

Abstract
Although membrane environment is known to boost drug metabolism by mammalian cytochrome P450s, the factors that stabilize the structural folding and enhance protein function are unclear. In this study, we use peptide-based lipid nanodiscs to “trap” the lipid boundaries of microsomal cytochrome P450 2B4. We report the first evidence that CYP2B4 is able to induce the formation of “raft” domains in a biomimetic of the endoplasmic reticulum. NMR experiments were used to identify and quantitatively determine the lipids present in nanodiscs. A combination of biophysical experiments and molecular dynamics simulations revealed a sphingomyelin binding region in CYP2B4. The protein-induced lipid raft formation increased the thermal stability of P450 and dramatically altered ligand binding kinetics of the hydrophilic ligand BHT. These results unveil membrane/protein dynamics that contribute to the delicate mechanism of redox catalysis in lipid membrane.
Keywords: Cytochrome P450, Nanodiscs, Membrane rafts, Lipid ordering, Membrane protein
Graphical Abstract
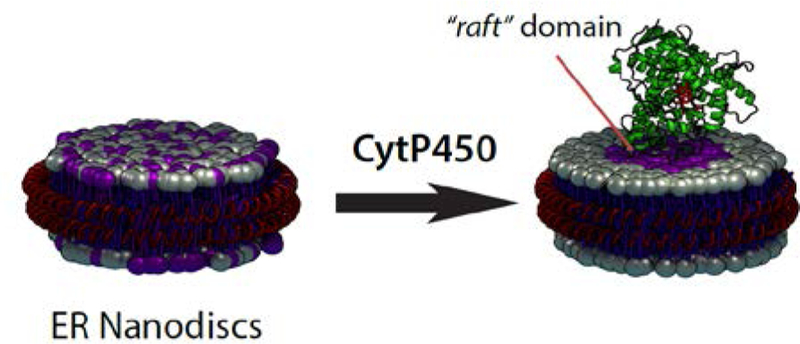
COMMUNICATION
Membrane environment boosts the drug metabolism by mammalian cytochrome P450s, but the factors that stabilize the structural folding and enhance their function are still elusive. By a novel application of peptide-nanodiscs, we discovered that CYP2B4 is able to induce the formation of “lipid raft” domains in a biomimetic of the endoplasmic reticulum. The protein-induced lipid raft increased P450’s thermal stability and dramatically altered the ligand binding kinetics of the hydrophilic ligand BHT.
Structure, function and regulation of membrane proteins are elicited through interaction with cell membrane.[1] A systematic “mapping” of lipid-protein interactions is one of the current challenges of molecular and cell biology,[1] and could represent a determinant advancement for understanding biological processes at membrane level. Indeed, more than 60% of drug targets are located at the cell surface and other membrane subcellular interfaces.[2] Drugs and xenobiotics detox functions are also regulated by membrane-anchored cytochrome P450s[3] located in the cytosolic side of the endoplasmic reticulum (ER); they have a broad catalytic activity which includes monooxygenation and dealkylation of a variety of ligands, including >70% of commercially available drugs, as well as xenobiotics;[4] several P450 isoforms are also involved in hormone synthesis and the arachidonic acid cascade.[3] The biophysical challenges posed by the lipid membrane, as well as its molecular intricacies, have impeded a molecular characterization of P450-lipid dynamics. There is consensus about the significance of the lipid membrane in P450’s catalytic activity:[5] it provides the landscape for electron transfer to occur, via physical interaction with the electron transferase CPR.[6] Membrane anchoring is one of the speculated mechanism that prevents P450 trafficking towards other subcellular compartments.[7] Also, lipid membrane is the main access pathway for hydrophobic substrates, playing a critical role in P450 drug-metabolism and pharmacokinetics.[8–12] There is mounting evidence that protein-protein interactions are also driven by cross-talking between transmembrane domains (TMD),[13–15] which further amplifies the role of lipids on P450’s structural stability and function. Thus, there is a need for new biophysical tools that can enlighten such presently precluded dynamics, and provide critical knowledge on P450’s function.
For membrane proteins, the information regarding subcellular organization in different membrane compartments is obtained through several methods, including indirect detergent-based assay (DRM), single-molecule imaging and spectroscopy,[16] and mass spectrometry.[17] Notwithstanding the critical role of membrane in P450 catalysis, only a few groups have addressed the fundamental questions regarding P450’s organization in ER. DRM experiments reported that P450 isoforms and CPR likely to co-exist in ER microdomains.[18] Nonetheless, given the controversies associated with DRM,[19] as well as the more recent definition of lipid rafts as small (<10 nm) and transient SM-rich domains,[20] new strategies are needed, particularly to reveal the underestimated role played by proteins in driving lipid segregation. Since P450 strongly interacts with lipid membrane via its TMD and FG-loop,[15, 21] we hypothesized that any protein-driven change in lipid composition could be trapped at the nanometric scale of a nanodisc.
The 4F peptide was able to form nanodiscs (Fig. S1) that retained the ER membrane composition (Table S1) after SEC purification (Fig. S1). Peptide-based nanodiscs have several advantages over membrane scaffolding protein nanodiscs (MSP) for studying membrane proteins. First, they allow detergent-free protein incorporation, which avoids post-reconstitution purification steps,[22–23] as shown by both size-exclusion chromatography (SEC, Fig. S3a) and dynamic light scattering (DLS, Figs. S1b and S3b-d). Second, the surrounding nanodisc’s belt consists of 4F peptide units (~20 per nanodisc);[22–23] compared to MSP nanodiscs, the size of peptide nanodisc is determined by the lipid:peptide ratio (Fig. S1b). At 1:0.75 w/w lipid:peptide ratio, the diameter of the nanodisc was 8 ± 1 nm, which is suitable for P450 monomerization.[23–24] When incorporated in 4F-nanodiscs, CYP2B4 showed a predominantly low-spin heme absorption spectrum (Fig. S1d), which is indicative of negligible interactions between the heme prosthetic group and the lipid components. Contrary to MSP nanodiscs, peptide-based nanodiscs allow size-rearrangement and lipid exchange between nanodiscs.[22] Our approach based on the hypothesis that the presence of a membrane protein in a fraction of nanodiscs would perturb the exchange equilibrium in a way that reflects the ability of the protein itself to recruit or exclude specific membrane components in its proximate boundaries.
CYP2B4 was reconstituted in excess 4F-nanodiscs (Fig.1a), the fraction containing the protein was subsequently purified by SEC (Fig. 1b), and lipids and cholesterol were quantified (Fig. 2a,b). The lipid composition uniquely reflects the perturbation induced by CYP2B4. The first observation is that, compared to the initial ER composition (“ER” in the rest of the manuscript), the lipid profiles were altered (Fig. 2b). Nanodiscs after lipid exchange, (“ERex”) were enriched of SM (~2.5-folds increase, p < 0.05) and cholesterol (~1-fold increase, p<0.001), and with a concomitant decrease of zwitterionic PE (~1-fold, p<0.01). Cytb5 – whose cytosolic domain poorly interact with the membrane[5] – was used as a control, and as expected it did not perturb the nanodiscs’s lipid composition (Fig. 2b). Lateral accumulation of saturated SM and cholesterol is indicative of raft-like liquid-ordered microdomain (lo) formation.[25] Indeed, protein-induced lipid domains in nanodiscs were corroborated by fluorescence correlation spectroscopy. For this, we evaluated the partition of the fluorescent lipid probe DiI C12 into nanodiscs as altered by the presence/absence of P450 (Figs. 2c and S4). When SM-rich domains were formed, a significant change in the G(0) autocorrelation function amplitude was observed (Fig. 2c and Table S2), indicating a different distribution of the fluorescent dye in presence of CYP2B4. Several control experiments performed on empty nanodiscs, as well as P450-reconstituted 4F-POPC nanodiscs (Fig. S4c) indicated that DiI C12 was not interacting with protein backbone, or entering the P450’s active site (Fig. S4d), making the heme-induced fluorescence quenching unlikely.
Figure 1. Peptide-based nanodiscs as a tool to unveil lipid-boundary regions of microsomal CYP2B4.
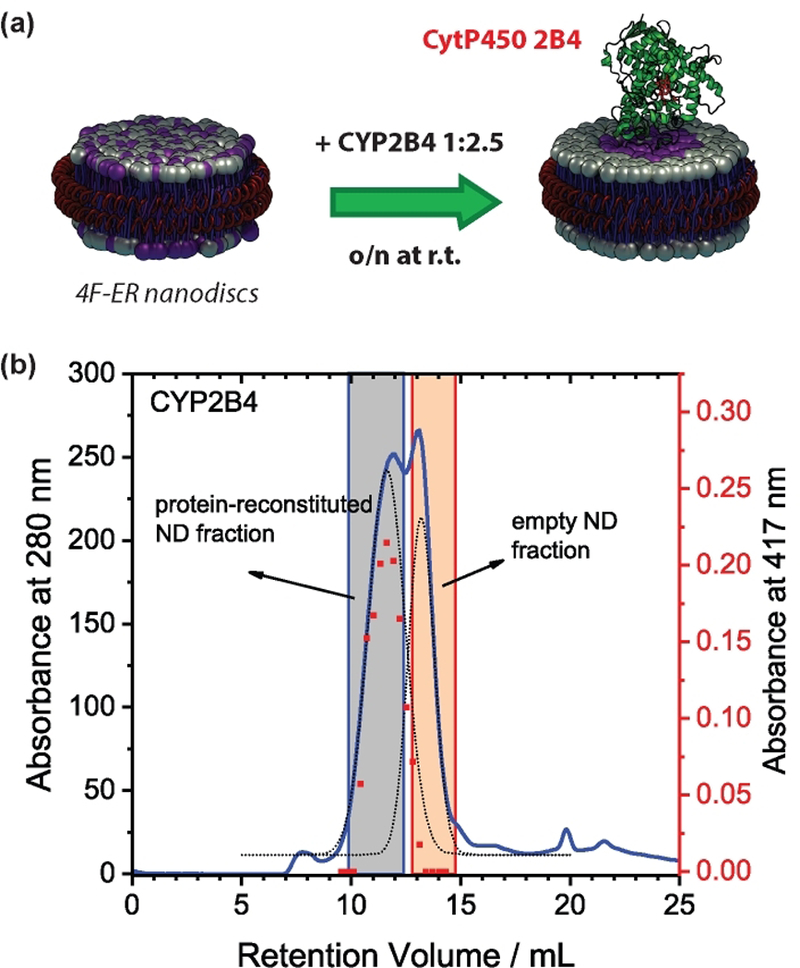
(a) Schematic of the lipid-exchange experiment. CYP2B4 was reconstituted with a 2.5-fold excess of empty nanodiscs and incubated overnight to allow lipid exchange. (b) SEC profiles showing two partially overlapping peaks, representing the reconstituted and empty fractions, respectively; the protein-containing fraction was also characterized by the absorbance at the Soret maximum (417 nm).
Figure 2. CYP2B4-induced liquid-ordered domain modulates drug affinity.
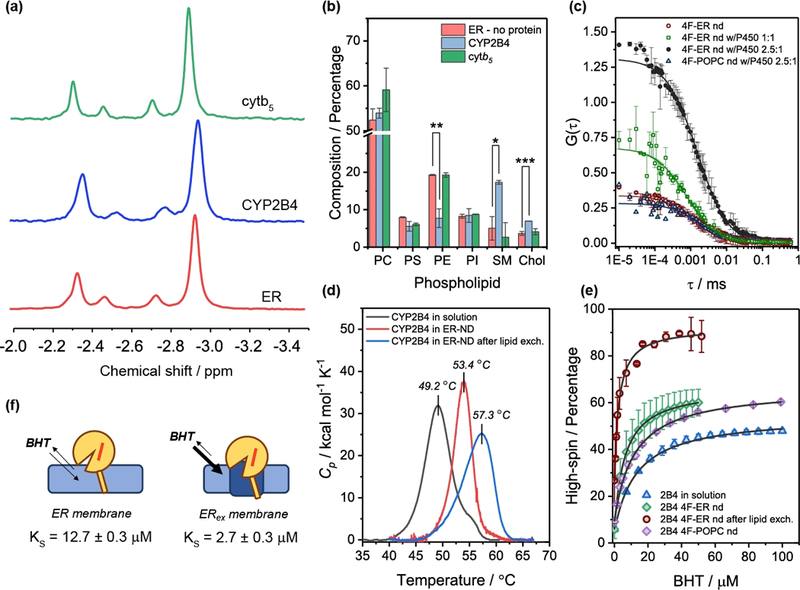
(a) 31P NMR on detergent-treated nanodiscs was used to assess the composition of the protein-containing fraction after overnight lipid exchange. (b) Lipids and cholesterol contents in empty, CYP2B4, and cytb5 4F-ER nanodiscs after lipid exchange, as measured by 31P NMR and GC-MS. Data are expressed as average ± standard deviation (n=3). *p<0.05, **p<0.01, ***p<0.001. (c) Fluorescence autocorrelation functions for nanodiscs. Scatter plot represent average ± standard deviation of two independent experiments; solid lines represent numerical fitting using a 3D correlation diffusion model. The lipid-probe DiI C12 differently partitioned in P450-containing nanodiscs, causing changes in the autocorrelation function. (d) DSC curves. (e) Protein-induced changes in lipid boundaries alter the affinity for BHT. (f) Schematic of lipid-induced modulation of ligand affinity for BHT.
Next, we investigated if an ordered region surrounding the protein was important to stability, structure and ligand-affinity of P450. A first striking evidence of a change in the membrane organization was the reduction in the nanodiscs size (~2.2 nm, p<0.05), likely indicating that the enrichment of SM and cholesterol in CYP2B4 caused the ERex bilayer to be more tightly “packed” (Fig. S5a). CD results indicated, compared to the protein in solution, no appreciable changes in the folding (Fig. S6a,b). Protein stability to thermal denaturation is an important parameter for assessing the effect of membrane on P450.[26] Compared to solution, CYP2B4 in 4F-ER nanodiscs resulted in an improved thermal stability with increased Tm (~+4°C) (Fig. 2d). The formation of the lo domain (ERex) further increased Tm (~+4°C), with ∆H increasing from 118 to 163 kcal mol−1 (p<0.05). Although a broader profile was observed for ERex, and could be interpreted in terms of opening of P450’s binding pocket, additional structural data are needed to confirm this interpretation; further studies are under progress which will be published in the future.
In P450, conformational shifts in the protein backbone are disclosed in the active site environment, by subtle but observable changes in the heme absorbance spectrum. In P450, the predominance of low-spin state is due to water as the sixth ligand,[27–28] and limiting the access of water to the active site can lead to high-spin equilibrium shift (Figs. S1d and 2e). These structural and dynamical fluctuations at the protein/lipid interface can potentially control the opening of access channels, playing a role in the recruitment of lipophilic substrates.[29–30] P450’s affinity for drugs is a critical step in determining their pharmacokinetics, since the intrinsic clearance of a pharmacophore is determined by its metabolic rate and its affinity for the metabolizer.[12, 31] We measured the spectral binding equilibrium constants (KS) by observing the ligand-induced spin shift in CYP2B4, when in solution or nanodiscs. We compared the effect of ligands, BHT and 4-CPI, which have different partitioning between membrane and bulk solution (their logP values are 5.54 and 1.99, respectively). UV-Vis titration with BHT led to several observations. First, in ERex nanodiscs, the initial high-spin population ([BHT]=0) was higher than in solution (in ER +15%, p < 0.05). Second, the equilibrium affinity KS increased by 4-fold from solution (12 μM) to ERex nanodiscs (3 μM), whereas in ER it was midway (8 μM). Third, the maximum high-spin fraction extrapolated by the sigmoidal binding curve showed a significant increase from solution (40%) to ER (60%); however, in ERex the lo domain surrounding the protein increased the population to ~100% high-spin. Similar experiments were performed with the more hydrophilic 4-CPI (Fig. S7), a tight type II binder, which coordinates to the heme iron via its N-1 atom in the imidazole ring. When in solution, CYP2B4 showed lower affinity for 4-CPI (KS = 0.2 μM, p<0.05), with a sigmoidal binding curve indicative of multiple binding events, as reported.[32] The reconstitution of CYP2B4 into nanodiscs attenuated the sigmoidal shape of the binding curve and increased the affinity by 5-fold. No significant differences were observed in the affinities among the lipid nature, with the KS values in the 0.04–0.07 μM range (Fig. S7). Hydrophobic ligands, such as BHT, are thought to partition into the membrane and reach the prosthetic group towards the 2a channel, situated between the FG loop which dips into the lipid membrane. On the other hand, hydrophilic ligands – such as 4-CPI – poorly interact with the membrane layer and are thought to enter the 2c channel located between the B’ helix/BC loop on the protein cytosolic domain.
Structural characterization of membrane proteins in a closely native lipid environment is not facile.[33] Alternatively, MD simulation on P450s embedded in homogeneous membrane mimetic,[9, 21, 34] has supplied insights into protein/lipid interfaces, despite not fully representing the physiological lipids. We performed both all-atom and coarse-grained (CG) simulations on CYP2B4 embedded in POPC, ER and ERex membranes. The initial CYP2B4’s TMD was oriented perpendicular to the bilayer surface for dynamics interpretation (Fig. 3a).[9] Under a 100 ns all-atom simulation, CYP2B4 showed no substantial alteration of protein folding around the catalytic site as revealed from its backbone RSMD. The CYP2B4 soluble domain depicted a constrained conformation with an average RMSD of 2.3 (ER), 2.0 (ERex) and 2.9 Å (POPC) in comparison to its full-length conformation. In contrast, the RMSD of Cα atoms of the full-length CYP2B4 were higher than 2.5 Å and the average RMSD were 3.1 (ER), 4.2 (ERex) and 3.6 Å (POPC). This suggested that TMD and lipid composition greatly modulate the overall CYP2B4 dynamics, and the lipid composition modulates the dynamics of both TMD and soluble domain.
Figure 3. Atomistic insights into the full-length CYP2B4 interaction with ER membrane.
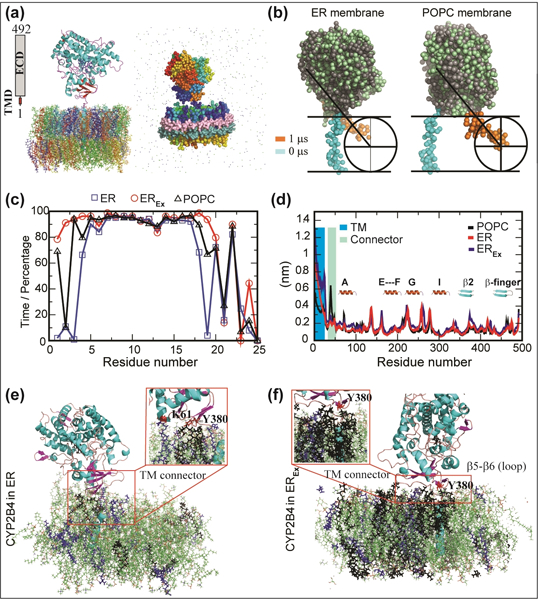
(a) All-atom (left) and coarse-grained (right) models of CYP2B4. Protein and lipids are shown as cartoon and stick, respectively, in the all-atom model, whereas as spheres and dotted spheres in the CG-MD models. (b) CG-MD snapshots showing the TMD orientation in POPC and ER nanodiscs at 0 (cyan) and 10 (orange) μs. (c) Percentage of helicity rise per residue in CYP2B4-TMD with simulation time. (d) Root mean square fluctuation of CYP2B4. TMD and linker connecting TMD and soluble domain are highlighted in blue and cyan, respectively. (e) MD snapshot illustrating the interaction between CYP2B4 and ER, and (f) SM and cholesterol-rich EREx. SM and POPS are shown in black and purple, respectively.
To gain further insight, we performed CG-MD at μs time scale. As anticipated from the all-atom MD, a comparatively high TMD dynamics was observed in POPC (Fig. 3b). The perpendicularly oriented TMD to bilayer surface ended with a tilt of ≈45º during 10 μs MD simulations for both ER and POPC membrane-nanodiscs. However, the translational motion of TMD (from center) was much larger in POPC and was restricted by the 4F-peptide belt. In contrast, the translational motion was well-restrained in ER membrane-nanodiscs during multi-microsecond MD calculation and was not found in close proximity to the belt (Fig.3b). Transient interaction between CYP2B4’s soluble domain and lipid membrane was consistently observed in all simulations. Simulations showed that in SM-enriched ER membrane (ERex), the M1-F20 is an α-helix along the entire course of simulation, whereas in both POPC and ER the M1-L5 residues were mostly disordered (Fig.3c). Monitoring the RMS fluctuation of backbone atoms of CYP2B4 in POPC, ER and ERex membranes (Fig. 3d), it was found that RMS>2.0 Å were observed for TMD, whereas the mobility of the proline-rich (P36-P49) loop was significant only in POPC. Loops connecting helix motifs in the soluble domain showed similar fluctuations over time. In the ER membrane, the FG-loop (bearing the F’ and G’ helices), as well as β2, β5 and β6 sheets (also known as β-finger) close to the C-terminus presented higher rigidity, due to an increased interaction with the membrane hydrophobic core (Fig. 3d).
All-atom MD simulations unveiled interactions between specific residues and lipids (Fig. 3). In ER, the soluble domain was found to interact with lipid head groups, in particular the FG-loops and the β-finger loop (Fig. 3e). Results also showed SM clustering around the TMD-connecting loop and the β-finger, corresponding to the region spanning the T375 to I382 chain. Further structure analysis showed H-bonding interaction between SM and Y380 (Fig.3e, insert). Simulations on ERex showed a dramatic increase of SM clustering, with additional specific interactions with the Pro-rich TM connecting loop (G28-L40). The TM domain’s α-helical character was calculated over the 100-ns simulation time (Fig. S8). Compared to ER, the enrichment of SM and cholesterol in ERex caused the helix to have an ideal angle (100°) along the entire simulation time (Fig.S8a). Finally, we calculated the number of H-bonds over simulations in both ER systems, as a tool to evaluate the interaction with polar groups of the lipids (Fig. S8b). As expected, the number of H-bonds was found to be substantially higher in SM-enriched ERex, indicating more interaction with the membrane surface. Sequence alignment shows that both TMD connecting region and β-finger are largely conserved among microsomal P450s (Fig. S9). Particularly, the β-finger has several conserved hydrophobic residues, including the 382Ile-Pro-Lys384 sequence, as well as the proline-rich connector domain. This leads us to hypothesize that other microsomal P450s can possess similar ability of specific interaction with lipid components.
In conclusion, this study reveals that P450s is highly dynamic, and able to interact and modify its surrounding lipid environment to enhance both function and stability. Protein-induced formation of SM-rich domain could facilitate P450 monomerization and stability. Recent experimental and computational evidences on the lateral organization of microsomal cytochrome P450 pointed to a strong interaction of both TMD and soluble domain with the lipid bilayer.[13, 15, 21, 29, 33] The results presented here also provide significant molecular insights into the factors governing P450 ligand affinity for hydrophobic molecules. Further, the innovative combination of peptide-based nanodiscs and 31P NMR experiments to quantitatively determine the different type of lipids constituting nanodiscs will be useful to study a variety of membrane proteins (like GPCR and pore-forming proteins[35]) and membrane-assisted amyloid aggregation.
Supplementary Material
Acknowledgements
This study was supported by NIH (GM084018 to A.R.). We thank Sarah Cox for help with TEM, Matthew Schweiss for assistance with GC-MS, the Biophysics SMART Center and Damon Hoff for assistance with Alba Confocal Microscope.
Footnotes
Supporting information is given via a link at the end.
References
- [1].Saliba AE, Vonkova I, Gavin AC, Nat Rev Mol Cell Biol 2015, 16, 753–761. [DOI] [PubMed] [Google Scholar]
- [2].Wymann MP, Schneiter R, Nat Rev Mol Cell Biol 2008, 9, 162–176. [DOI] [PubMed] [Google Scholar]
- [3].De Montellano PRO, Cytochrome P450: structure, mechanism, and biochemistry, Springer, New York, 2005. [Google Scholar]
- [4].Denisov IG, Makris TM, Sligar SG, Schlichting I, Chem Rev 2005, 105, 2253–2277. [DOI] [PubMed] [Google Scholar]
- [5].Barnaba C, Gentry K, Sumangala N, Ramamoorthy A, F1000Research 2017, 6, 662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Barnaba C, Martinez MJ, Taylor E, Barden AO, Brozik JA, J Am Chem Soc 2017, 139, 5420–5430. [DOI] [PubMed] [Google Scholar]
- [7].Szczesna-Skorupa E, Kemper B, Expert Opin Drug Metab Toxicol 2008, 4, 123–136. [DOI] [PubMed] [Google Scholar]
- [8].Wienkers LC, Heath TG, Nat Rev Drug Discov 2005, 4, 825–833. [DOI] [PubMed] [Google Scholar]
- [9].Jerabek P, Florian J, Martinek V, Phys Chem Chem Phys 2016, 18, 30344–30356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamada A, Shimizu N, Hikima T, Takata M, Kobayashi T, Takahashi H, Biochemistry 2016, 55, 3888–3898. [DOI] [PubMed] [Google Scholar]
- [11].Denisov IG, Grinkova YV, Baylon JL, Tajkhorshid E, Sligar SG, Biochemistry 2015, 54, 2227–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lewis DF, Jacobs MN, Dickins M, Drug Discov Today 2004, 9, 530–537. [DOI] [PubMed] [Google Scholar]
- [13].Yamamoto K, Caporini MA, Im SC, Waskell L, Ramamoorthy A, Sci Rep 2017, 7, 4116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Huang R, Yamamoto K, Zhang M, Popovych N, Hung I, Im SC, Gan Z, Waskell L, Ramamoorthy A, Biophys J 2014, 106, 2126–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yamamoto K, Durr UH, Xu J, Im SC, Waskell L, Ramamoorthy A, Sci Rep 2013, 3, 2538; K. Yamamoto, M. Gildenberg, S. Ahuja, S. C. Im, P. Pearcy, L. Waskell, A. Ramamoorthy, Sci Rep 2013, 3, 2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Barnaba C, Taylor E, Brozik JA, J Am Chem Soc 2017, 139, 17923–17934. [DOI] [PubMed] [Google Scholar]
- [17].Sezgin E, Levental I, Mayor S, Eggeling C, Nat Rev Mol Cell Biol 2017, 18, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Brignac-Huber L, Reed JR, Backes WL, Mol Pharmacol 2011, 79, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Lichtenberg D, Goni FM, Heerklotz H, Trends Biochem Sci 2005, 30, 430–436. [DOI] [PubMed] [Google Scholar]
- [20].Sezgin E, Levental I, Mayor S, Eggeling C, Nature Reviews Molecular Cell Biology 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Denisov IG, Shih AY, Sligar SG, J Inorg Biochem 2012, 108, 150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Midtgaard SR, Pedersen MC, Kirkensgaard JJ, Sorensen KK, Mortensen K, Jensen KJ, Arleth L, Soft Matter 2014, 10, 738–752. [DOI] [PubMed] [Google Scholar]
- [23].Ravula T, Barnaba C, Mahajan M, Anantharamaiah GM, Im SC, Waskell L, Ramamoorthy A, Chem Commun (Camb) 2017, 53, 12798–12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang M, Huang R, Ackermann R, Im SC, Waskell L, Schwendeman A, Ramamoorthy A, Angew Chem Int Ed Engl 2016, 55, 4497–4499. [DOI] [PubMed] [Google Scholar]
- [25].Simons K, Toomre D, Nat Rev Mol Cell Biol 2000, 1, 31–39. [DOI] [PubMed] [Google Scholar]
- [26].McClary WD, Sumida JP, Scian M, Paco L, Atkins WM, Biochemistry 2016, 55, 6258–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Hollingsworth SA, Batabyal D, Nguyen BD, Poulos TL, Proc Natl Acad Sci U S A 2016, 113, 8723–8728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Tripathi S, Li H, Poulos TL, Science 2013, 340, 1227–1230. [DOI] [PubMed] [Google Scholar]
- [29].Baylon JL, Lenov IL, Sligar SG, Tajkhorshid E, J Am Chem Soc 2013, 135, 8542–8551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Berka K, Paloncyova M, Anzenbacher P, Otyepka M, J Phys Chem B 2013, 117, 11556–11564. [DOI] [PubMed] [Google Scholar]
- [31].Nath A, Grinkova YV, Sligar SG, Atkins WM, J Biol Chem 2007, 282, 28309–28320. [DOI] [PubMed] [Google Scholar]
- [32].Zhao Y, Sun L, Muralidhara BK, Kumar S, White MA, Stout CD, Halpert JR, Biochemistry 2007, 46, 11559–11567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Monk BC, Tomasiak TM, Keniya MV, Huschmann FU, Tyndall JD, O’Connell JD 3rd, Cannon RD, McDonald JG, Rodriguez A, Finer-Moore JS, Stroud RM, Proc Natl Acad Sci U S A 2014, 111, 3865–3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Navratilova V, Paloncyova M, Berka K, Otyepka M, J Phys Chem B 2016, 120, 11205–11213. [DOI] [PubMed] [Google Scholar]
- [35].Weber DK, Yao S, Rojko N, Anderluh G, Lybrand TP, Downton MT, Wagner J, Separovic F, Biophys. J. 2015,108, 1987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.