

Abstract
Background
Juvenile amyotrophic lateral sclerosis (JALS) is a rare form of motor neuron disease and occurs before 25 years of age. Only a few cases of juvenile-onset ALS have been reported.
Material/Methods
To study genetic and clinicopathological features in Chinese patients with juvenile ALS, we retrospectively reviewed ALS patients in our hospital and screened out 2 patients with disease onset before the age of 25. Genetic analysis was carried out with next-generation sequencing (NGS) to identify ALS causative genes. Sanger sequencing was used to validate identified variants. The clinical, electrophysiological, and pathological data were summarized.
Results
A novel frameshift mutation c.1510dupG (p.G505Wfs*12) was found in Patient One using next-generation sequencing (NGS). Patient Two had a reported pathogenic mutation c.C1483T(p.R495X) with NGS. The mother of Patient Two carried the same mutation as her son and disease onset was at 1.5 years after the death of her son.
Conclusions
We identified a novel frameshift mutation associated with JALS. JALS and generally typical ALS, with the same FUS mutation, can appear in a family and present a phenomenon of anticipation. For diagnosis of central nervous system degeneration in adolescents with bulbar symptoms, great attention should be paid to JALS.
MeSH Keywords: Amyotrophic Lateral Sclerosis, Frameshift Mutation, RNA-Binding Protein FUS
Background
ALS is a progressive neurodegenerative disorder involving the upper and lower motor neurons of the brain and spinal cord. Approximately 90% of patients with ALS are sporadic (SALS), the remaining cases are familial (FALS). To date, there are 29 genes that are considered causes of ALS or are highly correlated with ALS [1]. Fused in sarcoma (FUS) mutations have been observed in patients with JALS, where symptoms start before 25 years old [2]. The FUS gene is located on chromosome 16p11.2, and consists of 14 introns and 15 exons, and encodes FUS protein of 526 amino acid residues that belongs to the multifunctional DNA/RNA-binding proteins. The FUS protein is ubiquitously expressed in the cell nucleus and cytoplasm, and continuously moves between the 2 areas. It is involved in cell proliferation, DNA repair, transcription regulation, RNA transport, and microRNA processing [3]. FUS mutations are responsible for about 5% of cases of familial ALS (FALS) and fewer than 1% of cases of sporadic ALS (SALS) [4]. JALS is a rare form of ALS that occurs before the age of 25 years, and features a heterogeneous onset region, variable progression, and variable survival time [5].
To date, 11 genes (SOD1, DDHD1, FUS, ALS2, SETX, BICD2, SPG11, CLEC4C, SIGMAR1, UBQLN2, ERLIN1) are considered causes of JALS or are highly correlated with JALS. The FUS gene mutation, with high clinical heterogeneity, is the most common pathogenic gene of JALS and is also the pathogenic gene of generally typical ALS. Clinical diagnosis of JALS is very difficult, and broad differential diagnosis need to be considered. These include Spinal Muscular Atrophy (SMA), Hereditary Spastic Paraplegia (HSP), and Hereditary Motor Neuronopathy (HMN/CMT). We used NGS to sequence the pathogenic genes from 2 cases with JALS with onset of bulbar symptoms, progressive aggravation of limb weakness, and rapid death from respiratory failure. Furthermore, clinical, electrophysiological, and skeletal muscle pathological data were retrospectively reported.
Material and Methods
Patients and clinical study
Two JALS patients were diagnosed by clinical manifestations and electrophysiology and the pedigree for 1 patient was studied. Clinical features and laboratory data of these patients were collected, including the pedigree analysis, electromyography (EMG) studies, and the results of histopathological examinations of muscle biopsy specimens. All patients provided written informed consent before skeletal muscle biopsy and gene detection.
Next-generation sequencing
DNA extraction and NGS library preparation
Genomic DNA (gDNA) was extracted from frozen muscle tissue specimens and peripheral blood (obtained from 2 patients and the biological parents of patient 2) with a DNA Kit (CoWin Biotech). Based on manufacturer’s standards, the minimum extraction amount of DNA was 3 μg. The size of the DNA library was 300–400 bp, including adapter sequences.
Target gene capture and NGS
ALS pathogenic gene was enriched with the GenCap capture probe (MyGenostics, Beijing, China). Biotinylated 100-mer oligo baits were designed to tile the target gene exon regions. The capture experiments were carried out according to the manufacturer’s standards. The enrichment paired-end libraries were sequenced with an Illumina HiSeq 2000 sequencer to acquire the reads of 100 bp.
Bioinformatics analysis
Filtered sequencing reads were aligned to each human reference genome sequence with the Burrows-Wheeler Alignment Program (http://bio-bwa.sourceforge.net/bwa.shtml). Uniquely-mapped reads were used for variation detection, while duplicated reads were deleted using Sequence Alignment/Map tools. Single-nucleotide variants (SNVs) and small insertions/deletions (Indels) were detected and genotyped using the GATK UnifiedGenotyper and GATK Indel Genotyper V2 respectively. Annotation of the variants, such as locations (exon, intron, and intergenic regions) and influence on protein coding (synonymous, missense, nonsense, and frameshift), were evaluated using RefSeq (hg19, from UCSC) and UCSC annotation. Then, the possible pathogenic mutations were predicted with IFT (http://sift.jcvi.org/), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), and MutationTaster tools (http://mutationtaster.org/).
Sanger sequencing
Sanger sequencing was used to verify the variants identified by NGS. The amplified fragments were directly sequenced with BigDye Terminator Cycle Sequencing Kits on an automated genetic analyzer (ABI3730; Applied Biosystems, Foster City, California). Sequence analysis was performed using Chromas software (http://technelysium.com.au/wp/chromas/) and compared with the reference sequences of ALS-related genes.
Muscle biopsy and histochemistry
Open muscle biopsy was performed in the biceps brachii muscles under local infiltration anesthesia. The specimen underwent liquid nitrogen isopentane cryopreservation and 7-μm-thick serial sections were prepared for histochemical staining and pathological analysis. Staining included hematoxylin and eosin (HE), nicotinamide adenine dinucleotide tetrazolium reductase (NADH-TR), succinic dehydrogenase (SDH), adenosine monophosphate deaminase, cytochrome coxidase (COX), acid phosphatase (ACID), and periodic acid-Schiff (PAS).
Results
Patients features
Patient One was a 14-year-old boy admitted to our hospital with a 7-month history of barylalia and weakness in bilateral legs and weakness of chewing and swallowing. The symptoms began with barylalia and weakness in the right leg. One month later, his left leg became weak. These symptoms progressively worsened. Over time, he struggled to climb stairs, squat, and stand. Four months later, his mastication weakened and he began to experience dysphagia symptoms. Five months later, he began to experience cervical muscle weakness and was unable to lift his head from the bed when lying down. As his disease progressed, he experienced a 5-kg weight loss. The patient was of below-average intelligence and his academic performance was poor. There was no history of similar disease in his family.
Neurologically, he was clear-minded but did not speak fluently. Tongue atrophy and fasciculation were noted on tongue protrusion. Muscle atrophy was noted in the bilateral thighs. The muscle strength of his bilateral legs was reduced. There were no obvious abnormalities in deep and shallow sensation. His biceps brachii reflex was symmetrical and active, his right knee tendon reflex was reduced, and the left knee tendon reflex was normal. His double-Hoffman sign and double-Babinski signs were positive. His Mini-Mental Status Examination (MMSE) score was 15 points, indicating moderately-impaired cognition.
His creatine kinase (CK) was elevated (587 U/L) and his remaining serum tests were normal. The pulmonary function test revealed his Tiffeneau-Pinelli index (FEV1/FVC) was 70% of predicted. Electrophysiological examination showed insertion, fibrillation, and positive phase potentials with widened duration and increased amplitude within the medulla oblongata, cervical spinal cord, thoracic spinal cord, and lumbar spinal cord, suggestive of extensive neurogenic damage. Brain magnetic resonance imaging (MRI) was normal. Pathological analysis of the right biceps brachii biopsy specimen showed neurogenic pathological changes. Patient 1 was diagnosed with ALS and treated with riluzole. His disease rapidly progressed and he died from respiratory failure 11 months after the onset of symptoms.
Patient Two (Figure 1) was a 17-year-old boy admitted to our neurological institute with an 18-month history of barylalia, dysphagia, progressive cervical, facial, and arm muscle weakness, and dyspnea. His symptoms began with barylalia and dysphagia. He developed cervical muscle weakness gradually. Ten months after onset of symptoms, he developed dyspnea and could not lie down. He developed a severe pulmonary infection and dyspnea and underwent a tracheotomy with mechanical ventilation at a local hospital. After this, his disease continued to progress and, over time, he found that lifting both arms was extremely difficult. He could not raise his head, open his mouth, or swallow, and demonstrated copious salivation. He sustained a 20-kg weight loss. Prior to symptom onset, the patient had good general health. There was no history of similar disease in his family when he was admitted to our hospital.
Figure 1.

Clinical image of Patient Two. (A) Nasogastric feeding and endotracheal intubation. (B, C) Atrophy of the tongue.
Internal medicine examination revealed emaciation, neck wound status-post tracheotomy, and noninvasive ventilator-assisted intermittent breathing. On neurological examination, he was clear-minded and nonverbal. He was unable to open his mouth, and demonstrated salivation and atrophy of the facial and limb muscles. The muscle strength of cervical and bilateral arms was reduced. There were no obvious abnormalities in deep and shallow sensation. His biceps brachii and knee tendon reflexes were symmetrical and reduced. Double-Hoffman sign and double-Babinski sign were negative.
His serum tests were normal, and pulmonary function testing revealed a FEV1/FVC that was 70% of predicted. Pulmonary CT was significant for right pneumonitis. Electrophysiological examination showed extensive neurogenic damage. His brain MRI was normal. Pathological analysis of the right biceps brachii biopsy specimen showed neurogenic pathological changes. The patient was diagnosed with ALS and treatment with riluzole. His disease progressed rapidly and he died from respiratory failure approximately 21 months after onset of symptoms.
Patient Three (Figure 2, I-1) was the mother of Patient Two (Figure 2, II-1). She was 46 years old, and carried the same mutation as her son, Patient Two. She was admitted to our neurological institute with a 2-month history of progressive limb weakness. Her symptoms began with right arm weakness. Combing her hair and washing her face became difficult. She later presented with right leg weakness and instability when walking, and intermittent dysphagia while drinking water. Fasciculation were observed in both limbs. One month later, she developed left-hand weakness.
Figure 2.
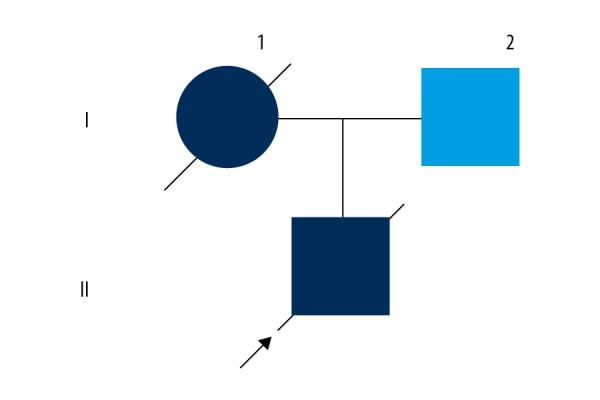
Pedigree of the Patient Two family. II-1: Patient Two; I-1: Patient Three.
On neurological examination, she was clear-minded, with dysfluent speech, tongue atrophy, and fasciculation. The muscle strength of bilateral arms was reduced. There were no obvious abnormalities of either deep or shallow sensation. The right knee tendon reflex was reduced and double-Hoffman sign and Babinski signs were negative.
All serum tests were normal. Pulmonary function testing revealed a FEV1/FVC that was 40% of predicted. Electrophysiological examination showed extensive neurogenic damage. Brain MRI was normal. Pathological analysis of the right biceps brachii biopsy specimen revealed neurogenic pathological changes. She was diagnosed with ALS and began treatment with riluzole. Her disease progressed rapidly and she died from respiratory failure approximately 12 months after the onset of symptoms. The clinical data of all 3 patients are summarized in Table 1.
Table 1.
Summary of our ALS patients with FUS mutations.
| Mutation | Sex | Age of onset | Site of onset | Phenotype | Region of involvement | Dementia | Duration (mo) | Family history | |
|---|---|---|---|---|---|---|---|---|---|
| Patient One | p.G505Wfs*12 | Male | 14 | Bulbar and Lower limbs | UMN and LMN | Four | Mental retardation | ≈11 | SALS |
| Patient Two | p.R495X | Male | 17 | Bulbar | LMN | Four | No | ≈21 | FALS |
| Patient Three | p.R495X | Female | 46 | Upper limbs | LMN | Four | No | ≈12 | FALS |
Disease duration was defined as the time from onset of the disease to death. ALS – amyotrophic lateral sclerosis; FALS – familial amyotrophic lateral sclerosis; SALS – sporadic amyotrophic lateral sclerosis; LMN – lower motor neuron; UMN – upper motor neuron.
Muscle pathology
The muscle biopsy specimens of these 3 patients showed acute-subacute neurogenic processes with increased fiber size variation. Small degenerating and necrotic fibers were observed, as were small angular fibers and atrophic muscle fiber groups. Connective tissue elements were slightly or moderately increased. Activities of mitochondrial enzymes (NADH, SDH, and COX) were partly decreased and typical target fibers could be seen. Acid phosphatase activity was increased in degenerating and atrophic fibers (Figure 3).
Figure 3.

Histochemical staining of skeletal muscle. (A) Patient One, sections stained for H&E. (B) Patient One, sections stained for NADH. (C) Patient One, sections stained for ACID. (D) Patient Two, sections stained for H&E. (E) Patient Two, sections stained for NADH. (F) Patient Two, sections stained for ACID. (G) Patient Three, sections stained for H&E. (H) Patient Three, sections stained for NADH. (I) Patient Three, sections stained for ACID. The black arrows point to target fibers.
Molecular genetic analyses
Patient One: A novel 1-bp duplication of a guanine in exon 14 of the FUS gene (c.1510dupG) (Figure 4A) was found. This heterozygous mutation was further investigated by Sanger sequencing in the patient. Unfortunately, because Patient One was adopted, we could not collect blood from his parents, and no DNA was available for further analysis of the mutation. Therefore, it was unclear if this mutation occurred de novo or if it was inherited from a healthy parent.
Figure 4.

Sequencing chromatograms of the p.G505Wfs*12 mutation of Patient One, and p.R495X mutation of Patient Two and his mother, Patient Three. (A) Sequencing chromatograms of the p.G505Wfs*12 mutation of Patient One. The arrow shows the position of a G duplication at nucleotide 1510 (c.1510dupG), and creates a premature stop codon (p.G505Wfs*12), which is supposed to produce a C-terminally truncated fused in sarcoma protein (the resulting protein would be 515 amino acids long). (B) Sequencing chromatograms of the p.R495X mutation of Patient Two. The arrow shows the position of a C-to-T transversion at nucleotide 1483 (c.1483C>T) that leads to production of a C-terminally truncated fused in sarcoma protein. (C) Sequencing chromatograms of the p.R495X mutation of Patient Three. The arrow shows the position of a C-to-T transversion at nucleotide 1483 (c.1483C>T), which leads to production of a C-terminally truncated fused in sarcoma protein.
Patient Two: The sequencing analysis revealed a truncating mutation at nucleotide 1483 with C-to-T transition in exon 14 of FUS (Figure 4B), resulting in premature amino acid termination translation p.R495X. This mutation was a known pathogenic mutation that causes ALS and JALS [5,6]. The mother of Patient Two carried the same mutation (Figure 4C), but she had no symptoms until age 46. The father did not have this mutation.
Discussion
Clinical phenotype
Among these 3 patients, 2 had JALS with bulbar involvement as the first symptom. In these 2 patients, the disease progressed rapidly and involved the limbs, and both died of respiratory failure, one at 11 months post-onset and one at 21 months post-onset. Patient One showed positive Hoffman and Babinski signs when he was admitted to our hospital. Patient Two and Patient Three only had clinical manifestations of lower motor neuron damage, and their pyramidal signs were negative. When Patient Three was 46 years old, her disease onset was characterized by limb weakness, which differed from JALS. In the latter stage of her disease, she exhibited bulbar signs and died from respiratory failure 12 months after onset of symptoms. A pathogenic mutation in the FUS gene was detected in Patient Three, and she was classified as generally typical ALS [7]. In this family, the son (Patient Two) had JALS, and the mother (Patient Three) had generally typical ALS, suggesting that the FUS gene mutation has high clinical heterogeneity. Even though the mother and son carried the same gene mutation, symptom onset occurred at a much earlier age in the son. This illustrates the phenomenon of anticipation, suggesting that additional genetic and/or environmental factors might play an important role in the pathogenesis of ALS with the FUS mutation [8]. Moreover, the electromyograms of the 3 patients involved 4 spinal cord segments, in accordance with the electrophysiological diagnostic criteria of ALS [9].
Mutations in the alsin (ALS2), senataxin (SETX) and spatacsin (SPG11) have been associated with slow-progressive JALS, but mutations in Cu/Zn superoxide dismutase 1 (SOD1) and FUS are reportedly associated with rapid-progressive JALS [5]. In the 2 patients with JALS, the bulbar symptom onset was consistent with the characteristics of JALS caused by FUS mutation, as previously reported by Liu et al. [2]. Early-stage bulbar involvement and rapid disease progression may be important clinical features of JALS caused by the FUS gene. In addition, Patient One exhibited mental retardation, with reduced scores on the MMSE; however, there was no atrophy of the cerebral cortex on cranial MRI. Although the clinical phenotype of the FUS gene mutation has been associated with ALS-frontotemporal dementia (ALS-FTD) [10], the intellectual impairment of Patient One warrants further study.
Pathology
Skeletal muscle biopsy is not necessary for diagnosing ALS, but it can provide evidence for clinical diagnosis and differential diagnosis. In our study, muscle biopsies of the 3 cases showed an acute-subacute neurogenic process. Histological features of neurogenic involvement included type grouping, muscle fiber atrophy, and target fibers. This is consistent with previous reports of ALS skeletal muscle pathologies [11–14]. Acid phosphatase activity was increased in degenerating and atrophic fibers under light microscopy, suggesting increased lysosomal enzyme activity in the cytoplasm. Autophagic vesicles and secondary lysosomes were found in electron microscopy, suggesting that apoptosis mechanisms play an important role in the pathogenesis of ALS [15]. Previous studies have shown that in patients with ALS and related animal models of skeletal muscle cells, mitochondrial reactive oxygen species and oxidative stress were increased, the ubiquitin proteasome and autophagy-lysosome systems were activated, leading to atrophy and degeneration of muscle fibers [16–18]. These changes are pathological mechanisms of persistent weakness and muscle atrophy in patients with ALS.
Genetics
To date, more than 100 mutations of FUS have been shown to be associated with ALS in the Amyotrophic Lateral Sclerosis Online Database (http://alsod.iop.kcl.ac.uk) and the Human Gene Mutation Database (HGMD professional). Most of these were associated with generally typical ALS. We found a novel frameshift mutation of FUS in Patient One. The novel c.1510dupG variant is located in the highly-conserved, extreme C-terminal of the FUS protein, where most of the mutations in FUS have been identified [19]. This duplication shifted the reading frame, and terminated duplication at position 516. The resulting protein consisted of 515 amino acids. Amino acids sequence of the wild-type FUS protein were very well-conserved across many species. Furthermore, this frameshift duplication was not observed in the Exome Sequencing Project (ESP) 6500 database and 1000 Genomes populations, indicating that this was not a common benign population variant. Therefore, we interpreted c.1510dupG as a pathogenic variant, according to the ACMG/AMP Variant Interpretation Standards and Guidelines [20]. This study enriches the pathogenic loci of JALS with the FUS mutation.
We detected a known nonsense mutation c.C1483T (P.R495X) in Patient Two and his mother (Patient Three). It is well known that the R495X variant was the first nonsense mutation in FUS and was predicted to generate a C-terminally truncated form of the FUS protein. The FUS mutation results in ALS, where the 2/3 mutation is located at the highly-conserved C-terminal of the FUS protein. The mutation sites of Patient One (c.1510dupG) and Patients Two and Three (c.C1483T) were close. All of these sites were involved in coding the highly-conserved C-terminal of the FUD protein. Gene mutation leads to C-terminal truncation of the FUS protein, resulting in loss of the nuclear localization sequence (NLS), redistribution of the FUS protein from the nucleus to the cytoplasm, dislocation and abnormal deposition of protein cytoplasm, and formation of basophilic inclusion bodies [21, 22]. Then, the abnormally-localized FUS protein is assembled into stress particles, which increase the stress response and cause pathogenicity, indicating the importance of the function of the mutant region [23,24].
Conclusions
We identified a novel frameshift mutation associated with JALS. JALS and generally typical ALS, with the same FUS mutation, can appear in a family and present the phenomenon of anticipation. NGS is recommended to achieve diagnostic accuracy when patients are suspected of demonstrating hereditary ALS. For diagnosis of central nervous system degeneration in adolescents with bulbar symptoms, great attention should be paid to JALS.
Footnotes
Source of support: Departmental sources
References
- 1.Chia R, Chiò A, Traynor BJ. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018;17:94–102. doi: 10.1016/S1474-4422(17)30401-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Liu ZJ, Lin HX, Liu GL, et al. The investigation of genetic and clinical features in Chinese patients with juvenile amyotrophic lateral sclerosis. Clin Genet. 2017;92:267–73. doi: 10.1111/cge.13015. [DOI] [PubMed] [Google Scholar]
- 3.Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet Neurol. 2010;9:995–1007. doi: 10.1016/S1474-4422(10)70195-2. [DOI] [PubMed] [Google Scholar]
- 4.Taylor JP, Brown RH, Jr, Cleveland DW. Decoding ALS: From genes to mechanism. Nature. 2016;539:197–206. doi: 10.1038/nature20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou ZY, Cui LY, Sun Q, et al. De novo FUS gene mutations are associated with juvenile-onset sporadic amyotrophic lateral sclerosis in China. Neurobiol Aging. 2013;34:1312.e1–e8. doi: 10.1016/j.neurobiolaging.2012.09.005. [DOI] [PubMed] [Google Scholar]
- 6.Waibel S, Neumann M, Rabe M, et al. Novel missense and truncating mutations in FUS/TLS in familial ALS. Neurology. 2010;75:815–17. doi: 10.1212/WNL.0b013e3181f07e26. [DOI] [PubMed] [Google Scholar]
- 7.Tsai MJ, Hsu CY, Sheu CC. Amyotrophic lateral sclerosis. N Engl J Med. 2017;377:1602. doi: 10.1056/NEJMc1710379. [DOI] [PubMed] [Google Scholar]
- 8.DeJesus-Hernandez M, Kocerha J, Finch N, et al. De novo truncating FUS gene mutation as a cause of sporadic amyotrophic lateral sclerosis. Hum Mutat. 2010;31:E1377–89. doi: 10.1002/humu.21241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brooks BR, Miller RG, Swash M, Munsat TL. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Mot Neuron Disord. 2000;1:293–99. doi: 10.1080/146608200300079536. [DOI] [PubMed] [Google Scholar]
- 10.Bott NT, Radke A, Stephens ML, Kramer JH. Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener Dis Manag. 2014;4:439–54. doi: 10.2217/nmt.14.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Llovera M, García-Martínez C, Agell N, et al. Ubiquitin gene expression is increased in human muscle undergoing neurogenic involvement. Neurochem Int. 1999;34:137–40. doi: 10.1016/s0197-0186(98)00080-1. [DOI] [PubMed] [Google Scholar]
- 12.Authier FJ, Chazaud B, Mhiri C, et al. Interleukin-1 expression in normal motor endplates and muscle fibers showing neurogenic changes. Acta Neuropathol. 1997;94:272–79. doi: 10.1007/s004010050703. [DOI] [PubMed] [Google Scholar]
- 13.Stewart HG, Mackenzie IR, Eisen A, et al. Clinicopathological phenotype of ALS with a novel G72C SOD1 gene mutation mimicking a myopathy. Muscle Nerve. 2006;33:701–6. doi: 10.1002/mus.20495. [DOI] [PubMed] [Google Scholar]
- 14.Yin F, Ye F, Tan L, et al. Alterations of signaling pathways in muscle tissues of patients with amyotrophic lateral sclerosis. Muscle Nerve. 2012;46:861–70. doi: 10.1002/mus.23411. [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N. Methods for monitoring autophagy. Int J Biochem Cell Biol. 2004;36:2491–502. doi: 10.1016/j.biocel.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Muller FL, Song W, Jang YC, et al. Denervation-induced skeletal muscle atrophy is associated with increased mitochondrial ROS production. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1159–68. doi: 10.1152/ajpregu.00767.2006. [DOI] [PubMed] [Google Scholar]
- 17.Powers SK, Wiggs MP, Duarte JA, et al. Mitochondrial signaling contributes to disuse muscle atrophy. Am J Physiol Endocrinol Metab. 2012;303:E31–39. doi: 10.1152/ajpendo.00609.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dobrowolny G, Aucello M, Rizzuto E, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab. 2008;8:425–36. doi: 10.1016/j.cmet.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Waibel S, Neumann M, Rosenbohm A, et al. Truncating mutations in FUS/TLS give rise to a more aggressive ALS-phenotype than missense mutations: A clinico-genetic study in Germany. Eur J Neurol. 2013;20:540–46. doi: 10.1111/ene.12031. [DOI] [PubMed] [Google Scholar]
- 20.Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–24. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kwiatkowski TJ, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. 2009;323:1205–8. doi: 10.1126/science.1166066. [DOI] [PubMed] [Google Scholar]
- 22.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis Type 6. Science. 2009;323:1208–11. doi: 10.1126/science.1165942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bosco DA, Lemay N, Ko HK, et al. Mutant FUS proteins that cause amyotrophic lateral sclerosis incorporate into stress granules. Hum Mol Genet. 2010;19:4160–75. doi: 10.1093/hmg/ddq335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ito D, Seki M, Tsunoda Y, et al. Nuclear transport impairment of amyotrophic lateral sclerosis-linked mutations in FUS/TLS. Ann Neurol. 2011;69:152–62. doi: 10.1002/ana.22246. [DOI] [PubMed] [Google Scholar]