

Abstract
Mammalian hepatitis B X-interacting protein (HBXIP) is an 18-kDa protein that regulates a large number of transcription factors such as TF-IID, E2F1, SP1, STAT3, c-Myc, and LXR by serving as an oncogenic transcription coactivator and plays an important role in the development of breast cancer. We previously showed that HBXIP as an oncoprotein could enhance the promoter activity of MDM2 through coactivating p53, promoting the MDM2 transcription in breast cancer. In this study we investigated the molecular mechanisms underlying the modulation of MDM2/p53 interaction by HBXIP in human breast cancer MCF-7 cells in vitro and in vivo. We showed that HBXIP could up-regulate MDM2 through inducing DNA methylation of miR-18b, thus suppressing the miR-18b expression, leading to the attenuation of p53 in breast cancer cells. In addition, HBXIP could promote the phosphorylation of MDM2 by increasing the level of pAKT and bind to pMDM2, subsequently enhancing the interaction between MDM2 and p53 for the down-regulation of p53 in breast cancer cells. In MCF-7 breast cancer xenograft nude mice, we also observed that overexpression of HBXIP promoted breast cancer growth through the miR-18b/MDM2 and pAKT/MDM2 pathways. In conclusion, oncoprotein HBXIP suppresses miR-18b to elevate MDM2 and activates pAKT to phosphorylate MDM2 for enhancing the interaction between MDM2 and p53, leading to p53 degradation in promotion of breast cancer growth. Our findings shed light on a novel mechanism of p53 down-regulation during the development of breast cancer.
Keywords: HBXIP, p53, MDM2, miR-18b, pAKT, human breast cancer, cell proliferation, MCF-7 cells, cancer xenograft nude mice.
Introduction
Tumor suppressor p53 is a key transcription factor, which activates or represses varieties of genes expression and has been documented in multiple biological processes [1–3]. It plays an important role in apoptosis, cell cycle arrest, metabolism, senescence and is a crucial regulator of therapeutic targets in cancer [4–6]. The expression of p53 is subject to a large number of regulations at the levels of transcription, posttranscription and translation [7]. The MDM2 protein, which is encoded by the mouse double-minute 2 (MDM2) gene, is a negative regulator of p53 and implicated in the abundance and subcellular localization of p53 [7, 8]. MDM2 is overexpressed in lots of human cancers, including lung cancer, melanoma, breast cancer, non-Hodgkin’s lymphoma, esophageal cancer, leukemia and sarcoma [9, 10]. Many studies have indicated that MDM2 can act as an E3 ligase to promote the proteasome degradation of p53 during cancer development [11–14]. Meanwhile, a variety of proteins in the cells, such as transcription factor p53, can up-regulate the MDM2 expression [15]. Yet, the underlying regulatory mechanism of MDM2-p53 circuit by different pathways in cancers still need to be further investigated.
The expression of MDM2 and MDM2-mediated p53 ubiquitination and degradation can be modulated in different ways [7, 16]. Overexpressed miR-18b is capable of reducing MDM2 and increasing p53 to regulate the MDM2–p53 pathway in melanoma cells [17]. MiR-18b is located at chromosome X in the cluster of miR-106a-363 and its aberrant expression is associated with a variety of human pathologies, such as multiple sclerosis, cardiac hypertrophy and hepatitis B virus chronic infection [18–20]. Moreover, miR-18b expression is also deregulated in human cancers and is highly expressed in ERα-negative as compared with ERα-positive clinical tumors [21]. The methylation takes great part in the regulation of miR-18b expression and MDM2–p53 pathway [17]. In addition, previous researches indicate that the phosphorylation of MDM2 by multiple stress-activated kinases including c-Abl, ATM, or AKT affects MDM2–p53 signaling and also significantly alters the function of p53 [22, 23]. Accumulating evidence has revealed that the protein kinase AKT is able to enhance the ubiquitination function of MDM2, leading to the down-regulation of p53 in breast cancer cells [24]. Nevertheless, the further mechanism of the pathway remains unknown.
Mammalian hepatitis B X-interacting protein (HBXIP), also known as LAMTOR5 [25], is an 18-kDa protein containing a leucine zipper at its C-terminal [26]. It was firstly identified via the interaction with hepatitis B virus X protein, resulting in the suppression of hepatitis B virus replication and modulating the duplication of centrosome in HeLa cells [27]. Our previous study has indicated that HBXIP as an oncopretein recruits the acetyltransferase p300 to p53 in the promoter of MDM2, promoting the MDM2 transcription in breast cancer [28]. It has been reported that HBXIP regulates a large number of transcription factors, such as TF-IID, E2F1, SP1, STAT3, c-Myc, and LXR by serving as an oncogenic transcription coactivator and plays an important role in the development of breast cancer [29–33]. However, the indirect regulatory mechanism of MDM2 by oncoprotein HBXIP in breast cancer remains unclear.
Here, we investigated the effect of HBXIP on modulation of MDM2–p53 pathway via two different cascades. Our data reveal that HBXIP suppresses miR-18b to increase the MDM2 expression and induces the phosphorylation of MDM2 to enhance the interaction of MDM2 with p53, leading to augmented p53 degradation in acceleration of breast cancer growth. The present study provides a new insight into the mechanism of p53 down-regulation mediated by HBXIP during the development of breast cancer.
Materials and methods
Cell culture and treatment
Breast cancer cell lines MCF-7, LM-MCF-7, (a metastatic subclone of MCF-7 breast cancer cell line) and MCF-7-HBXIP (stably transfected pCMV-HBXIP plasmid) were maintained in RPMI 1640 medium (Gibco, CA, USA). Dulbecco’s modified Eagle’s medium (Gibco) was used to culture human kidney epithelial HEK293T cells and human colon cancer cell line HCT116 p53−/−. All cell lines were maintained in heat inactivated 10% fetal bovine serum (FBS, Gibco), 100 mg/mL streptomycin as well as 100 U/mL penicillin at 37 °C and 5% CO2. All cell lines used in the manuscript were authenticated before the experiments.
Total RNA isolation, reverse-transcription PCR (RT-PCR) and real-time PCR
RNA was isolated from cells by TRIzol reagent (Invitrogen, USA). The PrimeScript reverse transcriptase Kit (TaKaRa Bio, China) was used to synthesize first-strand cDNA. To detect the expression of mature miR-18b, total RNA was polyadenylated by poly (A) polymerase (Ambion, Austin, TX, USA). The poly (A)-tailed total RNA and reverse transcription primer with ImPro-II Reverse Transcriptase (Promega, Madison, WI, USA) were used to perform reverse transcription, based on manufacturer’s instructions. The real-time PCR and RT-PCR were performed as described previously [34]. Primers used for detection of HBXIP, p53, MDM2, AKT, GAPDH cDNA, and miR-18b, U6 were described in Supplementary Table S1.
Plasmid construction and small interference
The pCMV-tag2B, siRNAs, pGL3-Basic vector, pRL-TK plasmid (Promega), pCMV-MDM2 and pCMV-HBXIP were kept in our laboratory. The constitutively active AKT (CA-AKT) was purchased from Upstate Biotechnology (Lake Placid, NY). The miR-18b mimics and anti-miR-18b mimics were obtained from Riobio Company (Guangzhou, China). For animal transplantation experiment, miR-18b expression plasmid was obtained also from Riobio Company (Guangzhou, China). The miR-18b promoter region was cloned into pGL3-Basic vector for generation of the pGL3-miR-18b construct. Primers used for pGL3-miR-18b cloning were described in Supplementary Table S1 according to previous study [17]. The nucleotide sequence of siRNA for AKT was subcloned into pSilencer 3.1-H1 neo vector for generating pSilencer-AKT. The siRNAs against HBXIP, p53, AKT, and control siRNAs were purchased from Riobio Company (Guangzhou, China). The sequences of the siRNAs were described previously [27, 35, 36].
Western blot analysis
Western blot analysis was performed according to manufacturer’s protocols. Primary antibodies used were rabbit anti-p53 (Proteintech Group, USA), rabbit anti-MDM2 (BOSTER, Wuhan, China), mouse anti-pMDM2 (Ser186) (Aviva Systems Biology, USA), rabbit anti-HBXIP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), mouse anti-pAKT (Abcam, Cambridge, UK), rabbit anti-AKT (BOSTER), rabbit anti-pPRAS40 (Thr246) (OmnimAbs, USA) and mouse anti-β-actin (Sigma, Aldrich, St. Louis, MO, USA). For related experiments, the LY294002 (a PI3K inhibitor) (MedChemExpress, USA) and MK-2206 (an AKT inhibitor) (MedChemExpress, USA) were used at 20 μM and 5 μM, respectively.
Luciferase reporter gene assay
The cells were cultured in 24-well plates and transfected with relative plasmids or siRNAs. Cell extracts were obtained through lysis buffer (Promega). The Dual-Luciferase Reporter Assay System (Promega) was used for luciferase reporter gene assay according to instructions for manufacturer. For related experiments, the methylation inhibitor, 5-aza-2’-deoxycytidine (Aza) (Sigma), was used at 5.0 μM.
Methylation-specific PCR
Promoter methylation of miR-18b gene was detected by methylation-specific PCR (MSP) as previously described [17]. Genomic DNA was subjected to bisulfite modification by using Epitect Bisulfite Kit (Qiagen, Germany) based on the protocol. MSP experiments were performed at least in duplicate.
Co-immunoprecipitation assay
The protocol for co-immunoprecipitation (co-IP) was indicated in previously published articles [37]. Lysates from the cells were incubated with relative antibodies and protein G-conjugated agarose beads for 2 h at 4 °C. The precipitates were washed at least six times with ice-cold lysis buffer, and then resolved using SDS-PAGE loading buffer followed by Western blot analysis.
Confocal microscopy
Confocal microscopy was performed as described previously [38]. The cells were fixed with paraformaldehyde, and permeabilized with 0.1% Triton X-100 in PBS. After blocking, primary antibodies and fluorophore-conjugated secondary antibody (DAKO) were used to incubate with cells at room temperature. After washing by PBS, slides were mounted with glycerol and observed under a confocal microscopy (Leica TCS SP5).
Analysis of cell proliferation
Proliferation of the cells was tested using 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) (Sigma) assay as previously described [39] and 5-ethynyl-2′-deoxyuridine (EdU) incorporation assay. EdU incorporation assay was carried out with the Cell-Light TM EdU imaging detection kit (RiboBio) based on the instructions for manufacturer.
Colony formation assay
For clonogenicity analysis, cells after transfection were cultured in 6-well plates with complete medium for 2–3 weeks. Colonies were fixed with methanol and stained with methylene blue.
Animal transplantation
4-week-old female BALB/c athymic nude mice were divided into four groups (Experiment Animal Center of Peking, China; each group, n = 5) and treated according to guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Based on the report [40], 1.7 mg of 17β-estradiol pellet (90 days release; Beijing Solarbio Science and Technology Co., China) was implanted into each nude mouse to provide the estrogen for the proliferation of MCF-7 cells. After implantation, MCF-7-pCMV + shControl, MCF-7-HBXIP + shControl, MCF-7-HBXIP + miR-18b (MCF-7-HBXIP cells transfected with 7.5 μg of miR-18b expression plasmids) and MCF-7-HBXIP + shAKT (MCF-7-HBXIP cells transfected with 7.5 μg of pSilencer-AKT) cells were harvested, suspended at 5 × 107 cells/mL with phosphate-buffered saline. The mouse mammary fat pads were injected with 0.2 mL of the cell suspensions. Beginning 5 days after injection, the growth of tumor was tested every 3 days. Tumor volume (V), quantified by testing the width (W) and length (L) using calipers, was calculated through the formula (W2 × L) × 0.5. After 30 days, the mice were sacrificed, and the tumors were excised and measured. All the studies were performed according to the guidelines established by the National Institutes of Health Guide for the Care and Use of Laboratory Animals and were performed according to the institutional ethical guidelines for animal experiment. All experiments were approved by the Institute Research Ethics Committee at Nankai University.
Statistical analysis
Each experiment was repeated at least three times. Statistical significance was assessed by comparing mean values ( ± SD) using a Student’s t test for independent groups and was assumed for P < 0.05 (*), P < 0.01 (**) and P < 0.005 (***).
Results
HBXIP up-regulates MDM2 through decreasing miR-18b in breast cancer cells
In the previous research, we reported that HBXIP could directly stimulate MDM2 transcription in regulation of the MDM2–p53 circuit via acting as a coactivator of p53 transcription factor [28]. Since HBXIP is a multifunctional oncogenic coactivator involved in numerous crosstalks between signaling pathways, we are interested in finding out whether HBXIP can modulate MDM2 or p53 levels by other ways in breast cancer cells. Then, we investigated the effect of HBXIP on MDM2 expression in breast cancer cells treated with p53 siRNA. Interestingly, when p53 was decreased by p53 siRNA in MCF-7 cells, HBXIP could still increase the mRNA level of MDM2 (Fig. 1a). Furthermore, we verified the result in a p53-deficient cell line, HCT116 p53−/− cells (Fig. 1b). These data demonstrated that HBXIP was capable of up-regulating MDM2 expression level in a p53 independent manner. It has been reported that the miR-18b is able to promote the reduction of MDM2 in melanoma cells [17]. Accordingly, we wondered whether HBXIP could up-regulate MDM2 via modulating miR-18b in breast cancer cells. As expected, we found that HBXIP could decrease the level of miR-18b by real-time PCR and increase the expression of MDM2 at the mRNA and protein levels in MCF-7 cells through RT-PCR and Western blot analysis (Fig. 1c). The similar result was obtained in HCT116 p53−/− cells (Supplementary Figure S1a). Meanwhile, knockdown of HBXIP in LM-MCF-7 cells (a metastatic subclone of MCF-7 cell line expressing high-level HBXIP [29]) or in MCF-7-HBXIP cells (stably transfected pCMV-HBXIP plasmid) showed the opposite results (Fig. 1d, Supplementary Figure S1b). Our data further revealed that overexpressed miR-18b could abolish the up-regulation of MDM2 mediated by HBXIP in the cells (Fig. 1e, Supplementary Figure S1c). However, anti-miR-18b could attenuate the inhibition of MDM2 mediated by HBXIP siRNA in LM-MCF-7 and MCF-7-HBXIP cells (Fig. 1f, Supplementary Figure S1d). Therefore, we conclude that HBXIP is capable of elevating MDM2 through bating miR-18b in breast cancer cells.
Fig. 1.

HBXIP up-regulates MDM2 through decreasing miR-18b in breast cancer cells. a The expression of HBXIP, p53, and MDM2 at the level of mRNA was examined by RT-PCR analysis in MCF-7 cells. b The mRNA level of HBXIP and MDM2 was examined by RT-PCR analysis in HCT116 p53−/− cells. c, d The expression of HBXIP and MDM2 at the levels of mRNA and protein was examined by RT-PCR and Western blot analysis in MCF-7 and LM-MCF-7 cells, respectively. The expression level of miR-18b was detected by real-time PCR analysis in MCF-7 and LM-MCF-7 cells, respectively. e, f The mRNA level of HBXIP and MDM2 was determined by RT-PCR analysis in MCF-7 and LM-MCF-7 cells, respectively. Each experiment was repeated at least three times. Student’s t test; *P < 0.05; **P < 0.01
HBXIP restrains p53 through enhancing MDM2 involving methylation of miR-18b promoter
Next, we tried to explore the underlying mechanism about the suppression of miR-18b induced by HBXIP. The core region of the miR-18b promoter [17, 41], termed as miR-18b-pro, was cloned into the pGL3-Basic plasmid for generating the construct of pGL3-miR-18b. We determined that the promoter activity of miR-18b was markedly decreased by HBXIP in MCF-7 cells (Fig. 2a), and the result was validated in a dose-dependent manner in MCF-7 and HEK293T cells (Fig. 2b ,c). Moreover, HBXIP siRNA enhanced the activity of miR-18b promoter in LM-MCF-7 cells (Fig. 2d). Thus, the results imply that the overexpression of HBXIP is able to suppress the promoter activity of miR-18b in breast cancer cells. Previous study has demonstrated that the promoter of miR-18b can be silenced by hypermethylation in cells [17]. Hence, we supposed that HBXIP might decrease miR-18b level by inducing methylation of miR-18b promoter and then performed methylation analysis of miR-18b-pro. Through the Meth Primer software (http://www.urogene.org/methprimer/) [42], we observed a large number of CpG-rich regions (Fig. 2e). Then, the CpG methylation of miR-18b promoter was observed in stably pCMV-HBXIP-transfected MCF-7 cells using methylation-specific PCR (MSP) analysis (Fig. 2f), suggesting that HBXIP could promote the methylation of miR-18b promoter in breast cancer cells. Furthermore, treatment of MCF-7 and HEK293T cells with 5-aza-2′-deoxycytidine (Aza), the DNA methylation inhibitor, reversed the suppression of miR-18b promoter activity mediated by HBXIP (Supplementary Figure S2a and b), supporting that HBXIP could suppress miR-18b through inducing methylation of miR-18b promoter in the cells. Western blot analysis further indicated that miR-18b could abolish the up-regulation of MDM2 and down-regulation of p53 mediated by HBXIP in MCF-7 cells (Fig. 2g), confirming that HBXIP up-regulated MDM2 and down-regulated p53 via modulation of miR-18b in the cells. Collectively, HBXIP can down-regulate p53 through up-regulating MDM2 involving inducing methylation of miR-18b promoter and decreasing miR-18b in breast cancer cells.
Fig. 2.

HBXIP restrains p53 through enhancing MDM2 involving methylation of miR-18b promoter. a–c The activities of miR-18b promoter (miR-18b-pro) were examined by luciferase reporter gene assay in MCF-7 and HEK293T cells transfected with pCMV-HBXIP plasmids. d Effect of HBXIP siRNA on miR-18b-pro activities was detected by luciferase reporter gene assay in LM-MCF-7 cells. e Schematic representation of methylation analysis of the miR-18b promoter, with CpG sites represented by vertical lines. f CpG island methylation in miR-18b promoter was examined by methylation-specific PCR (MSP). g The expression levels of HBXIP, MDM2, and p53 were detected by Western blot analysis in MCF-7 cells. Each experiment was repeated at least three times. Student’s t test; *P < 0.05; **P < 0.01
HBXIP is required for MDM2-induced p53 degradation
As we know, the E3 ubiquitin ligase activity of MDM2 requires other potent activators and numerous proteins are capable of interacting with MDM2 to promote the polyubiquitination of p53 mediated by MDM2 [7, 43–45]. Therefore, we speculated that HBXIP was involved in the MDM2-mediated p53 degradation through interacting with MDM2 in the cells. Co-IP analysis showed that HBXIP could bind to MDM2 in MCF-7 cells (Fig. 3a). The co-localization was further revealed by confocal microscopy (Fig. 3b), supporting that HBXIP could interact with MDM2 in breast cancer cells. Interestingly, silencing or overexpression of HBXIP could suppress or enhance the interaction between p53 and MDM2 in MCF-7 cells (Fig. 3c), implying that HBXIP was able to recruit p53 to MDM2 in the cells. In addition, Western blot analysis indicated that the HBXIP depletion could attenuate the inhibition of p53 mediated by MDM2 although MDM2 was overexpressed in MCF-7 cells (Fig. 3d), supporting that HBXIP was required for MDM2-mediated p53 reduction in breast cancer cells. The conclusion was further validated in MCF-7-HBXIP cells (Supplementary Figure S3). Thus, we summarize that HBXIP is required for the degradation of p53 mediated by MDM2 through recruiting p53 to MDM2.
Fig. 3.

HBXIP is required for MDM2-induced p53 degradation. a The interaction between HBXIP and MDM2 was detected by co-IP assay in MCF-7 cells in vivo. b The colocalization of MDM2 and Flag-HBXIP was examined in the MCF-7 cells by confocal microscopy. c The interaction of MDM2 with p53 was determined by co-IP assay in MCF-7 cells with HBXIP siRNA or pCMV-HBXIP treatment. d Effect of HBXIP siRNA on p53 was examined by Western blot analysis in MCF-7 cells when MDM2 was overexpressed. Each experiment was repeated at least three times
HBXIP enhances the interaction between p53 and MDM2 via promoting the phosphorylation of MDM2 mediated by AKT and binding to pMDM2
Taken a step further, we aimed to identify the mechanism by which HBXIP recruited p53 to MDM2. Previous study has proven that the protein kinase AKT is able to enhance MDM2-mediated ubiquitination and degradation of p53 by phosphorylation of Ser186 of MDM2 in breast cancer cells [24]. Given that HBXIP could activate AKT in breast cancer cells [46], we raised a presumption that HBXIP participated in MDM2-mediated degradation of p53 via modulating AKT-MDM2 signaling pathway. We firstly showed that HBXIP had no effect on the mRNA and protein levels of AKT in breast cancer cells as indicated by RT-PCR and Western blot analysis (Fig. 4a-d). Then, the result verified that overexpression of HBXIP could result in the increase of pAKT level and up-regulation of pMDM2 (Ser186) in MCF-7 cells in a dose-dependent manner (Fig. 4c). HBXIP siRNA led to the opposite result in LM-MCF-7 cells (Fig. 4d). The above data illustrated that HBXIP could increase the phosphorylation level of AKT as well as MDM2 in breast cancer cells. Furthermore, Western blot analysis revealed that the inactivation of AKT by treatment with LY294002, a PI3-kinase inhibitor, or AKT siRNA could impair the up-regulation of pMDM2 mediated by HBXIP in MCF-7 cells (Fig. 4e). Meanwhile, the HBXIP siRNA and MK-2206 (AKT inhibitor) could both decrease the level of pMDM2 in MCF-7-HBXIP cells (Fig. 4f). These results supported that HBXIP increased pMDM2 through up-regulating pAKT in the cells. Finally, co-IP analysis revealed that the inactivation of AKT using MK-2206 or LY294002 could suppress the HBXIP-enhanced interaction between p53 and MDM2 in MCF-7 cells (Fig. 4g, Supplementary Figure S4a), confirming that the HBXIP-induced recruitment of p53 to MDM2 required the phosphorylation of MDM2 mediated by AKT in the cells. Meanwhile, knockdown of HBXIP could also inhibit the interaction enhanced by constitutively active AKT (CA-AKT) in MCF-7 cells (Fig. 4h), indicating that the AKT-induced phosphorylation of MDM2 could enhance the interaction between p53 and MDM2 and the event required the participation of HBXIP. Additionally, we verified that the phosphorylation of MDM2 by AKT was required for HBXIP binding to MDM2 in MCF-7 cells (Fig. 4i, Supplementary Figure S4b). In conclusion, our data indicate that HBXIP enhances the interaction between p53 and MDM2 via promoting the phosphorylation of MDM2 mediated by AKT and binding to pMDM2 in breast cancer cells.
Fig. 4.

HBXIP enhances the interaction between p53 and MDM2 via promoting the phosphorylation of MDM2 mediated by AKT and binding to pMDM2. a, b The mRNA levels of HBXIP and AKT were detected by RT-PCR in MCF-7 and LM-MCF-7 cells. c, d The levels of HBXIP, total AKT, pAKT, and pMDM2 were examined by Western blot analysis in MCF-7 cells transfected with pCMV-HBXIP (or pCMV) and LM-MCF-7 cells transfected with HBXIP siRNA (or Control siRNA). e Effects of LY294002 (a PI3K inhibitor) or AKT siRNA on the levels of total AKT, pAKT, phosphorylated PRAS40 (Proline-rich AKT substrate 40 kDa) and pMDM2 were examined by Western blot analysis in MCF-7 cells when HBXIP was overexpressed. f Effects of MK-2206 (an AKT inhibitor) or HBXIP siRNA on the levels of total AKT, pAKT, pPRAS40, and pMDM2 were examined by Western blot analysis in MCF-7-HBXIP cells. g, h The interaction between MDM2 and p53 was determined by co-IP assay in MCF-7 cells treated with pCMV-HBXIP/MK-2206 or CA-AKT/HBXIP siRNA. i The interaction between HBXIP and MDM2 was detected by co-IP assay in MCF-7 cells treated with MK-2206. Each experiment was repeated at least three times
HBXIP promotes the proliferation of breast cancer cells by suppressing miR-18b and enhancing the interaction between MDM2 and p53
Next, we evaluated the effect of HBXIP on breast cancer cell proliferation mediated by miR-18b and HBXIP-enhanced interaction of MDM2-p53 in vitro. Since the preceding results revealed that HBXIP induced the interaction between p53 and MDM2 via activating AKT, the knockdown of AKT was thought to be an effective approach for blocking the MDM2-p53 interaction enhanced by HBXIP. Overall, the experiments were performed by using miR-18b/anti-miR-18b mimics and AKT siRNA/CA-AKT. As expected, the treatment with miR-18b or AKT siRNA could abolish the proliferation of MCF-7 cells enhanced by HBXIP by MTT assay (*P < 0.05, **P < 0.01, Student’s t test, Fig. 5a). The similar results were obtained by using EdU incorporation assay (*P < 0.05, **P < 0.01, Student’s t test, Fig. 5b). While, the treatment of anti-miR-18b or CA-AKT rescued the proliferation of LM-MCF-7 cells decreased by HBXIP siRNA (*P < 0.05, **P < 0.01, Student’s t test, Fig. 5c). Colony formation assay could repeat the above result in MCF-7 cells (Fig. 5d), suggesting that miR-18b and AKT pathway were required for the promotion of breast cancer cell proliferation mediated by HBXIP. Above all, we conclude that HBXIP promotes the proliferation of breast cancer cells by suppressing miR-18b and enhancing the interaction between MDM2 and p53.
Fig. 5.
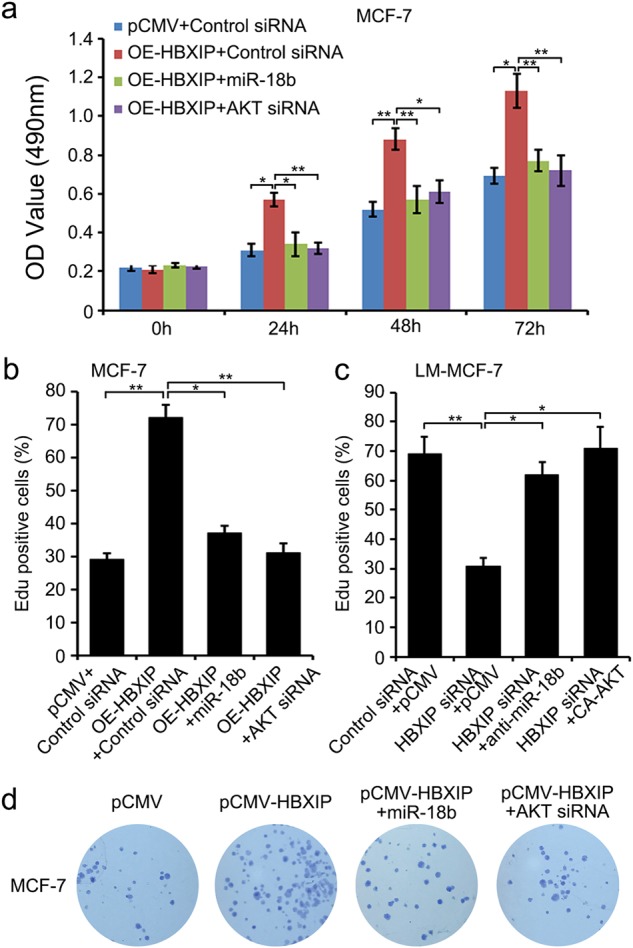
HBXIP promotes the proliferation of breast cancer cells by suppressing miR-18b and enhancing the interaction between MDM2 and p53. a The effect of miR-18b or AKT siRNA on HBXIP-enhanced cell proliferation was measured by MTT in MCF-7 cells. b The effect of miR-18b or AKT siRNA on HBXIP-enhanced cell proliferation was measured by EdU incorporation assay in MCF-7 cells. c The effect of anti-miR-18b or CA-AKT on cell proliferation suppressed by HBXIP siRNA was measured by EdU incorporation assay in LM-MCF-7 cells. d The effect of HBXIP with miR-18b or AKT siRNA on cell proliferation was detected by colony formation assay in MCF-7 cells. The data presented are from three independent experiments; Student’s t test; *P < 0.05; **P < 0.01
HBXIP accelerates the tumor growth through suppressing miR-18b and enhancing the interaction between MDM2 and p53 in mice
To further investigate the effect of HBXIP on tumor growth by modulation of miR-18b and MDM2–p53 interaction in vivo, we performed tumor formation assay. The increased volume and weight of tumors were observed in OE (overexpressed)-HBXIP group, whereas the OE-HBXIP combined with miR-18b or AKT knockdown (shAKT) group showed a significant reduction in tumor growth (*P < 0.05, **P < 0.01, Student’s t test, Fig. 6a-c). Meanwhile, the protein levels of HBXIP, MDM2, and p53 from tumor tissues of mice were validated using Western blot analysis (Fig. 6d), suggesting that HBXIP down-regulated p53 through up-regulating MDM2 involving suppressing miR-18b and modulating MDM2–p53 interaction. Overall, the results strongly suggest that HBXIP accelerates the tumor growth through suppressing miR-18b and enhancing the interaction between MDM2 and p53 in mice.
Fig. 6.

HBXIP accelerates the tumor growth through suppressing miR-18b and enhancing the interaction between MDM2 and p53 in mice. a Growth curve of tumors in nude mice. b Average weight of tumors. c Tumor images. d The relative expression levels of HBXIP, MDM2, and p53 in the tumor tissues from mice were detected by Western blot analysis. The data presented are from three independent experiments; Student’s t test; *P < 0.05; **P < 0.01
Discussion
Tumor suppressor p53 is a transcription factor, which can activate or repress varieties of genes expression and plays a critical role in multiple biological processes by acting as an important regulator of therapeutic target in cancer [47, 48]. The MDM2 protein is able to function as an E3 ligase to promote the p53 degradation during cancer development [13, 49]. However, the regulatory mechanism of MDM2 by different pathways and the underlying mechanism of p53 down-regulation mediated by MDM2 in cancers are still elusive. Our report has revealed that HBXIP can serve as a coactivator of p53 to stimulate the MDM2 transcription for the regulation of MDM2–p53 circuit in breast cancer [28]. In the current study, we concern whether HBXIP can modulate MDM2 and p53 levels by other ways in breast cancer cells.
We first found that HBXIP could up-regulate the expression level of MDM2 independent of p53 in cells. Since evidences demonstrated that the miR-18b could promote the reduction of MDM2 in melanoma cells [17, 50], we speculated that HBXIP up-regulated MDM2 through modulating miR-18b in breast cancer cells. As expected, we observed that HBXIP could decrease the level of miR-18b in breast cancer cells. Moreover, HBXIP enhanced MDM2 expression through decreasing miR-18b in the cells. Next, we aimed to clarify the mechanism of miR-18b suppression induced by HBXIP. Our results indicated that overexpressed HBXIP was able to decrease the miR-18b promoter activity in breast cancer cells. Previous research has indicated that the promoter of miR-18b can be silenced by hypermethylation in cells [17]. Interestingly, we revealed that HBXIP inhibited miR-18b through inducing methylation of miR-18b promoter in the cells. The expression of miR-18b is deregulated in varieties of human cancers, while the role of miR-18b in different cancers, especially in breast cancer, remains largely unexplored [51]. Here, we illustrated that the miR-18b was decreased by highly expressed HBXIP in MCF-7 cells, which was consistent with the previous research that miR-18b levels were significantly increased in most breast cancer cells but decreased in MCF-7 cells and normal breast tissues [52]. We first report that HBXIP down-regulates p53 through up-regulating MDM2 involving inducing methylation of miR-18b promoter and decreasing miR-18b in breast cancer cells.
Gene regulation is a complex network in cells that involves a number of regulators to have crosstalks with each other between different pathways [53, 54]. Hence, we are interested in other mechanism by which HBXIP modulates p53. Since the E3 ubiquitin ligase activity of MDM2 required other potent activators and numerous proteins could interact with MDM2 to promote the ubiquitination of p53 mediated by MDM2 [43, 45], we assumed that HBXIP was involved in the MDM2-mediated p53 degradation through interacting with MDM2. As expected, we verified that HBXIP participated in the degradation of p53 mediated by MDM2 by recruiting p53 to MDM2 in the cells. Previous study revealed that the protein kinase AKT was responsible for MDM2-mediated ubiquitination and degradation of p53 through Ser186 phosphorylation of MDM2 in breast cancer cells [24], but the deeper understanding for the event was still unclear. Additionally, we reported previously that HBXIP could activate AKT in breast cancer cells [46]. Accordingly, here we hypothesized that HBXIP took part in MDM2-mediated degradation of p53 via modulating AKT-MDM2 signaling pathway in the cells. As predicted, the result illustrated that HBXIP could enhance the interaction between p53 and MDM2 through promoting the phosphorylation of MDM2 mediated by AKT and binding to pMDM2 in breast cancer cells. Although AKT has been indicated to modulate the p53 degradation mediated by MDM2 [24], the relationship between pAKT/pMDM2 pathway and MDM2–p53 interaction was still elusive. In this study, we first revealed that the AKT-induced phosphorylation of MDM2 could enhance the interaction between p53 and MDM2 and the event required the participation of HBXIP in breast cancer cells. So far, we identified two HBXIP-mediated pathways for down-regulation of p53 in promotion of breast cancer growth. Additionally, we validated the relationship between the two pathways in breast cancer cells. The result demonstrated that AKT could not modulate miR-18b upon HBXIP in the cells (Supplementary Figure S4c), suggesting that HBXIP functioned through two independent pathways.
Growing evidences demonstrated that increased level of MDM2 or down-regulation of wild-type p53 was able to promote breast cancer progression [55, 56]. In function, we further approved that HBXIP promoted breast cancer development by suppressing miR-18b and enhancing the interaction between MDM2 and p53 in vivo and in vitro. Therefore, HBXIP can be taken as an important target of breast cancer.
The main approach for the stabilization of p53 is the decreased interaction between MDM2 and itself, resulting in the up-regulation of p53 [57]. But little is indicated about the mechanism of accelerating p53 down-regulation in cancers. In this study, we present a model showing that HBXIP enhances breast cancer cell proliferation by decreasing p53 in two pathways (Fig. 7): 1) HBXIP suppresses the expression of miR-18b to up-regulate MDM2 via inducing the CpG island methylation in miR-18b promoter, resulting in the reduction of p53; 2) HBXIP accelerates the p53 degradation through promoting the phosphorylation of MDM2 by increasing the level of pAKT and binding to pMDM2, which subsequently enhances the interaction between MDM2 and p53. Therefore, our findings provide new insights into the mechanism of p53 down-regulation in promotion of breast cancer growth.
Fig. 7.
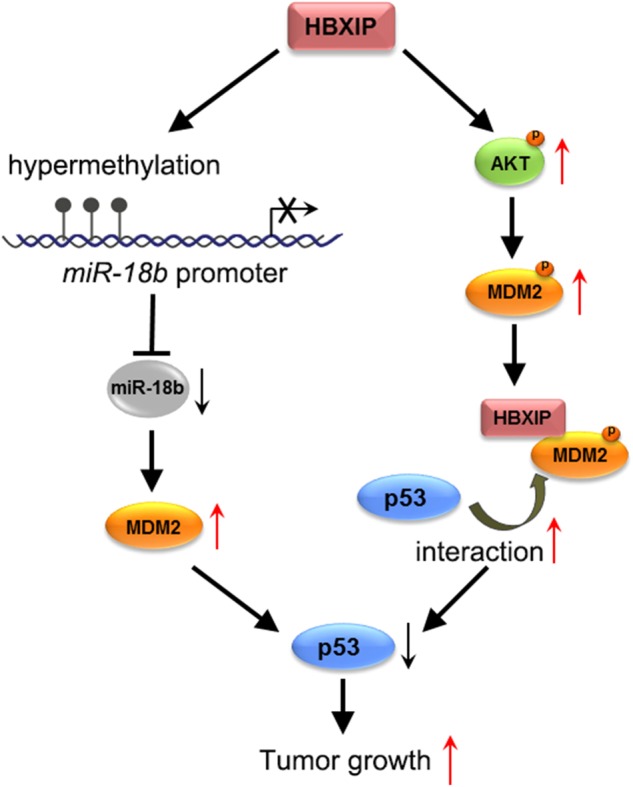
A model showing that the oncoprotein HBXIP down-regulates p53 through two pathways to promote the growth of breast cancer. HBXIP up-regulates MDM2 through suppressing miR-18b via inducing the CpG island methylation in miR-18b promoter, resulting in the reduction of p53; HBXIP enhances the p53 degradation through promoting the phosphorylation of MDM2 by increasing the level of pAKT and binding to pMDM2, which subsequently enhances the interaction between MDM2 and p53
Electronic supplementary material
Acknowledgements
This work was supported by grants from the National Basic Research Program of China (973 Program No. 2015CB553905), the National Natural Scientific Foundation of China (Nos. 81372186, 31670771), the Fundamental Research Funds for the Central Universities, Project of Prevention and Control of Key Chronic Non Infectious Diseases (No. 2016YFC1303401), CAMS Innovation Fund for Medical Sciences (CIFMS, 2017-I2M-3-019 and 2016-I2M-1-017), the PUMC Youth Fund and the Fundamental Research Funds for the Central Universities (No. 2017310027), the Tianjin Science and Technology Support Plan Project (TJKJZC, 14ZCZDSY00001).
Author contributions
HL, ZW, MJ, YS, X-lC, and QL performed the experiments. HL, R-pF, HS, and KY carried out the data analysis. HL, S-jF, W-yZ, L-hY designed the study. S-jF, W-yZ, and L-hY supervised the study. HL, S-jF, W-yZ, and L-hY wrote the manuscript with input from all authors.
Competing interests
The authors declare no competing interests.
Contributor Information
Sai-jun Fan, Email: fansaijun@irm-cams.ac.cn.
Wei-ying Zhang, Email: zhwybao@nankai.edu.cn.
Li-hong Ye, Email: yelihong@nankai.edu.cn.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0034-6) contains supplementary material, which is available to authorized users.
References
- 1.Wang L, Wu Q, Qiu P, Mirza A, McGuirk M, Kirschmeier P, et al. Analyses of p53 target genes in the human genome by bioinformatic and microarray approaches. J Biol Chem. 2001;276:43604–10. doi: 10.1074/jbc.M106570200. [DOI] [PubMed] [Google Scholar]
- 2.Mandriani B, Castellana S, Rinaldi C, Manzoni M, Venuto S, Rodriguez-Aznar E, et al. Identification of p53-target genes in Danio rerio. Sci Rep. 2016;6:32474. doi: 10.1038/srep32474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yao GD, Yang J, Li Q, Zhang Y, Qi M, Fan SM, et al. Activation of p53 contributes to pseudolaric acid B-induced senescence in human lung cancer cells in vitro. Acta Pharmacol Sin. 2016;37:919–29. doi: 10.1038/aps.2016.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amaral JD, Castro RE, Sola S, Steer CJ, Rodrigues CM. p53 is a key molecular target of ursodeoxycholic acid in regulating apoptosis. J Biol Chem. 2007;282:34250–9. doi: 10.1074/jbc.M704075200. [DOI] [PubMed] [Google Scholar]
- 5.Wiegering A, Matthes N, Muhling B, Koospal M, Quenzer A, Peter S, et al. Reactivating p53 and inducing tumor apoptosis (RITA) enhances the response of RITA-sensitive colorectal cancer cells to chemotherapeutic agents 5-fluorouracil and oxaliplatin. Neoplasia. 2017;19:301–9. doi: 10.1016/j.neo.2017.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Su LY, Shi YX, Yan MR, Xi Y, Su XL. Anticancer bioactive peptides suppress human colorectal tumor cell growth and induce apoptosis via modulating the PARP-p53-Mcl-1 signaling pathway. Acta Pharmacol Sin. 2015;36:1514–9. doi: 10.1038/aps.2015.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hollstein M, Hainaut P. Massively regulated genes: the example of TP53. J Pathol. 2010;220:164–73. doi: 10.1002/path.2637. [DOI] [PubMed] [Google Scholar]
- 8.Feng FY, Zhang Y, Kothari V, Evans JR, Jackson WC, Chen W, et al. MDM2 inhibition sensitizes prostate cancer cells to androgen ablation and radiotherapy in a p53-dependent manner. Neoplasia. 2016;18:213–22. doi: 10.1016/j.neo.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Momand J, Jung D, Wilczynski S, Niland J. The MDM2 gene amplification database. Nucl Acids Res. 1998;26:3453–9. doi: 10.1093/nar/26.15.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thomasova D, Mulay SR, Bruns H, Anders HJ. p53-independent roles of MDM2 in NF-kappaB signaling: implications for cancer therapy, wound healing, and autoimmune diseases. Neoplasia. 2012;14:1097–101. doi: 10.1593/neo.121534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kubbutat MH, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- 12.Sczaniecka M, Gladstone K, Pettersson S, McLaren L, Huart AS, Wallace M. MDM2 protein-mediated ubiquitination of numb protein: identification of a second physiological substrate of MDM2 that employs a dual-site docking mechanism. J Biol Chem. 2012;287:14052–68. doi: 10.1074/jbc.M111.303875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–7. doi: 10.1016/S0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 14.Ciemny MP, Debinski A, Paczkowska M, Kolinski A, Kurcinski M, Kmiecik S. Protein-peptide molecular docking with large-scale conformational changes: the p53–MDM2 interaction. Sci Rep. 2016;6:37532. doi: 10.1038/srep37532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Phelps M, Darley M, Primrose JN, Blaydes JP. p53-independent activation of the hdm2–P2 promoter through multiple transcription factor response elements results in elevated hdm2 expression in estrogen receptor alpha-positive breast cancer cells. Cancer Res. 2003;63:2616–23. [PubMed] [Google Scholar]
- 16.Dai MS, Lu H. Inhibition of MDM2-mediated p53 ubiquitination and degradation by ribosomal protein L5. J Biol Chem. 2004;279:44475–82. doi: 10.1074/jbc.M403722200. [DOI] [PubMed] [Google Scholar]
- 17.Dar AA, Majid S, Rittsteuer C, de Semir D, Bezrookove V, Tong S, et al. The role of miR-18b in MDM2-p53 pathway signaling and melanoma progression. J Natl Cancer Inst. 2013;105:433–42. doi: 10.1093/jnci/djt003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendell JT. miRiad roles for the miR-17-92 cluster in development and disease. Cell. 2008;133:217–22. doi: 10.1016/j.cell.2008.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tatsuguchi M, Seok HY, Callis TE, Thomson JM, Chen JF, Newman M, et al. Expression of microRNAs is dynamically regulated during cardiomyocyte hypertrophy. J Mol Cell Cardiol. 2007;42:1137–41. doi: 10.1016/j.yjmcc.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang ZZ, Liu X, Wang DQ, Teng MK, Niu LW, Huang AL, et al. Hepatitis B virus and hepatocellular carcinoma at the miRNA level. World J Gastroenterol. 2011;17:3353–8. doi: 10.3748/wjg.v17.i28.3353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leivonen SK, Makela R, Ostling P, Kohonen P, Haapa-Paananen S, Kleivi K, et al. Protein lysate microarray analysis to identify microRNAs regulating estrogen receptor signaling in breast cancer cell lines. Oncogene. 2009;28:3926–36. doi: 10.1038/onc.2009.241. [DOI] [PubMed] [Google Scholar]
- 22.Maya R, Balass M, Kim ST, Shkedy D, Leal JF, Shifman O, et al. ATM-dependent phosphorylation of Mdm2 on serine 395: role in p53 activation by DNA damage. Genes Dev. 2001;15:1067–77. doi: 10.1101/gad.886901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carr MI, Roderick JE, Zhang H, Woda BA, Kelliher MA, Jones SN. Phosphorylation of the Mdm2 oncoprotein by the c-Abl tyrosine kinase regulates p53 tumor suppression and the radiosensitivity of mice. Proc Natl Acad Sci USA. 2016;113:15024–9. doi: 10.1073/pnas.1611798114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K, et al. Akt enhances Mdm2-mediated ubiquitination and degradation of p53. J Biol Chem. 2002;277:21843–50. doi: 10.1074/jbc.M109745200. [DOI] [PubMed] [Google Scholar]
- 25.Bar-Peled L, Schweitzer LD, Zoncu R, Sabatini DM. Ragulator is a GEF for the rag GTPases that signal amino acid levels to mTORC1. Cell. 2012;150:1196–208. doi: 10.1016/j.cell.2012.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Melegari M, Scaglioni PP, Wands JR. Cloning and characterization of a novel hepatitis B virus x binding protein that inhibits viral replication. J Virol. 1998;72:1737–43. doi: 10.1128/jvi.72.3.1737-1743.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fujii R, Zhu C, Wen Y, Marusawa H, Bailly-Maitre B, Matsuzawa S, et al. HBXIP, cellular target of hepatitis B virus oncoprotein, is a regulator of centrosome dynamics and cytokinesis. Cancer Res. 2006;66:9099–107. doi: 10.1158/0008-5472.CAN-06-1886. [DOI] [PubMed] [Google Scholar]
- 28.Li H, Liu Q, Wang Z, Fang R, Shen Y, Cai X, et al. The oncoprotein HBXIP modulates the feedback loop of MDM2/p53 to enhance the growth of breast cancer. J Biol Chem. 2015;290:22649–61. doi: 10.1074/jbc.M115.658468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Q, Bai X, Li H, Zhang Y, Zhao Y, Zhang X, et al. The oncoprotein HBXIP upregulates Lin28B via activating TF II D to promote proliferation of breast cancer cells. Int J Cancer. 2013;133:1310–22. doi: 10.1002/ijc.28154. [DOI] [PubMed] [Google Scholar]
- 30.Liu F, You X, Wang Y, Liu Q, Liu Y, Zhang S, et al. The oncoprotein HBXIP enhances angiogenesis and growth of breast cancer through modulating FGF8 and VEGF. Carcinogenesis. 2014;35:1144–53. doi: 10.1093/carcin/bgu021. [DOI] [PubMed] [Google Scholar]
- 31.Li Y, Wang Z, Shi H, Li H, Li L, Fang R, et al. HBXIP and LSD1 scaffolded by lncRNA hotair mediate transcriptional activation by c-Myc. Cancer Res. 2016;76:293–304. doi: 10.1158/0008-5472.CAN-14-3607. [DOI] [PubMed] [Google Scholar]
- 32.Zhao Y, Li H, Zhang Y, Li L, Fang R, Li Y, et al. Oncoprotein HBXIP modulates abnormal lipid metabolism and growth of breast cancer cells by activating the LXRs/SREBP-1c/FAS signaling cascade. Cancer Res. 2016;76:4696–707. doi: 10.1158/0008-5472.CAN-15-1734. [DOI] [PubMed] [Google Scholar]
- 33.Zhou XL, Guo X, Song YP, Zhu CY, Zou W. The LPI/GPR55 axis enhances human breast cancer cell migration via HBXIP and p-MLC signaling. Acta Pharmacol Sin. 2018;39:459–71. doi: 10.1038/aps.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hu N, Zhang J, Cui W, Kong G, Zhang S, Yue L, et al. miR-520b regulates migration of breast cancer cells by targeting hepatitis B X-interacting protein and interleukin-8. J Biol Chem. 2011;286:13714–22. doi: 10.1074/jbc.M110.204131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shi X, Kachirskaia I, Yamaguchi H, West LE, Wen H, Wang EW, et al. Modulation of p53 function by SET8-mediated methylation at lysine 382. Mol Cell. 2007;27:636–46. doi: 10.1016/j.molcel.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Han S, Khuri FR, Roman J. Fibronectin stimulates non-small cell lung carcinoma cell growth through activation of Akt/mammalian target of rapamycin/S6 kinase and inactivation of LKB1/AMP-activated protein kinase signal pathways. Cancer Res. 2006;66:315–23. doi: 10.1158/0008-5472.CAN-05-2367. [DOI] [PubMed] [Google Scholar]
- 37.Marusawa H, Matsuzawa S, Welsh K, Zou H, Armstrong R, Tamm I, et al. HBXIP functions as a cofactor of survivin in apoptosis suppression. EMBO J. 2003;22:2729–40. doi: 10.1093/emboj/cdg263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Y, Zhao Y, Li H, Li Y, Cai X, Shen Y, et al. The nuclear import of oncoprotein hepatitis B X-interacting protein depends on interacting with c-Fos and phosphorylation of both proteins in breast cancer cells. J Biol Chem. 2013;288:18961–74. doi: 10.1074/jbc.M113.458638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shan C, Xu F, Zhang S, You J, You X, Qiu L, et al. Hepatitis B virus X protein promotes liver cell proliferation via a positive cascade loop involving arachidonic acid metabolism and p-ERK1/2. Cell Res. 2010;20:563–75. doi: 10.1038/cr.2010.49. [DOI] [PubMed] [Google Scholar]
- 40.Gonzalez L, Agullo-Ortuno MT, Garcia-Martinez JM, Calcabrini A, Gamallo C, Palacios J, et al. Role of c-Src in human MCF7 breast cancer cell tumorigenesis. J Biol Chem. 2006;281:20851–64. doi: 10.1074/jbc.M601570200. [DOI] [PubMed] [Google Scholar]
- 41.Yu X, Zhen Y, Yang H, Wang H, Zhou Y, Wang E, et al. Loss of connective tissue growth factor as an unfavorable prognosis factor activates miR-18b by PI3K/AKT/C-Jun and C-Myc and promotes cell growth in nasopharyngeal carcinoma. Cell Death Dis. 2013;4:e634. doi: 10.1038/cddis.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–31. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]
- 43.von der Chevallerie K, Rolfes S, Schierwater B. Inhibitors of the p53-Mdm2 interaction increase programmed cell death and produce abnormal phenotypes in the placozoon Trichoplax adhaerens (F.E. Schulze) Dev Genes Evol. 2014;224:79–85. doi: 10.1007/s00427-014-0465-0. [DOI] [PubMed] [Google Scholar]
- 44.Touqan N, Diggle CP, Verghese ET, Perry S, Horgan K, Merchant W, et al. An observational study on the expression levels of MDM2 and MDMX proteins, and associated effects on P53 in a series of human liposarcomas. BMC Clin Pathol. 2013;13:32. doi: 10.1186/1472-6890-13-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang X, Wang J, Jiang X. MdmX protein is essential for Mdm2 protein-mediated p53 polyubiquitination. J Biol Chem. 2011;286:23725–34. doi: 10.1074/jbc.M110.213868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu S, Li L, Zhang Y, Zhang Y, Zhao Y, You X, et al. The oncoprotein HBXIP uses two pathways to up-regulate S100A4 in promotion of growth and migration of breast cancer cells. J Biol Chem. 2012;287:30228–39. doi: 10.1074/jbc.M112.343947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer M, Steiner L, Engeland K. The transcription factorp53: not a repressor, solely an activator. Cell Cycle. 2014;13:3037–58. doi: 10.4161/15384101.2014.949083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hayashi Y, Tsujii M, Kodama T, Akasaka T, Kondo J, Hikita H, et al. p53 functional deficiency in human colon cancer cells promotes fibroblast-mediated angiogenesis and tumor growth. Carcinogenesis. 2016;37:972–84. doi: 10.1093/carcin/bgw085. [DOI] [PubMed] [Google Scholar]
- 49.He Y, Lian G, Lin S, Ye Z, Li Q. MDM2 inhibits axin-induced p53 activation independently of its E3 ligase activity. PLoS ONE. 2013;8:e67529. doi: 10.1371/journal.pone.0067529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jazirehi AR, Torres-Collado AX, Nazarian R. Role of miR-18b/MDM2/p53 circuitry in melanoma progression. Epigenomics. 2013;5:254. [PubMed] [Google Scholar]
- 51.Yoshimoto N, Toyama T, Takahashi S, Sugiura H, Endo Y, Iwasa M, et al. Distinct expressions of microRNAs that directly target estrogen receptor alpha in human breast cancer. Breast Cancer Res Treat. 2011;130:331–9. doi: 10.1007/s10549-011-1672-2. [DOI] [PubMed] [Google Scholar]
- 52.Fonseca-Sanchez MA, Perez-Plasencia C, Fernandez-Retana J, Arechaga-Ocampo E, Marchat LA, Rodriguez-Cuevas S, et al. MicroRNA-18b is upregulated in breast cancer and modulates genes involved in cell migration. Oncol Rep. 2013;30:2399–410. doi: 10.3892/or.2013.2691. [DOI] [PubMed] [Google Scholar]
- 53.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–66. doi: 10.1016/S0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 54.Macneil LT, Walhout AJ. Gene regulatory networks and the role of robustness and stochasticity in the control of gene expression. Genome Res. 2011;21:645–57. doi: 10.1101/gr.097378.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xia L, Paik A, Li JJ. p53 activation in chronic radiation-treated breast cancer cells: regulation of MDM2/p14ARF. Cancer Res. 2004;64:221–8. doi: 10.1158/0008-5472.CAN-03-0969. [DOI] [PubMed] [Google Scholar]
- 56.Wu CT, Lin TY, Hsu HY, Sheu F, Ho CM, Chen EI. Ling Zhi-8 mediates p53-dependent growth arrest of lung cancer cells proliferation via the ribosomal protein S7-MDM2-p53 pathway. Carcinogenesis. 2011;32:1890–6. doi: 10.1093/carcin/bgr221. [DOI] [PubMed] [Google Scholar]
- 57.Michael D, Oren M. The p53-Mdm2 module and the ubiquitin system. Semin Cancer Biol. 2003;13:49–58. doi: 10.1016/S1044-579X(02)00099-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.