

Abstract
A comprehensive evaluation of every patient with a bloodstream infection includes an attempt to identify the infectious source. Pathogens can originate from various places, such as the gut microbiome, skin, and external environment. Identifying the definitive origin of an infection would enable precise interventions focused on management of the source1,2. Unfortunately, hospital infection control practices are often informed by assumptions about the source of various specific pathogens; if these assumptions are incorrect they lead to interventions that do not decrease pathogen exposure3. Here, we develop and apply a streamlined bioinformatic tool, named StrainSifter, to match bloodstream pathogens precisely to a candidate source. We then leverage this approach to interrogate the gut microbiome as a potential reservoir of bloodstream pathogens in a cohort of hematopoietic cell transplantation recipients. We find that patients with Escherichia coli and Klebsiella pneumoniae bloodstream infections have concomitant gut colonization with these organisms, suggesting that the gut may be a source of these infections. We also find cases where classically non-enteric pathogens, such as Pseudomonas aeruginosa and Staphylococcus epidermidis, are found in the gut microbiome, thereby challenging existing informal dogma of these infections originating from environmental or skin sources. Thus, we present an approach to distinguish the source of various bloodstream infections, which may facilitate more accurate tracking and prevention of hospital-acquired infections.
Clinical management of infection involves the evaluation and elimination of infectious sources. Epidemiologically, bloodstream infections (BSI) are common in hospitalized patients and contribute substantially to patient morbidity and mortality4. Thus, identifying the source of BSIs is critical in both clinical care and hospital epidemiology. BSIs are particularly common in immunocompromised patients who are hospitalized for extended periods of time, such as hematopoietic cell transplantation (HCT) recipients5–7. Primary bloodstream infections with enteric organisms often arise as a result of translocation from the intestinal microbial reservoir across a damaged gastrointestinal barrier into the bloodstream8. In contrast, non-enteric commensal and environmental bacteria can access the bloodstream through intravenous lines and sites where skin epithelial integrity has been compromised. Existing methods for identifying the origins of bloodstream infections in HCT patients include pulsed-field gel electrophoresis and multi-locus sequence typing (MLST)9,10. Although rapid, affordable, and standardized across many organisms, these methods are not ideal for distinguishing bacterial strains. Yet, microbial pathogenicity and transmission depend in part on strain-level variability, as different strains of the same species can vary widely in their ability to cause disease11,12. Whole genome sequencing (WGS) has facilitated the exploration of strain-level determinants of virulence and has enabled precise tracking of pathogens11,13,14.
While comparisons of strain genomes have primarily been performed on bacterial isolates, newer computational tools (metaSNV, MIDAS, and StrainPhlAn)15–17 profile strain variation between metagenomes. These careful strain-level analyses allow us to understand when and how bacteria are transmitted and how they may change over time. However, bioinformatic tools have not been developed for identifying specific sources of infection by comparing disease-causing bacterial isolates to complex microbiome samples such as human stool. In this work, we present StrainSifter, a bioinformatics pipeline for matching pathogens to potential sources. We then apply this tool to compare bacterial strains between the gut and bloodstream in HCT patients, with the goal of better understanding the origin of BSI in this population.
We performed a retrospective cohort study of autologous and allogeneic HCT recipients at Stanford University Hospital. Weekly stool sampling was carried out for all subjects who consented to a tissue biobanking protocol between October 5, 2015 and June 9, 2017. Patients were included if a stool sample had been collected in the 30 days preceding an episode of BSI and if a bloodstream isolate meeting standard BSI criteria had also been saved18. Thirty patients (32 bloodstream isolates) met these criteria. We sequenced all bloodstream isolates as well as stool samples (n = 82) collected between 60 days before and 31 days after the date of BSI. Clinical characteristics of the cohort are listed in Table 1 (individual patient data in Table S1).
Table 1:
Cohort summary, n=30
| Baseline characteristics | N (%) | |
|---|---|---|
| Age (years) | ||
| ≤30 | 4 (13%) | |
| 31–40 | 4 (13%) | |
| 41–50 | 6 (20%) | |
| 51–60 | 4 (13%) | |
| 61–70 | 10 (33%) | |
| ≥71 | 2 (7%) | |
| Sex (%male) | 17 (57%) | |
| Underlying diagnosis | ||
| Lymphoma | 8 (27%) | |
| AML | 7 (23%) | |
| MDS/Myelofibrosis | 6 (20%) | |
| ALL | 5 (17%) | |
| CMMoL | 2 (7%) | |
| Other* | 2 (7%) | |
| Conditioning regimen | ||
| Myeloablative | 16 (53%) | |
| Reduced intensity | 9 (30%) | |
| Non-myeloablative | 5 (17%) | |
| Transplant source | ||
| Peripheral blood | 20 (67%) | |
| Bone marrow | 8 (27%) | |
| Double umbilical cord blood | 2 (7%) | |
| Type of donor | ||
| Autologous | 3 (10%) | |
| Allogeneic | 27 (90%) | |
| Matched related donor | 10 (37%) | |
| Matched unrelated donor | 14 (52%) | |
| Mismatched unrelated donor | 3 (11%) | |
| TPN within 30 days | 15 (50%) | |
| Antibiotics within 30 days** | 30 (100%) | |
| Fluoroquinolones | 26 (87%) | |
| Beta-lactams | 14 (47%) | |
| Carbapenems | 6 (20%) | |
| Vancomycin (IV) | 12 (40%) | |
| Bacteremia species, n=32 | 32 (100%) | |
| Gram-positive | Staphylococcus aureus | 8 (25%) |
| Methicillin-sensitive | 5 (63%) | |
| Methicillin-resistant | 3 (38%) | |
| Staphylococcus epidermidis | 4 (13%) | |
| Streptococcus mitis | 7 (22%) | |
| Enterococcus faecium | 2 (6%) | |
| Rothia mucilaginosa | 2 (6%) | |
| Gram-negative | Escherichia coli | 4 (13%) |
| Klebsiella spp. | 3 (9%) | |
| Klebsiella pneumoniae | 2 (67%) | |
| Klebsiella oxytoca | 1 (33%) | |
| Enterobacter cloacae | 1 (3%) | |
| Pseudomonas aeruginosa | 1 (3%) |
List of abbreviations: AML, acute myeloid leukemia; ALL, acute lymphoblastic leukemia; CMMoL, chronic myelomonocytic leukemia; MDS, myelodysplastic syndrome; TPN, total parenteral nutrition; IV, intravenous
Other=Paroxysmal nocturnal hemoglobinuria, testicular cancer
Select categories of antibiotics (antibiotics are not exclusive and do not add up to 100%)
We sequenced a median of two stool samples per patient (range 1–8), collected a median of nine days prior to BSI (range −58 to +31) (Figure S1, read counts in Tables S2, S3). Stool sequence data were taxonomically classified using the One Codex platform19. We observe the BSI species in the gut at a threshold of 0.1% or greater relative abundance for 15 of 32 (47%) unique organisms, 10 of which are of expected enteric origin (eight typically intestinal, two typically oral). One patient developed a BSI with two species, both of which were present in the stool above the threshold level (Table S4; full taxonomic classifications in Table S5).
We next investigated whether BSI organisms are present at a higher relative abundance in the gut prior to infection, as has been reported20,21. Of the 15 BSIs in which the organism was detected in the stool, we observe intestinal dominance by the BSI pathogen in two instances (Figure 1). In both cases, the BSI species are expected to be enteric in origin (Escherichia coli from patient 3; Enterococcus faecium from patient 25) (Figure 1, Table S4). In contrast, other enteric bacteria are poorly abundant (Klebsiella pneumoniae and Enterobacter cloacae from patient 2 at 2.8% and 0.6% relative abundance, respectively) (Figure 1, Table S4). All typically non-enteric organisms are poorly abundant (0.01–2%) or not detected in the gut prior to bloodstream infection (Pseudomonas aeruginosa from patient 19; Staphylococcus epidermidis from patient 13; Staphylococcus aureus from multiple patients) (Figure 1, Table S4). In the stool samples of several patients, we observe a high relative abundance of candidate pathogens that did not cause BSI in those individuals. Specifically, patient 14 experienced a K. pneumoniae BSI, yet stool samples at two timepoints are dominated by other potential pathogens: E. coli at 64% relative abundance nine days prior to BSI and E. faecium at 82% relative abundance 19 days after BSI (Table S5).
Figure 1. BSI pathogens are present in the gut microbiome at varying relative abundance prior to bloodstream infection.
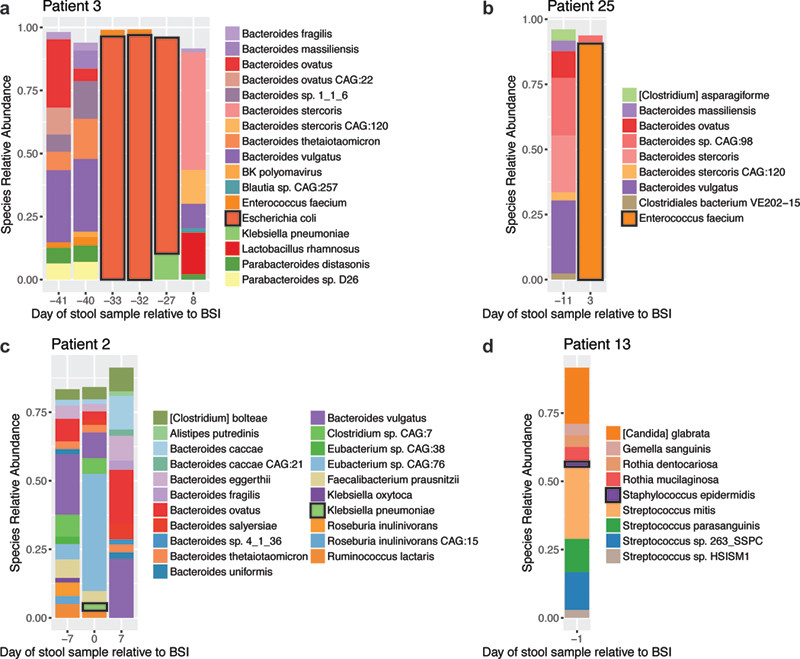
Relative abundance of microbial reads classified at the species level. Plots show species present at 1.5% relative abundance or greater and thus stacked bars do not necessarily add up to 100%. The BSI causing organism is outlined in black in the bar plot and figure legend for each panel. Timing of BSI and engraftment relative to HCT are available in Table S1. Domination by Escherichia coli (a) and Enterococcus faecium (b) occurs prior to bacteremia. Klebsiella pneumoniae (c) and Staphylococcus epidermidis (d) are present in the gut microbiome prior to BSI at relatively low abundance.
While taxonomic concordance suggests BSI organism presence in the gut microbiome, we sought to test this hypothesis with greater precision. To do so, we developed StrainSifter (Figure S2), a bioinformatic pipeline which detects whether an organism is present with sufficient abundance in short-read datasets, and outputs phylogenetic trees and single-nucleotide variant (SNV) counts between samples. We used StrainSifter to investigate the relatedness of strains of each BSI species in our metagenomes and isolates. Isolate reads were assembled into draft genomes using a short-read genome assembly tool (assembly statistics Table S6; CheckM assessment Table S7). We compared the phylogenetic relatedness of all BSI and stool strains in our sample collection to one another (Figure 2) and to publicly available data (Figure S3), and counted SNVs using Strainsifter (Tables S8, S9). Of note, none of the 30 patients included in our study had sufficient S. aureus in their stool samples to profile with StrainSifter, indicating that this organism likely infrequently colonizes the gut of HCT patients.
Figure 2. Gut and BSI strains from the same patient are more closely related than strains from different patients.
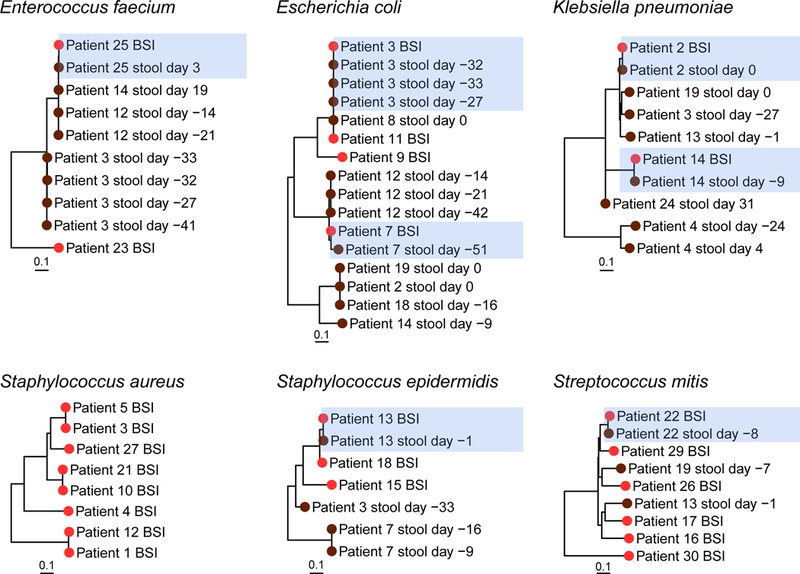
Phylogenetic relatedness between bacterial strains as assessed by StrainSifter. Branch tip colors indicate stool (brown) and bloodstream infection (BSI) (red) samples. Samples from the same patient are more closely phylogenetically related to each other (blue highlight) than to samples from other patients. Days given are relative to BSI. Phylogenetic trees for P. aeruginosa and E. cloacae are not shown, as these species are not observed with sufficient abundance in more than one gut metagenome. Of note, although patient 20’s BSI is classified as S. epidermidis, this strain does not meet the coverage requirements for inclusion in the S. epidermidis phylogenetic tree.
In general, we find that BSI and gut metagenomic strains from the same patient are more closely related than strains from unrelated patients. As expected, BSI and intestinal strains of typically enteric species such as E. coli (patients 3, 7), E. faecium (patient 25), K. pneumoniae (patient 2), and S. mitis (patient 22) are closely phylogenetically related (Figure 2) supporting the longstanding dogma that these organisms are gut-derived9,10. On one extreme, we observe zero SNVs between BSI and stool strains of patient 3 at timepoints 33, 32, and 27 days prior to BSI, indicating that the identical E. coli strain is present in the gut over a month before the onset of infection (Table S9). On the other extreme, we measured 259 SNVs between the E. coli BSI and the stool sample for patient 7. This surprising observation suggests the possibility of a population of closely-related strains, where the dominant strain is varying over time. Alternatively, the E. coli strain that resulted in BSI may have been acquired elsewhere.
Unexpectedly, we observe that gut and BSI strains are closely related in samples from the same patient for typically non-enteric taxa including S. epidermidis (patient 13) and P. aeruginosa (patient 19, not pictured) (Figure 2). We find one SNV (0.4 SNVs per megabase) between BSI and gut S. epidermidis strains of patient 13, indicating that the bloodstream strain is highly concordant with the strain found in the gut one day prior (Table S9). Further, we observe zero discriminating SNVs between identical strains of P. aeruginosa in both the blood and stool specimens. While P. aeruginosa can exist in the gut microbiome22, S. epidermidis is typically thought to originate from the skin23–26. As further evidence that S. epidermidis bacteremia was not clearly line-associated, patient 13’s blood cultures cleared within two days, despite retention of the line (Table S1). Interestingly, patient 7 did not develop a S. epidermidis BSI despite high relative abundance over two sequential timepoints (>60%) (Table S5). Finally, to compare WGS-based approaches to traditional strain typing, we performed in silico MLST (Table S10). In the four instances where an MLST type was resolved for both gut and BSI strains, results were concordant with StrainSifter.
For the gut microbiome to be a contributing source of pathogens, the organisms must be alive. However, it is not possible to ascertain whether these organisms are alive using StrainSifter. A surrogate for measurement of a living organism is the rate of DNA replication. We used an available bioinformatic tool27 to assess replication rates for 11 stool samples from 9 patients where gut and BSI strains were concordant and found that all had rates suggestive of active replication (Table S11).
We observe relatively few events of potential pathogen transmission between individuals despite overlapping hospital admissions during the 20-month study period, based on sequence-relatedness of bloodstream isolates or of candidate pathogens measured in the gut microbiome reservoir. For example, stool samples from patients 12 and 14 reveal E. faecium strains that differ by 49–76 SNVs (18–26 SNVs per megabase) relative to the bloodstream isolate of patient 25 (Table S8). Similarly, several S. aureus BSI strains appear related: 710 SNVs (250 per megabase) between BSIs from patients 10 and 21, 166 SNVs (58 per megabase) between patients 3 and 5, and 729 SNVs (263 per megabase) between patients 1 and 12. However, it is important to note that StrainSifter profiles the dominant strain in each sample. Thus, true transmission events may be missed if different strains dominate in different individuals.
Finally, we asked whether closely related strains from different patients are also functionally related. We compared computationally predicted (Figure 3, Table S12) and clinical antibiotic resistance (Table S13) for individual patient BSIs. We find that predicted and clinical antibiotic resistance results are highly concordant. As noted previously, E. coli bloodstream isolates from patients 3 and 11 are phylogenetically related, differing by relatively few SNVs (60 SNVs per megabase). Functional analysis reveals that patient 3’s BSI contains a gene encoding CTX-M, whereas patient 11’s BSI does not. CTX-M is an extended-spectrum beta-lactamase (ESBL), which confers resistance to most penicillins and cephalosporins. As predicted, clinical testing confirmed that patient 3’s BSI was resistant to most penicillins and cephalosporins, whereas patient 11’s BSI was not. In contrast, the E. coli BSI strain from patient 7 differs from that of patient 3 by 24,088 SNVs (7,390 SNVs per megabase), but demonstrates similar predicted and clinical ESBL activity, also likely conferred by CTX-M. Phylogenetically related S. aureus BSIs exhibit similar predicted and clinical phenotypes. For example, MecR1-mediated methicillin resistance is predicted and present in S. aureus BSIs 3 and 5, which are closely related (Figure 3, Tables S12, S13). S. aureus BSIs 1 and 12, which are closely related to each other but distant from BSIs 3 and 5, lack a gene encoding MecR1 and are methicillin-sensitive.
Figure 3. Antibiotic resistance gene predictions in bloodstream isolate genomes.
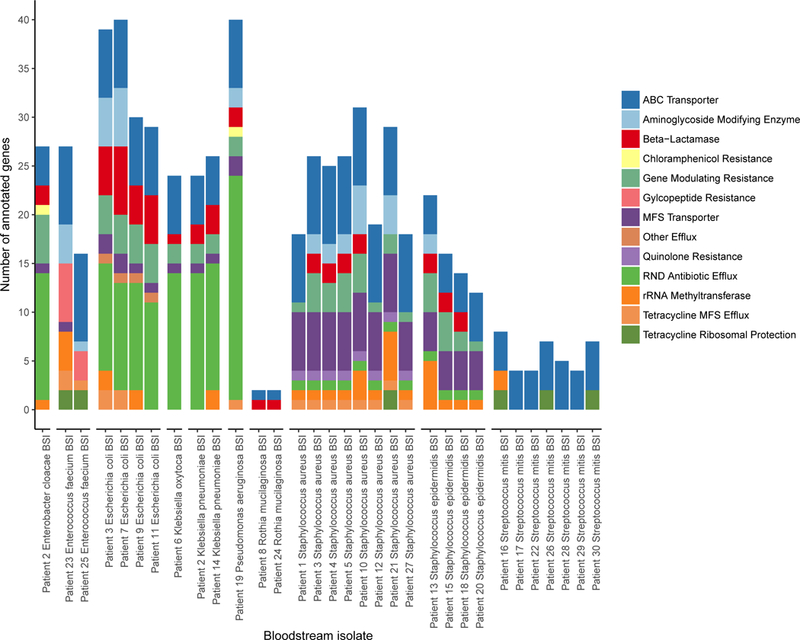
Antibiotic resistance genes predicted in bloodstream isolate draft genomes. Antibiotic resistance profiles are similar for different isolates of a given species. Of note, the Staphylococcus epidermidis isolate that was found to be concordant with a strain in the matching gut sample13, S. epidermidis BSI) has a larger number of predicted antibiotic resistance genes compared to the remaining S. epidermidis isolates.
In conclusion, a detailed analysis using StrainSifter allowed us to precisely and comprehensively identify the candidate source of various bloodstream infections. While there is great enthusiasm for the incorporation of WGS into real-time patient management, at present, challenges in sample preparation and sequencing turnaround-time limit the incorporation of such approaches into clinical care. Nevertheless, WGS is playing a growing role in hospital epidemiologic studies. Characterization of gut microbiome dynamics that occur prior to infection may help us precisely identify potential reservoirs of pathogens, thus enabling improved hospital infection prevention and management strategies.
The results presented are suggestive of a gut microbiome source for both enteric and non-enteric organisms. However, given that the present study sampled only stool microbiota, we cannot exclude the possibility of the same pathogenic strain colonizing multiple body sites from which the infection may have originated instead. Additionally, although StrainSifter can precisely identify shared variants between genomes and metagenomes, it is limited to profiling only the dominant strain of a given organism in a community. However, it has been shown that gut metagenomes frequently contain only one predominant strain of each species, so StrainSifter is likely to function well under many circumstances17.
In the future, we anticipate that high resolution WGS-based strain comparisons will facilitate discovery of additional instances where typically “non-enteric” organisms are found in the gut microbiome, a model supported here. This knowledge may complement the growing body of research on therapies to improve gut microbiota diversity and may inform attempts to bolster colonization resistance against pathogens. Further, more precisely identifying the origins of bloodstream infections may influence how hospitals and healthcare providers can most effectively work to prevent infections. With these powerful genomic tools, we anticipate that precision source identification and strain tracking will lead us to a new, sharpened model of infectious disease.
Methods
Cohort selection
A retrospective cohort study, approved by the institutional review board under the IRB protocol # 42053 (Principal investigator: Dr. Ami Bhatt), was performed at Stanford Hospital. Informed consent was obtained from all samples collected. At the time of cohort identification (July 2017), a stool biospecimen collection containing 964 stool samples from 402 patients was available for investigation. This collection consisted of convenience samples collected from autologous and allogeneic hematopoietic cell transplantation (HCT) patients at Stanford University Hospital between October 5, 2015 and June 9, 2017. Patients were included in this study if a stool sample had been collected within 30 days prior to an episode of bloodstream infection (BSI) for which a blood isolate was also available. From this final cohort, we sequenced all stool samples in our collection within 60 days prior to and 31 days after BSI.
Bloodstream isolate identification
Bloodstream isolates from HCT patients who received medical care at Stanford University Hospital were obtained from the Stanford Hospital Clinical Microbiology Laboratory. All isolates considered typical bloodstream pathogens by National Healthcare Safety Network (NHSN) guidelines were stored in a glycerol suspension at −80°C for up to 12 months17. Blood culture isolates considered to be skin-associated bacteria (including viridans group Streptococcus spp. and coagulase-negative Staphylococcus spp.) were saved if they were recovered in two or more blood culture sets as per NHSN criteria17. Isolates were identified by standard biochemical testing and matrix-assisted laser desorption and ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Bruker Daltonics).
Sample processing
Bacterial bloodstream isolates were plated on brain heart infusion agar with 10% horse blood. DNA was extracted from isolates using the Gentra Puregene Yeast/Bact. Kit per manufacturer’s instructions. Stool samples were collected and stored at 4°C for up to 24 hours prior to homogenization, aliquoting, and storage at −80°C. DNA was extracted from stool using the QIAamp DNA Stool Mini Kit (QIAGEN) per manufacturer’s instructions, with an initial bead-beating step prior to extraction using the Mini-Beadbeater-16 (BioSpec Products) and 1 mm diameter Zirconia/Silica beads (BioSpec Products). Bead-beating consisted of 7 rounds of alternating 30 second bead-beating bursts followed by 30 seconds cooling on ice. DNA concentration for all samples was measured using Qubit Fluorometric Quantitation (Life Technologies). DNA sequencing libraries from both isolates and stool were prepared using the Nextera XT DNA Library Prep Kit (Illumina) with isolates and stool microbiota libraries prepared at separate times following DNA decontamination of all lab surfaces and pipets (DNAZap, Ambion). Library concentration was measured using Qubit Fluorometric Quantitation (Life Technologies) and library quality and size distributions were analyzed with the Bioanalyzer 2100 (Agilent). Prepared libraries were multiplexed and subjected to 100 base pair paired-end sequencing on the HiSeq 4000 platform (Illumina).
Computational methods
WGS preprocessing
Sequence data were demultiplexed by unique barcodes (bcl2fastq v2.20.0.422, Illumina). Reads were deduplicated to remove PCR and optical duplicates using SuperDeduper v1.4 with the start location in the read at 5 base pairs (-s 5) and minimum length of 50 base pairs (-l 50)28. Deduplicated reads were trimmed using TrimGalore v0.4.4, a wrapper for CutAdapt v1.16, with a minimum quality score of 30 for trimming (-q 30), minimum read length of 50 (--length 50) and the “--nextera” flag to remove Illumina Nextera adapter sequences29,30. Draft genomes of bacterial isolates were assembled using SPAdes v3.11.031 with default parameters. Summary statistics for each BSI assembly were generated using ‘basic_assembly_stats.py’ from GAEMR v1.0.132. Draft genome completeness was assessed with CheckM v1.0.11 “lineage_wf”33. Draft genomes were filtered to remove contigs smaller than 1kb for downstream analyses.
Taxonomic classification
Gut metagenomic reads were taxonomically classified via the One Codex platform, a web-based tool for assigning read-level classifications based on unique k-mer signatures relative to a curated reference database (database v2017)19.
Phylogenetic tree building and variant identification with the StrainSifter pipeline
StrainSifter is a pipeline deployed as a Snakemake34 workflow packaged with conda, available at GitHub (https://github.com/bhattlab/strainsifter). Snakemake v5.1.4 and conda v4.5.9 were used in this manuscript. StrainSifter source code can be found in the Supplementary Information. Strainsifter contains modules for variant calling and phylogenetic tree building. StrainSifter accepts as input an assembled bacterial draft genome, designated as the reference, and two or more short read data sets (isolate or metagenomic), and can report a phylogenetic tree of input samples as well as pairwise SNV counts.
To build the phylogenetic trees reported in this manuscript, the most contiguous and complete genome from our isolate collection was chosen as the reference genome for each infectious species (based on clinical laboratory taxonomic classifications). For the variant counting reported herein, BSI isolate draft genomes were supplied to StrainSifter. For both analyses, all stool and BSI short read datasets were provided as input. We also used StrainSifter to evaluate the phylogenetic relatedness of our BSI strains to those available in a published database of pathogenic isolates from an intensive care setting (BioProject PRJNA267549)35. For both phylogeny and SNV-counting modules, preprocessed short reads are first aligned to the reference genome using the Burrows-Wheeler Aligner v0.7.1036. Alignments are filtered to include only high-confidence alignments with mapping quality of at least 60 using the “view” tool from the SAMtools suite (v1.7)37 (samtools view -b -q 60), and further filtered using BamTools “filter” (v2.4.0) to include only reads with the desired number or fewer mismatches (i.e. for five or fewer mismatches: bamtools filter -tag ‘NM:<=5’)38. For phylogenetic tree construction, reads with 5 or fewer mismatches were included; for determining strain single-nucleotide variants, reads were limited to 1 or fewer mismatches. Per-base coverage is calculated from each resulting BAM file using bedtools genomecov (v2.26.0) and processed with custom python scripts to identify samples meeting a minimum average coverage of 5X across at least 40% of the genome39,40. Only samples meeting the coverage requirement are continued through the pipeline. Pileup files are created from BAM files using SAMtools “mpileup”, and are analyzed using custom python scripts to identify bases occurring with at least 0.8 frequency at positions covered 5X or greater (Computational methods supplement). Only bases with a minimum phred score of >=20 are considered. Consensus sequences for each sample are created, wherein bases that cannot be confidently determined given the described parameters are called as “N”.
To create a phylogenetic tree, core positions are identified on a per-species basis, where core positions are defined as positions in the reference genome where a base could be confidently called for all samples meeting the coverage requirements. To generate phylogenetic trees, core positions with variants in at least one sample are identified and concatenated into one FASTA file per sample. FASTA files are aligned using MUSCLE v3.8.3141 and a maximum-likelihood phylogenetic tree is computed using FastTree v2.1.742. Phylogenetic trees are visualized in R using the ape v5.143, phangorn v2.4.044, and ggtree v1.10.545 packages. Pairwise SNVs are determined from the consensus sequences using a custom python script.
Synthetic multilocus sequence typing
Metagenomic short reads were assembled using metaSPAdes v3.11.046. Multilocus sequence typing (MLST) schemes and sequences were downloaded from the PubMLST database47. MLST gene sequences were aligned to metagenome assemblies using nucleotide BLAST v2.2.3148, and the top hit for each alignment was chosen based on E-value, percent identity, and alignment length. Only MLST sequences that were present in the metagenomic assembly with 100% identity across the entire length of the sequence were reported. MLST types generated by our in-house analysis were confirmed with the SRST2 synthetic multilocus sequence typing tool (v0.2.0)49.
Antibiotic resistance gene annotation
Putative protein sequences were identified in BSI draft genomes using Prodigal v2.6.350. Antibiotic resistance genes were annotated from protein sequences by searching the Resfams antibiotic resistance protein family database (v1.2)51 using hmmscan from the hmmer package with the “--cut_ga” and “--tblout” flags52.
Determination of bacterial replication rates within metagenomic samples
Bacterial replication rates were assessed using the iRep v1.10 software53. Gut metagenomic samples were aligned to the BSI draft genome from the same patient using StrainSifter as described above. The resulting BAM files were converted to SAM format using SAMtools “view” and the resulting SAM file and corresponding BSI draft genome were supplied to iRep as input for each sample.
Plots
Plots were generated using the R programming language (v3.4.0) using the ggplot2 v2.2.154, reshape2 v1.4.355, and dplyr v0.7.456 packages.
Reporting summary
Additional information on experimental design is available in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgements
We thank Joyce Kang for her assistance with stool sample processing, as well the other members of the Bhatt laboratory for providing feedback on the study design, bioinformatics pipeline, and manuscript revisions. We would like to thank Nick Greenfield and the One Codex team for help with using their platform. We appreciate Drs. Matthew Kelly, Chris Severyn, and Doyle Ward for their feedback on the manuscript. We would especially like to thank the patients and nurses on the Blood and Marrow Transplantation service for their enthusiastic participation in this project. This work was supported in part by National Science Foundation Graduate Research Fellowship (FBT), the National Institutes of Health, National Center for Advancing Translational Science, Clinical and Translational Science Award KL2 TR001083 and UL1 TR001085 (TMA). ASB was funded in part by the National Cancer Institute NIH K08 award, #CA184420, Damon Runyon Clinical Investigator Award, and Amy Strelzer Manasevit Award. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Competing financial interests
None noted.
Data and code availability
All sequencing datasets from the current study have been deposited in the National Center for Biotechnology Information Sequence Read Archive under the BioProject PRJNA477326. Accession numbers are listed in the Supplementary Information. StrainSifter and associated source code can be found at https://github.com/bhattlab/strainsifter.
References
- 1.Costa SF, Miceli MH & Anaissie EJ Mucosa or skin as source of coagulase-negative staphylococcal bacteraemia? Lancet Infect. Dis 4, 278–286 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Mermel LA et al. Clinical practice guidelines for the diagnosis and management of intravascular catheter-related infection: 2009 Update by the Infectious Diseases Society of America. Clin. Infect. Dis 49, 1–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Steinberg JP, Robichaux C, Tejedor SC, Reyes MD & Jacob JT Distribution of pathogens in central line-associated bloodstream infections among patients with and without neutropenia following chemotherapy: evidence for a proposed modification to the current surveillance definition. Infect. Control Hosp. Epidemiol 34, 171–175 (2013). [DOI] [PubMed] [Google Scholar]
- 4.Goto M & Al-Hasan MN Overall burden of bloodstream infection and nosocomial bloodstream infection in North America and Europe. Clin. Microbiol. Infect 19, 501–509 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Blennow O, Ljungman P, Sparrelid E, Mattsson J & Remberger M Incidence, risk factors, and outcome of bloodstream infections during the pre-engraftment phase in 521 allogeneic hematopoietic stem cell transplantations. Transpl. Infect. Dis 16, 106–114 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Gudiol C et al. Etiology, clinical features and outcomes of pre-engraftment and post-engraftment bloodstream infection in hematopoietic SCT recipients. Bone Marrow Transplant 49, 824–830 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Mikulska M et al. Blood stream infections in allogeneic hematopoietic stem cell transplant recipients: reemergence of Gram-negative rods and increasing antibiotic resistance. Biol. Blood Marrow Transplant 15, 47–53 (2009). [DOI] [PubMed] [Google Scholar]
- 8.See I et al. Impact of removing mucosal barrier injury laboratory-confirmed bloodstream infections from central line-associated bloodstream infection rates in the National Healthcare Safety Network, 2014. Am. J. Infect. Control 45, 321–323 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tancrede CH & Andremont AO Bacterial Translocation and Gram-Negative Bacteremia in Patients with Hematological Malignancies. J. Infect. Dis 152, 99–103 (1985). [DOI] [PubMed] [Google Scholar]
- 10.Samet A et al. Leukemia and risk of recurrent Escherichia coli bacteremia: genotyping implicates E. coli translocation from the colon to the bloodstream. Eur. J. Clin. Microbiol. Infect. Dis 32, 1393–1400 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snitkin ES et al. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc. Natl. Acad. Sci. U. S. A 108, 13758–13763 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lieberman TD et al. Genetic variation of a bacterial pathogen within individuals with cystic fibrosis provides a record of selective pressures. Nat. Genet 46, 82–87 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snitkin ES et al. Tracking a Hospital Outbreak of Carbapenem-Resistant Klebsiella pneumoniae with Whole-Genome Sequencing. Sci. Transl. Med 4, 148ra116–148ra116 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaysen A et al. Integrated meta-omic analyses of the gastrointestinal tract microbiome in patients undergoing allogeneic hematopoietic stem cell transplantation. Transl. Res 186, 79–94.e1 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Costea PI et al. metaSNV: A tool for metagenomic strain level analysis. PLoS One 12, e0182392 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nayfach S, Rodriguez-Mueller B, Garud N & Pollard KS An integrated metagenomics pipeline for strain profiling reveals novel patterns of bacterial transmission and biogeography. Genome Res 26, 1612–1625 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Truong DT, Tett A, Pasolli E, Huttenhower C & Segata N Microbial strain-level population structure & genetic diversity from metagenomes. Genome Res 27, 626–638 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.National Healthcare Safety Network Patient Safety Component Manual. Centers for Disease Control and Prevention. Available at: https://www.cdc.gov/nhsn/pdfs/validation/2016/pcsmanual_2016.pdf. (Accessed: 31st July 2018)
- 19.Minot SS, Krumm N & Greenfield NB One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. bioRxiv 027607 (2015). [Google Scholar]
- 20.Ubeda C et al. Vancomycin-resistant Enterococcus domination of intestinal microbiota is enabled by antibiotic treatment in mice and precedes bloodstream invasion in humans. J. Clin. Invest 120, 4332–4341 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taur Y et al. Intestinal domination and the risk of bacteremia in patients undergoing allogeneic hematopoietic stem cell transplantation. Clin. Infect. Dis 55, 905–914 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nesher L et al. Fecal colonization and infection with Pseudomonas aeruginosa in recipients of allogeneic hematopoietic stem cell transplantation. Transpl. Infect. Dis 17, 33–38 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Wade JC, Schimpff SC, Newman KA & Wiernik PH Staphylococcus epidermidis: an increasing cause of infection in patients with granulocytopenia. Ann. Intern. Med 97, 503–508 (1982). [DOI] [PubMed] [Google Scholar]
- 24.Rotstein C, Higby D, Killion K & Powell E Relationship of surveillance cultures to bacteremia and fungemia in bone marrow transplant recipients with Hickman or Broviac catheters. J. Surg. Oncol 39, 154–158 (1988). [DOI] [PubMed] [Google Scholar]
- 25.MacFie J et al. Gut origin of sepsis: a prospective study investigating associations between bacterial translocation, gastric microflora, and septic morbidity. Gut 45, 223–228 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Costa SF et al. Colonization and molecular epidemiology of coagulase-negative Staphylococcal bacteremia in cancer patients: a pilot study. Am. J. Infect. Control 34, 36–40 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Brown CT, Olm MR, Thomas BC & Banfield JF Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol 34, 1256–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Methods references
- 28.Petersen KR, Streett DA, Gerritsen AT, Hunter SS & Settles ML Super deduper, fast PCR duplicate detection in fastq files. in Proceedings of the 6th ACM Conference on Bioinformatics, Computational Biology and Health Informatics - BCB ‘15 (2015). doi:10.1145/2808719.2811568 [Google Scholar]
- 29.Martin M Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10 (2011). [Google Scholar]
- 30.Krueger, F. Trim Galore! Available at: http://www.bioinformatics.babraham.ac.uk/projects/trim_galore.
- 31.Bankevich A et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.GAEMR. Available at: https://www.broadinstitute.org/software/gaemr/. (Accessed: 19th April 2018)
- 33.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P & Tyson GW CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25, 1043–1055 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Köster J & Rahmann S Snakemake - A scalable bioinformatics workflow engine. Bioinformatics. 28, 2520–2522 (2012). [DOI] [PubMed] [Google Scholar]
- 35.Roach DJ et al. A Year of Infection in the Intensive Care Unit: Prospective Whole Genome Sequencing of Bacterial Clinical Isolates Reveals Cryptic Transmissions and Novel Microbiota. PLoS Genet 11, e1005413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li H & Durbin R Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25, 1754–1760 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li H et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnett DW, Garrison EK, Quinlan AR, Strömberg MP & Marth GT BamTools: a C++ API and toolkit for analyzing and managing BAM files. Bioinformatics 27, 1691–1692 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Costea PI et al. metaSNV: A tool for metagenomic strain level analysis. PLoS One 12, 1–9 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Touchon M et al. Organised genome dynamics in the Escherichia coli species results in highly diverse adaptive paths. PLoS Genet 5, e1000344 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Edgar RC MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price MN, Dehal PS & Arkin AP FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paradis E, Claude J & Strimmer K APE: Analyses of Phylogenetics and Evolution in R language. Bioinformatics 20, 289–290 (2004). [DOI] [PubMed] [Google Scholar]
- 44.Schliep KP phangorn: phylogenetic analysis in R. Bioinformatics 27, 592–593 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu G, Smith DK, Zhu H, Guan Y & Lam TT-Y ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data. Methods Ecol. Evol 8, 28–36 (2016). [Google Scholar]
- 46.Nurk S, Meleshko D, Korobeynikov A & Pevzner PA metaSPAdes: a new versatile metagenomic assembler. Genome Res 27, 824–834 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.PubMLST. Available at: https://pubmlst.org/. (Accessed: 20th April 2018)
- 48.Altschul SF, Gish W, Miller W, Myers EW & Lipman DJ Basic local alignment search tool. J. Mol. Biol 215, 403–410 (1990). [DOI] [PubMed] [Google Scholar]
- 49.Inouye M et al. SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 6, 90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hyatt D et al. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gibson MK, Forsberg KJ & Dantas G Improved annotation of antibiotic resistance determinants reveals microbial resistomes cluster by ecology. ISME J 9, 207–216 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.HMMER. Available at: http://hmmer.org. (Accessed: 22nd June 2018)
- 53.Brown CT, Olm MR, Thomas BC & Banfield JF Measurement of bacterial replication rates in microbial communities. Nat. Biotechnol 34, 1256–1263 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wickham H ggplot2: Elegant Graphics for Data Analysis. (Springer-Verlag; New York, 2009). [Google Scholar]
- 55.Wickham H Reshaping Data with the reshape Package. J. Stat. Softw 21, 1–20 (2007). [Google Scholar]
- 56.Hadley Wickham, Romain Francois, Lionel Henry, Kirill Müller. dplyr: A Grammar of Data Manipulation. (2017). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.