

Abstract
Microbes engineered to display heterologous proteins could be useful biotechnological tools for protein engineering, lignocellulose degradation, biocatalysis, bioremediation and biosensing. Bacillus subtilis is a promising host to display proteins, as this model Gram-positive bacterium is genetically tractable and already used industrially to produce enzymes. To gain insight into the factors that affect displayed protein stability and copy-number, we systematically compared the ability of different protease-deficient B. subtilis strains (WB800, BRB07, BRB08 and BRB14) to display a Cel8A-LysM reporter protein in which the Clostridium thermocellum Cel8A endoglucanase is fused to LysM cell wall binding modules. Whole-cell cellulase measurements and fractionation experiments demonstrate that genetically eliminating extracytoplasmic bacterial proteases improves Cel8A-LysM displays levels. However, upon entering stationary phase, for all protease-deficient strains, the amount of displayed reporter dramatically decreases, presumably as a result of cellular autolysis. This problem can be partially overcome by adding chemical protease inhibitors, which significantly increase protein display levels. We conclude that strain BRB08 is well-suited for stably displaying our reporter protein, as genetic removal of its extracellular and cell wall-associated proteases leads to the highest levels of surface accumulated Cel8A-LysM without causing secretion stress or impairing growth. A two-step procedure is presented that enables the construction of enzyme coated vegetative B. subtilis cells that retain stable cell-associated enzyme activity for nearly 3 days. The results of this work could aid the development of whole cell display systems that have useful biotechnological applications.
Keywords: Bacillus subtilis, protease-deficient, cell surface display, LysM
Introduction:
Bacteria engineered to display heterologous proteins have many potential biotechnological applications, including uses in protein engineering, biocatalysis, lignocellulose degradation, biosensing and bioremediation (Lee, Choi et al. 2003, Wernerus and Stahl 2004, Schuurmann, Quehl et al. 2014, Li and Tao 2015, Smith, Khera et al. 2015). Surface display methods are attractive because the microbe produces and displays the protein during growth, circumventing the need for time-consuming protein purification and immobilization procedures that are typically used to create protein coated materials (Garcia-Galan, Berenguer-Murcia et al. 2011, Mohamad, Marzuki et al. 2015, Hyeon, Shin et al. 2016). Moreover, surface-engineered bacteria that display cellulolytic enzymes can be used as consolidating bioprocessing microbes that convert abundant plant biomass into biofuels and other biocommodities (la Grange, den Haan et al. 2010, Olson, McBride et al. 2012, Huang, Anderson et al. 2014, Hyeon, Shin et al. 2016, Huang and Clubb 2017). Gram-positive bacteria may be particularly well-suited for displaying heterologous proteins due to the relatively simple structure of their cell envelope, which consists of a single membrane surrounded by a thick peptidoglycan wall. As a result, a number of groups have sought to engineer the surface proteome of Bacillus subtilis, a genetically tractable model Gram-positive bacterium that has an established role in industry and has generally recognized as safe status (GRAS) (Schallmey, Singh et al. 2004, Liu, Liu et al. 2013). B. subtilis is also capable of secreting large quantities of proteins, a requirement for densely displaying heterologous proteins on its surface. Devising methods to efficiently display proteins on vegetative B. subtilis is of significant interest, since unlike its spore form, these cells are metabolically active. Moreover, vegetative cells are significantly larger than spores, potentially enabling a greater number of proteins to be displayed.
Several approaches have been developed to display heterologous proteins on the surface of vegetative B. subtilis (Kobayashi, Toida et al. 2000, Desvaux, Dumas et al. 2006, Nguyen and Schumann 2006, Chen, Wu et al. 2008, Huang, Anderson et al. 2014, Huang and Clubb 2017). These methods either covalently or non-covalently attach the protein to the cell wall peptidoglycan after it has first been exported across the membrane through the Sec translocon. Covalent cell wall attachment is achieved by simultaneously expressing a protein containing a C-terminal LPXTG sorting signal sequence and a cysteine transpeptidase Sortase A enzyme that joins the protein to the Lipid II molecule, which is eventually incorporated into nascent peptidoglycan (Spirig, Weiner et al. 2011). This approach was first used to display α-amylase protein, which was surface attached by the Sortase A enzyme from Listeria monocytogenes (~240,000 proteins per B. subtilis cell) (Nguyen and Schumann 2006). Later, Liew et al. displayed ~47,300 copies of β-lactamase per cell, which were attached by the native B. subtilis YhcS sortase enzyme (Liew, Wang et al. 2012). Non-covalent attachment to the surface has also been achieved by fusing proteins to either LysM (lysin motif) (pfam 01476) or type II cell wall binding domains (pfam 04122) (Kobayashi, Toida et al. 2000, Chen, Wu et al. 2008). The highest levels of display were achieved by fusing the protein to LysM, with an estimated 1.1 X 108 β-lactamase proteins displayed per filamentous cell (Chen, Wu et al. 2008). While both non-covalent and covalent attachment methods have been demonstrated, non-covalent protein attachment methods are in principle simpler to use, as the cells only need to be engineered to express a fusion protein containing a cell wall binding domain.
For many biotechnological applications, the activity of proteins displayed on the surface of vegetative B. subtilis should be stable for long periods of time, at least several days if they are to be used as biocatalysts (Homaei, Sariri et al. 2013, Sirisha, Jain et al. 2016). While a number of display methods have been reported, the durability of displayed heterologous protein activity and how it is affected by extracytoplasmic proteases and solution conditions is not well known. In this study, we developed a B. subtilis protein display reporter system in which the Clostridum thermocellum Cel8A endoglucanase is fused to the B. subtilis LysM cell wall binding module. The effects of LysM positioning, extracytoplasmic proteases, and solution conditions on cell morphology, stress-response, displayed protein copy-number, and stability were determined. Reporter protein display was studied in B. subtilis strain 168, and protease-deficient strains WB800S, BRB07, BRB08 and BRB14 (Wu, Yeung et al. 2002, Pohl, Bhavsar et al. 2013). We show that fusion protein display has a profound impact on cell morphology depending on the host strain that is employed, with some of the highest levels of stable protein display obtained using strain BRB08. Interestingly, displayed enzyme activity is rapidly lost from the surface of B. subtilis cells, even when the cells are genetically modified to eliminate the production of extracytoplasmic proteases. However, this problem can be overcome by choosing solution conditions that maintain an energized cellular membrane, enabling stable, high-density protein display for over two days. The ability to produce stable enzyme-coated B. subtilis is an important step toward their practical use in biotechnological applications.
Materials and Methods:
Strains, plasmids, and cloning.
Bacterial strains and plasmids are listed in Table 1. WB800S was created by transforming WB800 with the pJL62 plasmid, which converted chloramphenicol resistance to spectinomycin resistance (Ledeaux and Grossman 1995). This was done to allow subsequent cloning with B. subtilis–E. coli shuttle plasmids that contained a chloramphenicol acetyltransferase gene. Genes encoding the Cel8A and LysM fusion proteins were introduced into B. subtilis using the pBL113 B. subtilis–E. coli shuttle vector derived from pRDC19 (Arigoni, Talabot et al. 1998), which integrates into the thrC locus. Shuttle vectors were constructed using standard molecular biology methods. The pE-SUMO vector (LifeSensors, Devault, PA, USA) was used to express Cel8A in E. coli for protein purification. The expression plasmid was constructed using standard methods and contains DNA encoding Cel8A that was altered using QuikChange to remove an internal BamHI site. The Cel8A expression plasmid was transformed into E. coli Rosetta (DE3) pLysS cells (Novagen) for protein expression. Primers used are listed in Table S1.
Table 1.
Strains and plasmids used in this study
| Strain | Description | Source |
|---|---|---|
| B. subtilis | ||
| 168 | trpC2 | BGSCa |
| 168 Cel8A | trpC2 thrC::pCel8A, ery | This work |
| 168 Cel8A-LysM | trpC2 thrC::pCel8A-LysM, ery | This work |
| 168 Cel8A-[L]-LysM | trpC2 thrC::pCel8A-[L]-LysM, ery | This work |
| BRB07 | trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr | Cobra Biologics (Pohl, Bhavsar et al. 2013) |
| BRB07 Cel8A |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr thrC::pCel8A, ery |
This work |
| BRB07 Cel8A-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr thrC::pCel8A-LysM, ery |
This work |
| BRB07 Cel8A-[L]-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr thrC::pCel8A-[L]-LysM, ery |
This work |
| BRB08 | trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA | Cobra Biologics (Pohl, Bhavsar et al. 2013) |
| BRB08 Cel8A |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA thrC::pCel8A, ery |
This work |
| BRB08 Cel8A-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA thrC::pCel8A-LysM, ery |
This work |
| BRB08 Cel8A-[L]-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA thrC::pCel8A-[L]-LysM, ery |
This work |
| BRB14 |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA, htrA, htrB |
Cobra Biologics (Pohl, Bhavsar et al. 2013) |
| BRB14 Cel8A |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA, htrA, htrB thrC::pCel8A, ery |
This work |
| BRB14 Cel8A-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA, htrA, htrB thrC::pCel8A-LysM, ery |
This work |
| BRB14 Cel8A-[L]-LysM |
trpC2, nprB, aprE, epr, bpr, nprE, mpr, vpr, wprA, htrA, htrB thrC::pCel8A-[L]-LysM, ery |
This work |
| WB800 |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg (cat) |
(Wu, Yeung et al. 2002) |
| WB800S |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc |
This work |
| WB800S Cel8A |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc thrC::pCel8A, ery |
This work |
| WB800S Cel8A-LysM |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc thrC::pCel8A-LysM, ery |
This work |
| WB800S Cel8A-[L]-LysM |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc thrC::pCel8A-[L]-LysM, ery |
This work |
| WB800S LysM-Cel8A |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc thrC::pLysM-Cel8A, ery |
This work |
| WB800S LysM-[L]-Cel8A |
trpC2, nprB::bsr, aprE, epr, bpr, nprE, mpr::ble, vpr, wprA::hyg, cat::spc thrC::pLysM-[L]-Cel8A, ery |
This work |
| E. coli | ||
| BL21 (DE3) |
fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 |
Novagen |
| BL21 (DE3) Cel8A |
fhuA2 [lon] ompT gal (λ DE3) [dcm] ∆hsdS λ DE3 = λ sBamHIo ∆EcoRI-B int::(lacI::PlacUV5::T7 gene1) i21 ∆nin5 pE-SUMO- Cel8A |
This work |
| Plasmid | ||
| pJL62 |
B. subtilis – E. coli shuttle vector that converts cam to scp; amp, tet |
(Ledeaux and Grossman 1995) |
| pBL113 |
B. subtilis – E. coli shuttle vector derived from pRDC19 (Arigoni, Talabot et al. 1998) that integrates into the thrC locus in the B. subtilis genome, amp, erm, IPTG inducible |
B. Lazazzera, UCLA (unpublished) |
| pCel8A | PhrC signal peptide (1–35) preceding His6 tagged Cel8A (32–394) in pBL113 |
This work |
| pCel8A-LysM | PhrC signal peptide (1–35) preceding His6 tagged Cel8A (32–394) and LysM domains (25–230) in pBL113 |
This work |
| pCel8A-[L]-LysM | PhrC signal peptide (1–35) preceding His6 tagged Cel8A (32–394), linker region from FnBPB (770–880) and LysM domains (25–230) in pBL113 |
This work |
| pLysM-Cel8A | PhrC signal peptide (1–35) preceding His6 tagged LysM domains (25–230) and Cel8A (32–394) in pBL113 |
This work |
| pLysM-[L]-Cel8A | PhrC signal peptide (1–35) preceding His6 tagged LysM domains (25–230), linker region from FnBPB (770–880) and Cel8A (32–394) in pBL113 |
This work |
| pE-SUMO |
E. coli plasmid for protein overexpression and purification, kan, IPTG inducible |
LifeSensors |
| pE-SUMO-Cel8A | Overexpression of the Cel8A (32–394) catalytic domain in pE-SUMO |
This work |
Table caption: amp, ble, bsr, cam, ery, hyg, kan, spc, and tet are antibiotic resistance markers for ampicillin, bleomycin, blasticidin S, chloramphenicol, erythromycin, hygromycin, kanamycin, spectinomycin, and tetracycline respectively.
BGSC, Bacillus Genetic Stock Center.
GenBank accession numbers are given in the materials and methods.
For gene integration, B. subtilis competent cells were created according to the following protocol (Anagnostopoulos and Spizizen 1961). B. subtilis lawn plates were grown overnight at room temperature. In the morning, cells were washed off of the lawn plates using Lawn Wash Buffer [15 mM (NH4)2SO4, 80 mM K2HPO4, 64 mM KH2PO4, 3.4 mM NaC3H5O(COO-)3, 1 mM MgSO4] and used to start a liquid culture of 25 mL SpC [15 mM (NH4)2SO4, 80 mM K2HPO4, 64 mM KH2PO4, 3.4 mM NaC3H5O(COO-)3, 1 mM MgSO4, 0.5% glucose, 0.2% yeast extract, 0.025% casamino acids, 0.04 mg/mL required amino acids] with an OD600 = 0.01 in a 250 mL flask, which grew at 37°C with shaking. OD600 was monitored until the cells had grown for two hours past the onset of stationary phase. At this point, 2.5 mL of cells were added to 22.5 mL SpII [15 mM (NH4)2SO4, 80 mM K2HPO4, 64 mM KH2PO4, 3.4 mM NaC3H5O(COO-)3, 3.5 mM MgSO4, 0.5% glucose, 0.1% yeast extract, 0.01% casamino acids, 0.04 mg/mL required amino acids]. After 90 minutes of growth, cells were collected by centrifugation. Cells were re-suspended in 2.5 mL of spent SpII media and mixed with glycerol to a final concentration of 10% for storage at −80°C. B. subtilis competent cells were transformed with the appropriate plasmids to produce the strains listed in Table 1. In large culture tubes, 100 uL competent cells were mixed with 100 uL Transformation Buffer [15 mM (NH4)2SO4, 80 mM K2HPO4, 64 mM KH2PO4, 3.4 mM NaC3H5O(COO-)3, 2 mM EGTA]. 5 uL of plasmid DNA was added to each tube. The tubes were incubated at 37°C with shaking for 20 minutes before plating on LB agar and antibiotic (spectinomycin: 100 μg/mL; erythromycin: 1 μg/mL; chloramphenicol: 5 μg/mL). Plates were incubated at 37°C overnight before colonies were picked, streak purified twice, patched to check for the correct phenotypes, and whole cell PCR product was sequenced (APEX Taq Red Master Mix, 2.0X; Genesee, San Diego, CA, USA).
Cel8A purification and standard curve creation.
Protein expression in E. coli Rosetta (DE3) pLysS cells containing the pE-SUMO-Cel8A plasmid was achieved using standard methods. Briefly, cells were grown to OD600 = 0.6, expression was induced by adding 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) and the cells were incubated with shaking for 16 hours at 17°C. After harvesting the cells by centrifugation, the pellet was sonicated in Resuspension Buffer (50 mM Tris, pH 8.0; 500 mM NaCl; 20 mM imidazole; 0.5% CHAPS; 1% Triton X-100; 10% glycerol; 2.5mM MgCl2) with added protease inhibitor cocktail powder (Sigma Aldrich P2714, St. Louis, MO, USA) and phenylmethanesulfonyl fluoride (Sigma Aldrich P7626). The lysis supernatant was passed through a HisPur cobalt column (ThermoFisher 89964, Waltham, MA, USA) and the resin washed with Wash Buffer (50 mM Tris, pH 8.0; 500 mM NaCl; 20 mM imidazole; 0.5% CHAPS; 1% Triton X-100; 10% glycerol). Cel8A was eluted from the column by adding Elution Buffer (50 mM Tris, pH 8.0; 500 mM NaCl; 150 mM imidazole; 0.5% CHAPS; 10% glycerol), concentrated, and the His6-SUMO tag was removed by adding ULP1 protease and incubation overnight in Cleavage Buffer (50 mM Tris, pH 7.0; 300 mM NaCl) at 4°C. The mixture was reapplied to the HisPur cobalt column to isolate untagged Cel8A and the pure protein was concentrated for use in enzyme assays. In order to generate a standard curve, purified Cel8A was assayed for cellulase activity using carboxymethylcellulose (CMC) as a substrate. Briefly, on ice, varying amounts of purified protein was added to a solution of 2.5% CMC dissolved in 20 mM Tris-HCl, pH 6.0, and brought to a final volume of 1 mL at a 2.0% CMC concentration. These samples were incubated at 37°C with shaking. After 1 hour, 0.5 mL of each sample was withdrawn and stored at −20°C. Enzyme activity was determined using the DNS method and the standard curve obtained by plotting the amount of Cel8A protein (mg) versus A575 (Miller 1959). The following linear dependence was obtained: y = 4112× – 0.1127, where y = A575, and x = protein (mg). The specific activity was determined to be 182 umol glucose x min−1 x mg−1.
Whole cell enzyme activity studies.
The amount of cell displayed Cel8A enzyme was determined using the CMC cellulase assay described above. Briefly, for each strain studied, cells from a glycerol stock were plated onto LB agar and antibiotic plates and incubated at 37°C to allow individual colonies to grow. Individual colonies were grown overnight at 37°C in 5 mL LB broth to saturation. The overnight culture was diluted to OD600 = 0.05 and cultured in 50 mL LB broth (with antibiotic) (the start of the culture is defined as the T = 0 time point). For cells cultured with protease inhibitor cocktail (PIC), Roche cOmplete mini EDTA-free PIC was also added to the media at 1 tablet per 10 mL media (Sigma Aldrich 11836170001, St. Louis, MO, USA.) (Westers, Westers et al. 2008). All cultures were grown at 37°C with shaking. When cultures grew to OD600 = 0.4, Cel8A production was induced by adding IPTG to a final concentration of 1 mM. This cell density typically occurred 2 to 2.5 hours after inoculation (T = 2 to 2.5 hours) and was found to be the optimal density for induction of Cel8A expression. At T = 5 and 10 hours, culture samples for activity analyses were collected and diluted in growth media to obtain OD600 = 2. A 1 mL sample of each culture was then centrifuged at 5000 × g for 3 minutes. The supernatant was collected and placed on ice, while the cell pellet was washed using 1 mL of 20 mM Tris-HCl, pH 6.0. The amount of cell-associated enzyme activity was determined by resuspending the washed cell pellet in 1 mL of 2.0% carboxymethylcellulose (CMC) dissolved in 20 mM Tris-HCl, pH 6.0. The cell-CMC mixture was incubated at 37°C with shaking for 1 hour, centrifuged at 20,000 × g for 10 minutes, and 0.5 mL of the supernatant removed and frozen at −20°C. The amount of reducing sugars generated by the cells was determined by the DNS method. Briefly, a thawed 0.5 ml sample was added to an equal volume of DNS reagent [1% 3,5 dinitrosalicylic acid, 1% NaOH, 0.2% phenol, 0.5% Na2SO3]. The mixture was vortexed, boiled for 10 minutes, and allowed to cool to room temperature. The absorbance at A575 was measured using a spectrophotometer (Shimadzu UV-1700 PharmaSpec, Kyoto, Japan) and the amount of cell-associated displayed enzyme determined using the aforementioned standard curve. A similar procedure was used to measure the amount of enzyme activity present in the culture supernatant. However, the volume of supernatant that was assayed was adjusted to ensure that the measured A575 obtained after addition of DNS was in the linear range of the spectrophotometer. Depending upon the culture assayed the following supernatants amounts were used: (i) 200 uL supernatant and 800 uL of 2.5% CMC; or (ii) 100 uL supernatant, 100 uL of 20 mM Tris-HCl, pH 6.0, and 800 uL of 2.5% CMC; or (iii) 40 uL supernatant, 160 uL of 20 mM Tris-HCl, pH 6.0, and 800 uL of 2.5% CMC.
The following procedure was used to determine the effects of additives on the stability of cell-associated cellulase activity. Cells were cultured in 40 mL LB with PIC tablets added as described above. Protein expression was induced at an OD600 = 0.4, the cells grown to T = 5 hours and then harvested by centrifugation. The cell pellets were washed twice with 30 mL water, and then resuspended in 30 mL CMC Buffer with additives (0.5% glycerol, 1.3% glucose, PIC, 0.5% glycerol + PIC, 1.3% glucose + PIC, or 75 mM sodium azide) to an OD600 = 0.6. At certain time intervals, OD600 was recorded to estimate the percentage of cells remaining and enzymatic activity assays were conducted as described above. Cell-associated enzymatic activity was normalized per volume, and cast as percent activity, with the activity of the pellet at the initial harvest immediately before resuspension with additives defined as 100%.
Cell fractionation, Western blotting, and microscopy.
A total of 200 mL of LB broth was inoculated to a starting OD600 = 0.05 with the relevant strain. Cultures were grown with shaking at 37°C, IPTG added at OD600 = 0.4 to 1 mM, and the cells were harvested after a total of 5 hours of growth (T = 5 hours). Cells were normalized to an OD600 = 333 in 0.5 mL. After centrifugation, the cell pellet and supernatant were prepared separately for immunoblot analysis. The supernatant containing secreted proteins was filtered through a 0.22 μm membrane to eliminate cell debris. Trichloroacetic acid was added to the supernatant to a final concentration of 10% to precipitate protein, and the solution was incubated on ice for 30 minutes, centrifuged at 20,000 × g for 30 minutes at 4°C, and re-suspended in 0.5 mL water (Link and LaBaer 2011). The cell pellet was washed with 25 mL Fractionation Wash Buffer [50 mM Tris-HCl, pH 7.6; 0.1% PMSF; 0.1% protease inhibitor cocktail, Sigma Aldrich P2714], prior to resuspension in 0.5 mL SET buffer [50 mM Tris-HCl, pH 7.6; 50 mM EDTA; 20% sucrose; 0.1% PMSF; 0.1% protease inhibitor cocktail, Sigma Aldrich: P2714] with 1 mg/mL lysozyme (Liew, Wang et al. 2012). The cell walls were digested for 30 minutes at 37°C, and the protoplast was pelleted by centrifugation at 20,000 × g for 30 minutes. The soluble cell wall sample was collected and saved in the freezer for later analysis. Protoplasts were resuspended in Fractionation Wash Buffer and 2X SDS-PAGE loading buffer to dilute them to a tenth of the concentration of the secreted and cell wall fractions for reasonable gel loadings, and boiled for 30 minutes to reduce the stickiness of the samples.
Samples were separated by SDS-PAGE using a NuPAGE Novex 4–12% Bis Tris 1.5 mm 15 well gel (Life Technologies NP0336BOX, Carlsbad, CA, USA). Samples were loaded such that each lane contains protein from approximately 2.5 × 108 cells and MagicMark XP Western Protein Standard (Life Technologies LC5602) was used as a molecular weight ladder. The gels were run at 200 V for 35 minutes in MES-SDS running buffer (Life Technologies NP0002). They were soaked briefly in water before transfer to a polyvinylidene difluoride (PVDF) membrane (Life Technologies iBlot2 System: IB24001, IB24002). The membrane was blocked in 20 mL TBST [20 mM Tris-HCl, pH 7.5; 500 mM NaCl, 0.05% Tween] and 5% Bovine Serum Albumin (BSA) overnight at 4°C on an orbital shaker. The membrane was then incubated with primary antibody for 1 hour [Life Technologies: anti-His mouse monoclonal antibody, MA121315, dilution of 10 uL in 10 mL TBST + 1% BSA; Abcam: anti-RpoB mouse monoclonal antibody, 8RB13, dilution of 10 uL in 10 mL TBST + 1% BSA], washed three times with 50 mL TBST, incubated with secondary antibody for 1 hour [Sigma Aldrich; anti-mouse anti-IgG linked to HRP, A9044, dilution of 2 uL in 10 mL TBST + 1% BSA], washed three times with 50 mL TBST, and then incubated with Western blot substrate. For more abundant or less abundant proteins, we used Pierce ECL Western Blotting Substrate [Life Technologies 32106] and SuperSignal West Pico Chemiluminescent Substrate [Life Technologies 34080], respectively. Membranes were visualized using a Konica Minolta SRX-101A.
Cells were observed by Differential Interference Contrast (DIC) microscopy using a Zeiss Axio Imager Z1 with 63X or 100X oil immersion objectives (Oberkochen, Germany). Cells were grown in the same conditions as mentioned above for the cellulase assay. They were harvested at T = 5 and 10 hours by centrifugation and the pellet was gently resuspended in PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4). Cover slips were coated with poly-L-lysine according the protocol described by the vendor (Sigma Aldrich P5899).
Quantitative RT-PCR for measuring gene expression.
Cells were grown in the same conditions as mentioned above for the whole cell enzyme activity studies and microscopy. 107 cells were harvested at T = 5 and T = 10 hours and briefly centrifuged, then re-suspended in RNAlater (ThermoFisher Scientific AM7020). After a one hour incubation in RNAlater at room temperature, cells were centrifuged, supernatant removed, and pellets frozen at −80°C. RNA was purified from frozen samples using the RiboPure RNA Purification Kit, bacteria (ThermoFisher Scientific AM1925) according to manufacturer guidelines. cDNA was generated in 96-well plates using the SuperScript III First-Strand Synthesis SuperMix for qRT-PCR (ThermoFisher Scientific 11752050). cDNA was used for dye-based quantitative polymerase chain reaction (qPCR) in a CFX connect Real-Time PCR detection system (BioRad 1855201, Hercules, CA, USA). qPCR was performed using the iTaq Universal Sybr Green Supermix (1725122). Primers used to generate cDNA and for qPCR are listed in Table S1. Data was analyzed using the 2−ΔΔCT method for relative quantification (Livak and Schmittgen 2001) using the DNA Gyrase B transcript as an internal control for RNA concentration and the respective gene (Cel8A-LysM, CssR, LiaR) from B. subtilis 168 Cel8A-LysM as a calibrator (i.e. all gene expression levels are normalized to B. subtilis 168 Cel8A-LysM RNA levels).
Accession numbers:
The accession numbers of the genes used in this study are as follows: Cel8A, GenBank K02088.1; amino acids 32–394; LysM, GenBank: U38819.1; amino acids 25–230; Linker, GenBank ABD31805.1; amino acids 770–880, Signal peptide, GenBank ZP_03590039.1; amino acids 1–35.
Results:
Construction of a Cel8A-LysM reporter system to quantify protein display.
We developed a reporter system to quantify heterologous protein display on the surface of vegetative B. subtilis cells. A series of proteins containing three LysM cell wall binding modules fused to the Cel8A endoglucanase from Clostridium thermocellum were expressed in B. subtilis strain 168 and several protease-deficient derivatives (Fig. 1). The LysM module is derived from the B. subtilis LytE protein and has previously been used to display β-lactamase enzymes on the cell surface, while the Cel8A enzyme was displayed because its enzymatic activity can readily be measured using a colorimetric dinitrosalicylic acid (DNS) reducing sugar assay (Miller 1959, Petre, Longin et al. 1981, Schwarz, Grabnitz et al. 1986, Chen, Wu et al. 2008). All proteins should be secreted by the microbe, as they contain an N-terminal signal peptide (SP) sequence derived from the B. subtilis PhrC protein. They also contain a hexa-histidine tag (His6) to enable detection by immunoblotting. Five protein expression constructs were characterized: (i) Cel8A-LysM, LysM fused to the C-terminus of Cel8A; (ii) LysM-Cel8A, LysM fused to the N-terminus of Cel8A; (iii) Cel8A-[L]-LysM, LysM fused to the C-terminus of Cel8A by a 110 amino acid linker segment derived from the Staphylococcus aureus FnBPB protein, (iv) LysM-[L]-Cel8A, Cel8A containing N-terminal LysM modules connected by the FnBPB linker and (v) Cel8A, which serves as a negative control because it does not contain LysM and therefore should not associate with the cell wall. The FnBPB polypeptide was used to connect the Cel8A and LysM proteins because it has previously been successfully employed to display proteins in B. subtilis, presumably because it enables heterologous enzymes to penetrate through the peptidoglycan (Strauss and Gotz 1996, Nguyen and Schumann 2006, Chen, Wu et al. 2008). Genes encoding the fusion proteins were integrated into the thrC locus on the B. subtilis genome. A list of the strains and plasmids described in this paper are presented in Table 1.
Figure 1:
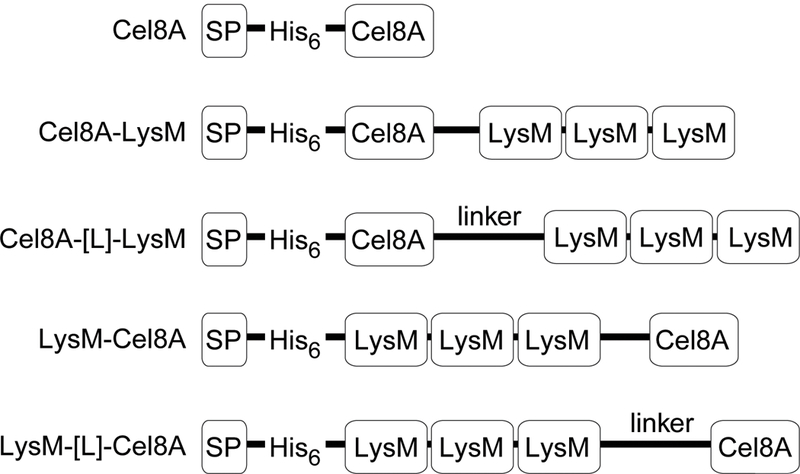
Schematic showing the recombinant proteins that were expressed in B. subtilis. Each protein contains a signal peptide from B. subtilis PhrC (SP), a hexahistidine tag (His6), the Clostridium thermocellum endoglucanase Cel8A catalytic domain (Cel8A), and the LysM cell wall binding domain from B. subtilis LytE protein (LysM). In some proteins the enzyme and cell wall binding domains are joined by a polypeptide linker derived from the Staphylococcus aureus FnBPB (linker). GenBank accession numbers are given in the materials and methods.
Fusion protein display levels were initially measured in laboratory strain B. subtilis 168 and strain WB800S in which eight extracytoplasmic proteases are genetically eliminated. WB800S is identical to strain WB800 originally constructed by Wu et al., but was further engineered to be resistant to spectinomycin rather than chloramphenicol to facilitate down-stream cloning efforts (Wu, Yeung et al. 2002). In WB800S, the genes encoding seven secreted feeder proteases (AprE, Epr, Bpr, Vpr, NprE, NprB, Mpr) and the cell wall associated protease (WprA) are deleted (Westers, Westers et al. 2004). Cells expressing each fusion protein were cultured, induced, and the cell-associated and secreted Cel8A activity was measured at mid-log phase (T = 5 hours) and at stationary phase (T = 10 hours). Strains 168 and WB800S expressing all constructs initially grew at similar rates, seemed to grow more slowly in late exponential phase, but achieved similar final densities, (representative growth data is shown for Cel8A-LysM in Fig. 2A). Only cells expressing proteins containing LysM fused to their C-termini (Cel8A-LysM and Cel8A-[L]-LysM) exhibited appreciable cell-associated Cel8A activity, with Cel8A-LysM displayed at the highest level (~18,000 per cell during exponential growth) (Fig. 2B). Higher cell-associated Cel8A-LysM and Cel8A-[L]-LysM activity is observed when the proteins are expressed in strain WB800S, presumably because of reduced proteolysis. In contrast, cells expressing LysM-Cel8A and LysM-[L]-Cel8A primarily secrete these enzymes into the growth medium and were not further characterized (data not shown).
Figure 2:
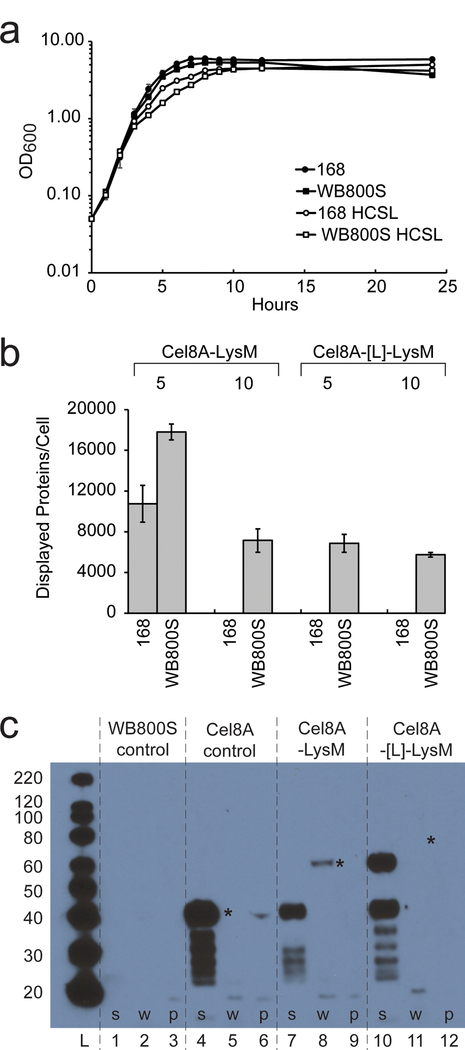
Reporter protein display in B. subtilis strains 168 and WB800S. (A) Representative growth curves of B. subtilis 168 (solid circles) and corresponding Cel8A-LysM expressing cells (open circles). Growth curves of parent WB800S and Cel8A-LysM expressing cells are also shown, open and solid squares, respectively. Protein expression only modestly decreases cellular growth. (B) Bar graph showing the amount of displayed reporter protein per cell sampled at mid-log (T = 5 hours) and stationary phase (T = 10 hours). Cel8A-LysM (left) and Cel8A-[L]-LysM (right) cells were expressed in wild-type 168 and protease-deficient WB800S strains. Per cell protein estimates were determined using a cellulase assay as described in the text. Genetically eliminating bacterial proteases increases display, but activity declines after the cells enter the stationary phase. (C) Western Blot of fractionated B. subtilis WB800S and cells expressing the Cel8A, Cel8A-LysM, and Cel8A-[L]-LysM reporter proteins. Cells were fractionated into secreted, cell wall, and protoplast fractions and reporter proteins detected using primary anti-His6 mouse monoclonal antibody, and secondary anti mouse Anti-IgG – HRP. ‘*’ indicates the expected location of the intact Cel8A, Cel8A-LysM, and Cel8A-[L]-LysM reporter proteins. A band for Cel8A-[L]-LysM in lane 11 is present, but is too faint to be seen in this image. Lanes are as follows: molecular weight ladder (L); lanes 1, 4, 7, 10 secreted fractions; lanes 2, 5, 8, 11 cell wall fractions; lanes 3, 6, 9, 12 protoplast fractions. The protoplast fraction was diluted by 10X relative to the other lanes.
Protein degradation reduces the amount of displayed reporter protein.
Whole-cell enzyme measurements suggest that the Cel8A-LysM and Cel8A-[L]-LysM fusions are not stably displayed, since lower per cell-associated enzyme activities are observed for cells at T = 10 versus T = 5 hours (Fig. 2B). To more precisely define the temporal dependence of protein display, cell-associated Cel8A-LysM and Cel8A-[L]-LysM activity was measured at various times following protein induction with IPTG at T = ~2 hours (OD600 = 0.4) (Fig. S2). Consistent with the two time-point data, higher display levels are observed in strain WB800S for both protein constructs; in both the 168 and WB800S strains, the number of Cel8A-LysM or Cel8A-[L]-LysM molecules per cell peaks during exponential growth and progressively declines as the cells enter stationary phase.
To better understand why surface enzyme activity is unstable, WB800S cells expressing the Cel8A, Cel8A-LysM, and Cel8A-[L]-LysM proteins were fractionated at T = 5 hours and the secreted, cell wall, and protoplast fractions were probed by Western blotting (Fig. 2C). As expected, the control strain that expresses Cel8A lacking the LysM modules primarily secretes this protein into the medium (lane 4), with little or no intact enzyme observed in the cell wall or protoplast fractions (lanes 5–6). In contrast, cells expressing the Cel8A-LysM fusion retain this protein in the cell wall fraction as evidenced by the presence of a band at ~63 kDa (lane 8). Cel8A-LysM likely undergoes partial proteolysis that causes it to be released from the cell surface, as an intense band is observed in the supernatant fraction at ~41 kDa that presumably corresponds to a His6-Cel8A degradation fragment that lacks the LysM modules (compare lane 7 to lane 4). The Western blotting data also explains why WB800S strains that produce Cel8A-LysM have more cell-associated activity than cells that produce Cel8A-[L]-LysM. This is because only a small amount of intact Cel8A-[L]-LysM protein is observed in the cell wall fraction of these cells, indicated by a very faint band at ~75 kDa (lane 11, marked with an “*”). In contrast, the majority of this longer construct is degraded, resulting in the release of two main degradation fragments into the medium; ~40 and 60 kDa fragments that presumably correspond to His6-Cel8A and a longer His6-Cel8A-[L] polypeptide, respectively. The notion that remaining proteases that have not been genetically eliminated act to reduce the amount of displayed proteins in WB800S is consistent with the presence of numerous lower molecular weight degradation fragments in the supernatants of all reporter-expressing cells (lanes 4, 7 and 10). Similar Western blotting analysis of fractionated WB800S cells that produce LysM-Cel8A and LysM-[L]-Cel8A revealed that they fail to display any intact protein, presumably because these protein constructs exhibit increased susceptibility to proteolysis (data not shown). Collectively, these data indicate that Cel8A proteins directly connected via their C-termini to LysM modules have increased stability as compared to Cel8A-[L]-LysM and they suggest that proteases present in both strains and/or cell wall turn-over reduces protein display levels as cells enter the stationary phase. Because strains expressing Cel8A-LysM yielded the highest levels of surface displayed protein, this reporter protein was characterized further.
Cel8A-LysM display in B. subtilis strains: WB800S, BRB07, BRB08 and BRB14.
Recently, Pohl et al. constructed several protease-deficient B. subtilis strains that may be better suited for industrial applications than WB800 (WB800S) because they genetically eliminate protease encoding genes using a markerless approach that does not introduce antibiotic resistance determinants (Wu, Yeung et al. 2002, Pohl, Bhavsar et al. 2013). To explore their utility in displaying heterologous proteins, we expressed the Cel8A-LysM reporter in strains BRB07, BRB08, and BRB14. Strain BRB07 eliminates the seven secreted scavenging proteases (AprE, Epr, Bpr, Vpr, NprE, NprB, Mpr); BRB08 eliminates the seven secreted proteases and the cell wall associated protease (WprA); and BRB14 is identical to BRB08, but also eliminates two membrane-associated proteases (HtrA, HtrB). Figure 3 shows representative growth data for all strains engineered to express either Cel8A or Cel8A-LysM (closed circle and square symbols, respectively). Strains 168, BRB07, BRB08, BRB14 and WB800S that express Cel8A protein with or without the LysM domain obtained similar OD600 values. Notably, strain BRB14 grew to lower final densities than the other strains. Parent strains grew at slightly higher rates than reporter-expressing strains, and again, BRB14 grew to lower densities (data not shown). This is consistent with previous studies that have shown that deletion of the quality control HtrA and HtrB membrane proteases adversely affects bacterial growth (Noone, Howell et al. 2001, Darmon, Noone et al. 2002). To determine the effects of protein expression and extracytoplasmic protease-deficiency on the cell morphology of parent strains and Cel8A-LysM expressing cells, strains were visualized using differential interference contrast (DIC) microscopy at mid-log (T = 5) and stationary phase (T = 10) (Fig. 4). Parent strains (WT) not engineered to express protein exhibited very similar, wild-type morphologies at both time points, as do strains 168 and WB800S expressing the Cel8A-LysM fusion protein (they form 1 to 2 rod shaped cells). In contrast, at T = 5, BRB14 cells expressing Cel8A-LysM formed a tangled mass of filamentous cells (Fig. 4). Subtle morphological changes are also observed in the related BRB07 and BRB08 strains induced to express Cel8A-LysM at T = 5, which form filaments that contain on average 3 to 4 cells. At stationary phase, cells expressing Cel8A-LysM exhibit morphologies that are generally similar to strain 168. However, the BRB-type strains, especially BRB14, have a somewhat curved and elongated shape as compared to strain 168. As discussed later in the text, these effects presumably result from changes in autolysin activity.
Figure 3:
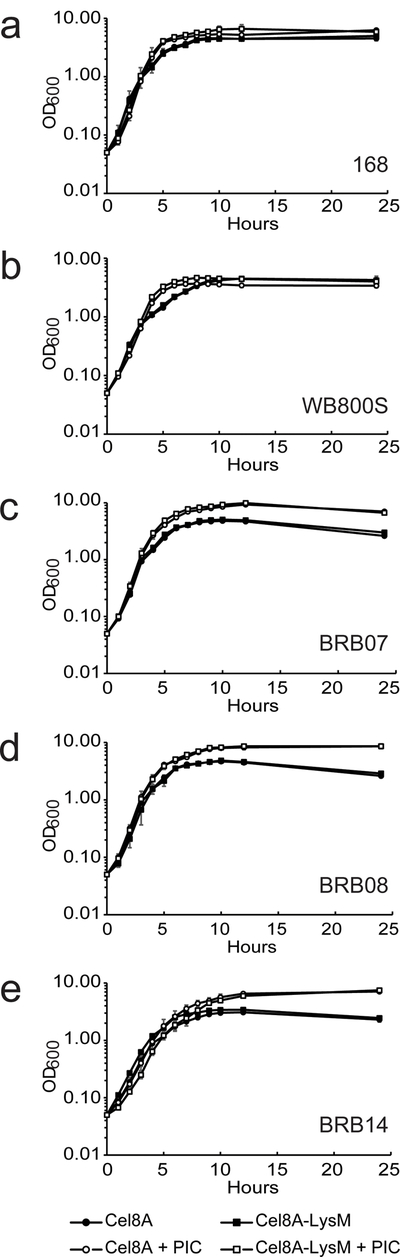
Effect of reporter protein expression and protease chemical inhibitors on cell growth. Panels show plots of OD600 as a function of time for the following cultures: (A) 168, (B) WB800S, (C) BRB07, (D) BRB08, and (E) BRB14. These cells express: Cel8A (solid circles), Cel8A in media containing protease inhibitor cocktail (PIC) (open circles), Cel8A-LysM (solid squares), Cel8A-LysM expressed in media containing PIC (open squares). Assays were performed in triplicate. Error bars reflect the standard deviation.
Figure 4:
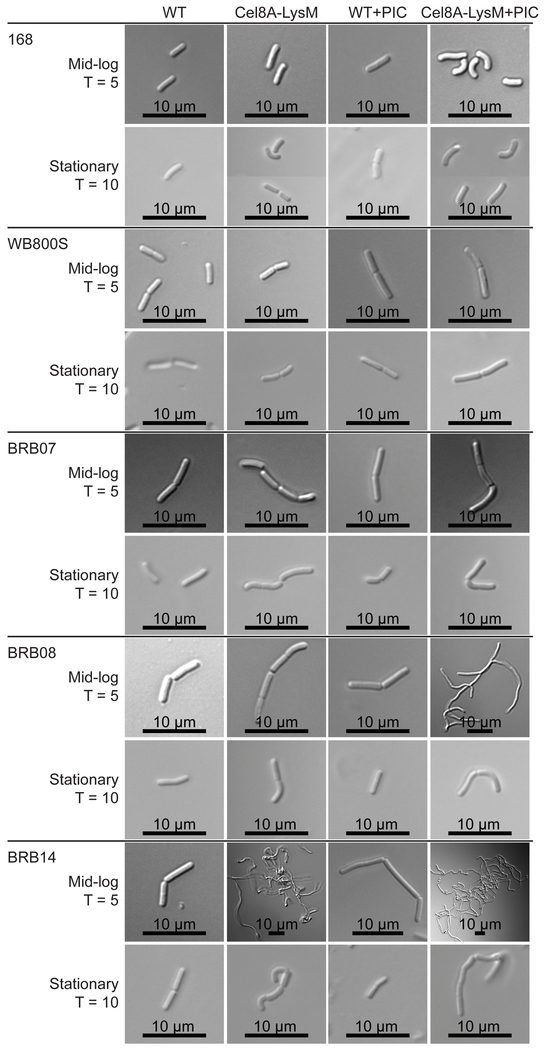
Differential interference contrast (DIC) microscopy of reporter protein expressing strains. Images were acquired for the parent strain (WT), cells expressing Cel8A-LysM (Cel8A-LysM), the parent strain grown in media containing a protease inhibitor cocktail (WT + PIC), and cells expressing the reporter protein grown in media containing a protease inhibitor cocktail (Cel8A-LysM + PIC). Images of mid-log and stationary phase cells were collected at T = 5 and T = 10 hours, respectively. Scale bar is presented at the bottom of each image.
For each Cel8A-LysM expressing strain, the amount of displayed and secreted enzyme activity was measured at mid-log (T = 5) and stationary phase (T = 10). The quantity of displayed protein is presented in two ways, either normalized to estimate the number of proteins displayed per cell (Fig. 5A, open bars), or as the total amount of cell-associated enzyme present in 100 ml of cell culture (Fig. 5B, open bars). The former analysis is valid for most strains, as they exhibit similar morphologies with strains BRB08 and BRB14 being notable exceptions as they form longer filaments under certain conditions at mid-log phase. As shown in Figure 5A, each strain displays 4900 – 18000 copies of Cel8A-LysM per cell at T = 5 hours. When assessed on a per cell level, WB800S exhibits the highest level of displayed reporter at mid-log. However, at stationary phase, this displayed activity is completely lost for all strains except for WB800S and BRB14, which retain proteins on their surface, albeit at reduced levels. Similar trends are observed when the data are normalized to account for differences in cell densities and morphologies, with all strains displaying enzyme at mid-log phase, but the levels decrease upon entering stationary phase (Fig. 5B, open bars). Extracytoplasmic protease expression is known to be upregulated upon entering stationary phase, however the decline in displayed reporter cannot be solely attributed to this effect, because all protease-deficient strains show declines in cell associated enzyme activity (Stephenson, Bron et al. 1999). Furthermore, the decline in cell-associated activity is not caused by decreases in fusion protein production, since for all strains the amount of secreted functional enzyme is larger for cell cultures collected at stationary phase as compared to cultures at mid-log phase (Fig. 5C, open bars). Thus, even though some residual Cel8A-LysM activity is retained on the surface of protease-deficient BRB14 and WB800S cells, genetically eliminating all known extracytoplasmic proteases (BRB14) does not prevent loss of displayed enzyme activity when cells are cultured in Luria-Bertani medium.
Figure 5:
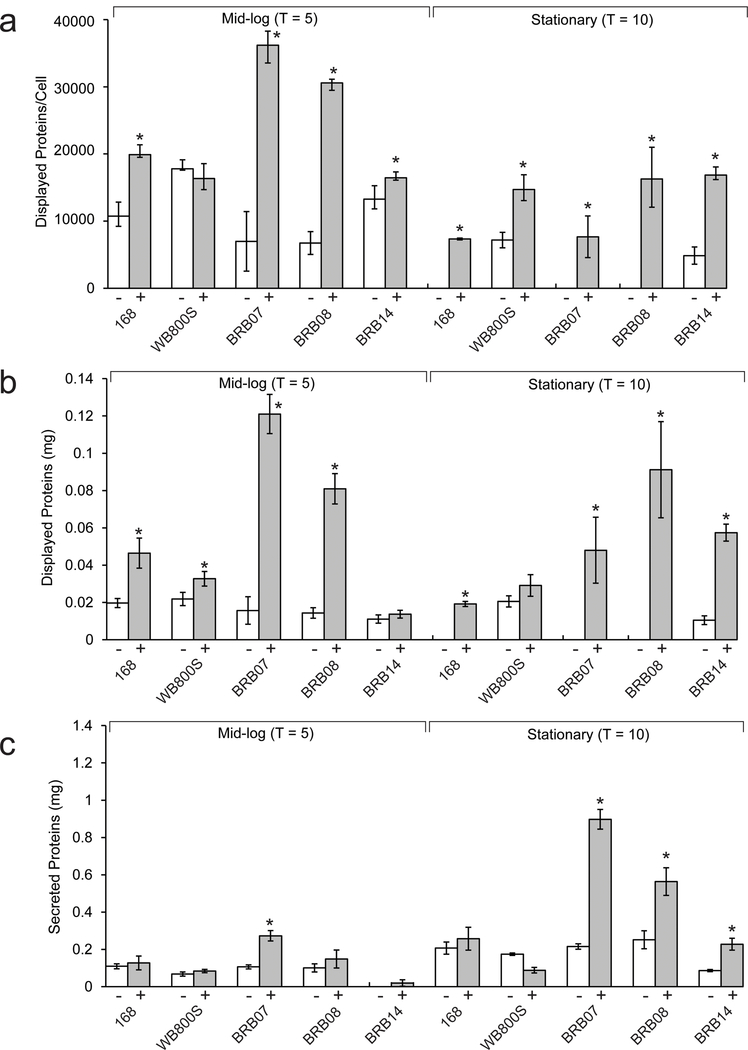
Quantification of displayed and secreted cellulase activity in different Cel8A-LysM expressing bacterial strains. Cellulase activity was determined for cultures at mid-log (T = 5 hours) and stationary phase (T = 10 hours), left and right side of each figure, respectively. The amount of measured cell-associated cellulase activity is represented as either: (A) the number of reporter proteins displayed per cell (normalized based on the culture’s OD600) or (B) the calculated number of milligrams of reporter protein displayed in a 100 mL culture of the cells. The latter activity is not normalized based on cell density. For each Cel8A-LysM expressing strain, activity measurements were made for cells grown in the absence (open bars, “−” symbol) or presence (filled bars, “+” symbol) of protease inhibitors in the growth media. Panel (C) reports on the amount of enzyme activity in the secreted fraction and is reported as milligrams of secreted reporter per 100 mL. Asterisks above the bars indicate that there is a statistically significant difference between the displayed cellulase for cultures grown in the presence and absence of protease inhibitors. The per cell estimates of displayed protein at mid-log are not accurate for strain BRB14 (both in the presence and absence of protease inhibitors) and BRB08 (in the presence of protease inhibitors). This is because these cells form filaments at mid-log when Cel8A-LysM is expressed. All assays were performed in triplicate, and error bars reflect the standard deviation.
Chemically inhibiting proteolysis increases the amount of displayed protein.
In the protease-deficient strains, cell-associated Cel8A-LysM activity declines upon entering the stationary phase (T = 10 hours), presumably because the reporter is degraded by an unidentified protease and/or lost from the cell surface as a result of cell wall turnover. To determine if residual protease activity was responsible for loss of the reporter, we measured display levels after culturing cells in growth media that was supplemented with a protease inhibitor cocktail (PIC) that chemically inhibits the activity of serine- and cysteine-type proteases (solid bars in Fig. 5). This cocktail was used because it had been previously employed by Westers et al. to study the B. subtilis exoproteome (Westers, Westers et al. 2008). For all strains, when the total amount of cell-associated activity is measured, significant improvements in display are observed when PIC is present (compare solid and open bars in Fig. 5B). This is especially noticeable in strains BRB07 and BRB08, since they respectively exhibit 7.6- and 5.8-fold increases in total displayed protein activity when PIC is present as compared to cell cultures that lack this additive. As described below, all strains exhibit generally similar morphologies at stationary phase in the presence of PIC, enabling the amount of protein displayed per cell to be compared (Fig. 5A, solid versus open bars). However, strain BRB14 is somewhat unique, since when these cells express Cel8A-LysM, they are more curved and elongated at stationary phase when they are grown with or without protease inhibitors. Normalization of the data reveals that strains WB800S, BRB08 and BRB14 display the highest amount of reporter protein per cell at stationary phase. Thus, deleting only the feeder proteases and WprA is sufficient to achieve maximal levels of per cell display when other, presumably intracellularly released proteases in the culture media are chemically inhibited. PIC in the culture media also increases Cel8A activity in the secreted fraction (Fig. 5C), consistent with previous studies that have shown that chemically inhibiting proteases stabilizes the B. subtilis exoproteome (Westers, Westers et al. 2008). We conclude that chemical protease inhibition can significantly increase the amount of heterologous protein displayed on the cell surface, even when the protein is expressed in B. subtilis strains that lack all known extracytoplasmic proteases. The chemical components of the cocktail presumably inhibit cytosolic proteases that are released as a result of cellular autolysis, which otherwise degrade surface attached Cel8A-LysM (Smith, Blackman et al. 2000).
Interestingly, adding protease inhibitors to the growth media enables both the parent and reporter expressing B. subtilis cultures to reach slightly higher optical densities, with the largest increases observed for strains BRB07, BRB08 and BRB14 (Fig. 3). A chemical component within the proprietary inhibitor mixture presumably aids bacterial growth as PIC increases the number of colony-forming units (CFUs) in cultures of BRB08 at T = 10 hours. (Fig. S3). However, the CFU data also indicate that when the BRB08 cells are rapidly growing during mid-log phase, OD600 measurements may overestimate bacterial growth. This is consistent with DIC images of reporter-expressing BRB08 cells at mid-log phase (T = 5 hours), which show in that in the presence of PIC the cells form filaments and clump (Fig. 4). Their morphology is similar to BRB14 cells at T = 5 hours expressing Cel8A-LysM cultured both in the presence or absence of PIC, which form even longer and more tangled filaments when PIC is present. More subtle PIC-dependent cell curvature occurs for other cells at T = 5 hours when they are expressing Cel8A-LysM, but with the exception of the BRB14 strain, these effects generally subside upon entering stationary phase and the cells appear to separate (T = 10 hours). For the isogenic BRB-type strains, the observed morphological differences highlight the importance of extracytoplasmic proteases in cell separation during exponential growth, as the Cel8A-LysM dependent effects become more pronounced as these enzymes are deleted or chemically inhibited.
Eliminating quality control proteases in BRB14 significantly increases cell membrane stress.
The protein expression induced cell-morphology differences in the isogenic BRB-strains (Fig. 4) may be caused by the accumulation of misfolded reporter protein in the membrane which alters autolysin function and induces cell stress. To investigate this issue, we used RT-qPCR to monitor the effect of protease inhibition and protein expression on membrane and cell envelope stress responses. Induction of the membrane stress response was determined by measuring cssR mRNA levels, which is part of the CssRS two-component response regulator that upregulates HtrA and HtrB protease levels during periods of secretory stress in B. subtilis (Hyyrylainen, Bolhuis et al. 2001, Westers, Westers et al. 2006, Marciniak, Trip et al. 2012). The effect of protein expression on cssR mRNA levels was determined by comparing the parent and Cel8A-LysM expressing strains (Fig. 6). BRB14 exhibits the highest level of stress (six-fold increases or greater in gene expression at mid-log phase) that, unlike the other strains, remains elevated in stationary phase. To probe the effect of chemical inhibition of extracytoplasmic proteases, we measured cssR RNA levels when the cells were cultured in media containing PIC. For nearly all parent strains, the presence of PIC increased cssR expression at both T = 5 and T = 10 hours (with some exceptions). Notably, strain BRB14 exhibited the highest levels of stress, with both parent and Cel8A-LysM cells showing significant upregulation of the cssRS operon when PIC was present. Previous studies have shown that protein secretion can also induce expression of the cell wall stress regulon which may protect the bacterium against oxidative stress or cell wall targeting antimicrobials (Mascher, Zimmer et al. 2004, Wolf, Kalamorz et al. 2010). Cell wall stress is detected by the LiaRS two-component response system and has been shown to cause upregulation of liaR. For each strain, we used RT-qPCR to monitor how liaR mRNA levels changed in response to Cel8A-LysM expression and the presence of protease inhibitors. In general, LiaR gene expression was unchanged, indicating that neither protease deficiency nor protein secretion activate stress response pathways related to cell wall stress (data not shown). Thus, we conclude that as compared to the other strains, BRB14 that lacks genes encoding all extracytoplasmic proteases exhibits significantly more secretion stress when expressing Cel8A-LysM or when it is cultured in media that contains protease inhibitors.
Figure 6:
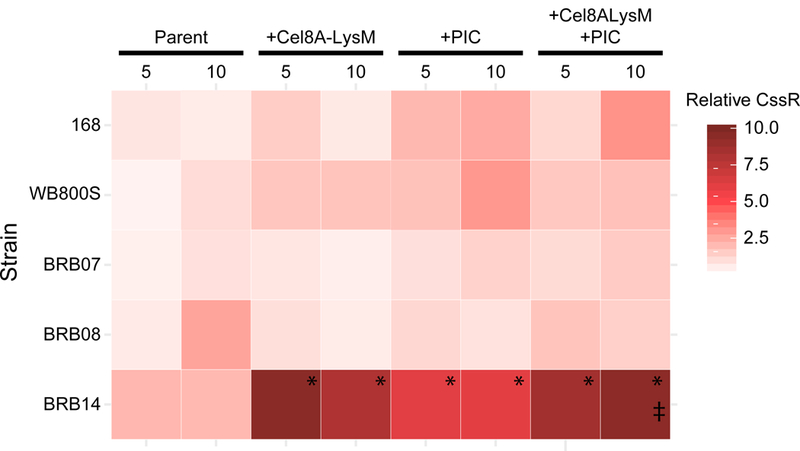
Heat map representing relative cssR expression levels in different strains and under different growth conditions. cssR expression levels in five strains were compared, and for each include the: parent strain (Parent), cells expressing the reporter (+ Cel8A-LysM), the parent strain cultured in the presence of protease inhibitors (+ PIC), and cells expressing the reporter cultured in the presence of protease inhibitors (+ Cel8A-LysM + PIC). Key indicates relative color association with the respective fold-change in cssR, relative to B. subtilis 168 + Cel8A-LysM. (*) Indicates strains experienced significant secretory stress relative to the parent strain (> 4 fold). (‡) Indicates strains experienced significant secretory stress as a result of Cel8A-LysM in the presence of PIC (> 4 fold), relative to the parent strain grown in the presence of PIC (i.e. Cel8A-LysM expression causes elevated secretory stress in strains already grown in the presence of PIC). Significance was determined using Student’s single-tail t-test, p<0.05. See materials and methods for controls.
Two-step procedure to create stable enzyme coated cells.
We explored conditions to produce stable Cel8A-LysM coated BRB08 cells, as this strain exhibits a high level of cell-associated activity at 10 hours when cultured with protease inhibitors and because reporter expression causes only modest levels of membrane stress (Figs. 5–6). Enzyme-coated cells were produced using a two-step procedure. First, BRB08 cells expressing Cel8A-LysM were grown in the presence of PIC and harvested at T = 5 hours. Cells were then washed and resuspended in buffer (20 mM Tris-HCl, pH 6.0) containing different additives. To determine how different solution conditions affected enzyme and cell stability, the cells were incubated at 37° C with shaking and the OD600 and cell-associated enzyme activity was determined at various times up to 2.5 days. When the cells are resuspended in buffer containing no additives, the OD600 steadily declines to ~35% of its original value by 66 hours (Fig. 7A). A concomitant decrease in cell-associated activity also occurs and is almost completely lost after 66 hours. The decline in OD600 is compatible with previous studies that have shown that B. subtilis undergoes autolysis when deprived of a carbon source, and suggests that the decline in cell-associated enzyme activity is caused by released cytosolic proteases that degrade Cel8A-LysM (Jolliffe, Doyle et al. 1981). This idea is supported by data collected using sodium azide as an additive. This small molecule is known to cause autolysis by decoupling oxidation from phosphorylation and in the assay leads to rapid decreases in cell-associated enzyme activity and OD600 (Fig. 7E) (Harold 1972, Jolliffe, Doyle et al. 1981). Furthermore, when PIC is included in the incubation buffer to prevent proteolysis, the cell-associated activity is stabilized, compatible with the idea that PIC inhibits released cytoplasmic proteases that degrade the reporter protein (Fig. 7D). We sought to prevent autolysis so as to obviate the need for adding PIC to the resuspended cells, as this is a proprietary commercial cocktail and its presence may also act to inhibit the activity of heterologous displayed enzymes. Jolliffe et al. demonstrated that the presence of glycerol or glucose in the culture media prevents cell lysis, presumably by maintaining the electrochemical gradient across the membrane bilayer (Jolliffe, Doyle et al. 1981). We therefore resuspended the enzyme coated cells in buffer containing either 0.5% glycerol or 1.3% glucose. In glycerol, approximately ~60% of the optical density is retained after 66 hours, and the displayed enzyme activity is very stable (Fig. 7C). Similar beneficial effects are observed when glucose is used as an additive, with enzyme activity maintained throughout the duration of the experiment (Fig. 7B). Adding PIC to buffers containing either the glycerol or glucose additives did not significantly improve cell-associated enzyme activity, suggesting that the additives are sufficient to maintain cell integrity and prevent the release of cytoplasmic proteases (not shown).
Figure 7:
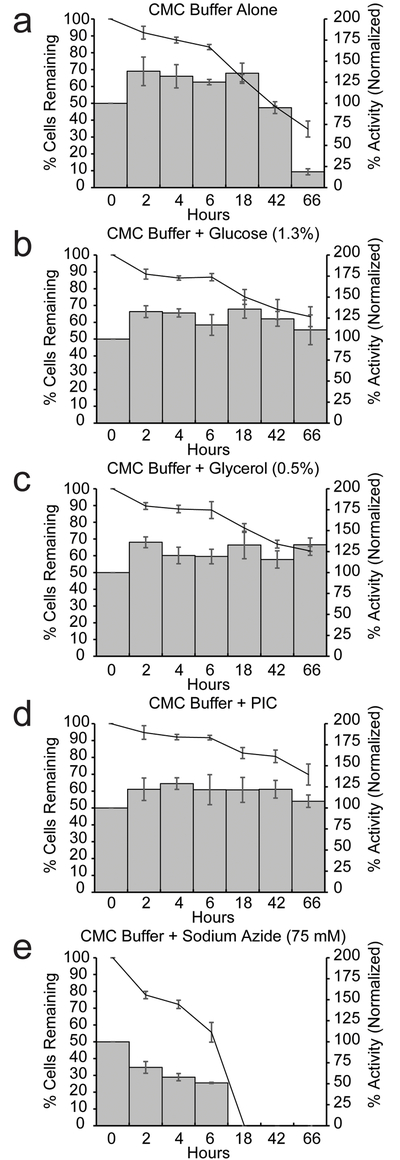
A two-step procedure stabilizes displayed Cel8A-LysM protein activity and decreases cell autolysis. Cells were prepared using the two-step procedure. Cell-associated activity at the time of harvest was determined. After incubation in buffers with additives, activity was normalized to the initial cell-associated activity at time of harvest. Cell-associated activity (bars) and percentage of remaining cells (lines) are shown. Cells were dissolved in: (A) CMC buffer, (B) CMC buffer containing 1.3% glucose, (C) CMC buffer containing 0.5% glycerol, (D) CMC buffer containing protease inhibitors, and (E) CMC buffer containing 25 mM sodium azide. Assays were performed in triplicate. The error bars are the standard deviation.
An immunoblot analysis of fractionated cells reveals that the two-step procedure stabilizes cell-associated Cel8A-LysM activity by limiting its proteolytic degradation (Fig. 8). In the first step of the procedure, protease inhibitors are added to the growth media to prevent the action of released intracellular proteases so that the cells are maximally decorated with intact enzyme. To demonstrate the effects of PIC on Cel8A-LysM display, protein expressing BRB08 cells were cultured in the presence or absence of PIC, harvested at T = 5 hours, and fractionated. For cells cultured without PIC, the full length 63 kD Cel8A-LysM protein correctly localizes to the cell wall (lane 8), but a large portion of the protein is proteolyzed and present in the secreted fraction as a ~40 kD fragment (lane 7). This fragment likely corresponds to Cel8A that contains the N-terminal His6 tag, as it is similar in molecular weight to the protein produced by BRB08 cells that express the Cel8A reporter. Interestingly, when PIC is added to the growth media the amount of cell wall associated Cel8A-LysM protein is similar (lane 11), but significantly lower amounts of degradation product is observed in the supernatant (lane 10). The presence of PIC in the growth media is clearly needed to prevent Cel8A-LysM degradation, as protein-expressing BRB08 cells grown in the presence of glycerol, but without PIC, shed degraded protein into the supernatant (lane 13). In the second step of the procedure, cells are resuspended in a buffer that contains either glycerol or glucose to prevent lysis of enzyme decorated cells. An immunoblot analysis substantiates that when glycerol is used as an additive in these suspensions, it prevents proteolytic degradation. Protein-coated cells produced using the first step of the procedure were dissolved in glycerol containing buffer, incubated at 37° C for 66 hours, and fractionated. Abundant, full-length cell wall-associated Cel8A-LysM predominates in the cell wall (lane 17), with no detectable degradation products shed from the cells into the supernatant (lane 16).
Figure 8:
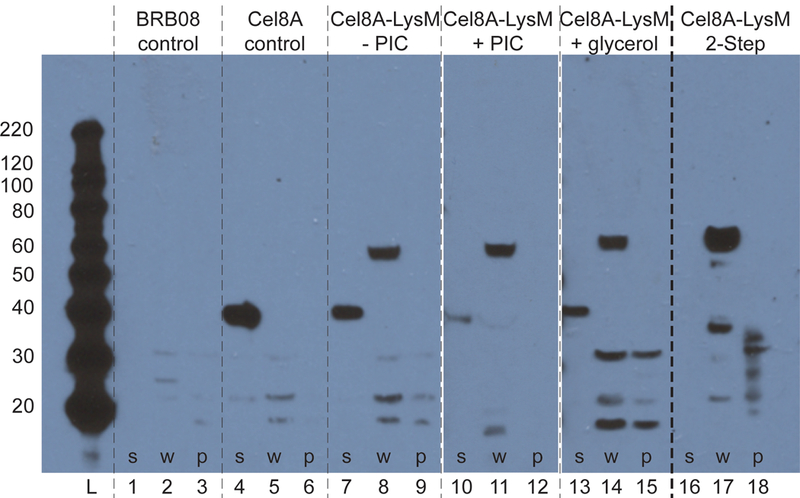
Western blot of fractionated B. subtilis BRB08 cells demonstrating stabilization of displayed Cel8A-LysM. Cells were fractionated into secreted, cell wall, and protoplast fractions and the presence of the reporter proteins detected using a primary anti-His6 mouse monoclonal antibody. Lanes 1–15 show the results of fractionating cells harvested at mid-log (T=5 hours): BRB08 parent strain (lanes 1–3); BRB08 expressing Cel8A (lanes 4–6); BRB08 expressing Cel8A-LysM in conventional growth media (lanes 7–9); BRB08 expressing Cel8A-LysM in growth media containing protease inhibitors (lanes 10–12); BRB08 expressing Cel8A-LysM in growth media containing 0.5% glycerol (lanes 13–15). Lanes 16–18 show the results of fractionating cells prepared using the two-step procedure that were incubated for 66 hours in stabilizing buffer containing 0.5% glycerol (corresponding activity for these cells is presented in figure 7C. Lanes are as follows: molecular weight ladder, lanes 1, 4, 7, 10, 13, 16 are secreted fractions; lanes 2, 5, 8, 11, 14, 17 are cell wall fractions; lanes 3, 6, 9, 12, 15, 18 are protoplast fractions. The protoplast fraction was diluted by 10X relative to the other lanes. The reporter proteins are expected to migrate on the gel at the following positions: Cel8A (41.3 kDa) and Cel8A-LysM (63.2 kDa).
Discussion:
Protein-displaying bacteria may be valuable biotechnological tools (Lee, Choi et al. 2003, Wernerus and Stahl 2004, Schuurmann, Quehl et al. 2014, Li and Tao 2015, Smith, Khera et al. 2015). Several research groups have engineered vegetative B. subtilis cells to display heterologous proteins because there are well-established methods to manipulate its genome, it is used industrially to produce biocommodities, it can secrete prodigious amounts of protein, and because it has generally recognized as safe status (Schallmey, Singh et al. 2004, Liu, Liu et al. 2013). However, factors that influence the density and stability of displayed heterologous proteins on vegetative cells are not well understood. Here we compared the ability of different protease-deficient B. subtilis strains to display a heterologous reporter protein that interacts with the peptidoglycan via a LysM module and we developed a two-step procedure to stably display the reporter for several days. To identify a suitable reporter to monitor protein display, we constructed a series of fusion proteins that contained LysM peptidoglycan binding modules fused to C. thermocellum Cel8A endoglucanase and we determined their display efficiencies using whole cell cellulase assays and Western blotting. Our results indicate that the positioning of the LysM modules relative to Cel8A, as well as the polypeptide segment that connects it to the Cel8A enzyme, are critical determinants for display and stability. The highest levels of stable display are achieved using a Cel8A-LysM reporter that contains three LysM modules appended directly to the C-terminus of Cel8A. Interestingly, in a study by Chen et al. they observed higher display levels when LysM was placed at the N-terminus of β-lactamase with a 55 amino acid linker, and You et al. displayed a protein scaffold on the cell surface using N-terminal LysM domains without an additional linker region (Chen, Wu et al. 2008, You, Zhang et al. 2012). It is interesting to note that in a covalent cell wall attachment system, a 123 amino acid linker derived from FnBPB was necessary to allow the protein of interest to extend to the outside of the cell wall (Nguyen and Schumann 2006), which may not be necessary for proteins fused to noncovalent binding motifs. Thus, when designing proteins for display, the optimal positioning of the LysM domains relative to the displayed protein presumably needs to be optimized on a case-by-case basis.
It is well known that B. subtilis produces at least ten extracytoplasmic proteases that degrade both secreted and cell wall associated proteins (Tjalsma, Antelmann et al. 2004, Harwood and Cranenburgh 2008). These include seven secreted “feeder” proteases, the cell wall associated WprA enzyme, and the membrane associated HtrA and HtrB proteases. To learn how these enzymes impact heterologous protein display, the Cel8A-LysM reporter was expressed in four different protease-deficient B. subtilis strains and the amount of displayed and secreted protein was quantified. The strains tested include wild type strain 168, which contains a full complement of extracellular and surface associated bacterial proteases, and four previously reported protease-deficient strains (BRB07, BRB08, BRB14 and WB800) (Wu, Yeung et al. 2002). The BRB-type strains were recently constructed and progressively delete proteases: BRB07 eliminates the seven feeder proteases; BRB08 eliminates the feeder proteases and WprA, and BRB14 eliminates all of the extracellular and cell surface associated proteases, including the membrane associated HtrA and HtrB enzymes (Pohl, Bhavsar et al. 2013). Strain WB800 was studied because it has been used extensively in the research literature (Wu, Yeung et al. 2002). WB800 and BRB08 eliminate the same set of enzymes, but unlike the BRB-type strains, the protease genes in WB800 are disrupted by introducing antibiotic resistant determinants. The abundance and stability of secreted heterologous proteins is reduced by the action of the extracytoplasmic proteases and is thought to depend upon the rate at which the protein folds on the outer-membrane surface, as well as the presence of protease sensitive sites within the final folded form of the protein (Stephenson and Harwood 1998, Jensen, Stephenson et al. 2000, Sarvas, Harwood et al. 2004, Pohl and Harwood 2010). Surprisingly, all strains are capable of displaying Cel8A-LysM when growing exponentially, but regardless of their exterior protease complement, the amount of cell-associated cellulase activity declines upon entering the stationary phase (Fig. 5). Displayed cellulase reporter activity is completely lost for strains 168, BRB07, and BRB08, while strains WB800S and BRB14 retain only ~40% activity. A more detailed temporal analysis of reporter display on WB800S cells reveals that surface activity peaks before the cells enter stationary phase and then declines significantly (Fig. S2). These findings were surprising, since prior studies have shown that when the seven secreted feeder proteases are deleted, only 0.15% of the extracellular protease activity remains (Ye, Yang et al. 1996), and that further reductions in activity are achieved by also deleting the wall-associated WprA protease (Stephenson and Harwood 1998).
We hypothesize that in all B. subtilis strains, reporter proteins are shed from the cell surface as a result of cellular autolysis which releases cytosolic proteases that degrade surface attached Cel8A-LysM (Jolliffe, Doyle et al. 1980). This idea is consistent with previous studies that demonstrated that B. subtilis autolyses during the stationary phase and that this process is accelerated in protease deficient strains (Jolliffe, Doyle et al. 1980, Stephenson, Bron et al. 1999, Krishnappa, Dreisbach et al. 2013). The greater propensity of protease-deficient strains to lyse is thought to be caused by increased autolysin activity, which is normally controlled by the major feeder proteases NprE and AprE (Stephenson, Bron et al. 1999). It is also substantiated by our findings. First, our immunoblot analyses of Cel8A-LysM expressing WB800S cells reveal that a Cel8A degradation product accumulates in the secreted fraction even though these cells lack all known secreted and wall associated bacterial proteases (Fig. 2C). Second, we have shown that significant gains in the amount of displayed Cel8A-LysM protein occurs when the strains are cultured in media that is supplemented with protease inhibitors (Fig. 5). Chemical protease inhibition has a very large effect, with all strains retaining displayed cellulase activity even after entering the stationary phase, and up to ~7-fold gains in cell-associated activity achieved at mid-log. This effect occurs in all protease-deficient strains, suggesting that the chemical inhibitors increase display in part by limiting the activity of cytosolic proteases that are released upon autolysis. Finally, we have shown that is possible to stabilize reporter protein display when the cells are resuspended in buffers containing additives that are known to limit autolysis (Figs. 7–8). On a per cell basis, strains WB800S, BRB08 and BRB14 display the greatest amount of reporter protein at stationary phase after being cultured with protease inhibitors (Fig. 5A). This suggests that knocking out the feeder proteases in BRB07 is insufficient to stabilize protein display and that other extracytoplasmic proteases must be removed. Interestingly, strain BRB14 produces the least amount of protein (sum of secreted and displayed activity), suggesting that the HtrA and HtrB membrane associated proteases are needed to secrete and display properly folded Cel8A-LysM. Overall, these data intimate that in both wild-type and protease deficient strains of B. subtilis, autolysis releases cytosolic proteases that degrade displayed proteins. However, protein loss can be greatly reduced by chemically inhibiting the proteases.
Reporter protein expression in strain BRB14 that lacks the full complement of extracytoplasmic proteases causes membrane stress and impairs proper cell separation (Fig. 6). During exponential growth in conventional media, BRB14 adopts an anomalous filamentous phenotype when Cel8A-LysM is expressed (Fig. 4), which is consistent with prior reports that these cells exhibited impaired growth (Pohl, Bhavsar et al. 2013). Interestingly, the other protease-deficient strains we tested do not form these filaments under similar conditions. As only strain BRB14 disrupts HtrA and HtrB function, we reason that these membrane associated proteins are required for proper cell separation when Cel8A-LysM is produced. In B. subtilis, LytC and LytD are the primary autolysins that cleave cell wall material at the nascent septum to enable cell separation during vegetative growth (Smith, Blackman et al. 2000). Notably, the phenotype of a lytC-lytD- mutant (Blackman, Smith et al. 1998) and Cel8A-LysM expressing BRB14 cells are generally similar, since both mutants form long chains during exponential growth that shorten when the cells enter the stationary phase. The HtrA and HtrB proteases are upregulated via the CssRS two component regulatory system as a result of secretion stress and are thought to clear misfolded exported proteins at the membrane-cell wall interface (Hyyrylainen, Bolhuis et al. 2001). We hypothesize that during exponential BRB14 cell growth, the lack of HtrA/HtrB activity causes misfolded Cel8A-LysM to accumulate in the membrane, disrupting the function of these autolysins by either preventing their proper localization to the septum or by changing the physiochemical properties of the septal cell wall material to make it less susceptible to cleavage. However, as growth slows when entering the stationary phase, residual autolysin activity at the septum is sufficient to resolve the cells into shorter chains. The effects on growth and morphology may in part also be caused by induction of the wall stress induced WalR operon, which is expected to interfere with cell-wall biosynthesis and induce the expression of HtrC, a membrane protease that is homologous to HtrA and HtrB (Fabret and Hoch 1998, Hyyrylainen, Bolhuis et al. 2001, Noone, Howell et al. 2001, Pohl, Bhavsar et al. 2013). To a lesser extent, other cell wall and secreted feeder proteases appear to compensate for lost HtrA and HtrB activity. This is because BRB08 cells that only contain the membrane proteases HtrA and HtrB also fail to separate properly when co-cultured with protease inhibitors, presumably because these chemicals inhibit HtrA and HtrB. However, strains that also produce WprA (BRB07) or WprA and the feeder proteases (168) do not form filaments when cultured with protease inhibitors, possibly because their extracytoplasmic proteolytic activity is not fully disrupted by the inhibitor cocktail enabling these enzymes to clear misfolded Cel8A-LysM from the surface that would otherwise adversely affect autolysin function. The important role of HtrA and HtrB in reporter protein display is supported by RT-qPCR measurements of cssR gene expression, which indicate that BRB14 cells expressing Cel8A-LysM exhibit the largest amount of secretion stress when grown in conventional media, relative to other protease-deficient strains (Fig. 6). Protein expression upregulates cssR expression 9-fold in BRB14 cells, whereas other strains exhibit substantially smaller, but sometimes significant up to 2 fold increases. This is consistent with proteomic studies that have shown that α-amylase expression in B. subtilis cells lacking the membrane proteases induces secretion stress, whereas eliminating the seven extracellular feeder proteases has less significant effects (Antelmann, Darmon et al. 2003). It is also consistent with results reported by Westers et al., who used a HtrB-LacZ fusion assay to determine that secretion stress is measurably increased in WB800 as a result of heterologous protein secretion (Westers, Westers et al. 2006). We also observed this stress induction in WB800S and other protease-deficient strains expressing Cel8A-LysM during mid-log phase, though we note that Westers et al. observed more significant stress in extra rich media than in Luria-Bertani Broth, and only assayed HtrB-LacZ activity after the onset of stationary phase. In general, chemically inhibiting proteases causes only minimal stress in all strains in the presence or absence of Cel8A-LysM expression. BRB14 is a notable exception since these cells, even when not expressing the reporter, are stressed by adding protease inhibitors (6-fold increase in cssR mRNA). This is likely a result of protease inhibitors exacerbating the accumulation of slowly- or mis-folding proteins at the membrane in the absence of HtrA and HtrB proteases. Display of Cel8A-LysM can be expected to coat the microbial cell wall. However, this does not appear to causing significant levels of cell wall stress, as RT-qPCR measurements revealed similar levels of liaR expression in all strains regardless of Cel8A-LysM expression or the presence of protease inhibitors. This finding is generally consistent with microarray studies reported by Marciniak et al., which showed that only some types of secreted proteins cause the liaFRS operon to be upregulated (Marciniak, Trip et al. 2012).
Our results indicate that strains WB800S and BRB08 differ markedly, even though they are derived from the same parent strain and lack the same set of proteases (Wu, Yeung et al. 2002, Pohl, Bhavsar et al. 2013). In particular, the cells behave differently when protease inhibitors are added to the growth media. When grown in media that lacks chemical protease inhibitors, WB800S displays a higher amount of reporter protein on its cell surface at both mid-log and stationary phase (Fig. 5). However, when chemical protease inhibitors are added, the amount of Cel8A-LysM displayed on BRB08 surpasses that of WB800S at both mid-log and stationary phase. Moreover, unlike WB800S, reporter-expressing BRB08 cells form filaments in the presence of chemical protease inhibitors (Fig. 4), and all BRB strains experience beneficial growth effects when protease inhibitors are present (Fig. 3). While it is beyond the scope of this study to identify the molecular basis of these discrepancies, they could stem from the way each strain was created. Notably, unlike the markerless and precise deletions made in the BRB strains, the protease genes in WB800 were disrupted by partial deletions removing the promoter, signal peptide, and part of the pro-region (Wang, Bruckner et al. 1989, He, Shyu et al. 1991) and by disrupting protease genes with the antibiotic resistance genes blasticidin, bleomycin and hygromycin (Wu, Yeung et al. 2002). The additional genetic burden of expressing these antibiotic resistance genes could be responsible for the slightly lower growth and protein production of WB800S. Additionally, strain WB800 may have acquired additional genetic modifications since it was first reported in 2002. This idea is supported by the fact that more recent studies using WB800 have shown that it is now resistant to chloramphenicol (Nguyen, Phan et al. 2011), even though this phenotype was not reported in the original strain (Wu, Yeung et al. 2002). Genome sequencing analyses are needed to elucidate the origin of the strain-specific differences.
To the best of our knowledge, our study is the first to determine how genetically and chemically eliminating all B. subtilis extracytoplasmic proteases affects heterologous protein display. However, other groups have studied how these parameters influence the abundance and stability of secreted heterologous proteins. Pohl et al. used the BRB strains to examine the effect of deleting extracytoplasmic proteases on the amount of secreted anthrax protective antigen (PA) (Pohl, Bhavsar et al. 2013). PA reached highest concentrations in the growth medium of BRB08, and similar to our findings with Cel8A-LysM, strain BRB14 produced lower amounts of heterologous PA. Interestingly, they found that levels of SdpC, a toxin that kills non-sporulating cells by collapsing the PMF and inducing autolysis was significantly increased in BRB07 and BRB08, as compared to B. subtilis 168. This supports our hypothesis that Cel8A-LysM display levels decrease during stationary phase due to cell lysis. Similar to our findings with surface displayed Cel8A-LysM, Westers et al., demonstrated that adding chemical protease inhibitors to the growth media and using strains that genetically deleted endogenous extracytoplasmic proteases jointly improved the production of protease-sensitive secreted proteins (Westers, Westers et al. 2008).
For many biotechnological applications, protein-coated B. subtilis cells will need to be stable for several days. We therefore developed a two-step procedure to stably coat BRB08 cells with Cel8A-LysM. BRB08 cells were used because they can be coated with proteins at high density and they eliminate the greatest number of proteases without causing large increases in membrane stress (Fig. 7). In the procedure, BRB08 cells expressing Cel8A-LysM are grown to mid-log phase in the presence of chemical protease inhibitors, and then harvested and dissolved in stabilizing buffer that contains either glycerol or glucose. These oxidizable carbon sources are believed to help maintain the electrochemical membrane gradient, which if lost may cause changes in bacterial surface chemistry that promote autolysin activity (Jolliffe, Doyle et al. 1981). The two-step procedure using either glycerol or glucose additives greatly increases the stability of protein coated B. subtilis cells, enabling the intact Cel8A-LysM reporter to be displayed for nearly three days (Fig. 7). Other additives may also prove useful in stabilizing displaying proteins, such as TiO2 nanoparticles that have recently been show to limit autolysis as well (McGivney, Han et al. 2017). Further rational modification of the protease deficient strains may also improve display and stability, as genetically eliminating the production of lytC and/or lytD (or σD that controls their expression) reduces cellular lysis (Blackman, Smith et al. 1998) and improves protein display (Kobayashi, Toida et al. 2000). However, these mutant strains adopt a filamentous phenotype during exponential growth that may exacerbated when the cells are further engineered to display proteins. Potentially, fine-tuning the levels of modulators of autolysin activity such as YoeB may prove useful to stabilize the cell wall and inhibit cell lysis (Salzberg and Helmann 2007). Significant gains in copy number and stability could also be achieved by displaying heterologous proteins that are more resistant to proteolysis than the Cel8A-LysM reporter. Finally, directed evolution approaches have enormous promise for creating surface optimized engineered microbes, which would have a plethora of potential biotechnological applications.
Supplementary Material
Acknowledgements:
We thank the Bacillus Genetic Stock Center (BGSC), Cobra Biologics, and Dr. Beth Lazazzera for providing plasmids and strains, and Dr. Peter Bradley for allowing us to use his microscope. This material is based upon work supported by the U.S. Department of Energy Office of Science, Office of Biological and Environmental Research program under Award Number DE-FC02-02ER63421. GL Huang was supported by a Ruth L. Kirschstein National Research Service Award GM007185.
References:
- Anagnostopoulos C, Spizizen J (1961) Requirements for transformation in Bacillus subtilis. J Bacteriol 81(5): 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antelmann H, Darmon E, Noone D, Veening JW, Westers H, Bron S, Kuipers OP, Devine KM, Hecker M, van Dijl JM (2003) The extracellular proteome of Bacillus subtilis under secretion stress conditions. Mol Microbiol 49(1): 143–156. [DOI] [PubMed] [Google Scholar]
- Arigoni F, Talabot F, Peitsch M, Edgerton MD, Meldrum E, Allet E, Fish R, Jamotte T, Curchod ML, Loferer H (1998) A genome-based approach for the identification of essential bacterial genes. Nat Biotechnol 16(9): 851–856. [DOI] [PubMed] [Google Scholar]
- Blackman SA, Smith TJ, Foster SJ (1998) The role of autolysins during vegetative growth of Bacillus subtilis 168. Microbiology 144(1): 73–82. doi: 10.1099/00221287-144-1-73 [DOI] [PubMed] [Google Scholar]
- Chen CL, Wu SC, Tjia WM, Wang CL, Lohka MJ, Wong SL (2008) Development of a LytE-based high-density surface display system in Bacillus subtilis. Microb Biotechnol 1(2): 177–190. doi: 10.1111/j.1751-7915.2007.00017.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmon E, Noone D, Masson A, Bron S, Kuipers OP, Devine KM, van Dijl JM (2002) A novel class of heat and secretion stress-responsive genes is controlled by the autoregulated CssRS two-component system of Bacillus subtilis. J Bacteriol 184(20): 5661–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvaux M, Dumas E, Chafsey I, Hebraud M (2006) Protein cell surface display in Gram-positive bacteria: from single protein to macromolecular protein structure. FEMS Microbiol Lett 256(1): 1–15. doi: 10.1111/j.1574-6968.2006.00122.x [DOI] [PubMed] [Google Scholar]
- Fabret C, Hoch JA (1998) A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapy. J Bacteriol 180(23): 6375–6383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Galan C, Berenguer-Murcia A, Fernandez-Lafuente R, Rodrigues RC (2011) Potential of different enzyme immobilization strategies to improve enzyme performance. Adv Synth Catal 353(16): 2885–2904. [Google Scholar]
- Harold FM (1972) Conservation and transformation of energy by bacterial membranes. Bacteriol Rev 36(2): 172–&. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood CR, Cranenburgh R (2008) Bacillus protein secretion: an unfolding story. Trends Microbiol 16(2): 73–79. doi: 10.1016/j.tim.2007.12.001 [DOI] [PubMed] [Google Scholar]
- He XS, Shyu YT, Nathoo S, Wong SL, Doi RH (1991) Construction and use of a Bacillus subtilis mutant deficient in multiple protease genes for the expression of eukaryotic genes. Ann NY Acad Sci 646: 69–77. [DOI] [PubMed] [Google Scholar]
- Homaei AA, Sariri R, Vianello F, Stevanato R (2013) Enzyme immobilization: an update. J Chem Biol 6(4): 185–205. doi: 10.1007/s12154-013-0102-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GL, Anderson TD, Clubb RT (2014) Engineering microbial surfaces to degrade lignocellulosic biomass. Bioengineered 5(2): 96–106. doi: 10.4161/bioe.27461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang GL, Clubb RT (2017) Progress towards engineering microbial surfaces to degrade biomass. Biomass Volume Estimation and Valorization for Energy. J. S. Tumuluru, InTech
- Hyeon JE, Shin SK, Han SO (2016) Design of nanoscale enzyme complexes based on various scaffolding materials for biomass conversion and immobilization. Biotechnol J 11(11): 1386–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyyrylainen HL, Bolhuis A, Darmon E, Muukkonen L, Koski P, Vitikainen M, Sarvas M, Pragai Z, Bron S, van Dijl JM, Kontinen VP (2001) A novel two-component regulatory system in Bacillus subtilis for the survival of severe secretion stress. Mol Microbiol 41(5): 1159–1172. [DOI] [PubMed] [Google Scholar]
- Jensen CL, Stephenson K, Jorgensen ST, Harwood C (2000) Cell-associated degradation affects the yield of secreted engineered and heterologous proteins in the Bacillus subtilis expression system. Microbiology 146 (10): 2583–2594. doi: 10.1099/00221287-146-10-2583 [DOI] [PubMed] [Google Scholar]
- Jolliffe LK, Doyle RJ, Streips UN (1980) Extracellular proteases modify cell wall turnover in Bacillus subtilis. J Bacteriol 141(3): 1199–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe LK, Doyle RJ, Streips UN (1981) The energized membrane and cellular autolysis in Bacillus subtilis. Cell 25(3): 753–763. [DOI] [PubMed] [Google Scholar]
- Kobayashi G, Toida J, Akamatsu T, Yamamoto H, Shida T, Sekiguchi J (2000) Accumulation of an artificial cell wall-binding lipase by Bacillus subtilis wprA and/or sigD mutants. FEMS Microbiol Lett 188(2): 165–169. [DOI] [PubMed] [Google Scholar]
- Krishnappa L, Dreisbach A, Otto A, Goosens VJ, Cranenburgh RM, Harwood CR, Becher D, van Dijl JM (2013) Extracytoplasmic proteases determining the cleavage and release of secreted proteins, lipoproteins, and membrane proteins in Bacillus subtilis. J Proteome Res 12(9): 4101–4110. doi: 10.1021/pr400433h [DOI] [PubMed] [Google Scholar]
- la Grange DC, den Haan R, van Zyl WH (2010) Engineering cellulolytic ability into bioprocessing organisms. Appl Microbiol Biotechnol 87(4): 1195–1208. doi: 10.1007/s00253-010-2660-x [DOI] [PubMed] [Google Scholar]
- Ledeaux JR, Grossman AD (1995) Isolation and characterization of kinC, a gene that encodes a sensor kinase homologous to the sporulation sensor kinases kinA and kinB in Bacillus subtilis. J Bacteriol 177(1): 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SY, Choi JH, Xu Z (2003) Microbial cell-surface display. Trends Biotechnol 21(1): 45–52. [DOI] [PubMed] [Google Scholar]
- Li PS, Tao HC (2015) Cell surface engineering of microorganisms towards adsorption of heavy metals. Crit Rev Microbiol 41(2): 140–149. doi: 10.3109/1040841X.2013.813898 [DOI] [PubMed] [Google Scholar]
- Liew PX, Wang CL, Wong SL (2012) Functional characterization and localization of a Bacillus subtilis sortase and its substrate and use of this sortase system to covalently anchor a heterologous protein to the B. subtilis cell wall for surface display. J Bacteriol 194(1): 161–175. doi: 10.1128/JB.05711-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, LaBaer J (2011) Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb Protoc 2011(8): 993–994. doi: 10.1101/pdb.prot5651 [DOI] [PubMed] [Google Scholar]
- Liu L, Liu YF, Shin HD, Chen RR, Wang NS, Li JH, Du GC, Chen J (2013) Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Appl Microbiol Biotechnol 97(14): 6113–6127. doi: 10.1007/s00253-013-4960-4 [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25(4): 402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Marciniak BC, Trip H, van-der Veek PJ, Kuipers OP (2012) Comparative transcriptional analysis of Bacillus subtilis cells overproducing either secreted proteins, lipoproteins or membrane proteins. Microb Cell Fact 11: 66. doi: 10.1186/1475-2859-11-66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mascher T, Zimmer SL, Smith TA, Helmann JD (2004) Antibiotic-inducible promoter regulated by the cell envelope stress-sensing two-component system LiaRS of Bacillus subtilis. Antimicrob Agents Chemother 48(8): 2888–2896. doi: 10.1128/AAC.48.8.2888-2896.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGivney E, Han LC, Avellan A, VanBriesen J, Gregory KB (2017) Disruption of autolysis in Bacillus subtilis using TiO2 nanoparticles. Scientific Reports 7 doi: 10.1038/srep44308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3): 426–428. [Google Scholar]
- Mohamad NR, Marzuki NHC, Buang NA, Huyop F, Wahab RA (2015) An overview of technologies for immobilization of enzymes and surface analysis techniques for immobilized enzymes. Biotechnol Biotechnol Equip 29(2): 205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Phan TT, Schumann W (2011) Analysis and application of Bacillus subtilis sortases to anchor recombinant proteins on the cell wall. AMB Express 1(1): 22. doi: 10.1186/2191-0855-1-22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Schumann W (2006) Establishment of an experimental system allowing immobilization of proteins on the surface of Bacillus subtilis cells. J Biotechnol 122(4): 473–482. doi: 10.1016/j.jbiotec.2005.09.012 [DOI] [PubMed] [Google Scholar]
- Noone D, Howell A, Collery R, Devine KM (2001) YkdA and YvtA, HtrA-like serine proteases in Bacillus subtilis, engage in negative autoregulation and reciprocal cross-regulation of ykdA and yvtA gene expression. J Bacteriol 183(2): 654–663. doi: 10.1128/JB.183.2.654-663.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson DG, McBride JE, Joe Shaw A, Lynd LR (2012) Recent progress in consolidated bioprocessing. Curr Opin Biotechnol 23(3): 396–405. doi: 10.1016/j.copbio.2011.11.026 [DOI] [PubMed] [Google Scholar]
- Petre J, Longin R, Millet J (1981) Purification and properties of an endo-beta-1,4-glucanase from Clostridium thermocellum. Biochimie 63(7): 629–639. [DOI] [PubMed] [Google Scholar]
- Pohl S, Bhavsar G, Hulme J, Bloor AE, Misirli G, Leckenby MW, Radford DS, Smith W, Wipat A, Williamson ED, Harwood CR, Cranenburgh RM (2013) Proteomic analysis of Bacillus subtilis strains engineered for improved production of heterologous proteins. Proteomics 13(22): 3298–3308. doi: 10.1002/pmic.201300183 [DOI] [PubMed] [Google Scholar]
- Pohl S, Harwood CR (2010) Heterologous protein secretion by Bacillus species from the cradle to the grave. Adv Appl Microbiol 73: 1–25. doi: 10.1016/S0065-2164(10)73001-X [DOI] [PubMed] [Google Scholar]
- Salzberg LI, Helmann JD (2007) An antibiotic-inducible cell wall-associated protein that protects Bacillus subtilis from autolysis. J Bacteriol 189(13): 4671–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarvas M, Harwood CR, Bron S, van Dijl JM (2004) Post-translocational folding of secretory proteins in Gram-positive bacteria. Biochim Biophys Acta 1694(1–3): 311–327. doi: 10.1016/j.bbamcr.2004.04.009 [DOI] [PubMed] [Google Scholar]
- Schallmey M, Singh A, Ward OP (2004) Developments in the use of Bacillus species for industrial production. Can J Microbiol 50(1): 1–17. doi: 10.1139/w03-076 [DOI] [PubMed] [Google Scholar]
- Schuurmann J, Quehl P, Festel G, Jose J (2014) Bacterial whole-cell biocatalysts by surface display of enzymes: toward industrial application. Appl Microbiol Biotechnol 98(19): 8031–8046. [DOI] [PubMed] [Google Scholar]
- Schwarz WH, Grabnitz F, Staudenbauer WL (1986) Properties of a Clostridium thermocellum Endoglucanase Produced in Escherichia coli. Appl Environ Microbiol 51(6): 1293–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirisha VL, Jain A, Jain A (2016) Enzyme immobilization: an overview on methods, support material, and applications of immobilized enzymes. Adv Food Nutr Res 79: 179–211. doi: 10.1016/bs.afnr.2016.07.004 [DOI] [PubMed] [Google Scholar]
- Smith MR, Khera E, Wen F (2015) Engineering novel and improved biocatalysts by cell surface display. Ind Eng Chem Res 54(16): 4021–4032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TJ, Blackman SA, Foster SJ (2000) Autolysins of Bacillus subtilis: multiple enzymes with multiple functions. Microbiology 146: 249–262. [DOI] [PubMed] [Google Scholar]
- Spirig T, Weiner EM, Clubb RT (2011) Sortase enzymes in Gram-positive bacteria. Mol Microbiol 82(5): 1044–1059. doi: 10.1111/j.1365-2958.2011.07887.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephenson K, Bron S, Harwood CR (1999) Cellular lysis in Bacillus subtilis; the affect of multiple extracellular protease deficiencies. Lett Appl Microbiol 29(2): 141–145. [Google Scholar]
- Stephenson K, Harwood CR (1998) Influence of a cell-wall-associated protease on production of alpha-amylase by Bacillus subtilis. Appl Environ Microbiol 64(8): 2875–2881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauss A, Gotz F (1996) In vivo immobilization of enzymatically active polypeptides on the cell surface of Staphylococcus carnosus. Mol Microbiol 21(3): 491–500. [DOI] [PubMed] [Google Scholar]
- Tjalsma H, Antelmann H, Jongbloed JD, Braun PG, Darmon E, Dorenbos R, Dubois JY, Westers H, Zanen G, Quax WJ, Kuipers OP, Bron S, Hecker M, van Dijl JM (2004) Proteomics of protein secretion by Bacillus subtilis: separating the “secrets” of the secretome. Microbiol Mol Biol Rev 68(2): 207–233. doi: 10.1128/MMBR.68.2.207-233.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang LF, Bruckner R, Doi RH (1989) Construction of a Bacillus subtilis Mutant-Deficient in 3 Extracellular Proteases. J Gen Appl Microbiol 35(6): 487–492. [Google Scholar]
- Wernerus H, Stahl S (2004) Biotechnological applications for surface-engineered bacteria. Biotechnol Appl Biochem 40(3): 209–228. doi: 10.1042/BA20040014 [DOI] [PubMed] [Google Scholar]
- Westers H, Westers L, Darmon E, van Dijl JM, Quax WJ, Zanen G (2006) The CssRS two-component regulatory system controls a general secretion stress response in Bacillus subtilis. FEBS J 273(16): 3816–3827. doi: 10.1111/j.1742-4658.2006.05389.x [DOI] [PubMed] [Google Scholar]
- Westers L, Westers H, Quax WJ (2004) Bacillus subtilis as cell factory for pharmaceutical proteins: a biotechnological approach to optimize the host organism. Biochim Biophys Acta 1694(1–3): 299–310. doi: 10.1016/j.bbamcr.2004.02.011 [DOI] [PubMed] [Google Scholar]
- Westers L, Westers H, Zanen G, Antelmann H, Hecker M, Noone D, Devine KM, van Dijl JM, Quax WJ (2008) Genetic or chemical protease inhibition causes significant changes in the Bacillus subtilis exoproteome. Proteomics 8(13): 2704–2713. doi: 10.1002/pmic.200800009 [DOI] [PubMed] [Google Scholar]
- Wolf D, Kalamorz F, Wecke T, Juszczak A, Mader U, Homuth G, Jordan S, Kirstein J, Hoppert M, Voigt B, Hecker M, Mascher T (2010) In-depth profiling of the LiaR response of Bacillus subtilis. J Bacteriol 192(18): 4680–4693. doi: 10.1128/JB.00543-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu SC, Yeung JC, Duan Y, Ye R, Szarka SJ, Habibi HR, Wong SL (2002) Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl Environ Microbiol 68(7): 3261–3269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye R, Yang L, Wong S (1996) Construction of protease-deficient Bacillus subtilis strains for expression studies: inactivation of seven extracellular protease and the intracellular LonA protease. Proc. International Symposium on Recent Advances in Bioindustry: 160–169.
- You C, Zhang XZ, Sathitsuksanoh N, Lynd LR, Zhang YH (2012) Enhanced microbial utilization of recalcitrant cellulose by an ex vivo cellulosome-microbe complex. Appl Environ Microbiol 78(5): 1437–1444. doi: 10.1128/AEM.07138-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.