

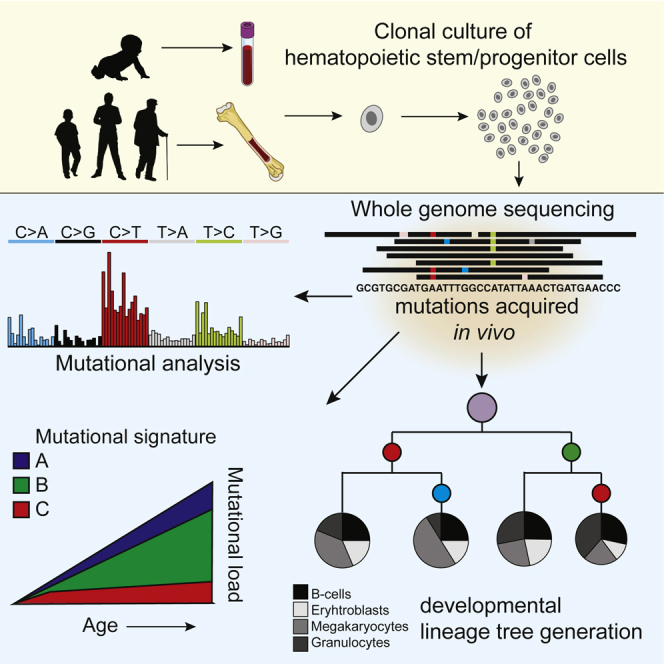
Summary
Mutation accumulation during life can contribute to hematopoietic dysfunction; however, the underlying dynamics are unknown. Somatic mutations in blood progenitors can provide insight into the rate and processes underlying this accumulation, as well as the developmental lineage tree and stem cell division numbers. Here, we catalog mutations in the genomes of human-bone-marrow-derived and umbilical-cord-blood-derived hematopoietic stem and progenitor cells (HSPCs). We find that mutations accumulate gradually during life with approximately 14 base substitutions per year. The majority of mutations were acquired after birth and could be explained by the constant activity of various endogenous mutagenic processes, which also explains the mutation load in acute myeloid leukemia (AML). Using these mutations, we construct a developmental lineage tree of human hematopoiesis, revealing a polyclonal architecture and providing evidence that developmental clones exhibit multipotency. Our approach highlights features of human native hematopoiesis and its implications for leukemogenesis.
Keywords: HSC, human hematopoiesis, somatic mutations, mutational processes, leukemia, developmental lineage tree
Graphical Abstract
Highlights
-
•
Base substitution rate is similar among human HSCs and MPPs
-
•
Mutations accumulate with 14 mutations per year per cell
-
•
Three signatures explain mutation spectra in HSC/MPPs and are also present in AML
-
•
Shared mutations allow construction of a developmental lineage tree
Osorio et al. report lifelong mutation accumulation in human hematopoietic stem and progenitor cells, which is explained by three distinct mutational signatures. Shared somatic mutations between cells of the same donor enable the construction of a developmental lineage tree and quantification of each branch to mature blood cell populations.
Introduction
Mature blood and immune cells are produced by the process of hematopoiesis, which is orchestrated by self-renewing hematopoietic stem and progenitor cells (HSPCs) in the bone marrow. As people age, clonal expansions of mutated stem cells within the blood more commonly occur, which is associated with increased risk of developing hematological malignancies (Genovese et al., 2014, Jaiswal et al., 2014, Xie et al., 2014). Somatic mutations are thought to gradually accumulate in the genomes of stem cells during life (Blokzijl et al., 2016). Most of these mutations will not have any functional consequences; however, some may render cells independent of specific external growth factors or provide insensitivity to intrinsic inhibitory signals, thereby promoting uncontrolled clonal expansion (Stratton et al., 2009). Although mutagenesis in HSPCs promotes dysfunctional hematopoiesis and leukemia (Rossi et al., 2008, Welch et al., 2012), the dynamics and mechanisms underlying mutation accumulation in these cells in human bone marrow are currently not well understood. In addition, clonal hematopoiesis is commonly observed in elderly and associated with an increased risk of hematologic cancers and death (Genovese et al., 2014, Jaiswal et al., 2014, Xie et al., 2014, Zink et al., 2017); however, the clonal composition of hematopoietic tissue within the normal human bone marrow has not been systematically determined.
Here, we assessed lifelong mutation accumulation in long-term engrafting hematopoietic stem cells (HSCs) and downstream multipotent progenitor cells (MPPs) by whole-genome sequencing (WGS) of clonally expanded primary cells from human bone marrow. By analyzing clones derived from donors of increasing age, we find that base substitutions gradually accumulate in a linear fashion from birth throughout adult life, which is driven by various endogenous mutational processes. One of these processes is specific for HSPCs when compared to other healthy organs and likely driven by mutagenic guanine analogs. Although HSPCs are believed to extensively divide during development (Bowie et al., 2006), the number of mutations at birth is limited. Using base substitutions, we constructed a developmental lineage tree revealing prenatal mutation rates, a polyclonal architecture of the hematopoietic tissue, and a multipotent but biased contribution of developmental lineages to adult tissue. Together, our approach highlights features of human hematopoiesis and its implications for hematopoietic disease.
Results
Cataloguing Somatic Mutations in Human HSCs and MPPs
We have previously shown that clonal cultures of primary cells can be used to characterize the dynamics of mutation accumulation during human life in individual tissue-specific stem cells (Blokzijl et al., 2016, Jager et al., 2018). To test whether a similar approach could be applied to hematopoietic stem cells, we used multiparameter flow cytometry to sort phenotypically defined long-term HSCs and MPPs obtained from human bone marrow biopsies (Notta et al., 2016) and then clonally expanded them to obtain sufficient DNA for WGS analysis (Figures 1A and S1). We also performed WGS of mesenchymal stem cells (MSCs) isolated from the same bone marrow in order to exclude germline variants. This procedure allowed us to catalog all the somatic mutations present in the original stem cell, which accumulated during the life of the cell. The majority of the somatic mutations in these cultures displayed a variant allele frequency (VAF) clustered around 0.5, which indicates that these mutations were shared by all cells in the culture and therefore present in the originally expanded stem cell (Figure S2). A smaller VAF peak could be observed around 0.2, which represents sub-clonal mutations that accumulated after the first cell division in vitro and are not shared by all cells in the cultures. These in vitro accumulated mutations are discarded based on the low VAF (Figure S2). We performed WGS on DNA from 18 HSCs/MPPs derived from adult marrow biopsies of 5 healthy donors, ranging from 26 to 63 years of age (Table S1). In addition, we sequenced 4 clones isolated from umbilical cord blood of 2 independent individuals to measure genome-wide somatic mutation load at birth. In total, we identified 11,082 base substitutions and 553 small insertions and deletions (indels). Independent validations using single-molecule molecular inversion probes (smMIPs) of a subset of the identified somatic mutations revealed an overall confirmation rate of approximately 91% (Tables S2 and S4B). We did not observe non-synonymous or truncating mutations in cancer driver genes for hematological neoplasms (Ju et al., 2017), excluding selective clonal outgrowth of cells in culture (Table S3).
Figure 1.

Determining Somatic Mutations in Hematopoietic Progenitors
(A) Schematic overview of experimental setup to catalog somatic mutations in single human blood progenitors. MSCs, mesenchymal stem cells; WGS, whole-genome sequencing.
(B) Average number of base substitutions in HSCs and MPPs (extrapolated to the whole autosomal genome) of the donor A. Error bars indicate SD. Each data point represents a single HSC or MPP clone. The p value indicates no statistical difference (NS) between the number of base substitutions in HSCs and MPPs (two-sided t test).
(C) Relative contribution of the indicated mutation types to the base substitution spectra in HSCs and MPPs. Error bars indicate SD. Each data point represents a single HSC or MPP clone.
(D) Average number of indels in HSCs and MPPs (extrapolated to the whole autosomal genome) of the donor A. Error bars indicate SD. Each data point represents a single HSC or MPP clone. The p values indicate no statistical difference (NS) between the number of indels in HSCs and MPPs (two-sided t test).
Long-term (LT)-HSCs and MPPs differ markedly in their ability to engraft long term in transplantation recipients (Notta et al., 2011, Oguro et al., 2013). Their proliferative histories and cell cycle control machinery are also extensively documented to be distinct (Foudi et al., 2009, Laurenti et al., 2015, Oguro et al., 2013, Passegué et al., 2005, Wilson et al., 2008). Notably, we found that the number and types of somatic mutations were highly similar between HSCs and MPPs (Figures 1B–1D). Our findings therefore suggest differences in self-renewal capacity and proliferation status do not affect genome-wide mutation accumulation in these populations. Nonetheless, cells of the same donor shared only a limited number of mutations (60 out of 11,082 base substitutions; Table S1), indicating that mutagenesis did occur independently during the lifetime of each assessed cell. Hereafter, we will refer to the HSCs and MPPs collectively as HSPCs, given their equivalent mutational profile.
Age-Related Mutation Accumulation in Human Blood Progenitors
A positive correlation (p < 0.05; t test linear mixed model) between the number of base substitutions and the age of the donors was observed (Figure 2A), indicating a gradual accumulation of this type of mutation during life. Base substitutions accumulated with an annual rate of 14.2 mutations per year (95% confidence intervals [CIs] are 6.1–22.4, respectively; Figure 2B), which remains very stable from birth throughout life. This observation indicates that the majority of mutations in adult HSPCs accumulated during life, and only a limited number of mutations (39.5; 95% CIs are 29.7–49.3, respectively; two-tailed t test) are acquired prenatally. This is surprising, considering the significant amount of cell proliferation that occurs during embryonic development and HSPC expansion in the fetal liver and the relative quiescent status of LT-HSCs in the adult marrow (Bowie et al., 2006). We detected a limited number of indels, which did not seem to correlate with the age of the donor (Figure 2C). Mutation spectra did not differ between donors (Figures 2D and 2E), suggesting that underlying causative mutagenic processes are equal among them.
Figure 2.

Age-Associated Mutation Accumulation in Human Blood Progenitors
(A) Correlation of the number of base substitutions accumulated per genome with age of the independent donors we assessed. Each data point represents a single clone. p value of the age effect in the linear mixed model is indicated above the plot (two-tailed t test). The sample size is 7 donors with a total of 22 clones sequenced (9 HSCs, 9 MPPs, and 4 cord blood progenitors). Linear mixed model was performed on the clones from adult bone marrow (5 donors; 18 clones). Dotted line indicates the extrapolation of this correlation to birth (age = 0). Subsequently, we confirmed this value at birth by performing WGS on umbilical cord blood samples (n = 4 clones) of 2 independent donors.
(B) Annual base substitution rate estimated by the linear mixed model in (A). Error bars represent the 95% confidence interval of the slope estimate.
(C) Correlation of the number of indels with age of the donors. p value of the age effect in the linear mixed model is indicated above the plot (two-tailed t test). Linear mixed model was performed on the clones from adult bone marrow. Dotted line indicates the extrapolation of this correlation to birth (age = 0).
(D) Relative contribution of the indicated mutation types to the base substitution spectra for each donor. The total number of base substitutions and assessed clones per donor is indicated.
(E) Relative contribution of the indicated mutation types to the total number of indels for each donor. The total number of indels and assessed clones per donor is indicated.
Taken together, our results provide insight into the mutation load in blood progenitors during life and at birth. Although accumulation of indels is stem cell dependent, the linear age-related accumulation of base substitutions and low inter- and intra-donor variability argues for continuously acting mutational processes during human life.
Processes Shaping the Tissue-Specific Mutational Landscape in HSPCs
We next aimed to examine the processes that underlie age-related mutation accumulation in HSPCs by analyzing mutation spectra and underlying signatures (Alexandrov et al., 2013a). We have previously reported that mutation spectra of human stem cells can vary extremely between tissues (Blokzijl et al., 2016). To compare the mutation spectra of HSPCs with stem cells of these other human tissues, we performed a principal-component analysis (PCA) using the contribution of the 6 different base substitution types in each cells while taking the direct 5′ and 3′ nucleotide context into account (Figure 3B). This PCA showed that stem cells cluster in a tissue-specific manner (Figures 3A and 3B), underscoring the notion that the underlying mutational processes can act in a tissue-specific manner. To identify these processes, we performed mutational signature analysis (Alexandrov et al., 2013b). The strongest factor in our PCA (PC1) separates cells with either high or low contribution of signatures 1 or 5 (Figure S3), which are signatures that were previously defined in a pan-cancer analysis (Alexandrov et al., 2013b) and reported to act in a “clock-like” manner. Mutation spectra in fast-cycling intestinal stem cells cluster toward signature 1. This signature has been attributed to spontaneous deamination of methylated cytosines into thymines (Alexandrov et al., 2013b) and likely reflects a cell-cycle-dependent mutational clock. Indeed, besides being the predominant mutational signature in fast cycling intestinal stem cells (Blokzijl et al., 2018), epithelial-derived cancers with a high cellular turnover also show high signature 1 mutation rates (Alexandrov et al., 2015). In contrast, mutation spectra in liver stem cells, thought to be slow dividing in vivo, cluster toward signature 5, for which the underlying process is still unknown (Alexandrov et al., 2015). Mutation spectra in adult HSPCs cluster more toward signature 5 than 1 (PC1), in line with the idea that HSPCs become quiescent postnatally (Abkowitz et al., 2002, Bowie et al., 2006). Indeed, signature 5 is the predominant contributor to the mutation spectra in adult HSPCs, whereas the contribution of signature 1 to the mutation spectra in these cells is minor (Figure 3C). Nonetheless, the number of mutations attributed to both signatures accumulate in a linear fashion with age (Figure 3C), indicating that also in these cells the underlying processes act in a clock-like manner. Interestingly, the mutation spectrum of the pooled umbilical cord blood clones clustered more toward signature 1 (Figure 3A), likely reflecting the higher division rate of HSPCs in utero (Bowie et al., 2006). Indeed, the relative contribution of signature 1 mutations in the umbilical cord blood clones was 13-fold higher compared with the adult HSPCs (Figure 3C).
Figure 3.

Signatures of Mutational Processes in HSPCs
(A) Principal-component analysis of 96-base substitution spectra. Spectra of single base substitutions sequenced clones (colored dots, 16 colon, 13 small intestine, 10 liver, 18 HSPCs, and 4 cord blood clones) and 96 profiles of signatures. Mutations for all four umbilical-cord-blood-derived samples were pooled together, as mutation load was low in these samples. Directions of signature contributions between samples are indicated on the x and y axis; see Figure S3.
(B) Total 96 mutational profiles for all mutations in the HSPC clones.
(C) Absolute contribution of each mutational signature type (extrapolated to the whole autosomal genome) plotted against the age of the donors. The observed absolute contributions of the signatures in the umbilical cord blood are plotted as triangles, and the numbers are indicated in the plot marked with CB. The p values of the age effects per tissue are shown (linear mixed model; two-tailed t test, excluding the cord blood data) with extrapolations of signature accumulation to the birth drawn in dotted lines.
(D) Transcriptional strand bias profile for all C > T transitions in HSPCs (pooled); ∗p < 0.05; two-sided Poisson test.
(E) Mean base substitution load per year per 10-MB genome of the indicated mutational signature types for the HSPC clones (n = 18) and the AML samples (n = 24). The total number of base substitutions and assessed samples per category is indicated. Error bars indicate SD. Each data point represents a single HSPC or AML sample.
The presence of a third pattern, reflecting a recently defined signature 32 (Inman et al., 2018), separates the mutation spectra in HSPCs from stem cells of the other human tissues (PC2; Figures 3A and S3). This observation indicates that an additional mutagenic process is active in the hematopoietic system, which is reflected by signature 32. The number of mutations attributed to signature 32 does not show a significant correlation with age, suggesting it is not constantly active during life. Nevertheless, we do observe presence of signature 32 in the umbilical cord blood clones (Figure 3C), suggesting it likely represents an endogenous mutagenic project. Signature 32 is characterized by C > T transitions with a preference for CpT or ApCp dinucleotides (Inman et al., 2018). Although the etiology of signature 32 is unknown, base substitutions specific for this signature were associated with a transcriptional strand bias in the HSPCs (Figures 3D and S4), consistent with activity of transcription-coupled repair (Pleasance et al., 2010). As C > T transitions are more present on the transcribed compared with the untranscribed strand (Figure 3D), our results argue that the lesion recognized by transcription-coupled repair is likely a guanine adduct. Thus, our data highlight unique specific mutational patterns and underlying mechanisms in HSPCs compared to other tissues.
Remarkably, the absolute contribution of all three signatures to the base substitution load in normal HSPCs was similar as those in a previously reported acute myeloid leukemia (AML) dataset (Welch et al., 2012; Figure 3E; p = 0.97; chi-square test; Figure S4B and S4C), indicating that the genesis of this malignancy does not necessarily require enhanced mutagenesis. Nevertheless, comparing somatic mutation load in 72 known driver genes for hematological neoplasms (Ju et al., 2017) revealed that the normal blood progenitors are depleted for potential cancer driver mutations (0 mutations in 72 genes out of 22 genomes; Table S3) compared to the AML samples (28 mutations in 72 genes out of 24 genomes; p < 0.05; one-sided binomial test; Welch et al., 2012). These potential driver mutations included indels (11 out of 28) and base substitutions (17 out of 28), of which 6 were C > T transitions (Welch et al., 2012). These results argue that, instead of altered mutagenesis, outgrowth of clones with stochastically acquired cancer-initiating mutations as a consequence of mutational processes active throughout life drives leukemogenesis (Alexandrov et al., 2015).
Construction of a Developmental Lineage Tree
Somatic mutations acquired during embryogenesis provide insight into developmental lineages and allow analysis of clonal contributions to adult cell populations (Behjati et al., 2014, Ju et al., 2017). Using the base substitutions identified in 10 WGS HSPCs from donor A, we determined genetic relatedness between these cells by assessing mutations that are shared between the different cells. In this analysis, somatic mutations that are shared between HSPCs of the same donor are indicative for a common ancestral cell. The more mutations two clones share, the later during development they separated. Using these shared mutations, we constructed a hypothetical developmental lineage tree (Figure 4A). Of note, the HSPCs shared very few mutations. As we obtain the somatic mutations by comparing to a donor-matched MSC sample, our analysis may be excluding base substitutions that were acquired during early embryonic development and also with low frequency present in this paired MSC sample. Therefore, we included mutations with sub-clonal evidence (variant allele frequency < 0.3) in the paired MSC control sample from this donor (STAR Methods). To evaluate the ability of our reconstructed map to explain lineage relationships in the bone marrow, we genotyped each base substitution defining a branch in 125 additional HSCs and MPP clones from the same donor (Table S4A). By genotyping these mutations, we could only attribute 81 out of 125 clones to the projected lineage map. This analysis indicated that our lineage tree was incomplete and we missed developmental branches. We reasoned that, by performing WGS in a bulk granulocyte sample of this donor, we would identify the somatic mutations to complete the tree. To this end, we aimed to find mutations with sub-clonal evidence in both the bulk granulocyte sample and the control MSC sample but that were absent in any of the WGS clones. This approach allowed us to identify two additional somatic mutations that defined an additional branch of the tree to which the remaining 44 progenitor clones could be attributed (Figure 4A). Thus, all analyzed HSCs and MPPs could be traced back to two cells likely arising in the first divisions of the human embryo. The number of mutations defining each branch indicated a rate of 1 base substitution per embryonic cell doubling, which is in line with previous reports (Ju et al., 2017, Lee-Six et al., 2018). If we assume that cell proliferation rate is stable during development, we estimated that, at birth, HSPCs have undergone approximately 40 divisions, based on the average number of base substitutions in the umbilical cord blood samples (Figure 4B).
Figure 4.

Reconstruction of the Development Lineage Tree and Branch-Specific Contributions to Different Blood Lineages
(A) Phylogenetic tree indicating the relatedness of the whole-genome sequenced clones. The different branches, which are defined by individual somatic base substitutions, are indicated with letters a–m. In the gray panel, the presence or absence of those base substitutions (y axes) in both the whole-genome sequenced clones and amplicon-based sequenced clones (x axes) are for HSCs (black) and MPPs (gray). The ɑ branch highlighted with red-dashed box showed asymmetrical contribution to HSCs and MPPs with a statistical significance (p < 0.05; permutation test).
(B) As a continuation of the phylogenetic tree, the number of their unique mutations in the WGS clones (black for HSCs; gray for MPPs) is shown by the length of their branches. The red horizontal line indicates the time of birth estimated based on the average number of base substitutions in the umbilical cord blood cells with the 95% confidence interval as gray box.
(C) Dendrogram depicting the correlations between the mature populations based on the hierarchical clustering of variant allele frequencies (VAFs) of the somatic mutations a–m.
Unequal Contribution of Embryonic Lineages to the Adult Hematopoietic Tissue
The reconstructed phylogeny revealed a pattern of asymmetric contribution of developmental branches to the adult hematopoietic stem cell and progenitor compartment, in line with previous observations in other tissues (Behjati et al., 2014, Ju et al., 2017). For instance, 65% of HSPCs are derived from branch a compared with branch b (p < 0.05; one-sided binomial test). Notably, this biased contribution was not as prevalent in the MSCs (Figure 4A), suggesting that such hematopoietic-specific asymmetry may have arisen later in development. Interestingly, we also observed biased contribution of some tree branches to the pools of HSCs and MPPs. For instance, branch α (Figure 4A) is significantly enriched for HSC clones (21 HSCs out of 37 clones; p < 0.05; permutation test). This finding might be suggestive of a non-overlapping developmental origin of some HSCs and MPPs, although we cannot rule out stochastic clonal amplification of these subsets postnatally.
Recent data have suggested that unilineage priming may occur in mice and humans at the level of HSC (Carrelha et al., 2018, Rodriguez-Fraticelli et al., 2018), and barcoding in the mouse embryo has suggested the presence of lineage-restricted HSCs (Pei et al., 2017). We wanted to assess the extent of developmental potency of the human developmental clones identified. To determine the contribution of each branch to mature hematopoietic tissue, we assayed for the presence of branch-specific base substitutions in sorted granulocytes, erythroblasts, pre-B cells, and megakaryocytes from the same bone marrow biopsy. Our data indicated that each developmental hematopoietic branch displayed some contribution to every single one of the assessed mature cell populations (Figure 4A). Of particular interest are base substitutions defining the last branching generation of the tree (branches l, m, and j), which are absent in MSCs, suggesting that these mutations arose during or after hematopoietic specification (considering that some MSCs are also mesodermally derived). Notably, these base substitutions also showed presence in most mature cell populations, underlining the multipotent nature of such developmental hematopoietic clones (Figure 4A; Notta et al., 2016). Hierarchical clustering of the different blood lineages by the contribution of each developmental branch to these populations revealed early branching of MSCs from mature blood lineages. Of these blood lineages, the megakaryocyte branch splits off more early, suggesting lineage differences between megakaryocytes and the other blood populations (Figure 4C), which is in line with recent observations in mice (Haas et al., 2015, Rodriguez-Fraticelli et al., 2018, Yamamoto et al., 2013).
Discussion
This study presents the survey of somatic mutation accumulation in normal HSPCs during human life and provides insight into the development and age-related mutagenesis of this tissue. We show that HSPCs after birth accumulate mutations at a stable rate of approximately 14 base substitutions per year. This rate is in the same range as previously reported for human neurons of the prefrontal cortex (Lodato et al., 2018) and approximately two-fold lower compared with stem cells of human colon, small intestine, liver, and neurons of the dentate gyrus (Blokzijl et al., 2016, Lodato et al., 2018). As human HSCs are thought to divide every 40 weeks (Catlin et al., 2011), our results suggest that they accumulate approximately 11 mutations per division. In contrast, our phylogenetic analysis shows that each of the developmental lineage branches, reflecting embryonic cell-doubling events (Ju et al., 2017), is defined by 1 mutation, suggesting that the per-division mutation rate is lower during development. Indeed, our data indicate that, at birth, HSPCs accumulate about 40 mutations, and these cells undergo many rounds of cell division during development (Bowie et al., 2006). In addition, we determined that the number and types of somatic mutations were highly similar between LT-HSCs and MPPs, even though these cell types are documented to differ extensively in their proliferative responses, cell cycle control machinery, and ability to produce long-term grafts in transplantation recipients (Foudi et al., 2009, Laurenti et al., 2015, Oguro et al., 2013, Wilson et al., 2008). Together, these observations suggest that differences in potency and self-renewal capacity might not be the most important determinant of somatic mutation load. It has been estimated that humans have about 50,000–200,000 active HSCs per person (Lee-Six et al., 2018), which would indicate that, in a lifetime of 80 years, approximately 60–240 million bases are mutated in the complete active stem cell pool. In line with this, our data suggest that no enhanced mutagenesis is needed to explain somatic base substitution load in AML. Mutational signature analysis indicated that the majority of base substitutions are generated by two processes that are age dependent and constantly active during life. One of these processes, reflected by signature 1, is thought to be driven by spontaneous deamination of methylated cytosines into thymines (Alexandrov et al., 2013b). As this signature has been found to act in a clock-like manner in cancers derived from normal epithelia with a high turnover (Alexandrov et al., 2015) as well as in fast cycling small intestinal and colon stem cells (Blokzijl et al., 2016), this signature may represent a cycle-dependent mutational clock. In line with this, the contribution of signature 1 in adult HSPCs, which are mostly quiescent after birth (Catlin et al., 2011), is minor, whereas at birth, the mutation spectra of HSPCs resemble fast-cycling human stem cells, potentially reflecting the massive proliferation of HSPCs in utero (Bowie et al., 2006). In contrast, the most predominant contributor of mutagenesis in adult HSPCs is signature 5, which can also act in a clock-like manner in cancers (Alexandrov et al., 2015) and in slowly cycling liver stem cells (Blokzijl et al., 2016) and likely reflects a cell-cycle-independent mutational clock. Presence of a third novel signature (signature 32) in the HSPCs defines the tissue specificity of the mutation spectra observed in the hematopoietic cells. This signature has been associated with azathioprine therapy (Inman et al., 2018), which can cause severe hematological toxicities (Karran and Attard, 2008). Of note, the transcriptional strand bias effect in the HSPCs can be only partially explained by the reported bias of signature 32 (Inman et al., 2018), suggesting that the underlying transcription blocking lesion might be slightly different from guanine metabolites that result from azathioprine treatment. To our best knowledge, none of the donors have been treated with azathioprine, arguing that the presence of this signature in healthy donors might reflect mutagenic action of endogenously generated guanine metabolites. Indeed, signature analysis of the pooled umbilical cord blood samples indicated that signature 32 mutations are already present at birth and thus acquired in utero. Finally, by using base substitutions, we were able to trace the developmental history of the hematopoietic stem and progenitor compartment in the bone marrow, demonstrating that asymmetrical contributions shape hematopoietic system ontogeny. Our study also provides support for the functional multipotent nature of early developmental hematopoietic clones. Thus, somatic mutations in blood progenitors provide a means to study lineage relationships of native human hematopoiesis.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-CD34 magnetic beads conjugated | Miltenyi Biotech | Cat# 130-046-703 |
| CD34-PB | BioLegend | Cat# 343512; RRID:AB_1877197 |
| Thy1/CD90-PE | BioLegend | Cat# 328110; RRID:AB_893433 |
| CD45RA-FITC | BioLegend | Cat# 304106; RRID:AB_314410 |
| CD49f-APC-Cy7 | BioLegend | Cat# 313628; RRID:AB_2616784 |
| CD38-PE-Cy7 | Thermo Fisher Scientific | Cat# 25-0388-42; RRID:AB_2573346 |
| CD66b-FITC | BioLegend | Cat# 305103; RRID:AB_314495 |
| CD11b-APC-Cy7 | BioLegend | Cat# 301341; RRID:AB_2563371 |
| CD19-PB | BioLegend | Cat# 302223; RRID:AB_493652 |
| CD42b-PE | BioLegend | Cat# 303905; RRID:AB_314385 |
| CD10-A700 | Thermo Fisher Scientific | Cat# 56-0106-41; RRID:AB_2574494 |
| CD41-PE-Cy7 | Thermo Fisher Scientific | Cat# 25-0419-41; RRID:AB_2573347 |
| CD235a-APC | Thermo Fisher Scientific | Cat# 17-9987-41; RRID:AB_2043824 |
| CD34-BV-421 | BioLegend | Cat# 343609; RRID:AB_11147951 |
| CD38-PE | BioLegend | Cat# 303505; RRID:AB_314357 |
| CD45RA-PerCP/Cy5.5 | BioLegend | Cat# 304121; RRID:AB_893358 |
| FITC anti-human Lineage Cocktail (CD3, CD14, CD16, CD19, CD20, CD56) | BioLegend | Cat# 348701; RRID:AB_1064401 |
| CD16-FITC | BioLegend | Cat# 302005; RRID:AB_314205 |
| CD11c-FITC | BioLegend | Cat# 301603; RRID:AB_314173 |
| Biological Samples | ||
| Whole bone marrow samples A,C,D,E | AllCells | ABM001 |
| Whole bone marrow sample B | Boston Children’s Hospital | N/A |
| Cord blood samples CB-1,CB-2 | University Medical Center Utrecht and the Wilhelmina Children’s Hospital | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant Human SCF | PeproTech | Cat#300-07 |
| Recombinant Human TPO | PeproTech | Cat#300-18 |
| Recombinant Human FLT3-L | PeproTech | Cat#300-19 |
| Recombinant Human IL-6 | PeproTech | Cat#200-06 |
| Recombinant Human IL-3 | PeproTech | Cat#160-01 |
| Deposited Data | ||
| Whole-genome sequence data from this article | This paper | European Genome-Phenome Archive (EGA; https://www.ebi.ac.uk/ega/home) Accession Number EGAS00001003068 |
| Oligonucleotides | ||
| Primers | This paper | Table S4 |
| Software and Algorithms | ||
| Whole genome sequencing read alignment and mutation calling pipeline | UMCU Genetics | https://github.com/UMCUGenetics/IAP |
| SNV Filtering pipeline | UMCU Genetics | https://github.com/UMCUGenetics/SNVFI |
| INDEL Filtering pipeline | This paper | https://github.com/ToolsVanBox/INDELFI |
| Mutational Patterns R package | Blokzijl et al., 2018 | N/A |
| Burrows-Wheeler Aligner v0.5.9 mapping tool | Li and Durbin, 2010 | N/A |
| SAMTOOLS | Li et al., 2009 | N/A |
| smMIP analysis script | This paper | https://github.com/ToolsVanBox/smMIPfil |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ruben van Boxtel (R.vanBoxtel@prinsesmaximacentrum.nl).
Experimental Model and Subject Details
Human bone marrow biopsies and umbilical cord blood
Sample B (26-year-old donor, male, white) was extracted at Boston Children’s Hospital, whereas samples A (33-year-old, male, white), C (55-year-old, female, hispanic), D (63-year-old, male, white), and E (41-year old, female, African American) were obtained from AllCells (Table S1). 10-20 mL of whole bone marrow aspirate were drawn into a 60cc syringe containing heparin (80 U/mL of BM) from a unique puncture in the posterior iliac crest. All the donors were healthy and did not show previous conditions. Patient’s informed consents were obtained by Boston Children’s Hospital and AllCells, respectively. Umbilical cord blood samples CB-1 (0-year old, female, ethnicity unknown) and CB-2 (0-year old, male, ethnicity unknown) was obtained at the University Medical Center Utrecht and the Wilhelmina Children’s Hospital. Informed consent was obtained and this study was approved by the ethical committee of University Medical Center Utrecht. No differences related with sex or gender of the samples were detected in this study.
Method Details
FACS
Erythrocytes were removed from the bone marrow samples using red blood cell lysis buffer. CD34-enrichment was performed using magnetic-assisted cell sorting with anti-CD34 magnetic beads (Miltenyi Biotech, 130-046-703). Different cell populations were purified through using FACSAria (Becton Dickinson) and MoFlo XDP (Beckman Coulter) equipment. The following combinations of cell surface markers were used to define cell populations(Notta et al., 2016). HSC: CD34+CD38-CD45RA-CD90+CD49f+; MPP: CD34+CD38-CD45RA-CD90-CD49f-; Granulocytes: CD66b+CD11b+CD19-CD10-CD235a-CD41-CD42b-; Erythroblasts: CD235a+CD66b-CD11b-CD19-CD10-CD41-CD42b-; pre-B cells: CD19+CD10+CD66b-CD11b-CD235a-CD41-CD42b-; Megakaryocyte progenitors: CD41+CD42b+CD66b-CD11b-CD19-CD10-CD235a-. Representative examples of sorted populations are shown in the Figure S1B. Flow cytometry data was analyzed using FlowJo software (Tree Star). Polyclonal mesenchymal stem cells (MSCs) cultures were established from a fraction of whole bone marrow samples after red blood cell lysis, cells were plated in tissue culture treated dishes in DMEM-F12 medium (GIBCO), supplemented with 10% FBS. MSCs were kept in culture for a week and medium was replaced each day to remove non-adherent cells. Umbilical cord blood mononuclear cells were isolated by density centrifugation over Lymphoprep (Stem Cell Technologies). Umbilical cord blood progenitors were sorted on the following cell markers: (CD34+, CD38-, CD45RA-, CD11c-, CD16-, Lin(CD3/14/19/20/56)-) and clonal cultures were established in the same manner as the HSPCs (below).
FACS antibodies
All antibodies were used at 1:100 dilution unless noted. Antibodies used for bone marrow isolation of HSPC and mature populations: CD34-PB (Biolegend, clone 581, 343512), Thy1/CD90-PE (Biolegend, clone 5E10, 328110), CD45RA-FITC (Biolegend, clone HI100, 304106), CD49f-APC-Cy7 (Biolegend, clone GoH3, 313628), CD38-PE-Cy7 (eBioscience, clone HB7, 25-0388-42), CD66b-FITC (Biolegend, clone G10F5, 305103), CD11b-APC-Cy7 (Biolegend, clone ICRF44, 301341), CD19-PB (Biolegend, clone HIB19, 302223), CD42b-PE (Biolegend, clone HIP1, 303905), CD10-A700 (eBioscience, clone eBioCB-CALLA, 56-0106-41), CD41-PE-Cy7 (eBioscience, Clone HIP8 25-0419-41), CD235a-APC (eBioscience, clone HIR2, 17-9987-41). Antibodies used for umbilical cord blood MPP isolation: CD34-BV-421 (Biolegend, Clone 561, 343609, 1:20), CD38-PE (Biolegend, Clone HIT2, 303505, 1:50), CD45RA-PerCP/Cy5.5(Biolegend, Clone HI100, 304121, 1:20), Lineage(CD3/CD14/CD19/CD20/CD56)-FITC(Biolegend, Clones UCHT1, HCD14, HIB19, HCD56, 348701, 1:20), CD16-FITC (Biolegend, Clone 3G8, 302005), CD11c-FITC (Biolegend, Clone 3.9, 301603, 1:20)
Establishment of clonal HSC/MPP cultures
HSCs and MPPs were first sorted into a collection tube and a second index sort was performed to seed single-cells into round-bottom 384-well plates. Cell were cultured in StemSpan SFEM medium supplemented with SCF (100 ng/mL), FLT3-L (100 ng/mL), TPO (50 ng/mL), IL-6 (20 ng/mL) and IL-3 (10 ng/mL) at 37°C, 5%CO2 for 3-4 weeks before collection.
Whole-Genome Sequencing and Read Alignment
DNA libraries for Illumina sequencing were generated by using standard protocols (Illumina) from 20 - 50 ng of genomic DNA isolated from the clonally expanded blood progenitors using DNeasy Blood & Tissue Kit (QIAGEN) according to manufacturer’s instructions. All samples were sequenced (2 × 150 bp) by using Illumina HiSeq X Ten sequencers to 30x base coverage. Sequence reads were mapped against human reference genome GRCh37 by using Burrows-Wheeler Aligner v0.5.9 mapping tool(Li and Durbin, 2010) with settings ‘bwa mem -c 100 -M’. Sequence reads were marked for duplicates by using Sambamba v0.4.732 and realigned per donor by using Genome Analysis Toolkit (GATK) IndelRealigner v2.7.2, and sequence read quality scores were recalibrated with GATK BaseRecalibrator v2.7.2. Full pipeline description and settings also available at: https://github.com/UMCUGenetics/IAP.
Mutation calling and filtering
Raw variants were multisample-called by using the GATK HaplotypeCaller v3.4-46 (DePristo et al., 2011) and GATK-Queue v3.4-46 with default settings and additional option ‘EMIT_ALL_CONFIDENT_SITES’. The quality of variant and reference positions was evaluated by using GATK VariantFiltration v3.4-46 with options ‘-snpFilterName LowQualityDepth -snpFilterExpression “QD < 2.0” -snpFilterName MappingQuality -snpFilterExpression “MQ < 40.0” -snpFilterName StrandBias -snpFilterExpression “FS > 60.0” -snpFilterName HaplotypeScoreHigh -snpFilterExpression “HaplotypeScore > 13.0” -snpFilterName MQRankSumLow -snpFilterExpression “MQRankSum < −12.5” -snpFilterName ReadPosRankSumLow -snpFilterExpression “ReadPosRankSum < −8.0” -cluster 3 -window 35’. To obtain high-quality somatic mutation catalogs, we applied postprocessing filters as described(Blokzijl et al., 2016). Briefly, we considered variants at autosomal chromosomes without any evidence from a paired control sample (MSCs isolated from the same bone marrow); passed by VariantFiltration with a GATK phred-scaled quality score ≥ 100 for base substitutions and ≥ 250 for indels; a base coverage of at least 20X in the clonal and paired control sample; no overlap with single nucleotide polymorphisms (SNPs) in the Single Nucleotide Polymorphism Database v137.b3730; and absence of the variant in a panel of unmatched normal human genomes (BED-file available upon request). We additionally filtered base substitutions with a GATK genotype score (GQ) lower than 99 or 10 in clonal or paired control sample, respectively. For indels, we filtered variants with a GQ score lower than 99 in both clonal and paired control sample and filtered indels that were present within 100 bp of a called variant in the control sample. In addition for both SNVs and INDELs, we only considered variants with a mapping quality (MQ) score of 60 and with a variant allele frequency of 0.3 or higher in the clones to exclude in vitro accumulated mutations (Blokzijl et al., 2016, Jager et al., 2018).
Principal component analysis
The occurrences of all 96-trinucleotide changes were counted for each HPSC and averaged per donor. In this analysis, we included, besides the blood progenitors, genome-wide mutation catalogs of individual adult stem cells of colon, small intestine and liver (Blokzijl et al., 2016). As for the umbilical cord blood samples mutational load was low, possibly affecting the outcome, all four umbilical cord blood-derived samples were pooled together. Principal component analysis was performed using the base R function prcomp.
Mutational profile and signature analysis
To identify most prominent signatures, which gives the largest separation to the clones in a plane in a principal component analysis, we extracted principal components 1 (PC1) and 2 (PC2), and separated positive and negative signals of the components. Using a in-house developed R package (MutationalPatterns) (Blokzijl et al., 2018), the patterns of the extracted components were compared to the COSMIC SigProfiler signatures (https://www.synapse.org/#!Synapse:syn11967914) and from their cosine similarities, Signature 1, 5 and 32 were selected. These three signatures were subsequently refitted to the adult HSPC data and the pooled umbilical cord blood mutational profiles. To determine contribution of signatures to the mutation load in AML, we obtained somatic mutation catalogs which were identified in the nonrepetitive portion of the genome (∼50% of the entire genome(Mardis et al., 2009)) of 24 AML samples(Welch et al., 2012). We determined the COSMIC SigProfiler signature contribution similarly as performed on HSPC data. To determine the transcriptional strand contribution and bias, we selected all point mutations that fall within gene bodies and checked whether the mutated C or T was located on the transcribed or non-transcribed strand.
Transcriptional strand bias analyses
We used a in house developed R package (MutationalPatterns)(Blokzijl et al., 2018) to determine transcriptional strand bias as described(Blokzijl et al., 2016). Transcriptional strand bias is calculated with the transcriptional single base substitution signatures obtained from the COSMIC Transcriptional Strand Signatures (https://www.synapse.org/#!Synapse:syn11967914).
Amplicon analysis of SNVs
DNA from the HSPC clones was extracted using QIAGEN DNeasy Blood and tissue kit (QIAGEN). A first amplicon-specific PCR was performed (primer sequences available upon request) using TruSeq Illumina adapters, then a second indexing PCR was performed. The DNA library was sequenced using the MiSeq reagent kit v2 500 cycles. Raw sequencing data was aligned against the human reference genome (hg19) using BWA-mem(Li and Durbin, 2010). The alignment data was compressed, sorted and indexed using SAMTOOLS(Li et al., 2009) and the per position sequencing information was extracted in pileup format requiring a minimum sequencing and mapping phred scores of 25 and 15 respectively. Finally, an in-house written perl script was used to calculate the read counts supporting both reference and variant alleles for each position of interest as well as to calculate variant allele frequencies.
smMIP analysis of SNVs
Clone-specific smMIPs were designed as described(Hiatt et al., 2013, Yu et al., 2018), (Table S4B). The genomic regions of interest were captured using 10ng for the indicated clones and 100 ng of genomic DNA for the matching granulocyte samples. UMI sequences were trimmed from sequenced smMIP reads and mapped to the human reference genome (hg19) using BWA-mem algorithm with -M option(Li and Durbin, 2010). For each UMI with at least 5 reads, the sequenced nucleotide at the mutation position were extracted for every read. When 70% or more of the reads with the same UMI had the same nucleotide, then the UMI was counted as valid as described(Yu et al., 2018) Mutation positions with less than 20 valid UMI’s were filtered out, and the rest mutations with higher than 0 VAF were counted as validated (Table S2). For bulk granulocyte data, the non-validated positions and positions with less than 20 valid UMI’s were excluded.
Construction of developmental lineage tree
We first constructed a developmental lineage tree by cataloguing somatic base substitutions, which were shared between the 10 whole-genome sequenced clones of donor A. To obtain base substitutions that were acquired during early embryonic development, we included mutations with sub-clonal (VAF < 0.3) evidence in the paired MSC control sample that were either clonally present or completely absent in the 10 clones. All of these shared base substitutions were manually inspected and false positive calls were excluded. To complete the tree, we whole-genome sequenced the bulk granulocyte samples and search for base substitutions that were sub-clonally present in the granulocytes and the paired MSC control sample without any evidence in the 10 whole-genome sequenced clones. We also considered mutations observed in clones and with sub-clonal evidence in the granulocytes. These mutations were also manually inspected and false positive calls excluded. For all of these early embryonic base substitutions primers were designed for amplicon-base re-sequencing in the mature populations and HSC and MPP clones as described above (Table S4A). In total, we sequenced 140 clones, 62 HSCs and 78 MPPs, from which we excluded 9 HSC and 6 MPP clones from the analysis, because they had less than 10x coverage at any of the selected substitutions or showed evidence for multiple base substitutions and could therefore not be assigned to a branch. A binary mutation table was created to summarize the shared base substitutions. To construct a heatmap with a lineage tree, lineage distances were calculated using binary method, clones were hierarchically clustered using average method and plotted using gplots package in R.
Quantification and Statistical Analysis
Sample and mutation numbers are indicated in the figures. Data are shown as mean ± standard deviation. For the slope estimation, the linear mixed model was used to take donor dependency into account and the p values are indicated in the figures. To assess statistical significance of mutation numbers between two groups, a two-sided t test was used after testing normality of data distribution using the Shapiro–Wilk test and equality of variances. To assess statistical significance of mutation spectra between two groups, a chi-square test was used. To assess statistical significance of enrichment or depletion of mutations in different genomic regions, the number of progenitor clones in different branches, or depletion of potential cancer-driver mutations in the normal blood progenitors, a one-sided binomial test was used. To assess statistical significance between distribution of HSCs and MPPs in different branches a permutation test was used.
Data and Software Availability
Code Availability
Mutation calling and filtering pipelines are available at https://github.com/UMCUGenetics/IAP, https://github.com/UMCUGenetics/SNVFI and https://github.com/ToolsVanBox/INDELFI, smMIP analysis script is available at https://github.com/ToolsVanBox/smMIPfil. The other scripts are available on request.
Data availability
The accession number for the whole-genome sequence data reported in this paper is EGA: EGAS00001003068.
Acknowledgments
The authors would like to thank the Hartwig Medical Foundation (Amsterdam, the Netherlands) for facilitating low-input whole-genome sequencing, P.J. Coffer for providing umbilical cord blood samples, and P.J. Campbell and D.C. Wedge for sharing scripts. This study was financially supported by an EMBO long-term fellowship to F.G.O. (ALTF 655-2016), an ERC starting grant (ERC2014-STG637904) to I.V., a VIDI grant of the Netherlands Organisation for Scientific Research (NWO) (no. 016.Vidi.171.023) to R.v.B., funding from Worldwide Cancer Research (WCR) (no. 16-0193) to R.v.B., and NIH grants HL128850-01A1 and P01HL13147 to F.D.C. F.D.C. is a scholar of the Howard Hughes Medical Institute and the Leukemia and Lymphoma Society.
Author Contributions
F.G.O. performed sample isolation and performed clonal expansions. F.G.O. and S.H.P. performed fluorescence-activated cell sorting (FACS). A.R.H., I.V., R.O., and R.v.B. performed bioinformatic analyses. A.R.H. and K.H. performed umbilical cord blood isolation and clonal expansion. M.V. and L.d.l.F. performed DNA isolations and library preparation. F.D.C. and R.v.B. designed the study and wrote the manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: November 27, 2018
Footnotes
Supplemental Information includes four figures and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.11.014.
Contributor Information
Fernando D. Camargo, Email: fernando.camargo@childrens.harvard.edu.
Ruben van Boxtel, Email: r.vanboxtel@prinsesmaximacentrum.nl.
Supplemental Information
HSPC clone, clone identifier (see Table S1), Chromosome of the variant, Position on the chromosome, Reference allele and mutated alternative allele, Mutation type of the variant, Gene in which variant is located, Amino acid change, allelic fraction of the variant.
(A) Variants and primer pairs used to sequence additional HSPC clones and mature blood populations for shared mutations in HSPCs of donor A. (B) Primer sequences used for smMIP validation of variants.
References
- Abkowitz J.L., Catlin S.N., McCallie M.T., Guttorp P. Evidence that the number of hematopoietic stem cells per animal is conserved in mammals. Blood. 2002;100:2665–2667. doi: 10.1182/blood-2002-03-0822. [DOI] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Campbell P.J., Stratton M.R. Deciphering signatures of mutational processes operative in human cancer. Cell Rep. 2013;3:246–259. doi: 10.1016/j.celrep.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A.J.R., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Børresen-Dale A.-L., Australian Pancreatic Cancer Genome Initiative. ICGC Breast Cancer Consortium. ICGC MMML-Seq Consortium. ICGC PedBrain Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Jones P.H., Wedge D.C., Sale J.E., Campbell P.J., Nik-Zainal S., Stratton M.R. Clock-like mutational processes in human somatic cells. Nat. Genet. 2015;47:1402–1407. doi: 10.1038/ng.3441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behjati S., Huch M., van Boxtel R., Karthaus W., Wedge D.C., Tamuri A.U., Martincorena I., Petljak M., Alexandrov L.B., Gundem G. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature. 2014;513:422–425. doi: 10.1038/nature13448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F., de Ligt J., Jager M., Sasselli V., Roerink S., Sasaki N., Huch M., Boymans S., Kuijk E., Prins P. Tissue-specific mutation accumulation in human adult stem cells during life. Nature. 2016;538:260–264. doi: 10.1038/nature19768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F., Janssen R., van Boxtel R., Cuppen E. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med. 2018;10:33. doi: 10.1186/s13073-018-0539-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie M.B., McKnight K.D., Kent D.G., McCaffrey L., Hoodless P.A., Eaves C.J. Hematopoietic stem cells proliferate until after birth and show a reversible phase-specific engraftment defect. J. Clin. Invest. 2006;116:2808–2816. doi: 10.1172/JCI28310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrelha J., Meng Y., Kettyle L.M., Luis T.C., Norfo R., Alcolea V., Boukarabila H., Grasso F., Gambardella A., Grover A. Hierarchically related lineage-restricted fates of multipotent haematopoietic stem cells. Nature. 2018;554:106–111. doi: 10.1038/nature25455. [DOI] [PubMed] [Google Scholar]
- Catlin S.N., Busque L., Gale R.E., Guttorp P., Abkowitz J.L. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–4466. doi: 10.1182/blood-2010-08-303537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foudi A., Hochedlinger K., Van Buren D., Schindler J.W., Jaenisch R., Carey V., Hock H. Analysis of histone 2B-GFP retention reveals slowly cycling hematopoietic stem cells. Nat. Biotechnol. 2009;27:84–90. doi: 10.1038/nbt.1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas S., Hansson J., Klimmeck D., Loeffler D., Velten L., Uckelmann H., Wurzer S., Prendergast Á.M., Schnell A., Hexel K. Inflammation-induced emergency megakaryopoiesis driven by hematopoietic stem cell-like megakaryocyte progenitors. Cell Stem Cell. 2015;17:422–434. doi: 10.1016/j.stem.2015.07.007. [DOI] [PubMed] [Google Scholar]
- Hiatt J.B., Pritchard C.C., Salipante S.J., O’Roak B.J., Shendure J. Single molecule molecular inversion probes for targeted, high-accuracy detection of low-frequency variation. Genome Res. 2013;23:843–854. doi: 10.1101/gr.147686.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman G.J., Wang J., Nagano A., Alexandrov L.B., Purdie K.J., Taylor R.G., Sherwood V., Thomson J., Hogan S., Spender L.C. The genomic landscape of cutaneous SCC reveals drivers and a novel azathioprine associated mutational signature. Nat. Commun. 2018;9:3667. doi: 10.1038/s41467-018-06027-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager M., Blokzijl F., Sasselli V., Boymans S., Janssen R., Besselink N., Clevers H., van Boxtel R., Cuppen E. Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat. Protoc. 2018;13:59–78. doi: 10.1038/nprot.2017.111. [DOI] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y.S., Martincorena I., Gerstung M., Petljak M., Alexandrov L.B., Rahbari R., Wedge D.C., Davies H.R., Ramakrishna M., Fullam A. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–718. doi: 10.1038/nature21703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P., Attard N. Thiopurines in current medical practice: molecular mechanisms and contributions to therapy-related cancer. Nat. Rev. Cancer. 2008;8:24–36. doi: 10.1038/nrc2292. [DOI] [PubMed] [Google Scholar]
- Laurenti E., Frelin C., Xie S., Ferrari R., Dunant C.F., Zandi S., Neumann A., Plumb I., Doulatov S., Chen J. CDK6 levels regulate quiescence exit in human hematopoietic stem cells. Cell Stem Cell. 2015;16:302–313. doi: 10.1016/j.stem.2015.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H., Øbro N.F., Shepherd M.S., Grossmann S., Dawson K., Belmonte M., Osborne R.J., Huntly B.J.P., Martincorena I., Anderson E. Population dynamics of normal human blood inferred from somatic mutations. Nature. 2018;561:473–478. doi: 10.1038/s41586-018-0497-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato M.A., Rodin R.E., Bohrson C.L., Coulter M.E., Barton A.R., Kwon M., Sherman M.A., Vitzthum C.M., Luquette L.J., Yandava C.N. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science. 2018;359:555–559. doi: 10.1126/science.aao4426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis E.R., Ding L., Dooling D.J., Larson D.E., McLellan M.D., Chen K., Koboldt D.C., Fulton R.S., Delehaunty K.D., McGrath S.D. Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notta F., Doulatov S., Laurenti E., Poeppl A., Jurisica I., Dick J.E. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- Notta F., Zandi S., Takayama N., Dobson S., Gan O.I., Wilson G., Kaufmann K.B., McLeod J., Laurenti E., Dunant C.F. Distinct routes of lineage development reshape the human blood hierarchy across ontogeny. Science. 2016;351:aab2116. doi: 10.1126/science.aab2116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H., Ding L., Morrison S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passegué E., Wagers A.J., Giuriato S., Anderson W.C., Weissman I.L. Global analysis of proliferation and cell cycle gene expression in the regulation of hematopoietic stem and progenitor cell fates. J. Exp. Med. 2005;202:1599–1611. doi: 10.1084/jem.20050967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W., Feyerabend T.B., Rössler J., Wang X., Postrach D., Busch K., Rode I., Klapproth K., Dietlein N., Quedenau C. Polylox barcoding reveals haematopoietic stem cell fates realized in vivo. Nature. 2017;548:456–460. doi: 10.1038/nature23653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pleasance E.D., Cheetham R.K., Stephens P.J., McBride D.J., Humphray S.J., Greenman C.D., Varela I., Lin M.-L., Ordóñez G.R., Bignell G.R. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–196. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Fraticelli A.E., Wolock S.L., Weinreb C.S., Panero R., Patel S.H., Jankovic M., Sun J., Calogero R.A., Klein A.M., Camargo F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature. 2018;553:212–216. doi: 10.1038/nature25168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi D.J., Jamieson C.H.M., Weissman I.L. Stems cells and the pathways to aging and cancer. Cell. 2008;132:681–696. doi: 10.1016/j.cell.2008.01.036. [DOI] [PubMed] [Google Scholar]
- Stratton M.R., Campbell P.J., Futreal P.A. The cancer genome. Nature. 2009;458:719–724. doi: 10.1038/nature07943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch J.S., Ley T.J., Link D.C., Miller C.A., Larson D.E., Koboldt D.C., Wartman L.D., Lamprecht T.L., Liu F., Xia J. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012;150:264–278. doi: 10.1016/j.cell.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A., Laurenti E., Oser G., van der Wath R.C., Blanco-Bose W., Jaworski M., Offner S., Dunant C.F., Eshkind L., Bockamp E. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135:1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- Xie M., Lu C., Wang J., McLellan M.D., Johnson K.J., Wendl M.C., McMichael J.F., Schmidt H.K., Yellapantula V., Miller C.A. Age-related mutations associated with clonal hematopoietic expansion and malignancies. Nat. Med. 2014;20:1472–1478. doi: 10.1038/nm.3733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto R., Morita Y., Ooehara J., Hamanaka S., Onodera M., Rudolph K.L., Ema H., Nakauchi H. Clonal analysis unveils self-renewing lineage-restricted progenitors generated directly from hematopoietic stem cells. Cell. 2013;154:1112–1126. doi: 10.1016/j.cell.2013.08.007. [DOI] [PubMed] [Google Scholar]
- Yu J., Antić Ž., van Reijmersdal S.V., Hoischen A., Sonneveld E., Waanders E., Kuiper R.P. Accurate detection of low-level mosaic mutations in pediatric acute lymphoblastic leukemia using single molecule tagging and deep-sequencing. Leuk. Lymphoma. 2018;59:1690–1699. doi: 10.1080/10428194.2017.1390232. [DOI] [PubMed] [Google Scholar]
- Zink F., Stacey S.N., Norddahl G.L., Frigge M.L., Magnusson O.T., Jonsdottir I., Thorgeirsson T.E., Sigurdsson A., Gudjonsson S.A., Gudmundsson J. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. doi: 10.1182/blood-2017-02-769869. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
HSPC clone, clone identifier (see Table S1), Chromosome of the variant, Position on the chromosome, Reference allele and mutated alternative allele, Mutation type of the variant, Gene in which variant is located, Amino acid change, allelic fraction of the variant.
(A) Variants and primer pairs used to sequence additional HSPC clones and mature blood populations for shared mutations in HSPCs of donor A. (B) Primer sequences used for smMIP validation of variants.
Data Availability Statement
The accession number for the whole-genome sequence data reported in this paper is EGA: EGAS00001003068.