

Abstract
Summary
The MetaboAnalyst web application has been widely used for metabolomics data analysis and interpretation. Despite its user-friendliness, the web interface has presented its inherent limitations (especially for advanced users) with regard to flexibility in creating customized workflow, support for reproducible analysis, and capacity in dealing with large data. To address these limitations, we have developed a companion R package (MetaboAnalystR) based on the R code base of the web server. The package has been thoroughly tested to ensure that the same R commands will produce identical results from both interfaces. MetaboAnalystR complements the MetaboAnalyst web server to facilitate transparent, flexible and reproducible analysis of metabolomics data.
Availability and implementation
MetaboAnalystR is freely available from https://github.com/xia-lab/MetaboAnalystR.
1 Introduction
Metabolomics aims to study all small compounds within a biological system. It complements other omics technologies in multi-omics characterization of biological systems, and is poised to play a significant role in precision medicine (Wishart, 2016). With the growing applications of metabolomics comes an urgent need for easy-to-use, open-source software tools that are able to analyze increasingly large and complex datasets, as well as to keep pace with rapidly evolving technological innovations. The open-source nature of such software will promote transparency and reproducibility in data analysis, as well as encourage academic collaboration by allowing different research groups to further extend the existing tools or incorporate them into new software pipelines.
A wide variety of Web or Galaxy-based tools exist for metabolomics data analysis (Gardinassi et al., 2017), such as MetaboAnalyst (Chong et al., 2018), XCMSOnline (Huan et al., 2017), Workflow4Metabolomics (Giacomoni et al., 2015), and Galaxy-M (Davidson et al., 2016). Among them, MetaboAnalyst is one of the most widely used tools for statistical and functional analysis of metabolomics data. Despite its user-friendliness, the web-based application comes with its inherent limitations. For instance, the comprehensive analysis options provided through the web interface make it difficult for users to reproduce their results when they re-analyze their data after a long time. To address this issue, MetaboAnalyst generates a comprehensive analysis report upon completion of each analysis session. However, not all commands and parameters are recorded. The web interface also presents significant constrains in terms of developing customized workflows and handling large data. As metabolomics is increasingly used across different fields and biological systems, data analysis is not ‘one size fits all’. To address the concerns with reproducibility, flexibility, scalability and transparency, we have developed a companion R package—MetaboAnalystR.
2 Implementation and features
MetaboAnalystR is written in the R language (Team, 2016). The development version is hosted on GitHub and the stable release will soon be available as an R package on CRAN. It builds upon the R code base from the web server, with extensive modifications to ensure functional compatibility across both the web and the R command line. To ease the learning process, we have completely revamped the MetaboAnalyst web interface to expose the underlying R commands during analysis. The R package and the web server have been extensively tested to ensure that the identical results will be generated. MetaboAnalystR conforms to the R package quality standards (Wickham, 2015), including comprehensive vignettes for each modules with detailed case studies. The analysis workflow is summarized in Figure 1.
Fig. 1.
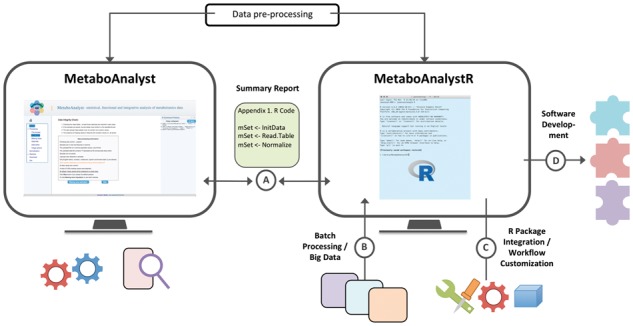
Main features of MetaboAnalystR package. The R command history generated on the web server can be directly used by MetaboAnalystR (A). Batch processing of metabolomics data can be accomplished using the R package (B). MetaboAnalystR can be integrated with other R packages (C). Its open-source nature will also facilitate further metabolomics software development (D)
2.1 Functionality
MetaboAnalystR consists of over 500 functions organized into 11 modules (statistical analysis, biomarker analysis, time-series analysis, power analysis, biomarker meta-analysis, enrichment analysis, pathway analysis, joint pathway analysis, network explorer, MS peaks to pathways and other utilities). MetaboAnalystR builds upon several R packages such as caret (Kuhn, 2008) for classification and performance evaluation, and ROCR (Sing et al., 2005) for visualizing biomarker performance. It also contains a high-performance implementation of the mummichog algorithm to infer pathway activities from m/z peak lists (Li et al., 2013). MetaboAnalystR utilizes the MetaboAnalyst knowledgebase, including compound libraries, pathway libraries, and metabolite set libraries. They will be downloaded from the central repository upon first request.
2.2 Reproducibility and transparency
The MetaboAnalyst web interface now features an R command history panel updated in real time during data analysis. Users can export this R script containing the R functions, parameters used, and the order in which they were executed. These commands can be copy-and-pasted into R or RStudio (Team, 2015) to reproduce identical results. The web interface coupled with R commands maximizes transparency underlying each analysis step, and will greatly help teach non-programmers in using MetaboAnalystR. Both the R package and the web server generate analysis reports for each module using Sweave (Leisch, 2002). We have updated the report template for all modules, which now contain detailed information surrounding each analysis step, followed by corresponding results, and the R command history.
2.3 Flexibility and scalability
Another key feature of MetaboAnalystR is its flexibility to allow users to perform their metabolomics data analysis. The R code from the command history and R package itself allows users to easily adjust the parameters or to modify the existing workflows. The modularity of MetaboAnalystR permits it to be easily integrated with other existing tools to develop custom metabolomics pipelines. For instance, MetaboAnalystR is currently interoperable with XCMS (xcms:::.write.metaboanalyst), and supports NetCDF, mzDATA and mzXML file formats. The support for mzTab (Griss, et al., 2014) will be added in a future release. Because the MetaboAnalyst public web server imposes a size restriction (50M), the R package will be of great use for users to both directly process and batch process larger datasets.
3 Case studies
To demonstrate the functionality, flexibility and scalability of the MetaboAnalystR package, we performed analyses on two sets of metabolomics data. The detailed discussions and comparisons are available on the GitHub under ‘Case Studies’.
4 Conclusion
Data analysis has become a major bottleneck in current metabolomics workflows. In-depth analysis of metabolomics data can be daunting to most researchers, and requires powerful and flexible software solutions. MetaboAnalystR complements the popular MetaboAnalyst web server by providing a comprehensive R package to facilitate flexible and reproducible metabolomics data analysis.
Funding
This work was supported by the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant and the Canada Research Chairs (CRC) program (to J. Xia). J. Chong is supported in part by the McGill Graduate Dean’s Award.
Conflict of Interest: none declared.
References
- Chong J. et al. (2018) MetaboAnalyst 4.0: towards more transparent and integrative metabolomics analysis. Nucleic Acids Res., 46, 486–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson R.L. et al. (2016) Galaxy-M: a galaxy workflow for processing and analyzing direct infusion and liquid chromatography mass spectrometry-based metabolomics data. Gigascience, 5, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardinassi L.G. et al. (2017) Bioinformatics tools for the interpretation of metabolomics data. Curr. Pharmacol. Rep., 3, 374–383. [Google Scholar]
- Giacomoni F. et al. (2015) Workflow4Metabolomics: a collaborative research infrastructure for computational metabolomics. Bioinformatics, 31, 1493–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griss J. et al. (2014) The mzTab data exchange format: communicating mass-spectrometry-based proteomics and metabolomics experimental results to a wider audience. Mol. Cell. Proteomics, 13, 2765–2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huan T. et al. (2017) Systems biology guided by XCMS Online metabolomics. Nat. Methods, 14, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn M. (2008) Caret package. J. Stat. Softw., 28, 1–26.27774042 [Google Scholar]
- Leisch F.S. (2002) Sweave: dynamic generation of statistical reports using literate data analysis. In Compstat 2002 - Proceedings in Computational Statistics. Physika Verlag, Heidelberg, Germany, pp. 575–580.
- Li S. et al. (2013) Predicting network activity from high throughput metabolomics. PLoS Comput. Biol., 9, e1003123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sing T. et al. (2005) ROCR: visualizing classifier performance in R. Bioinformatics, 21, 3940–3941. [DOI] [PubMed] [Google Scholar]
- Team,R. (2015) RStudio: Integrated Development for R. RStudio, Inc, Boston, MA. [Google Scholar]
- Team,R.C. (2016) R: A Language and Environment for Statistical Computing [Computer Software]. R Foundation for Statistical Computing, Vienna. [Google Scholar]
- Wickham H. (2015) R Packages: Organize, Test, Document, and Share Your Code. O'Reilly Media, Inc., Boston, Massachusetts, USA. [Google Scholar]
- Wishart D.S. (2016) Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov., 15, 473–485. [DOI] [PubMed] [Google Scholar]