

Abstract
The transition of differentiated Schwann cells to support of nerve repair after injury is accompanied by remodeling of the Schwann cell epigenome. The EED-containing polycomb repressive complex 2 (PRC2) catalyzes histone H3K27 methylation and represses key nerve repair genes such as Shh, Gdnf and Bdnf, and their activation is accompanied by loss of H3K27 methylation. Analysis of nerve injury in mice with a Schwann cell-specific loss of EED showed the reversal of polycomb repression is required and a rate limiting step in the increased transcription of Neuregulin 1 (type I), which is required for efficient remyelination. However, mouse nerves with EED-deficient Schwann cells display slow axonal regeneration with significantly low expression of axon guidance genes, including Sema4f and Cntf. Finally, EED loss causes impaired Schwann cell proliferation after injury with significant induction of the Cdkn2a cell cycle inhibitor gene. Interestingly, PRC2 subunits and CDKN2A are commonly co-mutated in the transition from benign neurofibromas to malignant peripheral nerve sheath tumors (MPNST’s). RNA-seq analysis of EED-deficient mice identified PRC2-regulated molecular pathways that may contribute to the transition to malignancy in neurofibromatosis.
Keywords: Schwann myelin chromatin nerve injury axon neurofibromatosis
Introduction
Schwann cells in the peripheral nervous system not only create myelin, but also become reprogrammed after nerve injury to support nerve regeneration. Soon after injury, repair cells derived from both myelinating and non-myelinating (Remak) Schwann cells create a structural and trophic environment that stimulates axon regeneration (Brosius Lutz and Barres 2014; Gomez-Sanchez et al. 2017; Jessen and Mirsky 2016). Repair Schwann cells activate autophagy and phagocytosis mechanisms to remove myelin debris, which inhibit axon regrowth and branching (Brosius Lutz et al. 2017; Gomez-Sanchez et al. 2015; Mukhopadhyay et al. 1994; Shen et al. 1998), and promote recruitment of macrophages that further facilitate myelin removal and regeneration (Cattin et al. 2015; Fischer et al. 2008; Niemi et al. 2013). Elongated Schwann cells distal to the injury site form Bands of Bungner, which serve as tracks for axonal regeneration (Arthur-Farraj et al. 2012; Gomez-Sanchez et al. 2017).
Such injury-responsive changes are mediated by a distinct transcriptional program that involves the transcription factor JUN (Arthur-Farraj et al. 2012). Many injury-induced genes encode intercellular signaling molecules such as sonic hedgehog (Shh), which is silenced throughout Schwann cell development prior to injury (Lin et al. 2015). Schwann cells in injured nerve induce genes encoding factors that promote axon survival and regeneration: e.g. brain- and glial-derived neurotrophic factors, Bdnf and Gdnf, nerve growth factor (Ngf), and leukemia inhibitory factor (Lif) (Boyd and Gordon 2003; Cafferty et al. 2001; Fontana et al. 2012; Li et al. 2015; Widenfalk et al. 2009). While Schwann cells are normally dependent on axonal neuregulin type III signaling in normal development (Michailov et al. 2004; Taveggia et al. 2005), neuregulin 1 (NRG1) type I is also secreted by Schwann cells shortly after injury, and is required for efficient remyelination (Stassart et al. 2013). In many cases, levels of these genes are low or absent in mature Schwann cells prior to injury.
Despite the striking adaptability of Schwann cells to damage, the clinical outcomes of human patients generally exhibit only partial recovery in many cases (Höke 2006; Lundborg 2000). One of the main reasons is that axons must regenerate over a relatively long distance, and Schwann cells more distal to injury sites gradually lose their ability to foster nerve regeneration (Jonsson et al. 2013; Ronchi et al. 2017; Sulaiman and Gordon 2009; Sulaiman and Gordon 2013), which could be in part due to reduced expression of neurotrophic factors like GDNF and BDNF (Eggers et al. 2010; Fontana et al. 2012; Höke et al. 2002; Li et al. 1997; Michalski et al. 2008; Sulaiman and Gordon 2009). Therefore, identifying the molecular mechanisms that enable rapid axon regeneration is important for improving therapeutic strategies for peripheral nerve damage.
While many studies of gene expression changes after nerve injury have been performed (Arthur-Farraj et al. 2012; Arthur-Farraj et al. 2017; Barrette et al. 2010; Clements et al. 2017; Nagarajan et al. 2002), somewhat less is known about the mechanisms governing the epigenomic transition of mature to repair Schwann cells. It has become clear that epigenomic changes are important for such reprogramming events, employing mechanisms of gene activation and derepression (Brügger et al. 2017; He et al. 2018; Hung et al. 2015; Jacob 2017; Ma and Svaren 2018). Our previous studies identified the association of trimethylation at Lys27 of histone H3 tail (H3K27me3) at promoters of many genes that become activated after peripheral nerve injury, and we found that the demethylation is required for full activation of some repair genes (Ma et al. 2016). H3K27 methylation is catalyzed by Polycomb Repressive Complex 2 (PRC2), comprising the lysine methyltransferase EZH1/2 and the nonredundant core subunits, suppressor of zeste 12 (SUZ12) and embryonic ectoderm development (EED). EED is not required during Schwann cell development and myelination (Ma et al. 2015), although there is Remak bundle disruption and hypermyelination in older EED deficient mice. Gene expression analyses, however, revealed a premature derepression of some injury-response genes in uninjured Eed cKO nerves. The findings suggested that Schwann cell EED and PRC2 normally repress the injury responsive-transcriptional program and therefore PRC2 could affect nerve regeneration. In addition to its role in injury, loss of PRC2 has been associated with disease progression of the Schwann cell-derived tumors in neurofibromatosis, caused by loss of the NF1 tumor suppressor. Inactivation of PRC2 occurs in the malignant form, known as malignant peripheral nerve sheath tumors (MPNST’s), as SUZ12 or EED genes encoding PRC2 subunits are mutated or deleted in a high proportion of MPNST’s (Cleven et al. 2016; De Raedt et al. 2014; Lee et al. 2014; Pekmezci et al. 2017; Zhang et al. 2014).
Given that PRC2 has been identified as a regulator of Schwann cell repair genes after injury (Ma et al. 2015; Ma et al. 2016), we used the Eed cKO model to determine how lack of PRC2 activity would affect nerve injury responses and the gene expression reprogramming that occurs in Schwann cells after injury.
Materials and Methods
Primer sequences and Antibodies.
The primers and antibodies are listed in Tables 1 and 2, respectively.
Table 1.
Primer sequences used for qRT-PCR and ChIP-qPCR experiments
| qRT-PCR primer sequence (rat) | ||
|---|---|---|
| Nrg1 type I | Forward | CCGCGTAGAGCGCTCATC |
| Reverse | CTTGCCTCTGCCTTCTTTGC | |
| qRT-PCR primer sequence (mouse) | ||
| Nrg1 type I | Forward | GGGAAGGGCAAGAAGAAGG |
| Reverse | TTTCACACCGAAGCACGAGC | |
| Nrg1 type III | Forward | ACTCAGCCACAAACAACAGAAAC |
| Reverse | GAAGCACTCGCCTCCATT | |
| Egr2 | Forward | TGCTAGCCCTTTCCGTTGA |
| Reverse | TCTTTTCCGCTGTCCTCGAT | |
| Mpz | Forward | CCCTGGCCATTGTGGTTTAC |
| Reverse | CCATTCACTGGACCAGAAGGAG | |
| Pmp22 | Forward | CACGGTCGGAGCATCAGG |
| Reverse | TCCTTGGAGGCACAGAACACT | |
| Mbp | Forward | GAGGAAGAGACAGCCGCTCTG |
| Reverse | CAGGATTCGGGAAGGCTGAG | |
| Sema4f | Forward | TGCTGACGGCGACCAAT |
| Reverse | TGGCTTTTTCCTGGGTGTTC | |
| Runx2 | Forward | ACCAAGTAGCCAGGTTCAAC |
| Reverse | GAGGATTTGTGAAGACTGTTATGG | |
| Olig1 | Forward | AGCGATGTAGTTGCTTGGGAT |
| Reverse | CTGGCTCTAAACAGGTGGGAT | |
| Fgf5 | Forward | AAAAGCCACCGGTGAAACC |
| Reverse | TCACTGGGCTGGGACTTCTG | |
| Shh | Forward | CAGCGACTTCCTCACCTTCCT |
| Reverse | AGCGTCTCGATCACGTAGAAGAC | |
| Gdnf | Forward | TCTCGAGCAGGTTCGAATGG |
| Reverse | AAGAACCGTCGCAAACTTTACC | |
| Bdnf | Forward | GGTATCCAAAGGCCAACTGA |
| Reverse | GCAGCCTTCCTTGGTGTAAC | |
| Jun | Forward | CGGCTACAGTAACCCTAAGATCCT |
| Reverse | GCCAGGTTCAAGGTCATGCT | |
| Epha5 | Forward | TGGTCAACAGCCAATTATTCTGA |
| Reverse | GCCCCCATCCACACATACC | |
| Cntf | Forward | TGCTGAGATTCCCATGTGATG |
| Reverse | TTGGAGATGGTGGCCTCTTT | |
| Ink4a/p16 | Forward | GAATCTCCGCGAGGAAAGC |
| Reverse | TGTCTGCAGCGGACTCCAT | |
| Arf/p19 | Forward | CACCGGAATCCTGGACCAGG |
| Reverse | CACCGTAGTTGAGCAGAAGAGCT | |
Table 2.
Antibodies used for immunohistochemistry, Western blots and ChIP
| Antibodies | Catalog number | Company |
|---|---|---|
| p(T308)-AKT | 13038 | Cell Signaling |
| p(S473)-AKT | 4060 | Cell Signaling |
| AKT | 4691 | Cell Signaling |
| pERK 1/2 | 4370 | Cell Signaling |
| ERK 1/2 | 4695 | Cell Signaling |
| H3K27me3 | AM39155 | Active Motif |
| JUN | SC-1694, H-79 | Santa Cruz |
| SOX10 | AF2864 | R & D Systems |
| Ki67 | Ab16667 | Abcam |
| NRG1 | MABN42 | Millipore |
| p19/ARF | sc-32748 | Santa Cruz |
Experimental animals and Nerve injury surgery.
All animal experiments were performed according to protocols approved by the University of Wisconsin Graduate School Animal Care and Use Committee. Eed-floxed mice (B6;129S1-Eedtm1Sho/J, RRID:IMSR_JAX:022727) were generated by backcrossing the flox allele for seven generations against the C57BL/6 genetic background and mated to mP0TOTA-Cre (B6N.FVB-Tg(Mpz-cre)26Mes/J, obtained from Jackson Laboratory, RRID: IMSR_JAX:017927). Mice were genotyped as described previously (Feltri et al. 1999; Xie et al. 2014). Samples collected from mice homozygous for floxed Eed served as control in this study. The sciatic nerves of adult Sprague-Dawley rats or mice at the age of 2 months were exposed and transected at the sciatic notch (Hung et al. 2015) or crushed 1 min using fine forceps. As a control, the contralateral limb also received a sham operation consisting of only a skin incision. The nerve tissues distal to the transection or crushed lesions, which were labeled with sterile black ink, and contralateral (sham) nerves were isolated for use in gene expression analysis, Western blotting, immunohistochemistry or ChIP experiments. For electron microscopy analysis, the sciatic nerve was analyzed 4 mm distal to the crushed lesion. Both male and female mice were used individually per sample at similar ratio between the floxed Eed and Eed cKO genotypes. Male rats were used in ChIP experiments after nerve injury surgery.
Electron microscopy and morphometric quantification.
Freshly dissected sciatic nerves were immersion fixed in a solution of 2.5% glutaraldehyde, 2.0% paraformaldehyde in 0.1 M sodium phosphate buffer, pH 7.4, overnight at 4°C. The nerves were then postfixed in 1% osmium tetroxide in the same buffer for 2 h at room temperature. Following OsO4 postfixation, the nerves were dehydrated in a graded ethanol series, and then further dehydrated in propylene oxide and embedded in Epon or Durcupan epoxy resin. Ultrathin transverse sections were contrasted with Reynolds lead citrate and 8% uranyl acetate in 50% ethanol. Images were obtained with a Philips CM120 electron microscope with an AMT BioSprint side-mounted digital camera at the UW Medical School Electron Microscope Facility. Densitometric quantification was performed using NIS-Elements 4.0. Three mice per genotype were analyzed, and statistical analyses were evaluated by one-way ANOVA in all the experiments.
Immunohistochemistry.
Freshly dissected nerves were embedded in Tissue-Tek OCT compound (Sakura Finetek) and snap frozen with liquid nitrogen. Longitudinal or transverse cryostat sections (14 μm) were air-dried for 5 min and fixed in 4% paraformaldehyde for 15 min. The sections were then blocked in PBS containing 5% donkey serum/1% BSA/3% Triton-X 100 for 1 h at room temperature. Primary antibody incubation was performed overnight at 4˚C in PBS containing 5% donkey serum/1% BSA/1% Triton-X 100 and secondary incubation was performed in PBS at room temperature for 1 h. Hoechst 33342 (1:5000 in PBS, Sigma) was applied to stain nuclei for 1 min. Three 4 min washes were performed in PBS after fixation and blocking, and in PBS containing 0.1% Tween20 after primary antibody incubation and nuclear staining. After coverslips were mounted using Fluoromount-G™ (SouthernBiotech), sections were examined on a confocal microscope (Nikon A1R-Si). Statistical analyses were evaluated by one-way ANOVA.
Western blot.
Freshly dissected nerves were snap frozen with liquid nitrogen and crushed. The nerves were then homogenized in lysis buffer (50 mM Tris-HCL at pH 6.8, 10% glycerol, 2% SDS, 10% β-mercaptoethanol, 50 mM NaF, 1 mM Na3VO4 and Protease Inhibitor Cocktail (Sigma, P8340)) using a motorized pellet pestle. Cells in culture were homogenized in 3X lysis buffer. After a 15 min incubation in ice, lysates were boiled at 95˚C for 3 min and centrifuged at 4˚C for 15 min. Subsequently, supernatants were collected and subjected to SDS-PAGE. After transfer to nitrocellulose membrane, proteins were blocked in TBST containing 5% nonfat dry milk for 1 h at room temperature. Primary and Secondary antibody incubations were performed in TBST containing 5% non-fat dried milk at 4˚C for overnight and at room temperature for 1 h, respectively. Three 5 min-washes were performed in TBST after the incubations. The membranes were scanned and quantitated with the Odyssey Infrared Imaging System (Li-Cor Biosciences). Statistical analyses were evaluated by one-way ANOVA.
Nerve explant cultures.
Adult male Sprague-Dawley rat sciatic nerves were cut into 3 mm segments and cultured in serum-free RPMI-1640 medium supplemented with penicillin/streptomycin in the presence of GSKJ4 (Tocris, Cat. No. 4594) or DMSO at 37 °C for 1 d. RNA was purified from the explanted nerves at the indicated timepoints.
Micrococcal nucleases (MNase) aided Chromatin immunoprecipitation (ChIP) in vivo.
Sciatic nerves were subjected to MNase-ChIP with an antibody targeting H3K27me3 as described previously (Ma et al. 2016) with a few changes. Instead of washing with RIPA buffer after the immunoprecipitation, ChIP samples were washed with washing buffer 1 (WB1, 50 mM Tris–HCl, pH7.5; 10 mM EDTA; 125 mM NaCl) once, WB2 (50 mM Tris–HCl, pH7.5; 10 mM EDTA; 250 mM NaCl) once, and WB3 (50 mM Tris–HCl, pH7.5; 10 mM EDTA; 500 mM NaCl) twice.
ChIP-seq.
Library preparation and sequencing was performed by the UW Biotechnology Center as described previously (Hung et al. 2015). Basecalling was performed using the standard Illumina Pipeline. Reads were mapped to the Rattus norvegicus genome rn5 using Bowtie (RRID:SCR_005476) to produce SAM files for further analysis. From the two biological replicates, we obtained 24,145,063 and 30,080,399 reads in input and 28,792,879 and 30,462,059 reads in H3K27me3 ChIP samples. Hypergeometric optimization of motif enrichment (HOMER, RRID:SCR_010881) (Heinz et al. 2010) was used to determine enriched binding regions for H3K27me3-ChIP relative to sequencing of an input chromatin sample. H3K27me3-occupied genes were defined by the presence of the histone modification around the transcriptional start site (±7 Kb) with HOMER peak-score ≥ 10 (Ma et al. 2016). The raw data files are deposited in National Center for Biotechnology Information Gene Expression Omnibus (GEO) under accession number GSE106994.
qRT-PCR.
RNA was isolated from sciatic nerves using RNeasy Lipid Tissue Mini Kit (Qiagen) according to the manufacturer’s directions. To prepare cDNA, 250 ng or 1 μg of total RNA of mouse or rat nerves, respectively, was used from each sample. qRT-PCR and data analysis were performed as described previously (Hung et al. 2012). qPCR was performed with two replicates per sample, i.e., two technical replicates. Statistical analyses were evaluated by one-way ANOVA.
RNA-seq.
>500 ng total RNA was used to generate RNA-seq libraries using the Illumina TruSeq Stranded total RNA sample preparation kit according to the manufacturer’s instructions. An average of 14 million reads per sample was obtained and mapped to the GRCm38/mm10 genome. Data were analyzed using CLC Bio Workbench to determine differentially regulated genes between uninjured and injured nerves in wild type and Eed cKO mice (p-value, < 0.05). The raw data files are deposited in National Center for Biotechnology Information Gene Expression Omnibus under accession number GSE106994.
Results
Analysis of Nerve Injury Responses in the Eed cKO
To explore the potential importance of EED in Schwann cell responses to nerve injury, we examined the repair process by electron microscopy at 14 d following sciatic nerve crush at the sciatic notches of 2 month old mice. Regenerating nerves initiate a program that includes myelin debris removal, axon regeneration and remyelination (Arthur-Farraj et al. 2012; Jessen and Mirsky 2016). Since our previous studies showed precocious activation of injury genes in the Eed cKO (Ma et al. 2016), we had anticipated that there may be accelerated regeneration. However, the ultrastructural analysis revealed that there were relatively fewer axons at the transverse sections 4 mm distal to the injury site in Eed cKO nerves compared to sections of control mice, which exhibited a substantial number of myelinated axons at 14 d after injury (Figure 1A, B). Importantly, the number of regenerated axons greater than 1 μm in diameter, which should eventually become remyelinated, were significantly reduced in Eed cKO nerves.
Figure 1. Schwann cell EED is required for timely axon regeneration.
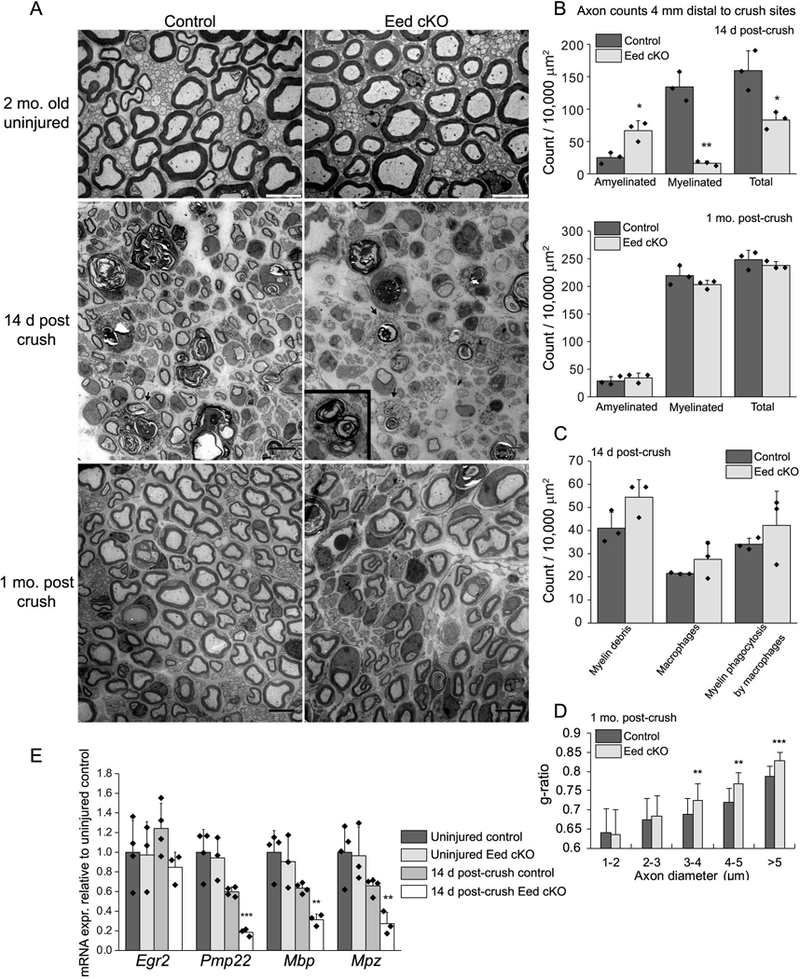
A, Electron micrographs of the sciatic nerves at the indicated time points after crush and uninjured nerves of Eed cKO mice and littermate controls. The arrows and inset show macrophages engulfing myelin debris. Scale bars of uninjured and injured nerve images are 5 μm and 8 μm, respectively. B, C, Myelinated and amyelinated axons (> 1μm in diameter), myelin debris, macrophages, and engulfed myelin by macrophages were counted in randomly selected fields that accounted for over 40% of an entire sciatic nerve cross section from each animal and normalized per surface area (10,000 μm2). Data: mean ± STDEV; **p < 0.005, *p < 0.05; n=3 per genotype (one-way ANOVA). D, For g-ratio analysis (axon diameter/diameter of myelinated fiber), the diameter of axon and outer diameter of myelinated fiber were measured on over 500 randomly selected fibers per genotype. Data: weighted mean ± pooled STDEV; ***p < 0.0005, **p < 0.005, *p < 0.05; n=3 per genotype and age (one-way ANOVA). E, qRT-PCR analysis was used to identify the expression level of myelin genes from 2 month Eed cKO and control sciatic nerves in uninjured condition or 14 day after crush. Expression levels were normalized with Gapdh. Asterisks indicate p-value between genotypes in the respective condition. Data: mean ± SD; **p < 0.005, ***p < 0.0005; n=4 for control and n=3 for Eed cKO (one-way ANOVA).
Clearance of myelin debris is a critical step because myelin, particularly myelin-associated glycoprotein (MAG), inhibits axon growth (Filbin 2003; Mukhopadhyay et al. 1994; Shen et al. 1998). Soon after injury, Schwann cells downregulate a myelin gene network and activate autophagy and phagocytic programs to carry out myelin clearance, which is further facilitated by recruited macrophages (Brosius Lutz et al. 2017; Gomez-Sanchez et al. 2015; Hirata and Kawabuchi 2002). Quantitation of myelin debris at 14 d post-injury did not reveal a significant difference in Eed cKO nerves compared to control nerves (Figure 1C). The number of infiltrating macrophages and engulfed myelin debris were also comparable. In addition, myelin breakdown was apparent 3 d post injury in Eed cKO nerves at levels comparable to control nerves (not shown), along with the expected decrease in myelin gene expression such as Mag, peripheral myelin protein 22 (Pmp22) and an essential transcription factor of myelination, Egr2/Krox20 (Figure 1-figure supplement 1).
To assess the regeneration affected by Eed deletion, we examined nerves at one month after crush injury (Figure 1A). Myelination in Eed cKO nerves appeared largely similar to control nerves at this later timepoint. The number of myelinated axons and, more importantly, the total number of axons greater than 1 μm in diameter were comparable between the two groups (Figure 1B), indicating that delayed axon regeneration did eventually recover. The analysis of g-ratios, the ratio of the axonal diameter to the diameter of outer myelin sheath, however, revealed persistently thinner myelin sheaths around axons greater than 3 μm in Eed cKO nerves (Figure 1D). In summary, the lower number of axons (> 1 μm) at 4 mm distal to crush sites at 14 d indicated that Eed cKO nerves have delayed axonal regeneration after injury.
Nerve remyelination involves re-activation of a number of myelin genes that are controlled by the EGR2/Krox20 transcription factor (Decker et al. 2006; Le et al. 2005; Topilko et al. 1994). To determine if there was a deficit in remyelination responses of Schwann cells, we examined levels of Egr2 and found that its level in the Eed cKO had recovered to a similar level as in control nerves (Figure 1E). However, the expression levels of myelin-associated genes such as Pmp22, Mbp, and Mpz of Eed cKO nerves were lower than that of control nerves, which reflects the overall reduced number of myelinated axons at 14 d after injury. Nonetheless, sciatic functional indexes and responses to the toe pinch test, which evaluate functional recovery of motor and sensory nerves, respectively, were comparable between control and Eed cKO mice, measured at 2 d and every week till the 4th week after crush injury (data not shown). In summary, these data indicate that PRC2 activity is required for timely axon regeneration during nerve repair process. Although remyelination eventually occurred in the Eed cKO, a lower level of myelin gene expression and thinner myelin sheaths (> 3 μm) at 14 days after injury is likely due to a delay in axon regeneration and subsequent myelination.
Control of Neuregulin-regulated Pathways by PRC2
We tested if EED affects activation of the ERK (extracellular signal-regulated kinase) pathway 1 d after injury, which becomes highly upregulated within few hours and regulates myelin breakdown at an early stage of nerve repair (Napoli et al. 2012), and lasts over 2 weeks after injury (Guertin et al. 2005; Sun et al. 2013; Yamazaki et al. 2009). The level of ERK activation was not statistically different in the Eed cKO compared to control (p value, ~ 0.09). Accordingly, RNA-seq analysis (described below) did not identify changes in ERK-dependent injury genes. Our previous analysis of uninjured nerves had revealed an increase in p-AKT in the cKO (Ma et al. 2015), and at 1 d after injury, there was a similar increase in AKT phosphorylation at Thr308 and Ser473, which are catalyzed by PDK1 and mTORC2, respectively (Andjelković et al. 1997; Sarbassov et al. 2005) (Figure 2). At 1 d after injury, there is little change in p-AKT in wild type mice (Norrmén et al. 2018; Ronchi et al. 2016).
Figure 2. Increased activation of AKT in Eed cKO nerves after injury.
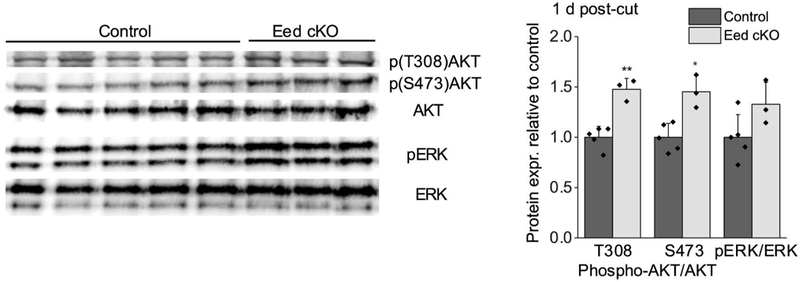
Western blot analysis of lysates from distal stumps of control and Eed cKO sciatic nerves 1 d after cut was performed using the indicated antibodies. n=5 for control and n=3 for Eed cKO nerves. Data: mean ± STDEV; **p < 0.005, *p < 0.05 (one-way ANOVA).
The AKT pathway is induced by neuregulin binding to the ERBB2/3 receptor in Schwann cells (Atanasoski et al. 2006b; Carroll et al. 1997; Fledrich et al. 2014; Guertin et al. 2005). Therefore, we decided to examine regulation of Neuregulin1 (Nrg1) type I, which is induced in Schwann cells within 24 hours after injury, and increases remyelination efficiency (Ronchi et al. 2016; Stassart et al. 2013). Interestingly, the ChIP-seq mapping of H3K27me3 in mature nerve (Ma et al. 2016) indicated that transcription start sites of Nrg1 type I and III are occupied by this repressive histone mark (Figure 3A), and ChIP-qPCR assays showed a decrease in H3K27me3 in the type I promoter after injury (Figure 3B), showing a correlation between methylation dynamics of H3K27 and Nrg1 type I gene activation. Gene expression analysis of Eed cKO nerves, which display a Schwann cell-specific loss of H3K27me3 (Ma et al. 2015), revealed derepression of Nrg1 type I in Eed cKO nerves at 2 months of age in the absence of injury (Figure 3C). In contrast, the Nrg1 type III transcript was not induced by injury in either control or Eed cKO nerves, as expected (Stassart et al. 2013).The increased level of Nrg1 type I in Eed cKO nerves was also observed by Western blot in uninjured nerves (Figure 3E), and could contribute to elevated AKT phosphorylation in the Eed cKO before and after injury (Ma et al. 2015).
Figure 3. Schwann cell Nrg1 type I is regulated by PRC2.
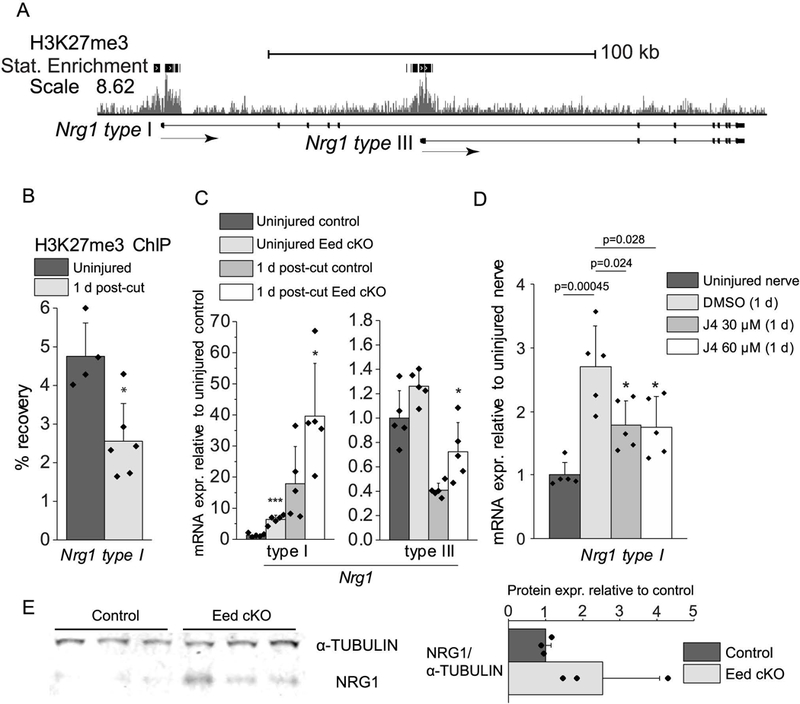
A, ChIP-seq mapping of H3K27me3 was performed in uninjured rat sciatic nerves. The transcription start site (TSS) is on the left. Statistical (Stat) Enrichment indicates regions with more sequencing reads than random chance. B, H3K27me3-ChIP assays were performed with distal stumps of rat sciatic nerves 1 d post-cut or sham surgery, and percent recovery relative to input was calculated by qPCR analysis. Data: mean ± SD; *p < 0.05; n=5 for sham and n=6 for 1 d post-cut (one-way ANOVA). C, qRT-PCR analysis was used to identify the expression level of Nrg1 type I and type III from 2 month Eed cKO and control sciatic nerves of uninjured condition or 1 day after cut. Expression levels were normalized with Gapdh. Data: mean ± SD; Asterisks indicate p-value between genotypes in the respective condition. *p < 0.05, ***p < 0.0005; n=5 per genotype and condition (one-way ANOVA). D, Rat sciatic nerve explants were cultured for 1 d in the presence of GSK-J4 at indicated concentrations or DMSO vehicle and subjected to qRT-PCR together with immediately frozen nerve segments after dissection (indicated as uninjured). Uninjured level of Nrg1 is set as 1. Expression levels were normalized to 18S rRNA. Data: mean ± SD; n=5 per condition (one-way ANOVA). E, Nerve lysates were obtained from uninjured control and Eed cKO nerves, and were blotted with an antibody to the extracellular domain of NRG1. The 30 kd band was normalized to α-tubulin, and the bar graph shows the average of the 3 replicates per condition.
In order to independently test if H3K27 trimethylation regulates induction of mRNA encoding NRG1 type I, we tested if the activity of H3K27 demethylases is required for the Nrg1 type I gene activation after injury. Peripheral nerve injury increases the protein expression of a H3K27 demethylase JMJD3/KDM6B in Schwann cells (Gomez-Sanchez et al. 2013). We employed a nerve explant model in which there is activation of injury-responsive signals and repair genes by incubation of nerve segments in culture media (Arthur-Farraj et al. 2012; Gomez-Sanchez et al. 2015; Ma et al. 2016; Shin et al. 2013). In this system, we did observe increased mRNA for Nrg1 Type I, but the GSK-J4 inhibitor of H3K27-demethylases JMJD3/KDM6B and UTX/KDM6A (Kruidenier et al. 2012), blocked the induction (Figure 3D). Our results collectively suggest that peripheral nerve injury promotes the activity of H3K27 demethylases that is required for full and timely activation of the Schwann cell Nrg1 type I transcript, which is repressed by H3K27me3 in uninjured nerves.
EED-mediated Transcriptional Regulation in Schwann Cells
To further investigate how PRC2 represses injury induced gene expression, we performed RNA-seq analysis of mouse sciatic nerves at 2 months of age. The use of RNA-seq compared to our previous microarray data (Ma et al. 2015) identified a significantly larger group of PRC2-regulated genes due to the inherent sensitivity of the RNA-seq analysis. We compared these expression data to a genome wide ChIP-seq profile of H3K27me3 in wildtype mature sciatic nerves to identify genes regulated by the EED-containing PRC2 complex. Our chromatin immunoprecipitation protocol was optimized to employ micrococcal nuclease digestion to produce mononucleosomal fragments (< 200 bp), to increase the resolution of ChIP-seq analysis. The analysis identified 4091 genes of peripheral nerves that were occupied by H3K27me3 around the transcription start site (± 7 kb), including 532 genes that are normally induced (> 2 fold) after injury, and revealed the H3K27me3 enrichment at silent or low-expressed genes (RPKM= < 1) (Figure 4A and Supporting Information Table 1). For example, H3K27me3 was highly enriched at a silenced gene of peripheral nerves, sonic hedgehog (Shh) (Arthur-Farraj et al. 2012; Lin et al. 2015), and over 73% of H3K27me3-occupied genes were expressed at very low levels (RPKM < 5). Interestingly, H3K27me3 was also abundant at some highly expressed genes, such as desert hedgehog (Dhh), a signaling molecule required for the structural and functional integrity of the peripheral nerves (Parmantier et al. 1999; Sharghi-Namini et al. 2006). This may be due to cell heterogeneity in sciatic nerves including endothelial cells and fibroblasts, although the majority of sciatic nerve (>75%) are Schwann cells (Joseph et al. 2004; Salonen et al. 1988).
Figure 4. H3K27me3-occupied silent genes induced in the Eed cKO.
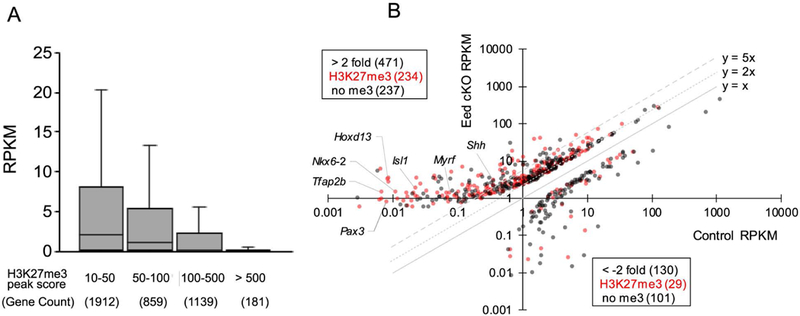
A, Box-and-Whisker plot shows RPKM distribution of genes in uninjured wildtype nerves, grouped by H3K27me3 peak score, which was determined by ChIP-seq analysis (n=2) using Hypergeometric optimization of motif enrichment (HOMER) (Heinz et al., 2010). B, Distribution of genes by RPKM values of indicated genotypes. Genes with the H3K27me3 score greater than 10 were indicated by red dots. RPKM, Reads Per Kilobase of transcript per Million mapped reads. RPKM values are averaged across three samples per genotypes (p < 0.05).
Comparing the Eed cKO to wild type littermate, there were 471 upregulated genes, including 85 injury genes, with greater than 2-fold change (p-value, < 0.05) (Figure 4B and Supporting Information Table 2). Approximately half (234) of the upregulated genes were associated with H3K27me3, including 48 injury genes. Among 196 genes with a high fold change (> 5-fold), the most highly induced genes were quite low in control nerves (RPKM < 1, 145 genes). Some of the more highly derepressed genes in Eed cKO nerves include fatty acid binding protein 7 (Fabp7/Bfabp), a marker of Schwann cell precursors and immature Schwann cells (Jacob et al. 2014; Kurtz et al. 1994), and protein tyrosine phosphatase receptor type Z 1 (Ptprz1), which regulates oligodendrocyte differentiation (Harroch et al. 2002; Kuboyama et al. 2015). 130 genes were decreased by the deletion (<2-fold), including 29 H3K27me3-associated genes. The function of H3K27me3 in gene activation has not been reported, however. In contrast, major myelin genes were largely not affected in Eed cKO nerves.
EED regulation of Injury-induced genes
Together with the ultrastructural analysis, the results showed that EED-mediated repression controls hundreds of genes, but appears dispensable for primary myelination as the phenotype of peripheral nerve is essentially normal at this age (Ma et al. 2015). In addition, Eed cKO resulted in derepression of only a subset of H3K27me3-associated injury genes. However, we reasoned that there may be other injury-induced genes in which loss of polycomb repression is not sufficient for activation but is nonetheless required. In other words, some polycomb-repressed genes may require additional injury responsive pathways, such as the transcription factor JUN-mediated regulation that is critically involved in reprogramming of Schwann cells for nerve repair (Arthur-Farraj et al. 2012; Hung et al. 2015).
To provide a more comprehensive view of injury responsive gene regulation and identify more EED-regulated genes, we performed RNA-seq analysis of control and Eed cKO nerves 1 d after nerve transection. This timepoint was chosen to show the transcriptome of injured nerves before significant infiltration by immune cells, such as macrophages, which migrate into peripheral nerve beginning at 3 d after injury (Hirata and Kawabuchi 2002). Previous studies have shown that the early repair response of Schwann cells promotes survival of injured neurons and axon regeneration through activation of neurotrophic factors and surface proteins, and elicits an innate immune response to further facilitate nerve regeneration (Boyd and Gordon 2003; Cafferty et al. 2001; Fontana et al. 2012; Hashimoto et al. 2008; Henderson et al. 1993; Martinez et al. 2015; Perrin et al. 2005; Rotshenker 2011).
In addition to the genes induced prior to injury in the Eed cKO (described above), the analysis revealed an additional 116 genes were upregulated in Eed cKO nerves only after injury, including 46 H3K27me3-associated genes, therefore identifying more EED-regulated genes in injury-induced pathways (Figure 5A). The list included 50 genes that normally become activated after injury, and 22 genes, such as Gap43, Sox2, Hmga2, Vgf, Wif1, and Esm1, were associated with H3K27me3. Many of this set normally become activated at later timepoints after injury (at 3–7 days, referred to as late injury genes), but are unchanged or even reduced at 1 d after injury in control mice (Figure 5C and Supporting Information Table 4) as previously reported for Bdnf (Ma et al. 2016). As discussed above, removal of PRC2 repression is presumably not sufficient for their activation, but their activation at this early time is accelerated by removal of H3K27me3. Therefore, loss of EED leads to augmented or premature induction of a number of genes in addition to those that are elevated in the uninjured Eed cKO nerves.
Figure 5. Polycomb activity regulates early transcriptional response after nerve injury.
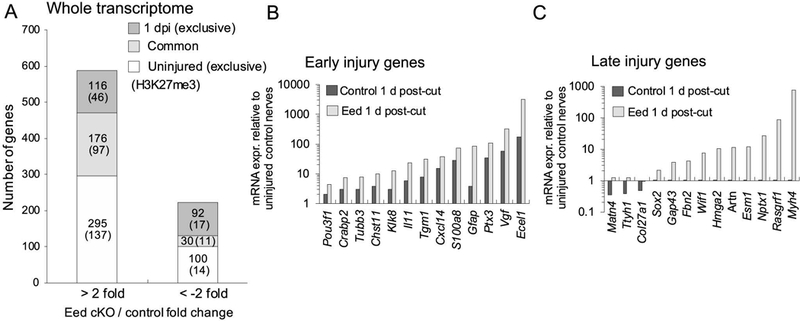
A, RNA-seq analysis identified a number of genes dysregulated by Eed cKO in uninjured or 1 d after injury conditions. Gene expression was determined by RNA-seq analysis of 3 samples per genotype and condition (p-value, < 0.05). The brackets indicate the number of genes associated with H3K27me3 around the transcription start site (±7 Kb). B, C, RNA-seq analysis was used to identify the expression level of early and late injury genes in Eed cKO nerves and control nerves at 1 d post-cut relative to uninjured control nerves. Note that the y-axis is on a log scale. Data: mean of n=3 with p-value, < 0.05 per genotype. The late injury genes were identified from microarray analysis of peripheral nerves 3, 5 or 7 d after injury (> 2 fold) (GEO accession: GSE22291, GSE38693, GSE33454) (Barrette et al., 2010; Arthur-Farraj et al., 2012; Kim et al., 2012)(Kim et al. 2012). See Supporting Information Table 3 and 4 for the complete list of genes analyzed in Figure 5.
Our analysis of wildtype transcriptome identified 845 genes that were induced greater than 2-fold (p-value, < 0.05) in distal stumps 1 d after injury, compared to contralateral uninjured nerves (Supporting Information Table 3). Consistent with previous studies, induction included a significant number of genes encoding secreted proteins, such as neurotrophic factors GDNF, FGF5, and NGF, in addition to molecules mediating signaling pathways such as TGF-β1 (Boyd and Gordon 2003; Cafferty et al. 2001; Fontana et al. 2012; Henderson et al. 1993; Meyer et al. 1992). Genes that promote macrophage recruitment and myelin debris clearance (e.g. Mcp1/Ccl2, Il1a, Il1b, Lif, Il11) were also induced at this time point (Napoli et al. 2012).
While we have previously demonstrated reduction of H3K27me3 on injury induced genes already at 1 day (Ma et al. 2016), we hypothesized that loss of EED could augment or accelerate induction of injury genes at this time point. Among genes activated at 1 d after injury, 38 genes were further upregulated in the Eed cKO (> 2 fold; p-value, < 0.05), including 18 genes associated with H3K27me3 (Figure 5B). For example, Vgf and Ecel1 became highly activated (> 50 fold, relative to uninjured control nerves) 1 d after injury. Eed cKO further elevated the induction of such genes by greater than 3-fold compared to injured control nerves. There are also genes that become repressed after injury, and the analysis showed that EED is required for injury-induced downregulation of a subset of such genes. However, this did not include myelin genes that normally are reduced after injury.
Polycomb Regulation of Axon Regeneration Genes
While we have initially focused on the effects of EED on early gene induction after injury, we also wished to analyze potential mechanisms for the delayed axon regeneration. Therefore, RNA-seq analysis of wildtype nerves was performed 14 d after crush injury, which revealed 1020 genes upregulated greater than 2 fold (p-value, < 0.05), compared to uninjured nerves. Comparison to the Eed cKO at 14 d after injury showed that a lack of EED led to a further upregulation of 203 genes (> 2 fold) and a downregulation of 100 genes (< −2 fold) in the overall transcriptome (Supporting Information Table 5). Only a small fraction of such genes was, however, part of the injury-responsive transcriptome of control nerves (Figure 6A).
Figure 6. The expression of critical axonal growth genes semaphorin 4F and ciliary neurotrophic factor is dependent on EED-mediated transcriptional regulation during nerve repair.
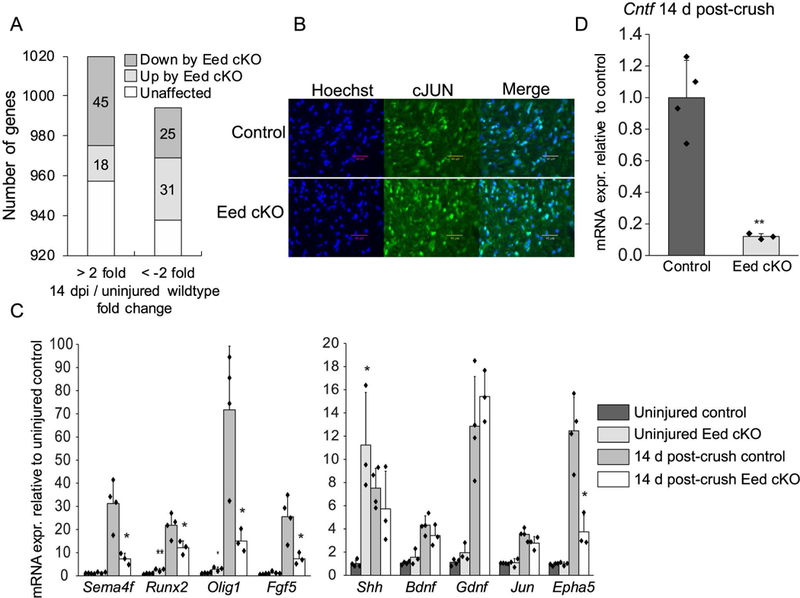
A, RNA-seq analysis identified a number of genes dysregulated by Eed cKO among genes differentially expressed during nerve regeneration at 14 d post-crush compared to uninjured nerves. See Supporting Information Table 2 and 5 for the list of dysregulated genes by Eed cKO in overall transcriptome and in the injury responsive transcriptome 14 d post-crush, respectively (n=3 per condition for control and n=2 for Eed cKO 14 d post-crush). B, Immunohistochemistry on transverse sections of distal stumps displays the JUN expression in nerves of indicated genotypes 5 d after cut. Scale bars, 40 μm. C, D, qRT-PCR analysis was used to identify the expression level of injury-responsive genes from 2 month Eed cKO and control sciatic nerves in uninjured condition or 14 day after crush. Expression levels were normalized with Gapdh. Data: mean ± SD; Asterisks indicate p-value between genotypes in the respective condition. *p < 0.05, **p < 0.005, ***p < 0.0005; n=4 for control and n=3 for Eed cKO (one-way ANOVA).
To better understand the phenotype of Eed cKO nerves, we compared our RNA-seq data with data from the analysis of injured nerves, in which Jun was deleted specifically in Schwann cells (Arthur-Farraj et al. 2012). The loss of the JUN transcription factor led to failures in axon regeneration but also in Bungner band formation and myelin debris clearance. Importantly, the Jun cKO array analysis identified a subset of injury genes implicated in nerve regeneration and provided a list of genes critical for axon regeneration. We also compared with gene expression analysis of injured nerves of WldS mice, in which axons degrade slowly, and Schwann cells therefore remain differentiated and incompetent to support repair process (Barrette et al. 2010). The comparison analysis did not yield a large overlap of genes that were commonly downregulated or upregulated between Eed cKO and Jun cKO studies. Such an observation was consistent with facts that the induction of JUN protein and mRNA in the Eed cKO was very similar to control, measured at 5 d and 14 d after injury, respectively (Figure 6B, C). Instead, we identified only 10 genes that were commonly downregulated in the Eed cKO and Jun cKO in injured nerves, including Sema4f, Epha5, Fgf5, Olig1, Btc, Mmp17, and Runx2 that were lower also in WldS-injured nerves compared to wildtype injured nerves (Figure 6C). Importantly, the critical role of semaphorin 4F transmembrane protein (SEMA4F) in axoglial interactions was demonstrated in a dorsal root ganglion neuron and Schwann cell co-culture system (Parrinello et al. 2008). EPHA5 has not been studied in the context of peripheral nerve injury, but it does affect axon guidance in retinal ganglion cells and spinal cord (Wang et al. 2016; Yue et al. 1999). The transcript level of other genes known for promoting axon regeneration and neuronal survival such as Shh, Bdnf and Gdnf (Boyd and Gordon 2003; Fontana et al. 2012; Hashimoto et al. 2008; Ma et al. 2016; Martinez et al. 2015) was largely unaffected at 14 d post-crush injury, even though EED loss caused derepression in uninjured nerves or premature induction (Figure 6C).
Furthermore, the analysis identified a significant decrease in ciliary neurotrophic factor (Cntf) (Figure 6D). The neuropoietic cytokine CNTF is primarily expressed by Schwann cells and substantially mediates axonal growth, demonstrated by markedly decreased number of regenerated axons in mouse models with genetic ablation of Cntf (Masu et al. 1993; Selvaraj et al. 2012; Simon et al. 2010).
EED Supports Injury-induced Schwann Cell Proliferation
One gene controlled by H3K27 trimethylation is the Cdkn2a tumor suppressor gene encoding the INK4A/p16 (cyclin-dependent kinase inhibitor) and ARF/p19 proteins (Bracken et al. 2007; Gomez-Sanchez et al. 2013). The p16 and p19 proteins are implicated in controlling Schwann cell over-proliferation during nerve regeneration after injury-induced expression (Atanasoski et al. 2006a; Gomez-Sanchez et al. 2013). Using transcript-specific primer sets for p16 and p19 transcripts in qRT-PCR, there is not much induction at 1 day after injury in control mice, but there is a premature induction of both transcripts 1 d after injury in the Eed cKO (Figure 7A). In addition, the two transcripts are modestly induced at 7d in control mice, but are greatly increased in the Eed cKO and remain elevated at 14 d after crush. Consistent with the regulation of Cdkn2a by PRC2, we also observed H3K27me3 over the promoters of both the p16 and p19 transcripts (not shown).
Figure 7. Eed cKO exhibits impaired proliferation in injured nerves.
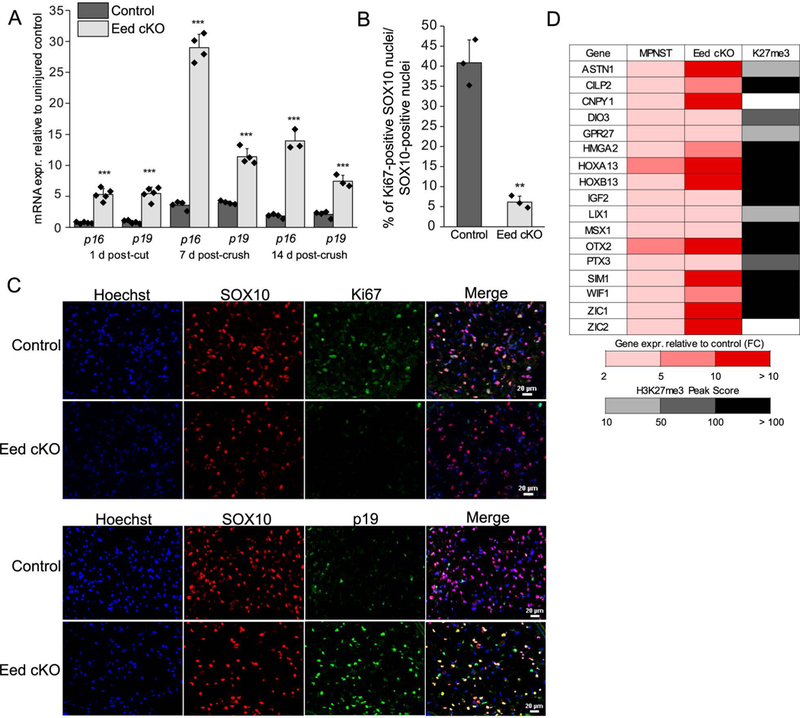
A, mRNA expression relative to uninjured control level set as 1 (not shown) at indicated time points was assessed by qRT-PCR with primer sequences specific to p16/Ink4a and p19/Arf transcripts of Cdkn2a. Expression levels were normalized with Gapdh. Data: mean ± SD; ***p < 0.0005; n=5 and n=4 per genotype and condition at 1 d post-cut and 7 d post-crush, respectively, and n=4 for control and n=3 for Eed cKO at 14 d-post crush (one-way ANOVA). B, C, The expression of a proliferation marker Ki-67 and p19/ARF among SOX10-positive nuclei at 5 d after denervation was assessed by immunohistochemistry on transverse sections of indicated genotypes. Scale bars, 20 μm. n=3 per genotype. Data: mean ± STDEV; **p < 0.005 (one-way ANOVA). D, Representative genes that were commonly upregulated in PRC2-deficient MPNSTs relative to non-deficient MPNSTs (> 3 fold, RNA-seq) (Lee et al., 2014) and Eed cKO nerves relative to wildtype nerves of uninjured or 1d, 14d post-injury conditions (> 2 fold, RNA-seq) are listed (FC, fold change). Gray shading indicates H3K27me3 occupancy of genes in peripheral nerves of uninjured or post-injury conditions. See Supporting Information Table 6 for expression fold changes by Eed cKO among PRC2-deficient MPNST genes and H3K27me3 peak score.
Since p16 and p19 control Schwann cell proliferation, we tested if injury-induced proliferation of Schwann cells after injury is affected in the Eed cKO by assessing the number of denervated Schwann cells with Ki67 expression 5 d after injury. Double labeling of Ki67 and the Schwann cell specific marker SOX10 showed that there was a significantly lower number of proliferating Schwann cells in Eed cKO nerves (Figure 7B, C). In addition, as predicted by the mRNA analyses, we see enhanced expression of the p19/ARF protein at the same timepoint. These data indicate that EED promotes proliferation of Schwann cells in the early response to injury by limiting induction of the p16 and p19 transcripts of the Cdkn2a gene, as predicted by earlier studies of H3K27 demethylation (Gomez-Sanchez et al. 2013). Previous studies have shown that Schwann cell proliferation after injury is offset by increased apoptosis, and that proliferation per se is not required for regeneration (Yang et al. 2008).
Comparison to NF1-associated MPNST gene expression profiles
PRC2 subunit mutations are often associated with CDKN2A mutations in development of malignant peripheral nerve sheath tumors. In one study, ~80% of ~50 MPNST’s lost CDKN2A expression, and ~75% of those tumors have mutations in either the EED or SUZ12 genes (Lee et al. 2014). To identify other EED-regulated genes that may be relevant to pathogenesis of MPNST, we compared genes upregulated in the Eed cKO with differentially expressed genes between PRC2-deficient and other MPNST’s. An RNA-seq analysis of MPNST’s identified 449 genes with a 3 fold or higher expression in the absence of SUZ12 or EED (Lee et al. 2014). The comparison analysis revealed that 40 such genes were upregulated in Eed cKO nerves either in uninjured or injured conditions (Figure 7D and Supporting Information Table 6). This includes Igf2 and homeobox transcriptional regulators, Hoxa13 and Hoxd13, which are highly overexpressed in cancers such as esophageal and gastric cancers (Gu et al. 2009; He et al. 2017). Our ChIP-seq analysis found that 193 genes among the 449 genes activated in PRC2-deficient human MPNST’s were occupied with H3K27me3 in rat peripheral nerve.
Discussion
Our studies of the Eed conditional knockout show that PRC2 function is dispensable for early postnatal myelination by Schwann cells, and that myelin is largely normal at two months of age (Ma et al. 2015). However, there is premature derepression of injury genes in the Eed cKO nerves, indicating that loss of H3K27me3 is an epigenomic switch involved in regulation of a subset of injury genes (Ma et al. 2016). Therefore, we decided to test if loss of EED may alter the nerve injury process and found that many of the early steps of repair process, termed Wallerian degeneration, appear to proceed normally in the Eed cKO. We found no significant differences in the amount of myelin debris and the number of infiltrating macrophages, suggesting that PRC2 was not required for clearance of myelin debris that can inhibit axon regeneration (Mukhopadhyay et al. 1994; Shen et al. 1998). Accordingly, our analysis of EED regulated genes did not identify known genes involved in autophagy or phagocytosis (with the notable exception of Megf10), nor did we see altered expression of genes required for macrophage infiltration (e.g. Ccl2/Mcp1). In addition, there was no impact on induction of the JUN transcription factor, which is important for several facets of nerve injury responses (Arthur-Farraj et al. 2012).
Given the augmented and early induction of some nerve injury genes, we did look for evidence of accelerated nerve injury processes, but early stages after nerve injury were apparently unaffected. We speculate the normal demethylation of H3K27, at least in young mice, may be sufficient to drive expression of adequate amounts of critical nerve repair genes. However, the ultrastructural analysis revealed that the function of Schwann cell EED is critical for timely axon regeneration, a function likely to be critical for efficient nerve regeneration since the survival and capacity of Schwann cells decrease at more distal sites that are chronically denervated (Benito et al. 2017; Eggers et al. 2010; Höke et al. 2002; Jessen and Mirsky 1999; Jonsson et al. 2013; Li et al. 1997; Michalski et al. 2008; Ronchi et al. 2017; Sulaiman and Gordon 2009).
The RNA-seq analysis together with H3K27me3-mapping revealed many injury-activated genes targeted by the PRC2 complex. Loss of polycomb repression was sufficient for a subset of injury genes to be activated in uninjured nerves (e.g., Shh, Gdnf, and Ngf). Activation of genes presumably involves coordination of loss of polycomb repression with activation of specific transcription factor pathways, and a substantial number of EED-repressed genes were identified only after injury. Some displayed a premature induction in injured Eed cKO nerves (Bdnf, Sox2, Artemin), indicating the needs of injury-signaling pathways in addition to loss of H3K27me3. It is striking that many injury-induced genes with H3K27me3 are efficiently induced within 24 hours of injury, and therefore the total number of PRC2-regulated genes may only become apparent from ongoing experiments examining the role of H3K27 demethylases in the injury response.
The slowed axon regeneration at 14 d prompted an examination of genes that could explain this phenotype. One potential cause is the substantial decrease in ciliary neurotrophic factor (Cntf). The neuropoietic cytokine CNTF is highly expressed by Schwann cells at the end of the first postnatal week, and studies demonstrated its neuroprotective and axon-growth promoting effect using animal models of neuropathy and peripheral nerve injury with genetic ablation of Cntf (Homs et al. 2011; Masu et al. 1993; Sendtner et al. 1991; Sendtner et al. 1992). In addition, the inhibition of CNTF by antibody-mediated receptor blocking significantly decreased the growth rate and number of regenerating axons (Vega-Meléndez et al. 2014). Likewise, exogenous CNTF improved axon regeneration after central and peripheral nerve injury (Homs et al. 2011; Hoyng et al. 2014; Müller et al. 2009; Vega-Meléndez et al. 2014). CNTF binding to neuronal receptors activates STAT3 that stimulates regeneration-associated genes and microtubule assembly, which play an important role in axonal stability and growth (Gu et al. 2016; Leibinger et al. 2013; Pellegrino and Habecker 2013; Selvaraj et al. 2012; Vigneswara et al. 2014). Furthermore, Eed cKO resulted in a significant downregulation of semaphorin 4F (Sema4f) that was greatly induced in injured control nerves. Depletion of SEMA4F in co-culture assays caused disruption of Schwann cell-axonal interactions (Parrinello et al. 2008). Injured Jun cKO nerves also exhibited a significant decrease in Sema4f expression and manifested slow axon regeneration (Arthur-Farraj et al. 2012). Therefore, deficient expression of Cntf and Sema4f may be responsible for delayed in axon regeneration in Eed cKO nerves during nerve repair.
Efficient remyelination of injured nerves requires the de novo activation of Nrg1 type I in denervated Schwann cells (Stassart et al. 2013). By analyzing the promoters of the type I and type III transcripts of Nrg1, we found that H3K27me3 mediates repression of Nrg1 and there is increased expression of the type I Nrg1 transcript in the uninjured Eed cKO nerves, indicating a mechanism by which type I Nrg1 is repressed in uninjured nerve. Interestingly, NRG1 increases the expression of nuc-ErbB3 (a nuclear variant of the NRG1 receptor, ErbB3), which has transcriptional activity by modulating H3K27me3 level in Schwann cells (Adilakshmi et al. 2011; Ness et al. 2016). However, nuc-ErbB3 likely has other activities since the phenotype and gene expression changes that we observe in the Eed knockout differ in several respects.
As has been shown in other systems (Chen et al. 2009; Conway et al. 2015; Ezhkova et al. 2011; He et al. 2012), PRC2 activity prevents inappropriate activation of lineage-defining transcription factors, as there was a low level activation of neuronal transcription factors (Pax6, Isl1, etc.). The derepressed level of these transcription factors was not sufficient to drive significant levels of neuronal gene expression, however. A number of homeobox genes (Hoxa7, Hoxa10, Hoxd13) and neural crest transcription factors (Pax3, Tfap2a, Tfap2b) that are expressed in early SC development (Balakrishnan et al. 2016; Doddrell et al. 2012) were also induced at a low level. Interestingly, a number of transcription factors involved in oligodendrocyte development (Nkx6.2, Nkx6.1, Myrf) were also somewhat elevated (Mitew et al. 2013), but did not drive induction of unique oligodendrocyte genes (Lopez-Anido et al. 2015) in the Eed cKO.
The polycomb pathway represses the Cdkn2a gene encoding cell cycle inhibitors INK4a/p16 and ARF/p19 (Biehs et al. 2013; Bracken et al. 2007; Chen et al. 2009; He et al. 2012; Voncken et al. 2003), and loss of EED or other PRC2 subunits resulted in derepression of Cdkn2a that was correlated to the proliferation-defective phenotype. Importantly, INK4a/p16 and ARF/p19 are implicated in controlling Schwann cell over-proliferation during nerve regeneration after injury-induced expression (Atanasoski et al. 2006a; Gomez-Sanchez et al. 2013). Eed cKO nerves display a slower rate of Schwann cell proliferation after injury with a premature and augmented expression of both transcripts for p16/Ink4a and p19/Arf, although this did not impair the regenerative process. Our observations are consistent with previous reports that nerve regeneration is not dependent upon Schwann cell proliferation (Kim et al. 2000; Yang et al. 2008).
Neurofibromatosis is a syndrome with Schwann cell-derived tumors caused by loss of either the NF1 or NF2 tumor suppressors (Carroll 2016). Loss of NF1 is associated with an increased risk that the disease progresses from benign neurofibromas to malignant peripheral nerve sheath tumors (MPNST’s). Interestingly, a very high percentage of MPNST’s have lost function of PRC2 through deletion/mutation of EED or SUZ12, or less frequent mutations of PRC2-associated proteins: e.g. AEBP2 and RBBP7 (De Raedt et al. 2014; Lee et al. 2014; Zhang et al. 2014). As shown here, proliferation in the absence of PRC2 function would be inhibited by induction of CDKN2A, but the PRC2 subunit mutations in MPNST’s are often associated with loss of function mutations in CDKN2A (Lee et al. 2014). The predominant co-occurrence of PRC2 alterations and CDKN2A mutation in MPNST’s with NF1 mutation suggests that these mutations coordinate to drive development of the malignant form of the disease. Furthermore, the findings of our study address some molecular pathways to MPNST’s mediated by the dysregulated polycomb repression. Previous studies have identified gene expression changes in MPNST’s with loss of PRC2 function (Lee et al. 2014), and we found some of the same changes in our Eed cKO mice. More specifically, Eed cKO led to an upregulation of Nrg1, which is also observed in neoplastic Schwann cells within human neurofibromas and MPNST’s (Stonecypher et al. 2005), and aberrant NRG1 signaling contributes to the pathogenesis of neurofibromas and MPNST’s in mouse models (Brosius et al. 2014; Gomez-Sanchez et al. 2013; Huijbregts et al. 2003; Kazmi et al. 2013). In addition, SEMA4F-deficiency was also identified in models of Neurofibromatosis type 1 and human neurofibromas, and appears to be a mechanism of loss of Schwann cell-axonal interaction, which contributes to tumorigenesis (Parrinello et al. 2008). It is interesting to note that mature myelinating Schwann cells are normally resistant to tumor formation in NF1 deficient mice, but after injury, neurofibromas develop at the wound site (Ribeiro et al. 2013). Thus, loss of polycomb repression after injury may contribute to neurofibroma development, but permanent loss of PRC2 may be required for transition to the MPNST stage. We did not find any evidence of neoplasia in EED deficient mice even after injury, presumably due to the induction of Cdkn2a and the lack of deregulated Ras signaling. A recent study used a shRNA screen to identify driver genes for MPNST development (Patel et al. 2016). Seven such genes were identified, and Meis1 was functionally validated. Another gene in this set, Pitx2, is highly upregulated in EED deficient nerves, and three of the seven have moderate to high levels of H3K27me3 on the promoters (Meis1, Pitx2, Prrx1), suggesting that loss of PRC2 function would play a role in their induction in MPNST’s.
Our study elucidated a novel regulation of repair Schwann cell genes mediated by the polycomb pathway and biological implications of EED deficiency in nerve regeneration and Schwann cell proliferation. The findings are of clinical importance and may lead to identification of therapeutic measures to facilitate peripheral axon regeneration and also to inhibit MPNST-pathogenesis with loss of the PRC2 subunits SUZ12 or EED.
Supplementary Material
Main Points.
Polycomb repressive complex 2 (PRC2) prevents premature induction of several Schwann cell injury genes, including neuregulin 1, type I.
Loss of PRC2 impairs Schwann cell proliferation after injury, and also leads to a delay in axon regeneration.
Acknowledgments
The authors thank the University of Wisconsin Biotechnology Center Gene Expression Center for providing RNA library preparation and the DNA Sequencing Facility for their sequencing services, and Stuart Orkin for generously providing the Eed fl/fl mice. This work was supported by the National Institutes of Health: NS075269 and NS100510 to JS and U54 core grant HD090256.
References
- Adilakshmi T, Ness-Myers J, Madrid-Aliste C, Fiser A, Tapinos N. 2011. A nuclear variant of ErbB3 receptor tyrosine kinase regulates ezrin distribution and Schwann cell myelination. J Neurosci 31:5106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. 1997. Role of translocation in the activation and function of protein kinase B. J Biol Chem 272:31515–24. [DOI] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Latouche M, Wilton DK, Quintes S, Chabrol E, Banerjee A, Woodhoo A, Jenkins B, Rahman M, Turmaine M and others. 2012. c-Jun reprograms Schwann cells of injured nerves to generate a repair cell essential for regeneration. Neuron 75:633–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arthur-Farraj PJ, Morgan CC, Adamowicz M, Gomez-Sanchez JA, Fazal SV, Beucher A, Razzaghi B, Mirsky R, Jessen KR, Aitman TJ. 2017. Changes in the Coding and Non-coding Transcriptome and DNA Methylome that Define the Schwann Cell Repair Phenotype after Nerve Injury. Cell Rep 20:2719–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atanasoski S, Boller D, De Ventura L, Koegel H, Boentert M, Young P, Werner S, Suter U. 2006a. Cell cycle inhibitors p21 and p16 are required for the regulation of Schwann cell proliferation. Glia 53:147–57. [DOI] [PubMed] [Google Scholar]
- Atanasoski S, Scherer SS, Sirkowski E, Leone D, Garratt AN, Birchmeier C, Suter U. 2006b. ErbB2 signaling in Schwann cells is mostly dispensable for maintenance of myelinated peripheral nerves and proliferation of adult Schwann cells after injury. J Neurosci 26:2124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balakrishnan A, Stykel MG, Touahri Y, Stratton JA, Biernaskie J, Schuurmans C. 2016. Temporal Analysis of Gene Expression in the Murine Schwann Cell Lineage and the Acutely Injured Postnatal Nerve. PLoS One 11:e0153256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrette B, Calvo E, Vallières N, Lacroix S. 2010. Transcriptional profiling of the injured sciatic nerve of mice carrying the Wld(S) mutant gene: identification of genes involved in neuroprotection, neuroinflammation, and nerve regeneration. Brain Behav Immun 24:1254–67. [DOI] [PubMed] [Google Scholar]
- Benito C, Davis CM, Gomez-Sanchez JA, Turmaine M, Meijer D, Poli V, Mirsky R, Jessen KR. 2017. STAT3 Controls the Long-Term Survival and Phenotype of Repair Schwann Cells during Nerve Regeneration. J Neurosci 37:4255–4269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biehs B, Hu JK, Strauli NB, Sangiorgi E, Jung H, Heber RP, Ho S, Goodwin AF, Dasen JS, Capecchi MR and others. 2013. BMI1 represses Ink4a/Arf and Hox genes to regulate stem cells in the rodent incisor. Nat Cell Biol 15:846–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyd JG, Gordon T. 2003. Glial cell line-derived neurotrophic factor and brain-derived neurotrophic factor sustain the axonal regeneration of chronically axotomized motoneurons in vivo. Exp Neurol 183:610–9. [DOI] [PubMed] [Google Scholar]
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, Theilgaard-Mönch K, Minucci S, Porse BT, Marine JC and others. 2007. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Genes Dev 21:525–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius Lutz A, Barres BA. 2014. Contrasting the glial response to axon injury in the central and peripheral nervous systems. Dev Cell 28:7–17. [DOI] [PubMed] [Google Scholar]
- Brosius Lutz A, Chung WS, Sloan SA, Carson GA, Zhou L, Lovelett E, Posada S, Zuchero JB, Barres BA. 2017. Schwann cells use TAM receptor-mediated phagocytosis in addition to autophagy to clear myelin in a mouse model of nerve injury. Proc Natl Acad Sci U S A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brosius SN, Turk AN, Byer SJ, Brossier NM, Kohli L, Whitmire A, Mikhail FM, Roth KA, Carroll SL. 2014. Neuregulin-1 overexpression and Trp53 haploinsufficiency cooperatively promote de novo malignant peripheral nerve sheath tumor pathogenesis. Acta Neuropathol 127:573–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brügger V, Duman M, Bochud M, Münger E, Heller M, Ruff S, Jacob C. 2017. Delaying histone deacetylase response to injury accelerates conversion into repair Schwann cells and nerve regeneration. Nat Commun 8:14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cafferty WB, Gardiner NJ, Gavazzi I, Powell J, McMahon SB, Heath JK, Munson J, Cohen J, Thompson SW. 2001. Leukemia inhibitory factor determines the growth status of injured adult sensory neurons. J Neurosci 21:7161–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SL. 2016. The Challenge of Cancer Genomics in Rare Nervous System Neoplasms: Malignant Peripheral Nerve Sheath Tumors as a Paradigm for Cross-Species Comparative Oncogenomics. Am J Pathol 186:464–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll SL, Miller ML, Frohnert PW, Kim SS, Corbett JA. 1997. Expression of neuregulins and their putative receptors, ErbB2 and ErbB3, is induced during Wallerian degeneration. J Neurosci 17:1642–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattin AL, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJ, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V and others. 2015. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 162:1127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Gu X, Su IH, Bottino R, Contreras JL, Tarakhovsky A, Kim SK. 2009. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev 23:975–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements MP, Byrne E, Camarillo Guerrero LF, Cattin AL, Zakka L, Ashraf A, Burden JJ, Khadayate S, Lloyd AC, Marguerat S and others. 2017. The Wound Microenvironment Reprograms Schwann Cells to Invasive Mesenchymal-like Cells to Drive Peripheral Nerve Regeneration. Neuron 96:98–114.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleven AH, Sannaa GA, Briaire-de Bruijn I, Ingram DR, van de Rijn M, Rubin BP, de Vries MW, Watson KL, Torres KE, Wang WL and others. 2016. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod Pathol 29:582–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conway E, Healy E, Bracken AP. 2015. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol 37:42–8. [DOI] [PubMed] [Google Scholar]
- De Raedt T, Beert E, Pasmant E, Luscan A, Brems H, Ortonne N, Helin K, Hornick JL, Mautner V, Kehrer-Sawatzki H and others. 2014. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 514:247–51. [DOI] [PubMed] [Google Scholar]
- Decker L, Desmarquet-Trin-Dinh C, Taillebourg E, Ghislain J, Vallat JM, Charnay P. 2006. Peripheral myelin maintenance is a dynamic process requiring constant Krox20 expression. J Neurosci 26:9771–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doddrell RD, Dun XP, Moate RM, Jessen KR, Mirsky R, Parkinson DB. 2012. Regulation of Schwann cell differentiation and proliferation by the Pax-3 transcription factor. Glia 60:1269–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eggers R, Tannemaat MR, Ehlert EM, Verhaagen J. 2010. A spatio-temporal analysis of motoneuron survival, axonal regeneration and neurotrophic factor expression after lumbar ventral root avulsion and implantation. Exp Neurol 223:207–20. [DOI] [PubMed] [Google Scholar]
- Ezhkova E, Lien WH, Stokes N, Pasolli HA, Silva JM, Fuchs E. 2011. EZH1 and EZH2 cogovern histone H3K27 trimethylation and are essential for hair follicle homeostasis and wound repair. Genes Dev 25:485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, D’Antonio M, Previtali S, Fasolini M, Messing A, Wrabetz L. 1999. P0-Cre transgenic mice for inactivation of adhesion molecules in Schwann cells. Ann N Y Acad Sci 883:116–23. [PubMed] [Google Scholar]
- Filbin MT. 2003. Myelin-associated inhibitors of axonal regeneration in the adult mammalian CNS. Nat Rev Neurosci 4:703–13. [DOI] [PubMed] [Google Scholar]
- Fischer S, Weishaupt A, Troppmair J, Martini R. 2008. Increase of MCP-1 (CCL2) in myelin mutant Schwann cells is mediated by MEK-ERK signaling pathway. Glia 56:836–43. [DOI] [PubMed] [Google Scholar]
- Fledrich R, Stassart RM, Klink A, Rasch LM, Prukop T, Haag L, Czesnik D, Kungl T, Abdelaal TA, Keric N and others. 2014. Soluble neuregulin-1 modulates disease pathogenesis in rodent models of Charcot-Marie-Tooth disease 1A. Nat Med 20:1055–61. [DOI] [PubMed] [Google Scholar]
- Fontana X, Hristova M, Da Costa C, Patodia S, Thei L, Makwana M, Spencer-Dene B, Latouche M, Mirsky R, Jessen KR and others. 2012. c-Jun in Schwann cells promotes axonal regeneration and motoneuron survival via paracrine signaling. J Cell Biol 198:127–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Carty L, Iruarrizaga-Lejarreta M, Palomo-Irigoyen M, Varela-Rey M, Griffith M, Hantke J, Macias-Camara N, Azkargorta M, Aurrekoetxea I and others. 2015. Schwann cell autophagy, myelinophagy, initiates myelin clearance from injured nerves. J Cell Biol 210:153–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Gomis-Coloma C, Morenilla-Palao C, Peiro G, Serra E, Serrano M, Cabedo H. 2013. Epigenetic induction of the Ink4a/Arf locus prevents Schwann cell overproliferation during nerve regeneration and after tumorigenic challenge. Brain 136:2262–78. [DOI] [PubMed] [Google Scholar]
- Gomez-Sanchez JA, Pilch KS, van der Lans M, Fazal SV, Benito C, Wagstaff LJ, Mirsky R, Jessen KR. 2017. After Nerve Injury, Lineage Tracing Shows That Myelin and Remak Schwann Cells Elongate Extensively and Branch to Form Repair Schwann Cells, Which Shorten Radically on Remyelination. J Neurosci 37:9086–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu YL, Gao GQ, Ma N, Ye LL, Zhang LW, Gao X, Zhang ZB. 2016. CNTF protects neurons from hypoxic injury through the activation of STAT3pTyr705. Int J Mol Med 38:1915–1921. [DOI] [PubMed] [Google Scholar]
- Gu ZD, Shen LY, Wang H, Chen XM, Li Y, Ning T, Chen KN. 2009. HOXA13 promotes cancer cell growth and predicts poor survival of patients with esophageal squamous cell carcinoma. Cancer Res 69:4969–73. [DOI] [PubMed] [Google Scholar]
- Guertin AD, Zhang DP, Mak KS, Alberta JA, Kim HA. 2005. Microanatomy of axon/glial signaling during Wallerian degeneration. J Neurosci 25:3478–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harroch S, Furtado GC, Brueck W, Rosenbluth J, Lafaille J, Chao M, Buxbaum JD, Schlessinger J. 2002. A critical role for the protein tyrosine phosphatase receptor type Z in functional recovery from demyelinating lesions. Nat Genet 32:411–4. [DOI] [PubMed] [Google Scholar]
- Hashimoto M, Ishii K, Nakamura Y, Watabe K, Kohsaka S, Akazawa C. 2008. Neuroprotective effect of sonic hedgehog up-regulated in Schwann cells following sciatic nerve injury. J Neurochem 107:918–27. [DOI] [PubMed] [Google Scholar]
- He A, Ma Q, Cao J, von Gise A, Zhou P, Xie H, Zhang B, Hsing M, Christodoulou DC, Cahan P and others. 2012. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ Res 110:406–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X, Zhang L, Queme LF, Liu X, Lu A, Waclaw RR, Dong X, Zhou W, Kidd G, Yoon SO and others. 2018. A histone deacetylase 3-dependent pathway delimits peripheral myelin growth and functional regeneration. Nat Med 24:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He YX, Song XH, Zhao ZY, Zhao H. 2017. HOXA13 upregulation in gastric cancer is associated with enhanced cancer cell invasion and epithelial-to-mesenchymal transition. Eur Rev Med Pharmacol Sci 21:258–265. [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38:576–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson CE, Camu W, Mettling C, Gouin A, Poulsen K, Karihaloo M, Rullamas J, Evans T, McMahon SB, Armanini MP. 1993. Neurotrophins promote motor neuron survival and are present in embryonic limb bud. Nature 363:266–70. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kawabuchi M. 2002. Myelin phagocytosis by macrophages and nonmacrophages during Wallerian degeneration. Microsc Res Tech 57:541–7. [DOI] [PubMed] [Google Scholar]
- Homs J, Ariza L, Pagès G, Udina E, Navarro X, Chillón M, Bosch A. 2011. Schwann cell targeting via intrasciatic injection of AAV8 as gene therapy strategy for peripheral nerve regeneration. Gene Ther 18:622–30. [DOI] [PubMed] [Google Scholar]
- Hoyng SA, De Winter F, Gnavi S, de Boer R, Boon LI, Korvers LM, Tannemaat MR, Malessy MJ, Verhaagen J. 2014. A comparative morphological, electrophysiological and functional analysis of axon regeneration through peripheral nerve autografts genetically modified to overexpress BDNF, CNTF, GDNF, NGF, NT3 or VEGF. Exp Neurol 261:578–93. [DOI] [PubMed] [Google Scholar]
- Huijbregts RP, Roth KA, Schmidt RE, Carroll SL. 2003. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. J Neurosci 23:7269–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung H, Kohnken R, Svaren J. 2012. The nucleosome remodeling and deacetylase chromatin remodeling (NuRD) complex is required for peripheral nerve myelination. J Neurosci 32:1517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung HA, Sun G, Keles S, Svaren J. 2015. Dynamic Regulation of Schwann Cell Enhancers after Peripheral Nerve Injury. J Biol Chem 290:6937–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höke A 2006. Neuroprotection in the peripheral nervous system: rationale for more effective therapies. Arch Neurol 63:1681–5. [DOI] [PubMed] [Google Scholar]
- Höke A, Gordon T, Zochodne DW, Sulaiman OA. 2002. A decline in glial cell-line-derived neurotrophic factor expression is associated with impaired regeneration after long-term Schwann cell denervation. Exp Neurol 173:77–85. [DOI] [PubMed] [Google Scholar]
- Jacob C 2017. Chromatin-remodeling enzymes in control of Schwann cell development, maintenance and plasticity. Curr Opin Neurobiol 47:24–30. [DOI] [PubMed] [Google Scholar]
- Jacob C, Lötscher P, Engler S, Baggiolini A, Varum Tavares S, Brügger V, John N, Büchmann-Møller S, Snider PL, Conway SJ and others. 2014. HDAC1 and HDAC2 control the specification of neural crest cells into peripheral glia. J Neurosci 34:6112–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. 1999. Why do Schwann cells survive in the absence of axons? Ann N Y Acad Sci 883:109–15. [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. 2016. The repair Schwann cell and its function in regenerating nerves. J Physiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson S, Wiberg R, McGrath AM, Novikov LN, Wiberg M, Novikova LN, Kingham PJ. 2013. Effect of delayed peripheral nerve repair on nerve regeneration, Schwann cell function and target muscle recovery. PLoS One 8:e56484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer D, Anderson DJ, Morrison SJ. 2004. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 131:5599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazmi SJ, Byer SJ, Eckert JM, Turk AN, Huijbregts RP, Brossier NM, Grizzle WE, Mikhail FM, Roth KA, Carroll SL. 2013. Transgenic mice overexpressing neuregulin-1 model neurofibroma-malignant peripheral nerve sheath tumor progression and implicate specific chromosomal copy number variations in tumorigenesis. Am J Pathol 182:646–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HA, Pomeroy SL, Whoriskey W, Pawlitzky I, Benowitz LI, Sicinski P, Stiles CD, Roberts TM. 2000. A developmentally regulated switch directs regenerative growth of Schwann cells through cyclin D1. Neuron 26:405–16. [DOI] [PubMed] [Google Scholar]
- Kim Y, Remacle AG, Chernov AV, Liu H, Shubayev I, Lai C, Dolkas J, Shiryaev SA, Golubkov VS, Mizisin AP and others. 2012. The MMP-9/TIMP-1 axis controls the status of differentiation and function of myelin-forming Schwann cells in nerve regeneration. PLoS One 7:e33664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruidenier L, Chung CW, Cheng Z, Liddle J, Che K, Joberty G, Bantscheff M, Bountra C, Bridges A, Diallo H and others. 2012. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 488:404–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuboyama K, Fujikawa A, Suzuki R, Noda M. 2015. Inactivation of Protein Tyrosine Phosphatase Receptor Type Z by Pleiotrophin Promotes Remyelination through Activation of Differentiation of Oligodendrocyte Precursor Cells. J Neurosci 35:12162–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtz A, Zimmer A, Schnütgen F, Brüning G, Spener F, Müller T. 1994. The expression pattern of a novel gene encoding brain-fatty acid binding protein correlates with neuronal and glial cell development. Development 120:2637–49. [DOI] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. 2005. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A 102:2596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Teckie S, Wiesner T, Ran L, Prieto Granada CN, Lin M, Zhu S, Cao Z, Liang Y, Sboner A and others. 2014. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet 46:1227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibinger M, Andreadaki A, Diekmann H, Fischer D. 2013. Neuronal STAT3 activation is essential for CNTF- and inflammatory stimulation-induced CNS axon regeneration. Cell Death Dis 4:e805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Terenghi G, Hall SM. 1997. Effects of delayed re-innervation on the expression of c-erbB receptors by chronically denervated rat Schwann cells in vivo. Glia 20:333–47. [DOI] [PubMed] [Google Scholar]
- Li S, Wang X, Gu Y, Chen C, Wang Y, Liu J, Hu W, Yu B, Ding F, Liu Y and others. 2015. Let-7 microRNAs regenerate peripheral nerve regeneration by targeting nerve growth factor. Mol Ther 23:423–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin HP, Oksuz I, Hurley E, Wrabetz L, Awatramani R. 2015. Microprocessor complex subunit DiGeorge syndrome critical region gene 8 (Dgcr8) is required for schwann cell myelination and myelin maintenance. J Biol Chem 290:24294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Anido C, Sun G, Koenning M, Srinivasan R, Hung HA, Emery B, Keles S, Svaren J. 2015. Differential Sox10 genomic occupancy in myelinating glia. Glia 63:1897–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundborg G 2000. A 25-year perspective of peripheral nerve surgery: evolving neuroscientific concepts and clinical significance. J Hand Surg Am 25:391–414. [DOI] [PubMed] [Google Scholar]
- Ma KH, Hung HA, Srinivasan R, Xie H, Orkin SH, Svaren J. 2015. Regulation of Peripheral Nerve Myelin Maintenance by Gene Repression through Polycomb Repressive Complex 2. J Neurosci 35:8640–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KH, Hung HA, Svaren J. 2016. Epigenomic Regulation of Schwann Cell Reprogramming in Peripheral Nerve Injury. J Neurosci 36:9135–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma KH, Svaren J. 2018. Epigenetic Control of Schwann Cells. Neuroscientist in press. [DOI] [PubMed] [Google Scholar]
- Martinez JA, Kobayashi M, Krishnan A, Webber C, Christie K, Guo G, Singh V, Zochodne DW. 2015. Intrinsic facilitation of adult peripheral nerve regeneration by the Sonic hedgehog morphogen. Exp Neurol 271:493–505. [DOI] [PubMed] [Google Scholar]
- Masu Y, Wolf E, Holtmann B, Sendtner M, Brem G, Thoenen H. 1993. Disruption of the CNTF gene results in motor neuron degeneration. Nature 365:27–32. [DOI] [PubMed] [Google Scholar]
- Meyer M, Matsuoka I, Wetmore C, Olson L, Thoenen H. 1992. Enhanced synthesis of brain-derived neurotrophic factor in the lesioned peripheral nerve: different mechanisms are responsible for the regulation of BDNF and NGF mRNA. J Cell Biol 119:45–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. 2004. Axonal neuregulin-1 regulates myelin sheath thickness. Science 304:700–3. [DOI] [PubMed] [Google Scholar]
- Michalski B, Bain JR, Fahnestock M. 2008. Long-term changes in neurotrophic factor expression in distal nerve stump following denervation and reinnervation with motor or sensory nerve. J Neurochem 105:1244–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitew S, Hay CM, Peckham H, Xiao J, Koenning M, Emery B. 2013. Mechanisms regulating the development of oligodendrocytes and central nervous system myelin. Neuroscience 276:29–47. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay G, Doherty P, Walsh FS, Crocker PR, Filbin MT. 1994. A novel role for myelin-associated glycoprotein as an inhibitor of axonal regeneration. Neuron 13:757–67. [DOI] [PubMed] [Google Scholar]
- Müller A, Hauk TG, Leibinger M, Marienfeld R, Fischer D. 2009. Exogenous CNTF stimulates axon regeneration of retinal ganglion cells partially via endogenous CNTF. Mol Cell Neurosci 41:233–46. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Le N, Mahoney H, Araki T, Milbrandt J. 2002. Deciphering peripheral nerve myelination by using Schwann cell expression profiling. Proc Natl Acad Sci U S A 99:8998–9003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napoli I, Noon LA, Ribeiro S, Kerai AP, Parrinello S, Rosenberg LH, Collins MJ, Harrisingh MC, White IJ, Woodhoo A and others. 2012. A central role for the ERK-signaling pathway in controlling Schwann cell plasticity and peripheral nerve regeneration in vivo. Neuron 73:729–42. [DOI] [PubMed] [Google Scholar]
- Ness JK, Skiles AA, Yap EH, Fajardo EJ, Fiser A, Tapinos N. 2016. Nuc-ErbB3 regulates H3K27me3 levels and HMT activity to establish epigenetic repression during peripheral myelination. Glia 64:977–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, Zigmond RE. 2013. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J Neurosci 33:16236–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norrmén C, Figlia G, Pfistner P, Pereira JA, Bachofner S, Suter U. 2018. mTORC1 Is Transiently Reactivated in Injured Nerves to Promote c-Jun Elevation and Schwann Cell Dedifferentiation. J Neurosci 38:4811–4828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parmantier E, Lynn B, Lawson D, Turmaine M, Namini SS, Chakrabarti L, McMahon AP, Jessen KR, Mirsky R. 1999. Schwann cell-derived Desert hedgehog controls the development of peripheral nerve sheaths. Neuron 23:713–24. [DOI] [PubMed] [Google Scholar]
- Parrinello S, Noon LA, Harrisingh MC, Wingfield Digby P, Rosenberg LH, Cremona CA, Echave P, Flanagan AM, Parada LF, Lloyd AC. 2008. NF1 loss disrupts Schwann cell-axonal interactions: a novel role for semaphorin 4F. Genes Dev 22:3335–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AV, Chaney KE, Choi K, Largaespada DA, Kumar AR, Ratner N. 2016. An ShRNA Screen Identifies MEIS1 as a Driver of Malignant Peripheral Nerve Sheath Tumors. EBioMedicine 9:110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekmezci M, Cuevas-Ocampo AK, Perry A, Horvai AE. 2017. Significance of H3K27me3 loss in the diagnosis of malignant peripheral nerve sheath tumors. Mod Pathol. [DOI] [PubMed] [Google Scholar]
- Pellegrino MJ, Habecker BA. 2013. STAT3 integrates cytokine and neurotrophin signals to promote sympathetic axon regeneration. Mol Cell Neurosci 56:272–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin FE, Lacroix S, Avilés-Trigueros M, David S. 2005. Involvement of monocyte chemoattractant protein-1, macrophage inflammatory protein-1alpha and interleukin-1beta in Wallerian degeneration. Brain 128:854–66. [DOI] [PubMed] [Google Scholar]
- Ribeiro S, Napoli I, White IJ, Parrinello S, Flanagan AM, Suter U, Parada LF, Lloyd AC. 2013. Injury signals cooperate with Nf1 loss to relieve the tumor-suppressive environment of adult peripheral nerve. Cell Rep 5:126–36. [DOI] [PubMed] [Google Scholar]
- Ronchi G, Cillino M, Gambarotta G, Fornasari BE, Raimondo S, Pugliese P, Tos P, Cordova A, Moschella F, Geuna S. 2017. Irreversible changes occurring in long-term denervated Schwann cells affect delayed nerve repair. J Neurosurg 127:1–14. [DOI] [PubMed] [Google Scholar]
- Ronchi G, Haastert-Talini K, Fornasari BE, Perroteau I, Geuna S, Gambarotta G. 2016. The Neuregulin1/ErbB system is selectively regulated during peripheral nerve degeneration and regeneration. Eur J Neurosci 43:351–64. [DOI] [PubMed] [Google Scholar]
- Rotshenker S 2011. Wallerian degeneration: the innate-immune response to traumatic nerve injury. J Neuroinflammation 8:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salonen V, Aho H, Röyttä M, Peltonen J. 1988. Quantitation of Schwann cells and endoneurial fibroblast-like cells after experimental nerve trauma. Acta Neuropathol 75:331–6. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. 2005. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307:1098–101. [DOI] [PubMed] [Google Scholar]
- Selvaraj BT, Frank N, Bender FL, Asan E, Sendtner M. 2012. Local axonal function of STAT3 rescues axon degeneration in the pmn model of motoneuron disease. J Cell Biol 199:437–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sendtner M, Arakawa Y, Stöckli KA, Kreutzberg GW, Thoenen H. 1991. Effect of ciliary neurotrophic factor (CNTF) on motoneuron survival. J Cell Sci Suppl 15:103–9. [DOI] [PubMed] [Google Scholar]
- Sendtner M, Schmalbruch H, Stöckli KA, Carroll P, Kreutzberg GW, Thoenen H. 1992. Ciliary neurotrophic factor prevents degeneration of motor neurons in mouse mutant progressive motor neuronopathy. Nature 358:502–4. [DOI] [PubMed] [Google Scholar]
- Sharghi-Namini S, Turmaine M, Meier C, Sahni V, Umehara F, Jessen KR, Mirsky R. 2006. The Structural and Functional Integrity of Peripheral Nerves Depends on the Glial-Derived Signal Desert Hedgehog. Journal of Neuroscience 26:6364–6376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen YJ, DeBellard ME, Salzer JL, Roder J, Filbin MT. 1998. Myelin-associated glycoprotein in myelin and expressed by Schwann cells inhibits axonal regeneration and branching. Mol Cell Neurosci 12:79–91. [DOI] [PubMed] [Google Scholar]
- Shin YK, Jang SY, Park JY, Park SY, Lee HJ, Suh DJ, Park HT. 2013. The Neuregulin-Rac-MKK7 pathway regulates antagonistic c-jun/Krox20 expression in Schwann cell dedifferentiation. Glia 61:892–904. [DOI] [PubMed] [Google Scholar]
- Simon CM, Jablonka S, Ruiz R, Tabares L, Sendtner M. 2010. Ciliary neurotrophic factor-induced sprouting preserves motor function in a mouse model of mild spinal muscular atrophy. Hum Mol Genet 19:973–86. [DOI] [PubMed] [Google Scholar]
- Stassart RM, Fledrich R, Velanac V, Brinkmann BG, Schwab MH, Meijer D, Sereda MW, Nave KA. 2013. A role for Schwann cell-derived neuregulin-1 in remyelination. Nat Neurosci 16:48–54. [DOI] [PubMed] [Google Scholar]
- Stonecypher MS, Byer SJ, Grizzle WE, Carroll SL. 2005. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 24:5589–605. [DOI] [PubMed] [Google Scholar]
- Sulaiman OA, Gordon T. 2009. Role of chronic Schwann cell denervation in poor functional recovery after nerve injuries and experimental strategies to combat it. Neurosurgery 65:A105–14. [DOI] [PubMed] [Google Scholar]
- Sulaiman W, Gordon T. 2013. Neurobiology of peripheral nerve injury, regeneration, and functional recovery: from bench top research to bedside application. Ochsner J 13:100–8. [PMC free article] [PubMed] [Google Scholar]
- Sun G, Li Z, Wang X, Tang W, Wei Y. 2013. Modulation of MAPK and Akt signaling pathways in proximal segment of injured sciatic nerves. Neurosci Lett 534:205–10. [DOI] [PubMed] [Google Scholar]
- Taveggia C, Zanazzi G, Petrylak A, Yano H, Rosenbluth J, Einheber S, Xu X, Esper RM, Loeb JA, Shrager P and others. 2005. Neuregulin-1 type III determines the ensheathment fate of axons. Neuron 47:681–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. 1994. Krox-20 controls myelination in the peripheral nervous system. Nature 371:796–799. [DOI] [PubMed] [Google Scholar]
- Vega-Meléndez GS, Blagburn JM, Blanco RE. 2014. Ciliary neurotrophic factor and fibroblast growth factor increase the speed and number of regenerating axons after optic nerve injury in adult Rana pipiens. J Neurosci Res 92:13–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigneswara V, Akpan N, Berry M, Logan A, Troy CM, Ahmed Z. 2014. Combined suppression of CASP2 and CASP6 protects retinal ganglion cells from apoptosis and promotes axon regeneration through CNTF-mediated JAK/STAT signalling. Brain 137:1656–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voncken JW, Roelen BA, Roefs M, de Vries S, Verhoeven E, Marino S, Deschamps J, van Lohuizen M. 2003. Rnf2 (Ring1b) deficiency causes gastrulation arrest and cell cycle inhibition. Proc Natl Acad Sci U S A 100:2468–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Galvao J, Beach KM, Luo W, Urrutia RA, Goldberg JL, Otteson DC. 2016. Novel Roles and Mechanism for Krüppel-like Factor 16 (KLF16) Regulation of Neurite Outgrowth and Ephrin Receptor A5 (EphA5) Expression in Retinal Ganglion Cells. J Biol Chem 291:18084–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widenfalk J, Wu W, Hao J, Person JK, Wiesenfeldt-Hallin Z, Risling M. 2009. Treatment of transected peripheral nerves with artemin improved motor neuron regeneration, but did not reduce nerve injury-induced pain behaviour. Scand J Plast Reconstr Surg Hand Surg 43:245–50. [DOI] [PubMed] [Google Scholar]
- Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C, Orkin SH. 2014. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell 14:68–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki T, Sabit H, Oya T, Ishii Y, Hamashima T, Tokunaga A, Ishizawa S, Jie S, Kurashige Y, Matsushima T and others. 2009. Activation of MAP kinases, Akt and PDGF receptors in injured peripheral nerves. J Peripher Nerv Syst 14:165–76. [DOI] [PubMed] [Google Scholar]
- Yang DP, Zhang DP, Mak KS, Bonder DE, Pomeroy SL, Kim HA. 2008. Schwann cell proliferation during Wallerian degeneration is not necessary for regeneration and remyelination of the peripheral nerves: axon-dependent removal of newly generated Schwann cells by apoptosis. Mol Cell Neurosci 38:80–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue Y, Su J, Cerretti DP, Fox GM, Jing S, Zhou R. 1999. Selective inhibition of spinal cord neurite outgrowth and cell survival by the Eph family ligand ephrin-A5. J Neurosci 19:10026–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wang Y, Jones S, Sausen M, McMahon K, Sharma R, Wang Q, Belzberg AJ, Chaichana K, Gallia GL and others. 2014. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat Genet 46:1170–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.