

Abstract
BACKGROUND:
Central nervous system Langerhans cell histiocytosis (CNS-LCH) brain involvement may include mass lesions and/or a neurodegenerative disease (LCH-ND) of unknown etiology. The goal of this study was to define the mechanisms of pathogenesis that drive CNS-LCH.
METHODS:
Cerebrospinal fluid (CSF) biomarkers including CSF proteins and extracellular BRAFV600E DNA were analyzed in CSF from patients with CNS-LCH lesions compared with patients with brain tumors and other neurodegenerative conditions. Additionally, the presence of BRAFV600E was tested in peripheral mononuclear blood cells (PBMCs) as well as brain biopsies from LCH-ND patients, and the response to BRAF-V600E inhibitor was evaluated in 4 patients with progressive disease.
RESULTS:
Osteopontin was the only consistently elevated CSF protein in patients with CNS-LCH compared with patients with other brain pathologies. BRAFV600E DNA was detected in CSF of only 2/20 (10%) cases, both with LCH-ND and active lesions outside the CNS. However, BRAFV600E+ PBMCs were detected with significantly higher frequency at all stages of therapy in LCH patients who developed LCH-ND. Brain biopsies of patients with LCH-ND demonstrated diffuse perivascular infiltration by BRAFV600E+ cells with monocyte phenotype (CD14+CD33+CD163+P2RY12−) and associated osteopontin expression. Three of 4 patients with LCH-ND treated with BRAF-V600E inhibitor experienced significant clinical and radiologic improvement.
CONCLUSION:
In LCH-ND patients, BRAFV600E+ cells in PBMCs and infiltrating myeloid/monocytic cells in the brain is consistent with LCH-ND as an active demyelinating process arising from a mutated hematopoietic precursor from which LCH lesion CD207+ cells are also derived. Therapy directed against myeloid precursors with activated MAPK signaling may be effective for LCH-ND.
Keywords: Langerhans cell histiocytosis, CNS neoplasms, neurodegeneration, osteopontin, BRAF-V600E
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a myeloid neoplasia characterized by lesions with pathologic CD1a+/ CD207+ myeloid dendritic cells among an inflammatory infiltrate,1 and somatic BRAFV600E mutations are identified in approximately 60% of all LCH lesions,2 with activating MAPK pathway mutations identified in almost all cases.3 Central nervous system manifestations of LCH (CNS-LCH) can include granulomatous parenchymal or pituitary mass lesions in approximately 25% of patients or a neurodegenerative disease (LCH-ND) in approximately 5% of patients.4–7 LCH-ND is a syndrome of progressive, often lethal neurodegeneration of unknown etiology that may arise decades after LCH is presumed to be cured.7–12 The mechanisms of pathogenesis of LCH-ND remain undefined, but it has been speculated to arise as an auto-immune phenomenon due to the observed presence of infiltrating T cells and lack of characteristic LCH CD1a+/CD207+ dendritic cells.8,13,14 The syndrome is identified radiologically by T2 and FLAIR intense lesions in the cerebellum (peduncles, dentate nuclei), basal ganglia, and/or brainstem.15,16 Symptoms include progressive tremors, ataxia, dysarthria, dysmetria, learning disabilities, and behavioral abnormalities.7 No standard therapy exists, but common practices include observation, immune suppression, and chemotherapy.17–20
The goal of this study was to perform a comprehensive and unbiased evaluation of biomarkers in patients with CNS-LCH to develop clinical tools to differentiate LCH from other neuropathological conditions, identify patients with LCH at risk for developing LCH-associated neurodegeneration, and define the mechanisms of pathogenesis to identify improved therapeutic strategies.
METHODS
Patients and Samples
This study was performed under protocols approved by the Baylor College of Medicine Institutional Review Board. Tissue specimens and clinical data were collected from subjects who had enrolled in an institutional biology study that ran from 2006 to 2016. All subjects and specimens meeting specified criteria were included in this study.
Inclusion of cerebrospinal fluid (CSF) in this study in the LCH group required biopsy-proven LCH from any site with evidence of CNS-LCH (ie, parenchymal mass lesion, pituitary mass lesion, and/or LCH-ND). CSF specimens were obtained from patients who had a lumbar puncture performed for clinical indications. LCH-ND was defined by characteristic magnetic resonance imaging (MRI) findings and/or neurologic changes in patients with a history of biopsy-proven LCH. Control cases were obtained from archived specimens. CSF from patients with acute lymphoblastic leukemia (ALL) in remission without a history of CNS leukemia were included as non-inflammatory controls. Similarly, archived CSF from children with active brain tumors (BTs), subjects with progressive neurodegenerative diseases (ND) other than LCH, and children with active untreated hemophagocytic lymphohistiocytosis (HLH) were used to control for general CSF responses to mass lesions, neurodegeneration, and hyperinflammation, respectively. Peripheral blood mononuclear cells (PMBCs) from all LCH subjects with available tissue were included in the peripheral mononuclear blood cell (PBMC) analyses, with the exception of subjects with isolated pituitary lesions. These were excluded from PBMC studies due to uncertainty of LCH disease activity at the time of blood sampling. Blood from LCH patients was obtained at various time points: pre-therapy (prior to chemotherapy) and post-therapy (>3 months since last chemotherapy). Medical records were analyzed retrospectively to correlate disease status of tissue samples and to evaluate clinical responses to therapy. Detailed longitudinal clinical courses of all subjects in this cohort treated with BRAF-V600E inhibitor therapy (n = 4) were reviewed. Patient and experimental details are summarized in Table 1 and Supporting Figure 1.
TABLE 1.
Clinical Characteristics
| Total LCH Cohort | Total | Neurodegeneration | Non- Neurodegeneration |
|---|---|---|---|
| Sample type | |||
| Patients with CSF | 16 | 5 | 11 |
| Patients with blood | 242 | 35 | 207 |
| Patients with CSF and blood |
23 | 23 | 0 |
| Biopsy tissue only | 4 | 2 | 2 |
| Biopsy tissue and blood |
4 | 0 | 4 |
| Biopsy tissue and blood and CSF |
1 | 1 | 0 |
| Lesion BRAFV600E | |||
| mutation status | |||
| BRAFV600E+ | 84 | 14 | 70 |
| BRAFV600E– | 56 | 1 | 55 |
| Unknown | 150 | 51 | 99 |
| Total | 290 | 66 | 224 |
| CSF studies: LCH | Discovery | Validation | P |
|---|---|---|---|
| Sex | 0.65 | ||
| Male | 14 | 11 | |
| Female | 6 | 8 | |
| Age, y | 0.86 | ||
| 0–3 | 2 | 1 | |
| 3–18 | 13 | 13 | |
| >18 | 5 | 5 | |
| LCH subtypes | 0.84 | ||
| Neurodegeneration (only) | 8 | 6 | |
| Neurodegeneration and CNS mass lesion |
6 | 7 | |
| CNS mass lesion (only) | 6 | 6 | |
| Total | 20 | 19 |
| CSF studies: ALL | Discovery | Validation | P |
|---|---|---|---|
| Sex | 1.00 | ||
| Male | 7 | 7 | |
| Female | 7 | 8 | |
| Age, y | 1.00 | ||
| 0–3 | 2 | 2 | |
| 3–18 | 12 | 13 | |
| >18 | 0 | 0 | |
| Total | 14 | 15 |
| CSF studies: BT | Discovery | Validation | P |
|---|---|---|---|
| Sex | 0.55 | ||
| Male | 5 | 7 | |
| Female | 8 | 5 | |
| Age, y | 1.00 | ||
| 0–3 | 3 | 2 | |
| 3–18 | 10 | 10 | |
| >18 | 0 | 0 | |
| Subtypes | 0.15 | ||
| Medulloblastoma | 5 | 8 | |
| Meningeal sarcoma | 1 | 0 | |
| Ependymoma | 3 | 0 | |
| Astrocytoma | 1 | 1 | |
| Germinoma | 2 | 0 | |
| Craniopharyngioma | 1 | 0 | |
| Pineal mass | 0 | 1 | |
| Teratoid rhabdoid tumor | 0 | 2 | |
| Total | 13 | 12 |
| CSF studies: HLH | HLH Full Dataset |
|---|---|
| Sex | |
| Male | 6 |
| Female | 3 |
| Age, y | |
| 0–3 | 1 |
| 3–18 | 8 |
| >18 | 0 |
| Total | 9 |
| CSF studies: ND controls | ND Full Dataset |
|---|---|
| Sex | |
| Male | 28 |
| Female | 8 |
| Unknown | 2 |
| Age, y | |
| 0–3 | 2 |
| 3–18 | 14 |
| >18 | 20 |
| Unknown | 0 |
| Subtypes | |
| X-linked adrenoleukodystrophy | 10 |
| Alzheimer’s disease | 15 |
| Active multiple sclerosis | 3 |
| Parkinson’s disease | 6 |
| Batten disease | 2 |
| Multiorgan failure | 1 |
| Other | 1 |
| Total | 38 |
| Peripheral blood studies: LCH |
Neurodegeneration | Non- Neurodegeneration |
|||||
|---|---|---|---|---|---|---|---|
| Sex | |||||||
| Male | 37 | 115 | |||||
| Female | 22 | 96 | |||||
| Age at diagnosis, y | |||||||
| 0–3 | 32 | 75 | |||||
| 3–18 | 20 | 86 | |||||
| >18 | 7 | 50 | |||||
| LCH subtypes | |||||||
| LR single lesion | 4 | 70 | |||||
| LR multiple lesion | 48 | 117 | |||||
| HR | 7 | 24 | |||||
| Lesion BRAFV600E
mutation status |
|||||||
| BRAFV600E+ | 11 | 67 | |||||
| BRAFV600E– | 1 | 52 | |||||
| Unknown | 47 | 92 | |||||
| Total | 59 | 211 | |||||
| Peripheral blood studies: BT | |
|---|---|
| Sex | 5 |
| Unknown | |
| Age, y | 5 |
| Pediatric | |
| Subtypes | 5 |
| Glioblastoma | |
| Lesion BRAFV600E | |
| mutation status | |
| BRAFV600E+ | 5 |
| BRAFV600E− | 0 |
| Unknown | 0 |
| Total | 5 |
Abbreviations: ALL, acute lymphoblastic leukemia; BT, brain tumor; CNS, central nervous system; CSF, cerebrospinal fluid; HLH, hemophagocytic lymphohistiocytosis; HR, high-risk; LCH, Langerhans cell histiocytosis; LR, low-risk; ND, neurodegenerative disease.
Determination of CSF Protein Levels
CSF was collected, spun at 1000g for 5 minutes, then frozen and stored as 250-μL aliquots at –80°C. None of the CSF samples analyzed underwent more than 2 freeze/ thaw cycles. Protein levels were determined using MagPix instrument (Luminex, Austin, TX) with kits (Supporting Table 1). The concentration of each analyte was measured by comparing with the protein standards.
Quality Control of CSF Expression Data
A total of 185 patient samples were used in the CSF expression analysis. A logarithmic (base 2) transformation was applied to the sample concentrations before quality control. To control the quality of the Luminex data, box plots containing the log concentrations of all the analytes in each of the samples were inspected to identify potential outlier samples in the experiment. No samples were rejected based on these criteria. The assays were organized so that the relative proportion of each class of sample (eg, LCH, BT, ALL, ND) was preserved as much as possible from plate to plate to minimize batch effects from the Luminex assays. Glial fibrillary acidic protein (GFAP) was analyzed using the GFAP ELISA kit (EMD Millipore, Billerica, Massachusetts) with a PowerWave XS2 (BioTek, Winooski, Vermont) plate reader, and total tau and phospho-tau were analyzed with the Magpix platform on a limited series of LCH and ALL CSF samples due to sample and volume availability.
Biomarker Discovery and Validation With CSF Protein Data
The CSF samples were partitioned into training and validation cohorts with comparable clinical parameters before the biomarker analysis. A Pearson’s chi-squared test was used to test whether the patient diagnosis/clinical parameters and the grouping of the samples were not significantly different from each other (P > .05). The resulting training and validation sets were then used to perform various class comparison analyses using analysis of variance. Biomarkers with false discovery proportion of 0.1 was considered significant. Post hoc pairwise analysis was performed with P < .01 considered significant.
The class comparison analysis was run using the ALL controls, BT controls, and LCH samples as predefined classes in the training cohort. A univariate parametric F-test was used with a confidence level of false discovery rate assessment at 80% and the maximum allowed proportion of false-positive analytes at 0.1. Pairwise significance was then calculated for the significantly differentially expressed analytes to show the pairs of classes with significantly different analyte expression at α < 0.01. Analytes that exhibited pairwise significance and increased or decreased expression in LCH samples relative to both ALL and BT controls were considered biomarkers of interest. These significant analytes were then tested in the validation cohort using a similar statistical procedure. The significant analytes identified in the validation cohort were reported (false discovery rate = 0.1) To ensure that the validated biomarkers were not due to a specific partitioning of the data, the samples in the training and validation sets were shuffled and regrouped 5 times. After each repartition, a Pearson’s chi-squared test was applied to ensure that no statistically significant biases were observed between the groupings and the clinical characteristics. The class comparison analysis was then performed again to identify validated biomarkers as described above. The percentage of cross-validation was calculated for each significant biomarker to ensure that they were also differentially expressed in other repartition datasets.
To determine whether the biomarkers discovered were due to general inflammatory responses, a class comparison analysis was performed after the initial analysis using samples with LCH and HLH controls as predefined classes using similar statistical parameters (ie, confidence level of false discovery rate assessment at 80% and the maximum allowed proportion of false-positive analytes at 0.1). Because of the number of available HLH control samples, the analysis was performed on all available samples. Lastly, unpaired t tests or F tests were used to compare osteopontin (OPN) and S100 calcium-binding protein B (S100B) in comparison with 2 or more sample groups (P < .05).
BRAFV600E Assay From PBMC and CSF
This quantitative polymerase chain reaction (qPCR) assay was performed with PBMC as described previously,1 with modifications for extracellular CSF DNA as detailed in the Supporting Methods. Fisher exact tests were performed to determine the significance of the detectable BRAFV600E in PBMC in various clinical groups, with P <.05 considered significant.
Evaluation of Clinical and Radiologic Responses to Therapy
The clinical courses of patients with LCH-ND with CSF and/or blood specimens in this study were reviewed. The clinical severity of neurodegeneration was evaluated using the ataxia rating scale.21 Radiographic ND was clinically defined by clinical MRI reports noting characteristic T2-FLAIR hyperintensity and were verified with central review. Staging and response to therapy of lesions outside the CNS were defined according to the Histiocyte Society’s LCH treatment guidelines.22 For subjects treated with BRAF-V600E inhibitors, significant clinical events, systemic disease activity, therapy (dose/schedule), changes in therapy, suspected toxicities, concurrent medications, responses of systemic lesions, radiologic responses of LCH-ND, and clinical responses (ataxia rating score) were obtained by chart review.
RESULTS
CSF Biomarker Analysis in CNS-LCH
OPN and S100B are candidate CSF biomarkers for LCH
To identify disease-specific CSF biomarkers in LCH, protein levels of 121 unique proteins associated with inflammation and/or neurodegeneration were assessed in CSF samples from 40 patients with CNS-LCH (12 CNS-LCH mass lesions only, 18 CNS-LCH mass lesions and LCH-ND, 10 LCH-ND only), 29 patients with ALL in remission and without CNS disease, and 25 patients with pediatric BTs (Table 1). The ALL samples were used to represent a noninflammatory control.
Of the analytes, only OPN was significantly increased in all LCH CSF samples when compared with BT and ALL controls. In contrast, S100B was significantly decreased in LCH CSF compared with BT controls, while there was no significant difference between S100B concentrations in CSF between the LCH and the ALL-control groups (P < .05) (Fig. 1A,B, Supporting Table 2A). To ensure that these biomarkers were not due to biased partitioning of the data, the sample grouping was shuffled using similar selection criteria to create new training and validation sets. The repartitioning analysis demonstrated similar results with significantly increased OPN in the LCH group relative to the BT and ALL control groups and decreased S100B in the LCH and ALL control groups relative to the BT group, with an 80% coefficient of variation in 5 iterations for both comparisons.
Figure 1.

Cerebrospinal fluid (CSF) osteopontin (OPN) and serum calcium binding protein B (S100B) concentrations differentiate central nervous system Langerhans cell histiocytosis (CNS-LCH) from other neoplastic, inflammatory, and neurodegenerative conditions. Dotplots demonstrate the relative concentration of OPN and S100B in (A) discovery and (B) validation series from acute lymphoblastic leukemia (ALL), brain tumor (BT), and LCH subjects. OPN and S100B expression is also illustrated from (C) hemophagocytic lymphohistiocytosis (HLH) and LCH subjects, (D) BT and LCH subjects with CNS tumors without LCH and neurodegenerative disease (LCH-ND), and (E) LCH subjects with ND [LCH (ND+)] and non-LCH ND (ND CTL) subjects. (F) CSF OPN concentration was compared between LCH-CNS categories and was significantly higher in patients with ND (ND+) and with LCH-ND and CNS-LCH mass lesions (Both) compared with LCH with mass lesion only (CNS Tumor). Red bars represent the mean values. *Statistically significant difference between groups.
OPN is elevated in LCH relative to HLH, a hyperinflammatory control
To identify the CSF proteins in LCH that are associated with general inflammatory responses, we performed a class comparison between the CSF samples from all patients with LCH and HLH (n = 9), a disease defined by extreme pathologic inflammation.23 Despite the increased concentration of other inflammatory proteins in HLH compared with LCH, OPN was significantly increased in the CSF of LCH patients compared with HLH (P < .05) (Fig. 1C, Supporting Table 2B).
OPN is elevated and S100B is decreased in LCH CNS tumors compared with other brain tumors
We then specifically tested whether OPN and S100B concentrations could distinguish the LCH cases with LCH-CNS mass lesions without LCH-ND from the BT controls. Similar to the LCH group-wide analysis, OPN was significantly increased, whereas S100B was significantly decreased in the LCH CNS mass lesion CSF cases compared with BT CSF (P < .05) (Fig. 1D, Supporting Table 2C).
OPN is significantly elevated in LCH-ND versus non-LCH ND CSF
The ability of the 2 biomarkers to differentiate LCH-ND from other neurodegenerative diseases was also tested. OPN was significantly increased, whereas S100B was significantly decreased in LCH-ND relative to the ND control group (n = 38) (Fig. 1E, Supporting Table 2D). Notably, OPN was also significantly elevated in CSF of patients with LCH-ND compared with patients with LCH-CNS mass lesions without ND (Fig. 1F).
LCH-ND is not associated with increased CSF GFAP or tau
In contrast to a previous report, CSF concentrations of GFAP, tau, and phospho-tau were not significantly increased in LCH-ND in this study compared with the ALL control or ND control groups.24 We analyzed the concentration of GFAP and tau (total and phospho-tau) in a subset of LCH and ALL cases from this series. We found the concentrations of GFAP, total tau, and phospho-tau in CSF were not significantly different between all cases of LCH with CNS involvement (n = 40 cases of LCH-ND, LCH-CNS mass, or both), control ND cases (n = 4), and control ALL cases (n = 22) (total tau, P = .81; phospho tau, P = .19; GFAP, P = .21). The volume required to perform these experiments limited the number of samples available for analysis, and insufficient sample was available to test NF-L.
OPN is highly expressed in both LCH-CNS mass lesions and LCH-ND
In addition to the CSF level, we examined the relative cellular expression of OPN: Immunoblotting and immunohistochemistry were performed on representative LCH-CNS biopsies, confirming abundant OPN expression in both LCH-CNS mass lesions and LCH-ND tissue relative to brain tumor and control brain. RNA expression of SPP1 (encoding osteopontin) was also detected across LCH lesions including a pituitary biopsy and LCH-ND biopsy. Immuno-histochemical analysis detected OPN expression in histiocytes and lymphocytes from non–CNS-LCH lesions25 and variable OPN expression in brain tumors.26 In contrast, S100B protein expression was higher in representative BT biopsies than LCH lesions as detected by immunoblotting (Fig. 2A-C).
Figure 2.

Cellular composition, relative expression of osteopontin (OPN) and serum calcium binding protein B (S100B), and BRAFV600E expression in central nervous system Langerhans cell histiocytosis (CNS-LCH) lesions and LCH with neurodegenerative disease (LCH-ND). (A) Immuno-histochemical analysis of CD207, VE-1 (identifies BRAF-V600E protein expression), OPN, and CD3 in representative tissue sections obtained from indicated biopsy specimens (original magnification 340). (B) Steady-state protein expression of OPN and S100B in representative LCH lesions and glioblastoma brain tumor biopsies. The blots were stripped and reprobed with glyceraldehyde 3-phosphate dehydrogenase (GADPH) to confirm equivalent loading. (C) Relative messenger RNA expression of the indicated transcripts (SPP1/OPN, blue; CD207, orange; CD1a, gray; CD3, black) as determined by quantitative polymerase chain reaction (qPCR) for LCH lesions from bone, LCH lesions from lungs, LCH with pituitary involvement (Pit), and LCH with early onset neurodegeneration (ND). Data were normalized to adjusted GAPDH expression and analyzed using the ΔCt method. *Complementary DNA level below the limit of detection by qPCR. (D) qPCR for BRAFV600E from genomic DNA (gray), and relative OPN (SPP1) expression (blue) from biopsies from various regions of whole brain autopsy from a patient with advanced LCH-ND. (E) Magnetic resonance images (T2 FLAIR) from the same patient obtained 2 years before death from progression of LCH-ND. The anatomic areas corresponding to the biopsy sections from qPCR are indicated.
Limited detection of extracellular BRAFV600E in LCH CSF
BRAFV600E has been readily detected in extracellular DNA from plasma and urine in patients with active disease.27 In this series, BRAFV600E was detectable by qPCR in CSF in only 2/20 cases (10%), both of whom had LCH-ND along with active lesions outside the CNS (Fig. 3).
Figure 3.
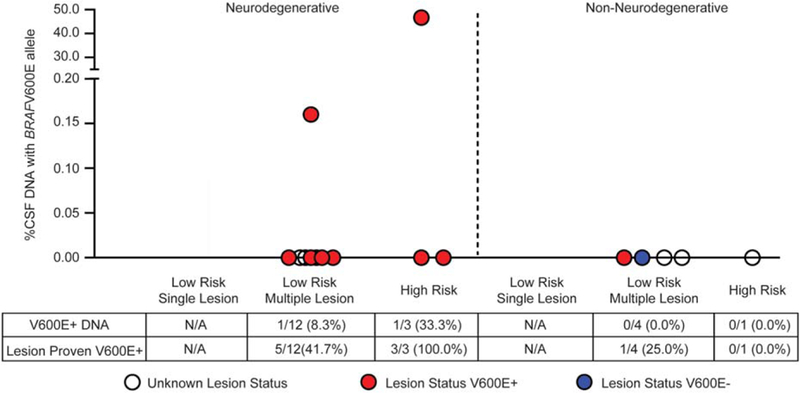
Investigation of BRAFV600E in cerebrospinal fluid (CSF) of patients with central nervous system Langerhans cell histiocytosis (LCH). Extracellular BRAFV600E DNA in the CSF was detected in only 2 of 20 patients, both with active LCH brain lesions and neurodegeneration.
Detection of BRAFV600E in Peripheral Blood in Patients With LCH-ND
To identify potential origins of LCH-ND precursor cells, we tested PBMCs from LCH patients with BRAFV600E+ lesions or with unknown mutation status. Whereas extracellular BRAFV600E was rarely detected in the CSF of patients with LCH-ND, PBMCs with BRAFV600E were frequently detected in all phases of therapy in patients who ultimately developed LCH-ND. In pre-therapy samples, PBMCs harboring the BRAFV600E mutation were identified in 59% (10/17) of patients who ultimately developed LCH-ND (3/9 with initial LCH limited to the CNS and 7/8 with active LCH beyond the CNS) versus 15% (21/139) of patients who did not develop LCH-ND. Detectable BRAFV600E in PBMCs was associated with risk of LCH-ND with a sensitivity of 0.59 and specificity of 0.86 (P < .0001) (Fig. 4A,E). In post-therapy blood collected at a time of systemic LCH relapse, BRAFV600E was detected in the PBMCs of 43% (6/14) of patients who developed LCH-ND and 7% (2/28) of patients who did not develop LCH-ND (sensitivity, 0.43; specificity, 0.93; P = .0105) (Fig. 4B,F). Most notably, in post-therapy PBMCs collected from patients with active LCH-ND without active systemic lesions, BRAFV600E was detected in 22% (8/36) versus 0% (0/22) of patients without active systemic LCH or LCH-ND (sensitivity, 0.22; specificity, 1; P = .0016) (Fig. 4C,G). All of the post-therapy non–LCH-ND, nonactive LCH controls had previously proven BRAFV600E+ lesions. The BRAFV600E allele was detected in PBMC from patients with LCH-ND in multiple myeloid lineages, as well as in lymphoid cells in some cases (Supporting Table 3).
Figure 4.

Identification of cells with BRAFV600E in peripheral blood of patients with non-neurodegenerative and Langerhans cell histiocytosis with neurodegenerative disease (LCH-ND) at specific time points. (A) BRAFV600E DNA was evaluated using quantitative polymerase chain reaction in peripheral blood mononuclear cells (PBMCs) in pre-chemotherapy non-neurodegenerative patients with active disease outside the central nervous system (CNS) (left), LCH-ND patients with no active disease outside the CNS (middle), and LCH-ND patients with active disease outside the CNS (right). PBMCs were evaluated for BRAFV600E in post-chemotherapy patients who were treated previously with systemic therapy at (B) relapse with active disease outside the CNS in non-neurodegenerative (left) and LCH-ND patients (right) and (C) in patients presumed to be cured with no active disease outside the CNS in both non-neurodegenerative (left) and LCH-ND (right) patients. (D) As controls, no BRAFV600E cells were detected in pre-chemotherapy PBMCs from LCH patients with known BRAF wild-type lesions (left) or in PBMCs from 5 pre-therapy pediatric glioblastoma patients with known BRAFV600E+ tumors (right). In panels A-D, the percentage of patients with BRAFV600E detected in PBMCs and the percentage of patients with known BRAFV600E lesion status are indicated below each graph. (E-G) Sensitivity, specificity, and positive and negative predictive values of BRAFV600E in PBMCs for LCH-ND are indicated. Paired analysis (P value) indicates statistical significance of BRAFV600E+ PBMCs between non-ND and LCH-ND groups.
As a control, BRAFV600E mutation was not detected in pre-therapy PBMCs of 5 patients with active BRAFV600E+ glioblastoma, nor in PBMC of 53 patients with BRAF wild-type LCH lesions (Fig. 4D).
BRAFV600E+ Cells in Brain Parenchyma of Patients With LCH-ND Localize to Areas of OPN Expression and Active Neurodegeneration
In 3 patients with extensive and progressive LCH-ND, brain biopsy or autopsy was performed. Immunohistochemistry identified perivascular cells concentrated in the white matter that stained with the VE-1 antibody that reacts with the BRAF-V600E protein in 2 diagnostic biopsies: 1 rapidly progressive new-onset LCH-ND (with BRAFV600E+ cells estimated at 7% by qPCR) and 1 long-standing LCH-ND (insufficient DNA for qPCR) (Fig. 2A,C). In an autopsy specimen from a third patient who died from progressive LCH-ND, qPCR identified enrichment of BRAFV600E+ cells in brainstem (13% of cells), including the pons (8%) and cerebellum (5%), with aggregates of perivascular VE-1+ cells in areas of active demyelination. Areas enriched for BRAFV600E+ cells also corresponded to characteristic areas of T2 hyperintensity illustrated in a brain MRI from the same patient (Fig. 2D,E). Expression of OPN was significantly increased in the LCH-ND brain biopsies samples compared with control brain, and regional expression of SPP1 (encoding OPN) correlated with increased BRAFV600E expression in the brain autopsy sections in the third patient (Fig. 2C). Whereas VE1+ cells were consistently identified in CNS-LCH mass lesion and LCH-ND cases from patients with BRAFV600E mutations, CD207 expression was decreased to absent in these CNS-LCH biopsies compared with typical CD207+ LCH lesions outside the CNS (Fig. 2A,C).
More extensive analysis of the whole brain formalin fixed paraffin embedded autopsy sections identified significant gliosis and active demyelination in the white matter of the most heavily affected regions with plump VE1+ mononuclear cells at the interface between the myelinated and demyelinated areas with residual naked axons (Luxol Fast Blue/PAS stain and Neurofilament immunostain not shown). There were also focal regions of VE1+ mononuclear cell aggregates noted in microscopic sections with active demyelination but lacking MRI T2 correlation (ie, temporal and frontal lobe). Representative images from the temporal lobe with white matter injury demonstrate VE1+ foci with CD14+CD33+CD163+ (hematopoietic myeloid/monocytic) cells in and around blood vessels with regional expression of MCP-1 (chemotaxis factor for myeloid cells). By contrast, P2RY12 (resident tissue microglia) staining was limited to physiologic microglia in the surrounding parenchyma but were relatively devoid within the VE1+ perivascular myeloid clusters (Fig. 5). CD207 staining was negative in the sections enriched with VE1+ cells (not shown).
Figure 5.
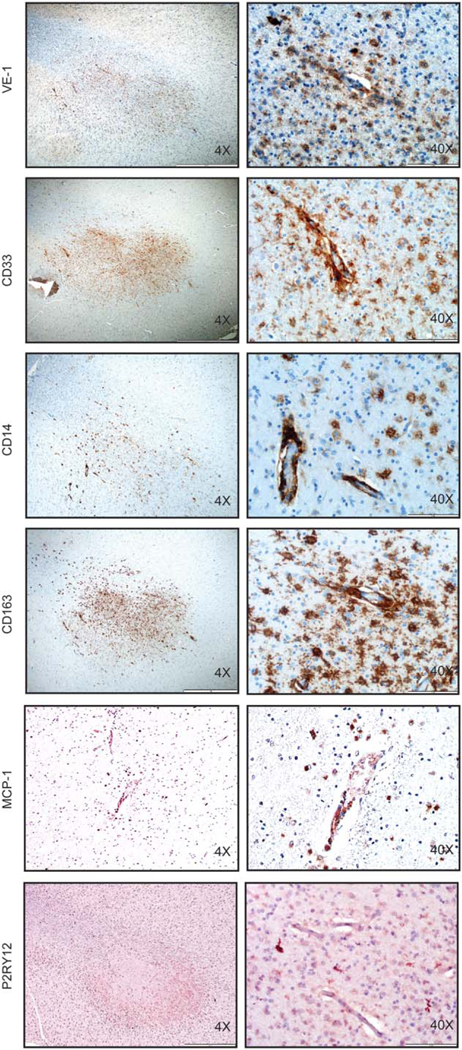
Phenotypic characterization of BRAFV600E-expressing cells in LCH-ND. (A) Immuno-histochemical analysis of VE-1 (identifies BRAFV600E protein), CD33 (identifies myeloid/ monocytic cells), CD14 (identifies monocytes), CD163 (identifies monocytes/macrophages), MCP-1(attracts monocytes), and P2RY12 (identifies resident microglia) in representative tissue sections obtained from autopsy brain specimens of LCH-ND from a patient who died from progressive Langerhans cell histiocytosis with neurodegenerative disease, demonstrating perivascular white matter infiltration by BRAFV600E+ (VE1+) cells with monocyte phenotype (CD14+CD33+CD163+P2RY12–). Original magnification ×40 and ×400.
Clinical and Radiologic Responses in Patients With LCH-ND to Chemotherapy and BRAFV600E Inhibition
Patients with LCH-ND in this series were treated with a variety of therapies, with clinical improvement in some patients receiving cytotoxic chemotherapy, including cytarabine or clofarabine. BRAFV600E in PBMC became undetectable in many patients after treatment with myelotoxic chemotherapy (Supporting Figure 2), Fig. 6). Four patients with clinical deterioration despite chemotherapy were treated with BRAF inhibitors. In 3 patients, 1 with early-onset LCH-ND and 2 with symptoms >2 years, MRI changes and clinical status (measured by ataxia rating score) initially improved with BRAF-V600E inhibition (vemurafenib or dabrafenib). The first patient achieved complete clinical response after 16 months on therapy. The second patient had continued improvement (partial clinical response) after 21 months on therapy. The third patient had initial improvement (partial clinical response), then experienced episodes of deterioration and improvement. A fourth patient with more than a decade of progressive LCH-ND and severe neurologic impairments who received vemurafenib for 2 months before stopping due to rash and arthralgias, followed by dabrafenib (for 9 months) and dabrafenib/trametinib (for 4 months), continued to have progressive clinical neurological deficits with stable brain MRI (Fig. 6, Supporting Tables 4A-D).
Figure 6.

Radiographic and clinical responses of LCH-ND to chemotherapy and MAPK pathway inhibition. (A) Magnetic resonance images (T2 FLAIR) of patients with LCH-ND at onset of neurodegenerative disease (ND) (top panel) before BRAFV600E inhibitor therapy (middle panel) and on BRAFV600E inhibitor therapy at the time of best response or longest duration (if there was no response on therapy) (bottom panel). Time in months/years indicates time between each image. Radiologic and clinical responses at the time of the MRI image at best response are reported as complete response (CR), partial response (PR), stable disease (SD), or progressive disease (PD). Three out of 4 patients with progressive disease on chemotherapy had clinical CR or PR and radiologic PR to BRAF inhibition. One patient with long-standing ND showed periods of stable radiologic and clinical disease but experienced clinical and radiologic progression overall despite BRAF inhibition. (B) Corresponding clinical and molecular responses for each patient. Clinical symptoms are reported using the ataxia rating score (0–100), with best response on BRAF-V600E inhibition indicated by an “x.” The molecular response (%BRAFV600E+ PBMC) was measured over serial blood samples while on cytotoxic chemotherapy (black bar) or BRAF-V600E inhibition (blue bar). Detailed clinical courses and radiologic interpretation are outlined in the clinical response forms (Supporting Table 4A-D).
DISCUSSION
OPN in CNS-LCH Disease
In this series, OPN levels in CSF differentiated patients with LCH-CNS mass lesions and LCH-ND from other CNS neoplastic and inflammatory conditions. Among LCH-CNS patients, OPN levels in CSF were higher in those with LCH-ND than in those with LCH-CNS mass lesions without LCH-ND. SPP1 expression was highly elevated in tissue brain biopsies from patients with LCH-ND and correlated with regions of enriched BRAFV600E+ cells, consistent with previous observations that SPP1 is one of the most highly up-regulated genes in non–CNS-LCH lesions.25 OPN has several roles on immune regulation and is produced by activated T helper 1 cells as well as dendritic cells.28,29 Increased concentrations of OPN have been described in the CSF, plasma, and brain tissue of patients with several other NDs.30 In multiple sclerosis, CSF OPN levels correlate with disease activity.31,32 The severity of disease in mouse models of multiple sclerosis is attenuated with targeted disruption of SPP1, which results in decreased migration of activated T cells to the brain.33 OPN may therefore represent a novel biomarker and possible therapeutic target for CNS-LCH.
Hematopoietic Cell of Origin in LCH-ND
We have previously identified PBMC and myeloid dendritic cell precursors harboring the BRAFV600E mutation in patients with HR LCH lesions, but not in patients with isolated LR LCH lesions with the BRAFV600E mutation.34 We therefore investigated the presence of BRAFV600E+ precursor cells in circulation in LCH-ND patients and identified a significantly higher frequency of BRAFV600E+ PBMCs at all stages of therapy in patients who went on to develop LCH-ND compared with those who did not. Additionally, we identified BRAFV600E+ cells in brain parenchyma of patients with LCH-ND, with regional enrichment in brainstem and cerebellum, areas typically noted to have characteristic hyperintensity on MRI, along with expression of BRAF-V600E in areas of active demyelination. Inability to detect BRAFV600E in PBMC in some subjects with LCH-ND may occur due to elimination of the circulating clone by myelotoxic chemotherapy and/or restriction to brain tissue. Some patients may also have alternative mutations, though 14/ 15 (93%) of the LCH-ND patients in this study with proven genotype had BRAFV600E+ peripheral LCH lesions, with similar frequency reported by Heritier et al35 Notably, 1 patient continued to have relatively high levels of BRAFV600E+ PBMC 16 months after starting BRAF-V600E inhibitor therapy despite achieving nearly normal clinical symptoms and normalized MRI (Fig. 6, Supporting Table 4A). BRAF-V600E inhibition may therefore block effects of MAPK hyperactivation without elimination of the pathogenic clone.
Earlier studies demonstrating biopsies with infiltrating CD8+ T cells without CD207+ histiocytes interpreted LCH-ND to represent an autoimmune or paraneoplastic phenomenon.8,36 However, data from this study suggest this process is mediated through migration of BRAFV600E+ (or other activating mutation) hematopoietic precursors to specific regions of the brain via perivascular accumulation and parenchymal infiltration. The activated BRAFV600E+ myeloid/monocytic cells are associated with regional parenchymal white matter injury, gliosis and demyelination and a leukoencephalopathic pattern of neurodegeneration along with infiltrating lymphocytes as described previously.8,13,14,37
Common origins of monocyte-derived inflammatory microglia and myeloid dendritic cells indicate the potential for aberrant differentiation of myeloid precursors driven by activated ERK to result in neuropathology in LCH-ND.38,39 Microglia are resident myeloid cells of the CNS that may arise from yolk sac during gestation or from infiltrating mononuclear cells that can mediate innate and adaptive immune responses. Myeloid markers CD14, CD33, and CD163 are expressed on hematopoietic myeloid and monocytic cells. P2RY12 is a specific resident microglial marker,40 unlike CD68, a pan-lysosomal marker, which can be expressed on a wide range of cell types including both activated microglia and hematopoietic myeloid/monocytic cells. The expression of CD14/ CD33/CD163 and lack of P2RY12 expression on VE1+ aggregates and perivascular cells in areas of active demyelination in a patient with progressive LCH-ND is consistent with a hematopoietic origin for these cells. An alternative hypothesis that has been raised could be that LCH-ND arises from fetal yolk sac progenitors.39 However, the typical timing of LCH-ND following LCH lesions at other sites, presence of BRAFV600E+ PBMC in patients with LCH-ND, but not in patients without LCH-ND or other active lesions, and perivascular concentration of BRAF-V600E+ cells with myeloid/monocyte phenotype strongly support a hematopoietic origin for LCH-ND-associated infiltrating myeloid cells. We therefore propose a model in which a hematopoietic clone causing the original LCH lesions may persist (or reemerge) after presumed cure and serve as a reservoir for future LCH-ND further explaining the lack of CD207+ cells in such foci. Although we interpret the results of this study to be consistent with hematopoietic origin, it remains possible that yolk sac–derived resident microglia with activating MAPK pathway gene mutations may also play a role in neurodegeneration in some patients with histiocytic disorders.39
The LCH-ND patients in this study represent a historically large cohort for this rare condition. Due to real-time enrollment, subjects presented at various stages of disease progression and treatment course. Lineage analysis of the BRAFV600E+ cells in circulation may be affected by previous treatment with myeloablative chemotherapy. However, the wide range of lineages harboring the BRAFV600E mutation is consistent with patients with LCH-ND acquiring the mutation at an early hematopoietic precursor stage. Prospective collection of blood samples on all LCH patients treated on clinical trials may be helpful to precisely identify which lineages of cells with BRAFV600E (or alternative MAPK pathway mutations) are associated with subsequent development of LCH-ND.
Clinical Implications of a Hematopoietic Clone With MAPK Pathway Activation in LCH-ND: Surveillance and Therapy
Our data support a model of LCH-ND not as a reactive autoimmune or paraneoplastic process, but rather an active neurodegenerative process driven by common BRAFV600E+ myeloid precursors that are shared with systemic LCH lesion CD207+ cells. Elevated OPN in CSF likely results from pathologic ERK activation of these infiltrating myelomonocytic cells that drive neurodegeneration (or from LCH lesion dendritic cells in the case of CNS mass lesions). Therefore, clinical strategies for patients with LCH-ND should 1) investigate for the presence of BRAFV600E (or other LCH lesion mutations) in PBMCs and elevated OPN levels in CSF and 2) design therapy protocols to selectively eliminate LCH-ND precursors and/or impair the function of infiltrating mutated monocytes. Current strategies to identify the development of LCH-ND are inconsistent and imprecise, typically involving periodic MRI and clinical examinations of variable frequency driven by local practice preference. Objective measures of disease activity would greatly support diagnosis and evaluation of response to therapy for patients with LCH-ND, for whom early treatment is critical for therapeutic response.17 In this study, all patients with BRAFV600E+ PBMCs developed LCH-ND despite not having active LCH lesions after initial therapy, suggesting that the mutation in PBMCs is a very specific means of detecting risk of LCH-ND. However, sensitivity of BRAFV600E+ PBMCs for LCH-ND in heavily pretreated patients without active lesions was only 22%, realizing that chemotherapy has the potential to clear BRAFV600E+ PBMCs from circulation in these patients. By comparison, the sensitivity of BRAFV600E+ PBMCs from pre-therapy patients at risk of ultimately developing LCH-ND in this series was 59% (with uncertain lesion genotype in most of these cases).
Therapies aimed at controlling inflammation may improve symptoms but may not achieve cure if LCH-ND myeloid precursors persist in circulation. In this series, moderate doses of cytarabine or clofarabine were associated with elimination of BRAFV600E+ PBMCs and clinical improvement in some patients. In 3 out of 4 severe cases of LCH-ND that progressed despite chemotherapy, BRAF-V600E inhibition was associated with improved clinical and radiologic status. In patients with long-standing disease, the potential benefit of chemotherapy or targeted MAPK inhibition is less certain. Early detection of LCH-ND and initiation of therapy is therefore critical to prevent irreversible brain injury. Future studies including lesion genotype and prospective serial CSF and blood analysis will be helpful to validate the clinical utility of elevated CSF OPN and BRAFV600E+ PBMCs to identify patients at risk for LCH-ND. Clinical trials are required to determine the safety and efficacy of chemotherapy and MAPK pathway inhibition for patients with LCH-ND.
Supplementary Material
Acknowledgments
FUNDING SUPPORT
The TXCH Histiocytosis Program is supported by a research grant from the HistioCure Foundation. Additional support was received from National Institutes of Health (NIH) grants R01 CA154489 (to M.M. and C.E.A.), CA154947 (to M.M. and C.E.A.), and UL1TR001857 (to J.P.); NIH SPORE in Lymphoma (grant P50CA126752 to C.E.A.); the NIH Alex’s Lemonade Stand Foundation Young Investigator Grant (to R.C.); the American Society of Hematology Scholar Award (to R.C.); and the Stand Up to Cancer–St. Baldrick’s Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT1113) (to N.A. and D.W.P.). This article was a collaborative project with support from St. Baldrick’s Foundation, which sponsors the North American Consortium for Histiocytosis Research (K.L.M., A.K., C.R.-G., P.C., D.W.P., M.M., C.E.A.).
Footnotes
This collaborative study was conceived during discussions at the Nikolas Symposium, sponsored and organized by the Kontayannis family and is dedicated to the memory of Marian Malone, pediatric pathologist of Great Ormond Street Hospital. We also thank Lori Schmitt for technical support.
CONFLICT OF INTEREST DISCLOSURES
The authors made no disclosures.
REFERENCES
- 1.Berres ML, Allen CE, Merad M. Pathological consequence of misguided dendritic cell differentiation in histiocytic diseases. Adv.Immunol 2013;120:127–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Badalian-Very G, Vergilio JA, Degar BA et al. Recurrent BRAF mutations in Langerhans cell histiocytosis. Blood 2010;116:1919–1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chakraborty R, Burke TM, Hampton OA, et al. Alternative genetic mechanisms of BRAF activation in Langerhans cell histiocytosis. Blood 2016;128:2533–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Donadieu J, Rolon MA, Thomas C, et al. Endocrine involvement in pediatric-onset Langerhans’ cell histiocytosis: a population-based study. J Pediatr 2004;144:344–350. [DOI] [PubMed] [Google Scholar]
- 5.Grois N, Potschger U, Prosch H, et al. Risk factors for diabetes insipidus in Langerhans cell histiocytosis. Pediatr Blood Cancer 2006;46:228–233. [DOI] [PubMed] [Google Scholar]
- 6.Dunger DB, Broadbent V, Yeoman E, et al. The frequency and natural history of diabetes insipidus in children with Langerhans-cell histiocytosis. N Engl J Med 1989;321:1157–1162. [DOI] [PubMed] [Google Scholar]
- 7.Grois N, Fahrner B, Arceci RJ, et al. Central nervous system disease in Langerhans cell histiocytosis. J.Pediatr 2010;156:873–881. [DOI] [PubMed] [Google Scholar]
- 8.Grois N, Prayer D, Prosch H, Lassmann H. Neuropathology of CNS disease in Langerhans cell histiocytosis. Brain 2005;128:829–838. [DOI] [PubMed] [Google Scholar]
- 9.Laurencikas E, Gavhed D, Stalemark H, et al. Incidence and pattern of radiological central nervous system Langerhans cell histiocytosis in children: a population based study. Pediatr Blood Cancer 2011;56: 250–257. [DOI] [PubMed] [Google Scholar]
- 10.Shioda Y, Adachi S, Imashuku S, Kudo K, Imamura T, Morimoto A. Analysis of 43 cases of Langerhans cell histiocytosis (LCH)-induced central diabetes insipidus registered in the JLSG-96 and JLSG-02 studies in Japan. Int J Hematol 2011;94:545–551. [DOI] [PubMed] [Google Scholar]
- 11.The French LCH Study Group. A Multicenter retrospective survey of LCH: 348 cases observed between 1983 and 1993. Arch Dis Child 1993;75:17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wnorowski M, Prosch H, Prayer D, Janssen G, Gadner H, Grois N. Pattern and course of neurodegeneration in Langerhans cell histiocytosis. J Pediatr 2008;153:127–132. [DOI] [PubMed] [Google Scholar]
- 13.Grois N, Tsunematsu Y, Barkovich AJ, Favara BE. Central nervous system disease in Langerhans cell histiocytosis. Br J Cancer Suppl 1994;23:S24–S28. [PMC free article] [PubMed] [Google Scholar]
- 14.Grois NG, Favara BE, Mostbeck GH, Prayer D. Central nervous system disease in Langerhans cell histiocytosis. Hematol Oncol Clin North Am 1998;12:287–305. [DOI] [PubMed] [Google Scholar]
- 15.Martin-Duverneuil N, Idbaih A, Hoang-Xuan K, et al. MRI features of neurodegenerative Langerhans cell histiocytosis. Eur Radiol 2006; 16:2074–2082. [DOI] [PubMed] [Google Scholar]
- 16.Prayer D, Grois N, Prosch H, Gadner H, Barkovich AJ. MR imaging presentation of intracranial disease associated with Langerhans cell histiocytosis. AJNR Am J Neuroradiol 2004;25:880–891. [PMC free article] [PubMed] [Google Scholar]
- 17.Allen CE, Flores R, Rauch R, et al. Neurodegenerative central nervous system Langerhans cell histiocytosis and coincident hydrocephalus treated with vincristine/cytosine arabinoside. Pediatr Blood Cancer 2010;54:416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Idbaih A, Donadieu J, Barthez MA, et al. Retinoic acid therapy in “degenerative-like” neuro-langerhans cell histiocytosis: a prospective pilot study. Pediatr Blood Cancer 2004;43:55–58. [DOI] [PubMed] [Google Scholar]
- 19.Imashuku S, Okazaki N’, Nakayama M, et al. Treatment of neurodegenerative CNS disease in Langerhans cell histiocytosis with a combination of intravenous immunoglobulin and chemotherapy. Pediatr Blood Cancer 2008;50:308–311. [DOI] [PubMed] [Google Scholar]
- 20.Imashuku S, Shioda Y, Tsunematsu Y, Imamura T, Morimoto A. VCR/AraC chemotherapy and ND-CNS-LCH. Pediatr Blood Cancer 2010;55:215–216. [DOI] [PubMed] [Google Scholar]
- 21.Trouillas P, Takayanagi T, Hallett M, et al. International Cooperative Ataxia Rating Scale for pharmacological assessment of the cerebellar syndrome. The Ataxia Neuropharmacology Committee of the World Federation of Neurology. J Neurol Sci 1997;145:205–211. [DOI] [PubMed] [Google Scholar]
- 22.Gadner H, Minkov M, Grois N, et al. Therapy prolongation improves outcome in multi-system Langerhans cell histiocytosis. Blood 2013;121:5006–5014. [DOI] [PubMed] [Google Scholar]
- 23.Jordan MB, Allen CE, Weitzman S, Filipovich AH, McClain KL. How I treat hemophagocytic lymphohistiocytosis. Blood 2011;118:4041–4052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gavhed D, Akefeldt SO, Osterlundh G, et al. Biomarkers in the cerebrospinal fluid and neurodegeneration in Langerhans cell histiocytosis. Pediatr Blood Cancer 2009;53:1264–1270. [DOI] [PubMed] [Google Scholar]
- 25.Allen CE, Li L, Peters TL, et al. Cell-specific gene expression in Langerhans cell histiocytosis lesions reveals a distinct profile compared with epidermal Langerhans cells. J.Immunol 2010;184:4557–4567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Coppola D, Szabo M, Boulware D, et al. Correlation of osteopontin protein expression and pathological stage across a wide variety of tumor histologies. Clin Cancer Res 2004;10:184–190. [DOI] [PubMed] [Google Scholar]
- 27.Hyman DM, Diamond EL, Vibat CR, et al. Prospective blinded study of BRAFV600E mutation detection in cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2015;5:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cantor H, Shinohara ML. Regulation of T-helper-cell lineage development by osteopontin: the inside story. Nat Rev Immunol 2009;9: 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shinohara ML, Jansson M, Hwang ES, Werneck MB, Glimcher LH, Cantor H. T-bet-dependent expression of osteopontin contributes to T cell polarization. Proc Natl Acad Sci USA 2005;102:17101–17106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Brown A Osteopontin: a key link between immunity, inflammation and the central nervous system. Transl Neurosci 2012;3:288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Braitch M, Constantinescu CS. The role of osteopontin in experimental autoimmune encephalomyelitis (EAE) and multiple sclerosis (MS). Inflamm Allergy Drug Targets 2010;9:249–256. [DOI] [PubMed] [Google Scholar]
- 32.Chowdhury SA, Lin J, Sadiq SA. Specificity and correlation with disease activity of cerebrospinal fluid osteopontin levels in patients with multiple sclerosis. Arch Neurol 2008;65:232–235. [DOI] [PubMed] [Google Scholar]
- 33.Hur EM, Youssef S, Haws ME, Zhang SY, Sobel RA, Steinman L. Osteopontin-induced relapse and progression of autoimmune brain disease through enhanced survival of activated T cells. Nat Immunol 2007;8:74–83. [DOI] [PubMed] [Google Scholar]
- 34.Berres ML, Lim KP, Peters T, et al. BRAF-V600E expression in precursor versus differentiated dendritic cells defines clinically distinct LCH risk groups. J Exp Med 2014;211:669–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heritier S, Emile JF, Barkaoui MA, et al. BRAF mutation correlates with high-risk Langerhans cell histiocytosis and increased resistance to first-line therapy. J Clin Oncol 2016;34:3023–3030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Imashuku S, Arceci RJ. Strategies for the prevention of central nervous system complications in patients with Langerhans cell histiocytosis: the problem of neurodegenerative syndrome. Hematol Oncol Clin North Am 2015;29:875–893. [DOI] [PubMed] [Google Scholar]
- 37.Kepes J Histiocytosis X. Handbook of Neurology New York: Elsevier; 1979. [Google Scholar]
- 38.Ginhoux F, Greter M, Leboeuf M, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 2010;330:841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mass E, Jacome-Galarza CE, Blank T, et al. A somatic mutation in erythro-myeloid progenitors causes neurodegenerative disease. Nature 2017;549:389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Butovsky O, Jedrychowski MP, Moore CS, et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat.Neurosci 2014;17:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.