, a recombinant human parathyroid hormone (1–34), are anabolic drugs that are administered subcutaneously and have been shown to not only increase bone formation markers but also increase bone resorption markers [59–68]. Parathyroid hormone therapy can cause increased BMD at the lumbar spine, femoral neck, and total hip [9,59–64,69–72], as well as reduced risk of both vertebral and nonvertebral fractures [59,60,64,73]. Additionally, one study suggested that the decreased risk of nonvertebral fractures could last for 30 months after discontinuation of therapy [74]. Parathyroid hormone therapy is generally well tolerated [61–63,74].
Hormone therapy
Estrogen or estrogen plus progestogen therapy are designed to prevent osteoporosis and treat menopausal symptoms instead of treating osteoporosis, despite the fact that risk of vertebral fractures and hip fracture was reduced in the Women’s Health Initiative trials [7,75,76]. Although the incidence of adverse events was increased [2,7,76], some experts held the view that benefits over risks with initiation near menopause [77]. In the USA and most European countries, the lowest effective dose and shortest duration within the first few years of menopause was recommended for treatment of menopausal symptoms [2,7]. Of note, a guideline suggested that “Menopausal estrogen or estrogen plus progestogen therapy or raloxifene should not be used to treat women with osteoporosis” [4].
Denosumab
Denosumab, also known as AMG 162, is a fully human monoclonal antibody that binds to RANKL with high affinity, mimicking the effects of OPG on RANKL. Therefore, denosumab can block the differentiation of osteoclasts, resulting in inhibition of osteoclast-mediated bone resorption.
In the phase 1 clinical trial, a single dose of AMG 162 caused a decrease in bone resorption turnover markers, and this decrease was long-lasting. These effects were dose-dependent and reversible, and no serious adverse events were observed [78].
In the phase 2 clinical trial lasting 2 years [8,79], denosumab was administered subcutaneously to postmenopausal women with low BMD. Doses were administered either every 3 months (6, 14, or 30 mg) or every 6 months (14, 60, 100, or 210 mg), and bone gain was evaluated. Compared with alendronate (70 mg, weekly), the administration of denosumab at 30 mg every 3 months or 60 mg every 6 months resulted in similar increases of BMD at the lumbar spine and total hip. There was no significant difference in adverse events or serious adverse events compared to the placebo [8,79]. Because adherence for clinical use is likely higher for infrequent treatments, subcutaneous administration of 60 mg every 6 months was chosen for the subsequent clinical trial [9,10,69,70,80,81].
The Fracture Reduction Evaluation of Denosumab in Osteoporosis Every 6 Months (FREEDOM) was a phase 3 clinical trial [10,80–83] in which postmenopausal women who had a T score of −2.5 to −4.0 at the lumbar spine or total hip were enrolled and divided into 2 groups: the long-term group (10 years denosumab) and the crossover group (transition from 3 years’ placebo to 7 years’ denosumab). In the first 3 years, the risks of new radiographic vertebral fracture, hip fracture, and nonvertebral fracture were reduced compared with placebo (68%, 40%, and 20%, respectively) [81]. In the subsequent 7 years in the long-term group, the yearly incidence of new vertebral fracture and nonvertebral fractures was similar to the low rate observed during the first 3 years in the long-term group [10]. The study showed that denosumab could progressively increase BMD for up to 10 years, possibly without plateau. However, yearly bone gain peaked during year 1 and gradually attenuated throughout the course of the trial. The incidence of adverse events gradually decreased, suggesting that denosumab could be tolerated. The rate of serious adverse events, including osteonecrosis of the jaw and atypical femoral fracture, was stable during the course of the study.
In the DATA study, BMD in postmenopausal women with osteoporosis was evaluated in response to denosumab (60 mg every 6 months, administered subcutaneously), teriparatide (20 μg daily), or both drugs in combination. Administration of the 2 drugs in combination resulted in greater BMD increases at the lumbar spine, femoral neck, and total hip than either drug alone [69,70].
In the subsequent DATA-Switch study [9], women were divided into 3 groups: denosumab to teriparatide, teriparatide to denosumab, and combined therapy to denosumab. In both the teriparatide to denosumab group and the combined therapy to denosumab group, BMD continued to increase at the lumbar spine, femoral neck, and total hip. The increase was greater in the teriparatide to denosumab group than the combined therapy to denosumab group. However, BMD underwent a transient decrease and then reversed in the denosumab to teriparatide group. Bone gain was significantly lower at the total hip and the femoral neck in the denosumab to teriparatide group than in the other groups [9].
Romosozumab
Romosozumab, formerly known as AMG 785, is a monoclonal antibody that binds sclerostin. Romosozumab can increase bone formation and decrease bone resorption when administered subcutaneously.
In the phase 1 clinical trial, a single dose of AMG 785 increased bone formation markers while decreasing bone resorption markers and promoted the gain of BMD at the lumbar spine and total hip in 85 days. Its effects were dose-dependent. During the short period treatment, AMG 785 was generally well tolerated [84].
In a phase 2 clinical trial, the bone gain effects of romosozumab administered monthly (70, 140, or 210 mg) or at 3-month intervals (140 or 210 mg) for 1 year were evaluated in postmenopausal women with low BMD [11]. Romosozumab (210 mg monthly) showed significantly better efficacy in increasing BMD at the lumbar spine, total hip, and femoral neck when compared to placebo, alendronate (70 mg weekly), or teriparatide (20 μg daily), with increases of 11.3%, 4.1%, and 3.7%, respectively. A monthly dose of 210 mg has been used in some phase 3 clinical trials due to its superior BMD gains [72,85]. Interestingly, the effects of romosozumab on bone gain attenuated over time after 6 months, which could explain why romosozumab was used for only 1 year in other clinical trials [12,72,85,86]. There was no significant difference in adverse events with romosozumab compared with other groups.
Interestingly, this phase 2 clinical trial [11] used quantitative computed tomography (QCT) to further examine bone gain in response to romosozumab compared with teriparatide and placebo [71]. In the previous study, romosozumab demonstrated significant increases in BMD compared with teriparatide and placebo [11]. In this QCT analysis [71], romosozumab increased the integral volumetric BMD and bone mineral content (BMC) significantly at the lumbar spine and total hip compared with teriparatide and placebo, consistent with previous BMD results evaluated by DXA [11]. Additionally, romosozumab resulted in significant increases in trabecular/cortical volumetric BMD at the lumbar spine and in trabecular volumetric BMD at the total hip compared with placebo. Significant increases in cortical BMC at the lumbar spine were also observed compared with placebo. Compared with teriparatide, there were significant increases in cortical volumetric BMD at the lumbar spine, trabecular volumetric BMD at the total hip, and cortical BMC at the lumbar spine and total hip. Notably, romosozumab resulted in an increase from baseline in cortical volumetric BMD at the total hip, whereas teriparatide showed a decrease. These findings demonstrated an advantage of romosozumab over teriparatide in promoting bone gain in the trabecular and cortical compartments [71].
ARCH (the Active-Controlled Fracture Study in Postmenopausal Women with Osteoporosis at High Risk) was a phase 3 clinical trial that compared sequential romosozumab and alendronate therapy to alendronate alone [85]. It found that 1-year romosozumab (210 mg monthly) followed by 2 years alendronate (70 mg weekly) caused a significantly greater increase in BMD at the lumbar spine and total hip compared with 3 years of alendronate alone. In the romosozumab followed by alendronate group, the risk of new vertebral fracture, nonvertebral fractures, hip fracture, and clinical fractures was lower by 48%, 19%, 38%, and 27%, respectively, compared with the alendronate alone group. The ability to lower the risk of new vertebral fracture is especially important [85]. Overall adverse events and serious adverse events were balanced between the 2 groups. However, there were more serious cardiovascular adverse events in the sequential treatment group, including cardiac ischemic events and cerebrovascular events, which were not observed in the phase 2 clinical trial [11]. The incidence of osteonecrosis and atypical femoral fracture was similar in the subsequent 2 years [85].
In the Fracture Study in Postmenopausal Women with Osteoporosis (FRAME) trial, a phase 3 clinical trial, postmenopausal women who had a T score of −2.5 to −3.5 at the total hip or femoral neck received a monthly subcutaneous injection of romosozumab (210 mg) or placebo for 12 months, and then both groups transitioned to subcutaneous injections of denosumab (60 mg, administered every 6 months) for another 12 months [12]. At 12 months, the risks of new vertebral fracture and clinical fractures were significantly lower with romosozumab, at 73% and 36%, respectively [12]. At 24 months, the risk of vertebral facture was lowered by 75% with romosozumab. After the transition, denosumab continued increasing the BMD at the lumbar spine, total hip, and femoral neck, suggesting that sequential administration of romosozumab and denosumab provides improved consolidation of BMD [12]. The incidence of adverse events, including serious cardiovascular events, appeared similar in the 2 groups. It could not be determined whether the occurrence of osteonecrosis of the jaw and atypical femoral fracture were related to treatment in the romosozumab group, because the underlying diseases of patients were complicated [12].
In another phase 3 clinical trial, the effects of subcutaneous injection of romosozumab (210 mg monthly) or teriparatide (20 μg, daily) on BMD were compared among women with osteoporosis who had taken oral bisphosphonate for at least 3 years or alendronate for 1 year before screening [72]. After transitioning from bisphosphonate treatment, the increases in BMD at the lumbar spine, total hip, and femoral neck were significantly higher in the romosozumab group compared with the baseline and the teriparatide group. QCT showed that romosozumab significantly increased the integral cortical volumetric BMD at the total hip compared with teriparatide and placebo. In addition, significant increases in the integral volumetric BMC were observed with romosozumab compared with teriparatide, with greater gains in the cortical compartment in comparison to the trabecular compartment [72]. Notably, the gain in hip strength estimated by finite element analysis was consistent with the increase in BMD. The study also showed that teriparatide significantly increased the areal BMD at the lumbar spine, whereas BMD was reduced at the total hip and femoral neck. QCT showed that teriparatide significantly increased the trabecular volumetric BMD, whereas cortical volumetric BMD was decreased. The percentage change in the trabecular volumetric BMD was higher than in the cortical volumetric BMD. Interestingly, the change in the integral volumetric BMD at the total hip was not significant at 12 months, indicating that cortical bone played a more important role in contributing to maintaining BMD at the total hip. The total hip and femoral neck are skeletal sites with a high proportion of cortical bone, whereas the lumbar spine has a high proportion of trabecular bone. A decrease in cortical volumetric BMD at the total hip with teriparatide was also observed in a previous study [71]. Some studies have suggested that the catabolic effect of teriparatide contributes to increased cortical porosity [72,87–89]. The overall incidence of adverse events was balanced between the 2 groups, and the serious adverse events in these groups were judged to have no relationship with the treatment [72].
Surprisingly, a significant increase in the incidence of serious cardiovascular events was observed in the ARCH trial [85]. This could be due to compensatory elevation of Dkk-1 [90–92], because sclerostin and Dkk-1 have been associated with ischemic stroke [93]. However, Saag et al. measured the level of Dkk-1 in ovariectomized cynomolgus monkeys and found no significant change in those treated with romosozumab for 1 year [94]. Romosozumab might reduce the risk of cardiovascular disease [95], which is in contrast to the results seen in the ARCH study [85]. Notably, the fact that sclerostin is exclusively secreted by osteocytes supports the prediction that anti-sclerostin therapy could be safe [13]. Whether romosozumab could increase the risk of cardiovascular disease is not yet clear, and this potential risk requires further study [91,94].
Therapeutic Method
Single therapy
Among these drugs, romosozumab has the strongest effects on bone gain, ranging from 11.3% to 13.7% at the lumbar spine [11,12,85]. This large effect is similar to the effect of 5 years of denosumab administration [80,83], 10 years of alendronate administration [47], or 2 years of teriparatide administration [63,70,96].
Concurrent combination therapy
Combination therapy trials are done mostly on parathyroid hormone with concurrent denosumab, zoledronic acid, or alendronate. Combination therapy with teriparatide and denosumab showed greater efficacy for the gain of BMD at the lumbar spine, total hip, and femoral neck than either used alone [69,70], demonstrating a synergistic effect between teriparatide and denosumab. However, the studies were of more complexity, because some patients had received antiresorptive therapies prior to the combination therapy, which might have influenced the therapeutic effects [69,70]. Interestingly, although concomitant therapy of teriparatide and zoledronic acid resulted in greater bone gain than either agent alone at the lumbar spine, total hip, or femoral neck, there was no significant difference in the gain of BMD at the lumbar spine between teriparatide and the combination therapy [97]. No bone gain advantage was observed from the combination of teriparatide and alendronate over either drug alone at the lumbar spine or the femoral neck [98]. Combined therapy with alendronate and parathyroid hormone (1–84) resulted in no significant improvement in BMD over either drug alone at the lumbar spine, total hip, or femoral neck [66–68]. These results suggest that alendronate impairs the ability of parathyroid hormone to increase BMD.
Sequential therapy
Patients receiving teriparatide after transitioning from bisphosphonate therapy underwent a transient reduction in BMD at the total hip and the femoral neck, which was subsequently reversed. The overall beneficial effects at the lumbar spine, total hip, and femoral neck were not superior to those in patients previously untreated with bisphosphonates [62,63,72]. However, patients transitioning from parathyroid hormone (1–84) to alendronate showed continuous increases in BMD at the lumbar spine, total hip, and femoral neck compared with placebo [65,68], and the effect was similar to that from 2 years of treatment with teriparatide [63]. Moreover, the volumetric BMD in trabecular bone at the spine and hip was significantly higher than in those transitioning to placebo [68]. Although the gain in the volumetric BMD in cortical bone did not significantly differ between the 2 groups, the BMC and bone mass increased in the alendronate-treated group, indicating that alendronate may provide more bone strength [68].
Interestingly, patients transitioning from denosumab to teriparatide also showed a transient BMD decrease at the lumbar spine, total hip, and femoral neck [9]. The bone-gaining effects of teriparatide were similar to or lower than those previously untreated with denosumab [9]. However, a greater bone gain effect of denosumab after transitioning from teriparatide was observed compared to denosumab alone [9].
Surprisingly, patients transitioning from alendronate treatment to romosozumab treatment demonstrated a continuous increase in BMD at the lumbar spine, total hip, and femoral neck [72]. However, previous alendronate treatment seemed to attenuate the bone gain effects of romosozumab when compared with the results of other studies [11,12,85]. Transition from romosozumab to alendronate showed an increase in BMD at the lumbar spine and total hip in the first year, with a slight decrease in the following year [85]. Continuous increases in BMD at the lumbar spine and the total hip were observed with sequential treatment of romosozumab to denosumab [12]. Interestingly, this increase was slightly lower than the therapeutic effect of denosumab alone [8,12,69,70].
In conclusion, for parathyroid hormone, when both adherence and bone gain in clinical settings are considered, sequential therapy may be a good choice. An anabolic agent followed by an antiresorptive agent may achieve the best outcome in treating osteoporosis [7,99].
Conclusions
Several agents are now available in clinical practice for the management of osteoporosis. With recent advances in understanding of the RANK/RANKL/OPG pathway and the canonical Wnt/β-catenin pathway, the mechanisms of osteoporosis have been further elucidated. Romosozumab, an antagonist of sclerostin, has the strongest effects on bone gain among existing drugs. However, because its efficacy is gradually attenuated, it is usually administered for only 1 year. Denosumab, an antagonist of RANKL, continuously increases BMD without a plateau over 10 years of administration. Moreover, denosumab has been shown to be safe for long-term administration. For long-term administration and the greatest therapeutic efficacy, anabolic agents followed by antiresorptive agents may be superior for the treatment of osteoporosis.
Abbreviations
- Dkk-1
Dickkopf-related protein 1
- DXA
dual-energy x-ray absorptiometry
- LDL
low-density lipoprotein
- LRP5
LDL receptor-related protein co-receptor 5
- LRP6
LDL receptor-related protein co-receptor 6
- M-CSF
macrophage colony-stimulating factor
- OPG
osteoprotegerin
- QCT
quantitative computed tomography
- RANK
receptor activator of NF-κB
- RANKL
receptor activator of NF-κB ligand
Footnotes
Source of support: This work was supported by the National Key R&D Program of China (2017YFB0403801)
Conflict of interest
None.
References
- 1.Siris ES, Miller PD, Barrett-Connor E, et al. Identification and fracture outcomes of undiagnosed low bone mineral density in postmenopausal women: Results from the National Osteoporosis Risk Assessment. JAMA. 2001;286:2815–22. doi: 10.1001/jama.286.22.2815. [DOI] [PubMed] [Google Scholar]
- 2.Kanis JA, McCloskey EV, Johansson H, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2013;24:23–57. doi: 10.1007/s00198-012-2074-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qaseem A, Forciea M, McLean R, Denberg T. Treatment of low bone density or osteoporosis to prevent fractures in men and women: A clinical practice guideline update from the American College of Physicians. Ann Intern Med. 2017;166:818–39. doi: 10.7326/M15-1361. [DOI] [PubMed] [Google Scholar]
- 4.Cotts K, Cifu AS. Treatment of osteoporosis. JAMA. 2018;319:1040–41. doi: 10.1001/jama.2017.21995. [DOI] [PubMed] [Google Scholar]
- 5.Ensrud KE, Crandall CJ. Osteoporosis. Ann Intern Med. 2017;167:ITC17–32. doi: 10.7326/AITC201708010. [DOI] [PubMed] [Google Scholar]
- 6.Rachner TD, Khosla S, Hofbauer LC. Osteoporosis: Now and the future. Lancet. 2011;377:1276–87. doi: 10.1016/S0140-6736(10)62349-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cosman F, de Beur SJ, LeBoff MS, et al. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014;25:2359–81. doi: 10.1007/s00198-014-2794-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McClung MR, Lewiecki EM, Cohen SB, et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–31. doi: 10.1056/NEJMoa044459. [DOI] [PubMed] [Google Scholar]
- 9.Leder BZ, Tsai JN, Uihlein AV, et al. Denosumab and teriparatide transitions in postmenopausal osteoporosis (the DATA-Switch study): Extension of a randomised controlled trial. Lancet. 2015;386:1147–55. doi: 10.1016/S0140-6736(15)61120-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bone HG, Wagman RB, Brandi ML, et al. 10 years of denosumab treatment in postmenopausal women with osteoporosis: Results from the phase 3 randomised FREEDOM trial and open-label extension. Lancet Diabetes Endocrinol. 2017;5:513–23. doi: 10.1016/S2213-8587(17)30138-9. [DOI] [PubMed] [Google Scholar]
- 11.McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370:412–20. doi: 10.1056/NEJMoa1305224. [DOI] [PubMed] [Google Scholar]
- 12.Cosman F, Crittenden DB, Adachi JD, et al. Romosozumab treatment in postmenopausal women with osteoporosis. N Engl J Med. 2016;375:1532–43. doi: 10.1056/NEJMoa1607948. [DOI] [PubMed] [Google Scholar]
- 13.van Bezooijen RL, Roelen BA, Visser A, et al. Sclerostin is an osteocyte-expressed negative regulator of bone formation, but not a classical BMP antagonist. J Exp Med. 2004;199:805–14. doi: 10.1084/jem.20031454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poole KE, van Bezooijen RL, Loveridge N, et al. Sclerostin is a delayed secreted product of osteocytes that inhibits bone formation. FASEB J. 2005;19:1842–44. doi: 10.1096/fj.05-4221fje. [DOI] [PubMed] [Google Scholar]
- 15.Nakashima T, Hayashi M, Fukunaga T, et al. Evidence for osteocyte regulation of bone homeostasis through RANKL expression. Nat Med. 2011;17:1231–34. doi: 10.1038/nm.2452. [DOI] [PubMed] [Google Scholar]
- 16.Capulli M, Paone R, Rucci N. Osteoblast and osteocyte: Games without frontiers. Arch Biochem Biophys. 2014;561:3–12. doi: 10.1016/j.abb.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 17.Canalis E. Wnt signalling in osteoporosis: mechanisms and novel therapeutic approaches. Nat Rev Endocrinol. 2013;9:575–83. doi: 10.1038/nrendo.2013.154. [DOI] [PubMed] [Google Scholar]
- 18.Nagy V, Penninger JM. The RANKL-RANK story. Gerontology. 2015;61:534–42. doi: 10.1159/000371845. [DOI] [PubMed] [Google Scholar]
- 19.Baron R, Rawadi G. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology. 2007;148:2635–43. doi: 10.1210/en.2007-0270. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Zhang Y, Kang H, et al. Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J Biol Chem. 2005;280:19883–87. doi: 10.1074/jbc.M413274200. [DOI] [PubMed] [Google Scholar]
- 21.Kawano Y, Kypta R. Secreted antagonists of the Wnt signalling pathway. J Cell Sci. 2003;116:2627–34. doi: 10.1242/jcs.00623. [DOI] [PubMed] [Google Scholar]
- 22.Robinson JA, Chatterjee-Kishore M, Yaworsky PJ, et al. Wnt/beta-catenin signaling is a normal physiological response to mechanical loading in bone. J Biol Chem. 2006;281:31720–28. doi: 10.1074/jbc.M602308200. [DOI] [PubMed] [Google Scholar]
- 23.Robling AG, Niziolek PJ, Baldridge LA, et al. Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J Biol Chem. 2008;283:5866–75. doi: 10.1074/jbc.M705092200. [DOI] [PubMed] [Google Scholar]
- 24.Tu X, Rhee Y, Condon KW, et al. Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone. 2012;50:209–17. doi: 10.1016/j.bone.2011.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brunkow ME, Gardner JC, Van Ness J, et al. Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am J Hum Genet. 2001;68:577–89. doi: 10.1086/318811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23:860–69. doi: 10.1359/jbmr.080216. [DOI] [PubMed] [Google Scholar]
- 27.Li X, Ominsky MS, Warmington KS, et al. Sclerostin antibody treatment increases bone formation, bone mass, and bone strength in a rat model of postmenopausal osteoporosis. J Bone Miner Res. 2009;24:578–88. doi: 10.1359/jbmr.081206. [DOI] [PubMed] [Google Scholar]
- 28.Ominsky MS, Vlasseros F, Jolette J, et al. Two doses of sclerostin antibody in cynomolgus monkeys increases bone formation, bone mineral density, and bone strength. J Bone Miner Res. 2010;25:948–59. doi: 10.1002/jbmr.14. [DOI] [PubMed] [Google Scholar]
- 29.Glass DA, 2nd, Bialek P, Ahn JD, et al. Canonical Wnt signaling in differentiated osteoblasts controls osteoclast differentiation. Dev Cell. 2005;8:751–64. doi: 10.1016/j.devcel.2005.02.017. [DOI] [PubMed] [Google Scholar]
- 30.Otero K, Shinohara M, Zhao H, et al. TREM2 and β-catenin regulate bone homeostasis by controlling the rate of osteoclastogenesis. J Immunol. 2012;188:2612–21. doi: 10.4049/jimmunol.1102836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yasuda H, Shima N, Nakagawa N, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kong YY, Yoshida H, Sarosi I, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–23. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- 33.Lacey DL, Timms E, Tan HL, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–76. doi: 10.1016/s0092-8674(00)81569-x. [DOI] [PubMed] [Google Scholar]
- 34.Lacey D, Tan H, Lu J, et al. Osteoprotegerin ligand modulates murine osteoclast survival in vitro and in vivo. Am J Pathol. 2000;157:435–48. doi: 10.1016/S0002-9440(10)64556-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simonet W, Lacey D, Dunstan C, et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–19. doi: 10.1016/s0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- 36.Bucay N, Sarosi I, Dunstan C, et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–68. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Capparelli C, Morony S, Warmington K, et al. Sustained antiresorptive effects after a single treatment with human recombinant osteoprotegerin (OPG): A pharmacodynamic and pharmacokinetic analysis in rats. J Bone Miner Res. 2003;18:852–58. doi: 10.1359/jbmr.2003.18.5.852. [DOI] [PubMed] [Google Scholar]
- 38.Kostenuik P, Bolon B, Morony S, et al. Gene therapy with human recombinant osteoprotegerin reverses established osteopenia in ovariectomized mice. Bone. 2004;34:656–64. doi: 10.1016/j.bone.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 39.Eghbali-Fatourechi G, Khosla S, Sanyal A, et al. Role of RANK ligand in mediating increased bone resorption in early postmenopausal women. J Clin Invest. 2003;111:1221–30. doi: 10.1172/JCI17215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bekker P, Holloway D, Nakanishi A, et al. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16:348–60. doi: 10.1359/jbmr.2001.16.2.348. [DOI] [PubMed] [Google Scholar]
- 41.Fisher JE, Rodan GA, Reszka AA. In vivo effects of bisphosphonates on the osteoclast mevalonate pathway. Endocrinology. 2000;141:4793–96. doi: 10.1210/endo.141.12.7921. [DOI] [PubMed] [Google Scholar]
- 42.Martins CA, Leyhausen G, Volk J, Geurtsen W. Effects of alendronate on osteoclast formation and activity in vitro. J Endodont. 2015;41:45–49. doi: 10.1016/j.joen.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 43.Black DM, Cummings SR, Karpf DB, et al. Randomised trial of effect of alendronate on risk of fracture in women with existing vertebral fractures. Fracture Intervention Trial Research Group. Lancet. 1996;348:1535–41. doi: 10.1016/s0140-6736(96)07088-2. [DOI] [PubMed] [Google Scholar]
- 44.Rizzoli R, Greenspan SL, Bone G, 3rd, et al. Two-year results of once-weekly administration of alendronate 70 mg for the treatment of postmenopausal osteoporosis. J Bone Miner Res. 2002;17:1988–96. doi: 10.1359/jbmr.2002.17.11.1988. [DOI] [PubMed] [Google Scholar]
- 45.Yan Y, Wang W, Zhu H, et al. The efficacy and tolerability of once-weekly alendronate 70 mg on bone mineral density and bone turnover markers in postmenopausal Chinese women with osteoporosis. J Bone Miner Metab. 2009;27:471–78. doi: 10.1007/s00774-009-0057-7. [DOI] [PubMed] [Google Scholar]
- 46.Liberman UA, Weiss SR, Broll J, et al. Effect of oral alendronate on bone mineral density and the incidence of fractures in postmenopausal osteoporosis. The Alendronate Phase III Osteoporosis Treatment Study Group. N Engl J Med. 1995;333:1437–43. doi: 10.1056/NEJM199511303332201. [DOI] [PubMed] [Google Scholar]
- 47.Bone HG, Hosking D, Devogelaer JP, et al. Ten years’ experience with alendronate for osteoporosis in postmenopausal women. N Engl J Med. 2004;350:1189–99. doi: 10.1056/NEJMoa030897. [DOI] [PubMed] [Google Scholar]
- 48.Chesnut CH, 3rd, Skag A, Christiansen C, et al. Effects of oral ibandronate administered daily or intermittently on fracture risk in postmenopausal osteoporosis. J Bone Miner Res. 2004;19:1241–49. doi: 10.1359/JBMR.040325. [DOI] [PubMed] [Google Scholar]
- 49.Reginster J, Minne HW, Sorensen OH, et al. Randomized trial of the effects of risedronate on vertebral fractures in women with established postmenopausal osteoporosis. Vertebral Efficacy with Risedronate Therapy (VERT) Study Group. Osteoporos Int. 2000;11:83–91. doi: 10.1007/s001980050010. [DOI] [PubMed] [Google Scholar]
- 50.Harris ST, Watts NB, Genant HK, et al. Effects of risedronate treatment on vertebral and nonvertebral fractures in women with postmenopausal osteoporosis: A randomized controlled trial. Vertebral Efficacy With Risedronate Therapy (VERT) Study Group. JAMA. 1999;282:1344–52. doi: 10.1001/jama.282.14.1344. [DOI] [PubMed] [Google Scholar]
- 51.Black DM, Schwartz AV, Ensrud KE, et al. Effects of continuing or stopping alendronate after 5 years of treatment: the Fracture Intervention Trial Long-term Extension (FLEX): A randomized trial. JAMA. 2006;296:2927–38. doi: 10.1001/jama.296.24.2927. [DOI] [PubMed] [Google Scholar]
- 52.Bauer DC, Schwartz A, Palermo L, et al. Fracture prediction after discontinuation of 4 to 5 years of alendronate therapy: The FLEX study. JAMA Intern Med. 2014;174:1126–34. doi: 10.1001/jamainternmed.2014.1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Black DM, Delmas PD, Eastell R, et al. Once-yearly zoledronic acid for treatment of postmenopausal osteoporosis. N Engl J Med. 2007;356:1809–22. doi: 10.1056/NEJMoa067312. [DOI] [PubMed] [Google Scholar]
- 54.Eriksen EF, Diez-Perez A, Boonen S. Update on long-term treatment with bisphosphonates for postmenopausal osteoporosis: A systematic review. Bone. 2014;58:126–35. doi: 10.1016/j.bone.2013.09.023. [DOI] [PubMed] [Google Scholar]
- 55.Boonen S, Reginster JY, Kaufman JM, et al. Fracture risk and zoledronic acid therapy in men with osteoporosis. N Engl J Med. 2012;367:1714–23. doi: 10.1056/NEJMoa1204061. [DOI] [PubMed] [Google Scholar]
- 56.Greenspan SL, Perera S, Ferchak MA, et al. Efficacy and safety of single-dose zoledronic acid for osteoporosis in frail elderly women: A randomized clinical trial. JAMA Intern Med. 2015;175:913–21. doi: 10.1001/jamainternmed.2015.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reid IR, Brown JP, Burckhardt P, et al. Intravenous zoledronic acid in postmenopausal women with low bone mineral density. N Engl J Med. 2002;346:653–61. doi: 10.1056/NEJMoa011807. [DOI] [PubMed] [Google Scholar]
- 58.Hue TF, Cummings SR, Cauley JA, et al. Effect of bisphosphonate use on risk of postmenopausal breast cancer: results from the randomized clinical trials of alendronate and zoledronic acid. JAMA Intern Med. 2014;174:1550–57. doi: 10.1001/jamainternmed.2014.3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lindsay R, Nieves J, Formica C, et al. Randomised controlled study of effect of parathyroid hormone on vertebral-bone mass and fracture incidence among postmenopausal women on oestrogen with osteoporosis. Lancet. 1997;350:550–55. doi: 10.1016/S0140-6736(97)02342-8. [DOI] [PubMed] [Google Scholar]
- 60.Neer R, Arnaud C, Zanchetta J, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344:1434–41. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- 61.Orwoll E, Scheele W, Paul S, et al. The effect of teriparatide human parathyroid hormone (1–34) therapy on bone density in men with osteoporosis. J Bone Miner Res. 2003;18:9–17. doi: 10.1359/jbmr.2003.18.1.9. [DOI] [PubMed] [Google Scholar]
- 62.Boonen S, Marin F, Obermayer-Pietsch B, et al. Effects of previous antiresorptive therapy on the bone mineral density response to two years of teriparatide treatment in postmenopausal women with osteoporosis. J Clin Endocrinol Metab. 2008;93:852–60. doi: 10.1210/jc.2007-0711. [DOI] [PubMed] [Google Scholar]
- 63.Obermayer-Pietsch BM, Marin F, McCloskey EV, et al. Effects of two years of daily teriparatide treatment on BMD in postmenopausal women with severe osteoporosis with and without prior antiresorptive treatment. J Bone Miner Res. 2008;23:1591–600. doi: 10.1359/jbmr.080506. [DOI] [PubMed] [Google Scholar]
- 64.Hodsman AB, Bauer DC, Dempster DW, et al. Parathyroid hormone and teriparatide for the treatment of osteoporosis: A review of the evidence and suggested guidelines for its use. Endocr Rev. 2005;26:688–703. doi: 10.1210/er.2004-0006. [DOI] [PubMed] [Google Scholar]
- 65.Rittmaster R, Bolognese M, Ettinger M, et al. Enhancement of bone mass in osteoporotic women with parathyroid hormone followed by alendronate. J Clin Endocrinol Metab. 2000;85:2129–34. doi: 10.1210/jcem.85.6.6614. [DOI] [PubMed] [Google Scholar]
- 66.Black DM, Greenspan SL, Ensrud KE, et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–15. doi: 10.1056/NEJMoa031975. [DOI] [PubMed] [Google Scholar]
- 67.Finkelstein JS, Hayes A, Hunzelman JL, et al. The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med. 2003;349:1216–26. doi: 10.1056/NEJMoa035725. [DOI] [PubMed] [Google Scholar]
- 68.Black D, Bilezikian J, Ensrud K, et al. One year of alendronate after one year of parathyroid hormone (1–84) for osteoporosis. N Engl J Med. 2005;353:555–65. doi: 10.1056/NEJMoa050336. [DOI] [PubMed] [Google Scholar]
- 69.Tsai JN, Uihlein AV, Lee H, et al. Teriparatide and denosumab, alone or combined, in women with postmenopausal osteoporosis: the DATA study randomised trial. Lancet. 2013;382:50–56. doi: 10.1016/S0140-6736(13)60856-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Leder BZ, Tsai JN, Uihlein AV, et al. Two years of Denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): A randomized controlled trial. J Clin Endocrinol Metab. 2014;99:1694–700. doi: 10.1210/jc.2013-4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Genant HK, Engelke K, Bolognese MA, et al. Effects of romosozumab compared with teriparatide on bone density and mass at the spine and hip in postmenopausal women with low bone mass. J Bone Miner Res. 2017;32:181–87. doi: 10.1002/jbmr.2932. [DOI] [PubMed] [Google Scholar]
- 72.Langdahl B, Libanati C, Crittenden D, et al. Romosozumab (sclerostin monoclonal antibody) versus teriparatide in postmenopausal women with osteoporosis transitioning from oral bisphosphonate therapy: a randomised, open-label, phase 3 trial. Lancet. 2017;390:1585–94. doi: 10.1016/S0140-6736(17)31613-6. [DOI] [PubMed] [Google Scholar]
- 73.Kendler D, Marin F, Zerbini C, et al. Effects of teriparatide and risedronate on new fractures in post-menopausal women with severe osteoporosis (VERO): A multicentre, double-blind, double-dummy, randomised controlled trial. Lancet. 2017 doi: 10.1016/S0140-6736(17)32137-2. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 74.Prince R, Sipos A, Hossain A, et al. Sustained nonvertebral fragility fracture risk reduction after discontinuation of teriparatide treatment. J Bone Miner Res. 2005;20:1507–13. doi: 10.1359/JBMR.050501. [DOI] [PubMed] [Google Scholar]
- 75.Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women’s Health Initiative randomized controlled trial. JAMA. 2002;288:321–33. doi: 10.1001/jama.288.3.321. [DOI] [PubMed] [Google Scholar]
- 76.Komm BS, Morgenstern D, A Yamamoto L, Jenkins SN. The safety and tolerability profile of therapies for the prevention and treatment of osteoporosis in postmenopausal women. Expert Rev Clin Pharmacol. 2015;8:769–84. doi: 10.1586/17512433.2015.1099432. [DOI] [PubMed] [Google Scholar]
- 77.Langer R, Manson J, Allison M. Have we come full circle – or moved forward? The Women’s Health Initiative 10 years on. Climacteric. 2012;15:206–12. doi: 10.3109/13697137.2012.666916. [DOI] [PubMed] [Google Scholar]
- 78.Bekker PJ, Holloway DL, Rasmussen AS, et al. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19:1059–66. doi: 10.1359/JBMR.040305. [DOI] [PubMed] [Google Scholar]
- 79.Lewiecki EM, Miller PD, McClung MR, et al. Two-year treatment with denosumab (AMG 162) in a randomized phase 2 study of postmenopausal women with low BMD. J Bone Miner Res. 2007;22:1832–41. doi: 10.1359/jbmr.070809. [DOI] [PubMed] [Google Scholar]
- 80.Papapoulos S, Chapurlat R, Libanati C, et al. Five years of denosumab exposure in women with postmenopausal osteoporosis: results from the first two years of the FREEDOM extension. J Bone Miner Res. 2012;27:694–701. doi: 10.1002/jbmr.1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Cummings S, San Martin J, McClung M, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med. 2009;361:756–65. doi: 10.1056/NEJMoa0809493. [DOI] [PubMed] [Google Scholar]
- 82.Bone HG, Chapurlat R, Brandi ML, et al. The effect of three or six years of denosumab exposure in women with postmenopausal osteoporosis: Results from the FREEDOM extension. J Clin Endocrinol Metab. 2013;98:4483–92. doi: 10.1210/jc.2013-1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Papapoulos S, Lippuner K, Roux C, et al. The effect of 8 or 5 years of denosumab treatment in postmenopausal women with osteoporosis: Results from the FREEDOM Extension study. Osteoporos Int. 2015;26:2773–83. doi: 10.1007/s00198-015-3234-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Padhi D, Jang G, Stouch B, et al. Single-dose, placebo-controlled, randomized study of AMG 785, a sclerostin monoclonal antibody. J Bone Miner Res. 2011;26:19–26. doi: 10.1002/jbmr.173. [DOI] [PubMed] [Google Scholar]
- 85.Saag K, Petersen J, Brandi M, et al. Romosozumab or alendronate for fracture prevention in women with osteoporosis. N Engl J Med. 2017;377:1417–27. doi: 10.1056/NEJMoa1708322. [DOI] [PubMed] [Google Scholar]
- 86.Evenepoel P, D’Haese P, Brandenburg V. Romosozumab in postmenopausal women with osteopenia. N Eng J Med. 2014;370:1664. doi: 10.1056/NEJMc1402396. [DOI] [PubMed] [Google Scholar]
- 87.Ma YL, Zeng QQ, Chiang AY, et al. Effects of teriparatide on cortical histomorphometric variables in postmenopausal women with or without prior alendronate treatment. Bone. 2014;59:139–47. doi: 10.1016/j.bone.2013.11.011. [DOI] [PubMed] [Google Scholar]
- 88.Tsai J, Uihlein A, Burnett-Bowie S, et al. Comparative effects of teriparatide, denosumab, and combination therapy on peripheral compartmental bone density, microarchitecture, and estimated strength: the DATA-HRpQCT Study. J Bone Miner Res. 2015;30:39–45. doi: 10.1002/jbmr.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Macdonald H, Nishiyama K, Hanley D, Boyd S. Changes in trabecular and cortical bone microarchitecture at peripheral sites associated with 18 months of teriparatide therapy in postmenopausal women with osteoporosis. Osteoporos Int. 2011;22:357–62. doi: 10.1007/s00198-010-1226-1. [DOI] [PubMed] [Google Scholar]
- 90.Lewis J, Schousboe J, Prince R. Romosozumab versus alendronate and fracture risk in women with osteoporosis. N Engl J Med. 2018;378:194–95. doi: 10.1056/NEJMc1714810. [DOI] [PubMed] [Google Scholar]
- 91.Saag K, Petersen J, Grauer A. Romosozumab versus alendronate and fracture risk in women with osteoporosis. N Engl J Med. 2018;378:195–96. doi: 10.1056/NEJMc1714810. [DOI] [PubMed] [Google Scholar]
- 92.Tsourdi E, Rachner T, Hofbauer L. Romosozumab versus alendronate and fracture risk in women with osteoporosis. N Engl J Med. 2018;378:195. doi: 10.1056/NEJMc1714810. [DOI] [PubMed] [Google Scholar]
- 93.He X, Wang E, Bao Y, et al. High serum levels of sclerostin and Dickkopf-1 are associated with acute ischaemic stroke. Atherosclerosis. 2016;253:22–28. doi: 10.1016/j.atherosclerosis.2016.08.003. [DOI] [PubMed] [Google Scholar]
- 94.Song G, Lee Y. Romosozumab versus alendronate and fracture risk in women with osteoporosis. N Engl J Med. 2018;378:194. doi: 10.1056/NEJMc1714810. [DOI] [PubMed] [Google Scholar]
- 95.Hirsch C. In postmenopausal women with osteoporosis, romosozumab followed by alendronate reduced fractures vs. alendronate alone. Ann Intern Med. 2018;168:JC3. doi: 10.7326/ACPJC-2018-168-2-003. [DOI] [PubMed] [Google Scholar]
- 96.Eastell R, Nickelsen T, Marin F, et al. Sequential treatment of severe postmenopausal osteoporosis after teriparatide: final results of the randomized, controlled European Study of Forsteo (EUROFORS) J Bone Miner Res. 2009;24:726–36. doi: 10.1359/jbmr.081215. [DOI] [PubMed] [Google Scholar]
- 97.Cosman F, Eriksen E, Recknor C, et al. Effects of intravenous zoledronic acid plus subcutaneous teriparatide [rhPTH(1–34)] in postmenopausal osteoporosis. J Bone Miner Res. 2011;26:503–11. doi: 10.1002/jbmr.238. [DOI] [PubMed] [Google Scholar]
- 98.Finkelstein JS, Wyland JJ, Lee H, Neer RM. Effects of teriparatide, alendronate, or both in women with postmenopausal osteoporosis. J Clin Endocrinol Metab. 2010;95:1838–45. doi: 10.1210/jc.2009-1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hofbauer LC, Rachner TD. More DATA to guide sequential osteoporosis therapy. Lancet. 2015;386:1116–18. doi: 10.1016/S0140-6736(15)61175-8. [DOI] [PubMed] [Google Scholar]