

Abstract
The robust synthetic flexibility of metal-organic frameworks (MOFs) offers a promising class of tailorable materials, for which, the ability to tune specific physicochemical properties is highly desired. This is achievable only through a thorough description of the consequences for chemical manipulations both in structure and dynamics. Magic angle spinning solid-state NMR spectroscopy offers many modalities in this pursuit, particularly for dynamic studies. Herein, we employ a separated-local-field NMR approach to show how specific intra-framework chemical modifications to MOF UiO66 heavily modulate the dynamic evolution of the organic ring moiety over several orders of magnitude.
Keywords: NMR Spectroscopy, Metal-organic frameworks, Dynamics, Density Functional Theory, Separated-Local-Field
Graphical Abstract
Intra-framework ring rotations in metal-organic are sensitively detected by dipolar dephasing over the rotor period in magic angle spinning solid-state NMR experiments.
Metal-organic frameworks (MOFs) are constructed from inorganic clusters bridged by organic linkers and possess ideal architectures for gas storage and mixture separation as well as generating recent interest as nano-devices and molecular machines.[1] In comparison to traditional inorganic porous materials, MOFs are far more dynamic/flexible due to the incorporation of organic ligands.[2–4] A key advantage of MOFs is the synthetic ease of chemical modification to the organic moieties without change of framework structure, known as isoreticular synthesis. This allows for many avenues of functional modification including adsorptive selectivity based on favorable ligand-guest interactions[5], changes in pore aperture for separations[6,7], and shifts in optical behavior[8]. An additional feature of MOFs, which has captured much attention, is the prevalence of framework dynamics which can lead to dramatic structural shifts such as breathing, swelling, and subnetwork displacements.[9,10] While deformations in the metal cluster have been implicative of dynamic events, the nature of the linker and its functionality is a major contributor to framework dynamics,[10] and play a significant role in determining macroscopic functions such as gas storage and separation, ferroelectricity, spin crossover and luminescence in a variety of MOFs.[11–17] As such, establishing relationships between local dynamics driven by the organic ligands and their chemical modification is of general interest for materials design in MOFs.[17,18]
Though the structural picture of MOFs is most informed by analysis with X-ray diffraction, this yields an essentially static picture, failing to capture the dynamic aspects of the framework. However, dynamic measures of crystalline materials are challenging due to the potential of multiple timescales of coordinated motions in the solid-state. Many dynamic studies have employed solid-state NMR spectroscopy (ssNMR) through 2H-lineshape analysis to characterize the rotational motion of simple p-phenylene rings within the MOF framework.[15,19–24] Separated-local-field (SLF) solid-state NMR is an alternative, attractive means of dynamic characterization as it utilizes the heteronuclear dipolar coupling as a proxy for molecular dynamics, does not require isotopic labeling, and can be used to characterize motions over a broad dynamic range with atomic resolution.[25]
In this communication, a combination of computation and the DIPSHIFT[26,27] (Dipolar chemical SHIFT correlation) SLF methodology is employed to characterize ligand dynamics in MOFs; details on the preparation, characterization and calculations are given in the Supporting Information (Figures S1-S6 and Tables S1-S2). For typical heteronuclear dipole-dipole couplings such as 13C-1H and 15N-1H, the experiment is sensitive to a dynamic range spanning 0.1μs-10ms (see Figure 1).[28] Though a number of MOFs are suitable for this approach, we focus on UiO-66[29] and its derivatives (see Figure 2) due to its wide range of potential applications as well as its high thermal and water stability. We demonstrate that the dynamics of the ligands in the framework are heavily impacted by ligand functionalization; specifically, that the variable dynamic behavior arises from differences in interactions with the local chemical environment and is highly responsive to temperature.
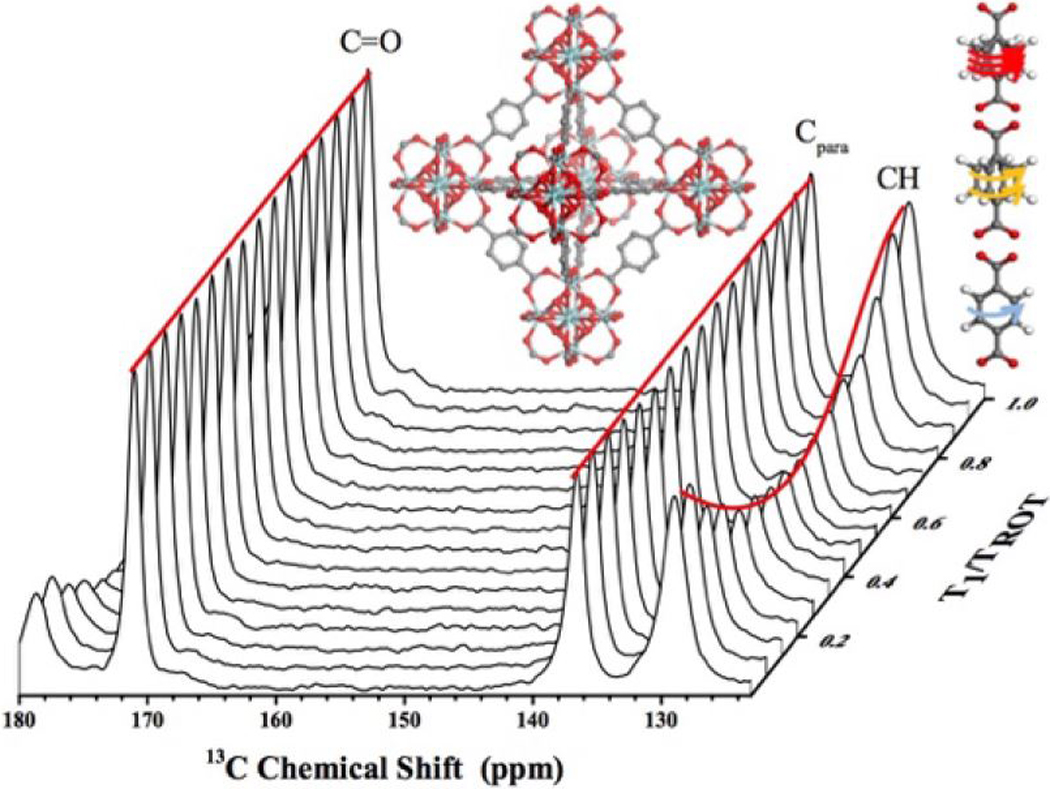
Figure 1. DIPSHIFT curves as a function of dynamic rate on NMR timescale.
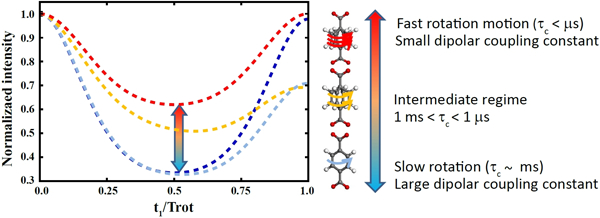
The lines are model fits assuming diffusive anisotropic motions under the Anderson-Weiss approximation[28,30] from UiO-66 data at various temperatures (see SI). The curves correspond to estimated correlation times (τc) of ms or the rigid limit (dark blue), ~350 μs (light blue), ~5 μs (yellow), and the fast limit < μs (red). In the present study, DIPSHIFT curves were measured using the pulse sequence shown in Figure S7 and analyzed as explained in the Supporting Information. t1 is the duration of evolution of nuclear spin magnetization under the effect of a combination of 13C-1H dipolar coupling and 13C chemical shift (see Figures S7 and S8) and Trot is the sample rotation period.
Figure 2. Structure of MOFs.
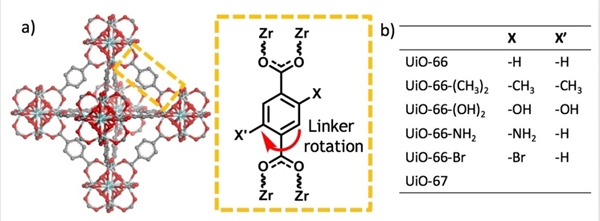
a) UiO-66 structure and scheme of linker rotation. b) A list of UiO type structures investigated in this study.
Terephthalic acid is the most commonly used linker for the construction of MOFs. The phenylene units are symmetric and comprised of four equivalent aromatic 13C-1H bonds making it a suitable candidate for DIPSHIFT measurements. Thus, UiO-66 (Zr6(μ3-O)4(μ3-OH)4(terephthalate)6) (see Figure 2a) was investigated first to understand the dynamic behavior of a simple phenylene unit. Using the 13C-1H bond on the phenylene ring as the local field pair, DIPSHIFT measurements were performed on UiO-66 at 23 °C (Figure 3a) resulting in a heteronuclear dipolar coupling constant (DCH) of 16.4 ±0.2 kHz. The exhibited DIPSHIFT curves feature well-depths that are roughly proportional to the strength of the C-H dipolar coupling and can be fit with standard MAS equations to extract the coupling constants (see Figures 3, and S9-S15).[31,32] The experimentally determined DCH value is smaller than the theoretical value for a rigid 13C-1H bond (~22 kHz), which is due to dynamic averaging resulting from the motional modes, namely ring rotations and librations, available to p-phenylene within the framework.1 Given the timescales detected by this approach, the primary event detected is associated with the ring flip although smaller, more rigorous librational modes are also expected to contribute to a lesser degree. By adopting an appropriate model, a correlation time for the motional ring flip can be estimated, which is found to be approximately a few milliseconds falling in “slow” regime dynamics for UiO-66 at 23 °C (see SI) and is confirmed by higher temperature experiments (vide infra). This result aligns with reports on MIL-53[19] and MOF-5[33] both containing p-phenylene units in a sterically unhindered environment. It is notable that the DIPSHIFT data is symmetric around half the rotor period for UiO-66 at 23 °C, which is a model-free confirmation of slower motional time scales assuming the dynamics are not already in the fast limit. Asymmetries arising from intensity decay due to transverse relaxation effects during the encoding time occur when the molecular motion is accelerated (e.g. temperature induced) to timescales coinciding with the inverse of the coupling constant, known as the ‘intermediate motional regime,’ as depicted in Figure 1.[28]
Figure 3. Effect of substituents on the mobility of phenylene group measured from DIPSHIFT experiments at 23 °C.
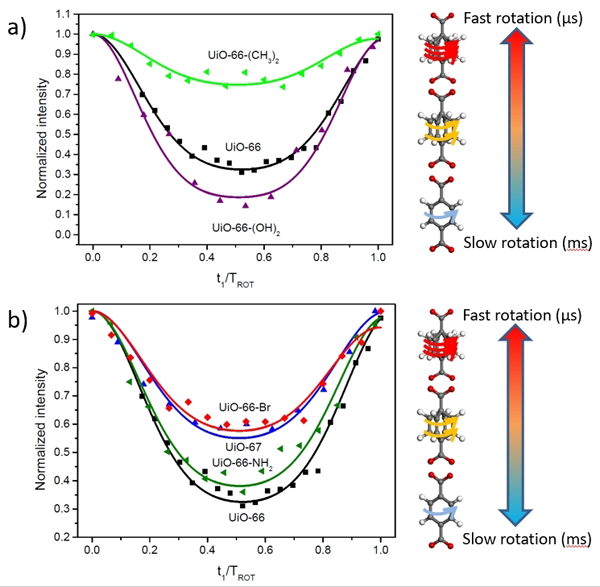
a) UiO-66, UiO-66-(OH)2 and UiO-66-(CH3)2; b) UiO-66, UiO-66-NH2, UiO-66-Br and UiO-67. The symbols are the experimental data points, while the curves are simulated from the fits of the dipolar coupling constants (see details in SI section 3; Figures S9-S15).
After establishing the effectiveness of DIPSHIFT based characterization in the UiO-66 sample, we next turned our investigation to ligand functionalized UiO-66 MOFs. Methyl and hydroxyl groups are among the most commonly used functionalities and are particularly relevant for applications such as separations and catalysis.[34,35] The bulkiness of the methyl group and the hydrogen bonding potential of the hydroxyl groups make them a diametric pair to study their effects on rotational dynamics. Indeed, experiments on UiO-66-(OH)2 (Zr6(μ3-O)4(μ3-OH)4(2,5-dihydroxyterephthalate)6) and UiO-66-(CH3)2 (Zr6(μ3-O)4(μ3-OH)4(2,5-dimethylterephthalate)6) resulted in dramatically different DIPSHIFT curves from one another (Figure 3). UiO-66-(OH)2 gives a higher DCH value (~19.0 ±0.5 kHz) than the parent UiO-66 (DCH ~16 kHz) indicating reduced mobility; on the other hand, the measured DCH value for UiO-66-(CH3)2 is only 10 kHz. This stark reduction of the C-H dipolar coupling constant for UiO-66-(CH3)2 evidences much faster rotary motion than UiO-66, placing it well into the fast regime (τc < 1 μs). These simple substitutions cause dramatic changes to the ligand framework interaction which induces ~4 orders of magnitude difference in the dynamic rotation. These same trends hold at different temperatures: UiO-66-(OH)2, the least dynamic of the three MOFs, must be heated to ~105 °C (60 °C higher than UiO-66) to achieve rotational times on the order of 100 μs (see SI). In contrast, the 10 kHz value for the methyl substituent is similar to the plateau value observed for UiO-66 in the fast regime (τc < μs) at 155 °C (~ 11 kHz). Indeed, when UiO-66-(CH3)2 is cooled to 6 °C, an asymmetric curve is detected (see Figure S12), suggesting that it is in a comparable dynamic regime to UiO-66 at temperatures ~50 °C lower.
To complement the experimental results, DFT simulations on a model complex were utilized as a tool to understand the details of rotational dynamics. In the case of UiO-66, a model of Zr6O4(OH)4(COOH)11(COOC6H5) bearing a single phenyl rotor was constructed and its geometry was optimized. A relaxed potential energy surface scan was performed with the phenyl ring rotation through its principle axis. A rotational energy barrier of 43.5 kJ/mol was obtained. Despite the simplicity of the model, the obtained activation energy is similar to the one found for MOF-5[36] and MIL-53[19]. As with UiO-66, Zr-oxo complexes were constructed and calculations were also performed on models with hydroxyl and methyl functional groups. A high activation energy of 75.3 kJ/mol was found for the model Zr6O4(OH)4(COOH)11(COOC6H5-o-OH). This is expected to dramatically slow down the ring dynamics which supports the experimental data. A closer inspection of the model shows that the hydroxyl group hydrogen bonds with the carboxylate group, an interaction that must be broken upon rotation (Figure 4). The hydrogen bonding explains the high energy barrier and suggests that it is the dominant factor leading to the experimentally observed high C-H dipolar coupling constant and slow rotational dynamics in UiO-66-(OH)2. On the other hand, the model for UiO-66-(CH3)2 (Zr6O4(OH)4(COOH)11(COOC6H5-o-CH3)) shows a lower activation energy of 33.5 kJ/mol. This reduced energy barrier is attributed to repulsive interactions between the bulky methyl group and the cluster. The simulated energy barrier results are fully consistent with the experimental observations for rotational frequencies: UiO-66-(o-OH)2<< UiO-66<<UiO-66-(o-CH3)2. The model above, however, does not account for geometric restrictions and electronic effects imposed on the rotors from Zr6 clusters at both ends. To simulate these effects, two spatially fixed clusters held at crystallographic distances and bridged by a linker were constructed; the relaxed potential energy scan was performed with a semi-empirical method (SI Section 2). The energy barrier was then found using DFT single point energy calculations on the highest and lowest energy conformations determined from the relaxed torsional scan. Activation energies with this approach resulted in 65.77 kJ/mol, 23.39 kJ/mol and 91.71 kJ/mol for models of UiO-66, UiO-66-(CH3)2 and UiO-66-(OH)2, respectively. These results are consistent with the results of the simpler model and further support the influence of local sterics and electronic effects in determining linker dynamics.
Figure 4. DFT simulation of rotational dynamics in MOFs.
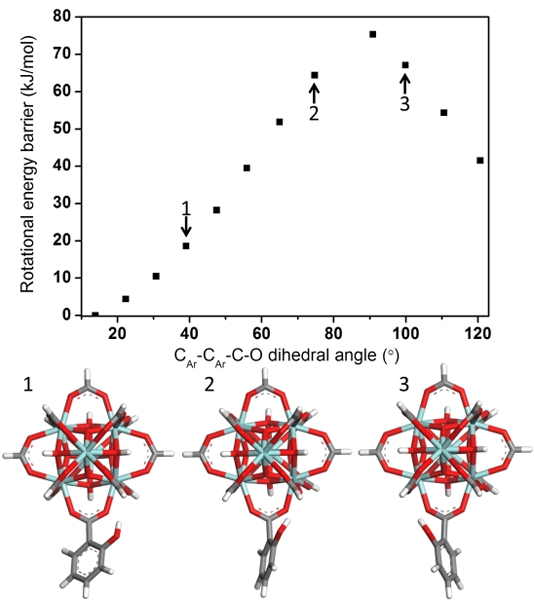
Rotational energy barrier calculation results and snap shot of the model complexes incorporating a single cluster with CAr-CAr-C-O at 40°, 75° and 100°. Coordinates and energies for all models used in the calculations are given in SI.
UiO-66-Br (Zr6(μ3-O)4(μ3-OH)4(2-bromoterephthalate)6) and UiO-66-NH2 (Zr6(μ3-O)4(μ3-OH)4(2-aminoterephthalate)6) were also synthesized to test the influence of linker dynamics from another bulky substituent other than a methyl group and a potential hydrogen bond donor. The measured DCH for UiO-66-Br is closer to UiO-66 at 15 kHz but exhibits a slight asymmetry in the curve (Figure 3b)2. Despite the higher value than UiO-66-(CH3)2, the temperature behavior seen in Figure S13 of UiO-66-Br also shows that it is in the fast regime limit at room temperature. This suggests that the bromine atom also induces a steric effect which lowers the rotational energy barrier similar to the function brought by methyl groups, but is not as extreme given the higher fast limit value. UiO-66-NH2 shows a DCH value of 15.3 ±0.3 kHz, value close to UiO-66. However, the temperature behavior is quite constant in a similar temperature range (see SI Figure S14). This behavior could be interpreted as a combination of effects: a balance between a steric effect and the hydrogen bonding potential of the NH2 group.
Changing the length of the linker while maintaining the topology, known as isoreticular synthesis, is an important strategy that has been used to increase the surface area of MOFs without altering the topology.[37] Thus UiO-67 (Zr6(μ3-O)4(μ3-OH)4(biphenyl-4,4’-dicarboxylate)6) was examined for its larger pore window (~9–11 Å) compared to the parent structure UiO-66 (~5–7 Å). The measured DCH for UiO-67 is 12.4 ±0.3 kHz, which is smaller than that obtained from UiO-66. The lowering of the rotational energy barrier is attributed to the junction of the biphenyl ring, which has a much lower frictional constraint vs the Zr cluster interaction. At higher temperature, the depth of the curve decreases substantially, but no intermediate dynamics are detected (Figure S15). This is possible in the fast limit regime when the reorientational angle of motion increases; it is plausible that the increased length and biphenyl junction could contribute additional bending modes to facilitate such behavior.
In summary, DIPSHIFT solid-state NMR experiments performed on a series of Zr MOFs with various structural modifications showed different ligand dynamics. The presence of functional groups such as methyl or hydroxyl groups can greatly speed or slow down the rotational motion of the linker in the Zr MOFs. Insights from DFT simulation reveals that such dynamics changes arise from the local chemical interactions such as hydrogen bonding or steric repulsion. It is also found that isoreticular structure expansion and factors such as temperature also influence the ligand dynamics. The insights gained are relevant to applications, where dynamics of the ligands are heavily involved, such as gas separations and kinetically limited processes such as molecular sieving.
Supplementary Material
Acknowledgements:
This study was supported by the United States Department of Energy (DE-SC0004888 to A.J.M), the National Institute of Health (GM084018 to A.R.) and funds to purchase the 600 MHz and to upgrade the 400 MHz solid-state NMR spectrometer (to A.R.). J.T.D would like to thank the support from the National Science Foundation Graduate Research Fellowship under Grant No. DGE #1256260. We also thank Alexey Krushelnitsky for his help and informative discussion.
Footnotes
Motions occurring at much faster rates than the coupling constants will result in a “pre-averaged” tensor producing a reduced coupling constant. The motions responsible for the pre-averaging are on distinctly faster time-scales than the motions detected in the experiment.
This value is larger than UiO-67, despite similar curve depths in the plot because UiO-66-Br measurements were completed at higher spinning frequencies (see SI).
Supporting information for this article is given via a link at the end of the document.
References
- [1].Chang Z, Yang DH, Xu J, Hu TL, Bu XH, Adv. Mater. 2015, 27, 5432–5441. [DOI] [PubMed] [Google Scholar]
- [2].Inukai M, Fukushima T, Hijikata Y, Ogiwara N, Horike S, Kitagawa S, J. Am. Chem. Soc. 2015, 137, 12183–12186. [DOI] [PubMed] [Google Scholar]
- [3].Horike S, Shimomura S, Kitagawa S, Nat. Chem. 2009, 1, 695–704. [DOI] [PubMed] [Google Scholar]
- [4].Horike S, Matsuda R, Tanaka D, Matsubara S, Mizuno M, Endo K, Kitagawa S, Angew. Chemie Int. Ed. 2006, 45, 7226–7230. [DOI] [PubMed] [Google Scholar]
- [5].Huang Y, Qin W, Li Z, Li Y, Dalt. Trans. 2012, 41, 9283. [DOI] [PubMed] [Google Scholar]
- [6].Schneemann A, Henke S, Schwedler I, Fischer RA, ChemPhysChem 2014, 15, 823–839. [DOI] [PubMed] [Google Scholar]
- [7].Horcajada P, Salles F, Wuttke S, Devic T, Heurtaux D, Maurin G, Vimont A, Daturi M, David O, Magnier E, et al. , J. Am. Chem. Soc. 2011, 133, 17839–17847. [DOI] [PubMed] [Google Scholar]
- [8].Hendrickx K, Vanpoucke DEP, Leus K, Lejaeghere K, Van Yperen-De Deyne A, Van Speybroeck V, Van Der Voort P, Hemelsoet K, Inorg. Chem. 2015, 54, 10701–10710. [DOI] [PubMed] [Google Scholar]
- [9].Schneemann A, Bon V, Schwedler I, Senkovska I, Kaskel S, Fischer RA, Chem. Soc. Rev. 2014, 43, 6062–6096. [DOI] [PubMed] [Google Scholar]
- [10].Férey G, Serre C, Chem. Soc. Rev. 2009, 38, 1380. [DOI] [PubMed] [Google Scholar]
- [11].Rodríguez-Velamazán JA, González MA, Real JA, Castro M, Muñoz MC, Gaspar AB, Ohtani R, Ohba M, Yoneda K, Hijikata Y, et al. , J. Am. Chem. Soc. 2012, 134, 5083–5089. [DOI] [PubMed] [Google Scholar]
- [12].Coudert FX, Chem. Mater. 2015, 27, 1905–1916. [Google Scholar]
- [13].Setaka W, Yamaguchi K, Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 9271–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tayi AS, Kaeser A, Matsumoto M, Aida T, Stupp SI, Nat. Chem. 2015, 7, 281–294. [DOI] [PubMed] [Google Scholar]
- [15].Moreau F, Kolokolov DI, Stepanov AG, Easun TL, Dailly A, Lewis W, Blake AJ, Nowell H, Lennox MJ, Besley E, et al. , Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 3056–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Cadiau A, Adil K, Bhatt PM, Belmabkhout Y, Eddaoudi M, Science 2016, 353, 137–40. [DOI] [PubMed] [Google Scholar]
- [17].Férey G, Chem. Soc. Rev. 2008, 37, 191–214. [DOI] [PubMed] [Google Scholar]
- [18].Slater AG, Cooper AI, Science (80-. ). 2015, 348, 988. [DOI] [PubMed] [Google Scholar]
- [19].Kolokolov DI, Jobic H, Stepanov AG, Guillerm V, Devic T, Serre C, Férey G, Angew. Chemie - Int. Ed. 2010, 49, 4791–4794. [DOI] [PubMed] [Google Scholar]
- [20].Kolokolov DI, Stepanov AG, Guillerm V, Serre C, Frick B, Jobic H, J. Phys. Chem. C 2012, 116, 12131–12136. [Google Scholar]
- [21].Kolokolov DI, Stepanov AG, Jobic H, J. Phys. Chem. C 2014, 118, 15978–15984. [Google Scholar]
- [22].Comotti A, Bracco S, Valsesia P, Beretta M, Sozzani P, Angew. Chemie - Int. Ed. 2010, 49, 1760–1764. [DOI] [PubMed] [Google Scholar]
- [23].Sutrisno A, Huang Y, Solid State Nucl. Magn. Reson. 2013, 49–50, 1–11. [DOI] [PubMed] [Google Scholar]
- [24].Khudozhitkov AE, Kolokolov DI, Stepanov AG, Bolotov VA, Dybtsev DN, J. Phys. Chem. C 2015, 119, 28038–28045. [Google Scholar]
- [25].Hester RK, Ackerman JL, Neff BL, Waugh JS, Phys. Rev. Lett. 1976, 36, 1081–1083. [Google Scholar]
- [26].Hong M, Gross JD, Griffin RG, J. Phys. Chem. B 1997, 101, 5869–5874. [Google Scholar]
- [27].Munowitz MG, Griffin RG, Bodenhausen G, Huang TH, J. Am. Chem. Soc. 1981, 103, 2529–2533. [Google Scholar]
- [28].deAzevedo ER, Saalwachter K, Pascui O, de Souza AA, Bonagamba TJ, Reichert D, J. Chem. Phys. 2008, 128, 104505. [DOI] [PubMed] [Google Scholar]
- [29].Cavka JH, Jakobsen S, Olsbye U, Guillou N, Lamberti C, Bordiga S, Lillerud KP, J. Am. Chem. Soc. 2008, 130, 13850–13851. [DOI] [PubMed] [Google Scholar]
- [30].Hirschinger J, Concepts Magn. Reson. Part A 2006, 28A, 307–320. [Google Scholar]
- [31].Duer MJ, in Solid-State NMR Spectrosc. Princ. Appl, Blackwell Science Ltd, Oxford, UK, 2002, pp. 111–178. [Google Scholar]
- [32].Schmidt-Rohr K, Spiess HW, Multidimensional Solid-State NMR and Polymers, 2012. [Google Scholar]
- [33].Gould SL, Tranchemontagne D, Yaghi OM, Garcia-garibay MA, J. Am. Chem. Soc. 2008, 130, 3246–3247. [DOI] [PubMed] [Google Scholar]
- [34].Wee LH, Alaerts L, Martens JA, De Vos D, in Met. Fram., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, pp. 191–212. [Google Scholar]
- [35].Barea E, Turra F, Rodriguez Navarro JA, in Met. Fram., Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany, 2011, pp. 69–97. [Google Scholar]
- [36].Gould SL, Tranchemontagne D, Yaghi OM, Garcia-Garibay MA, J. Am. Chem. Soc. 2008, 130, 3246–3247. [DOI] [PubMed] [Google Scholar]
- [37].Yaghi OM, O’Keeffe M, Ockwig NW, Chae HK, Eddaoudi M, Kim J, Nature 2003, 423, 705–714. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.