

Abstract
Escin, a triterpene saponin isolated from horse chestnut seed, has been used to treat encephaledema, tissue swelling and chronic venous insufficiency. Recent studies show that escin induces cell cycle arrest, tumor proliferation inhibition and tumor cell apoptosis. But the relationship between escin-induced DNA damage and cell apoptosis in tumor cells remains unclear. In this study, we investigated whether and how escin-induced DNA damage contributed to escin-induced apoptosis in human colorectal cancer cells. Escin (5–80 μg/mL) dose-dependently inhibited the cell viability and colony formation in HCT116 and HCT8 cells. Escin treatment induced DNA damage, leading to p-ATM and γH2AX upregulation. Meanwhile, escin treatment increased the expression of p62, an adaptor protein, which played a crucial role in controlling cell survival and tumorigenesis, and had a protective effect against escin-induced DNA damage: knockdown of p62 apparently enhanced escin-induced DNA damage, whereas overexpression of p62 reduced escin-induced DNA damage. In addition, escin treatment induced concentration- and time-dependent apoptosis. Similarly, knockdown of p62 significantly increased escin-induced apoptosis in vitro and produced en escin-like antitumor effect in vivo. Overexpression of p62 decreased the rate of apoptosis. Further studies revealed that the functions of p62 in escin-induced DNA damage were associated with escin-induced apoptosis, and p62 knockdown combined with the ATM inhibitor KU55933 augmented escin-induced DNA damage and further increased escin-induced apoptosis. In conclusion, our results demonstrate that p62 regulates ATM/γH2AX pathway-mediated escin-induced DNA damage and apoptosis.
Keywords: escin, p62, DNA damage, ATM/γH2AX pathway; apoptosis, colorectal cancer; HCT116 cells; HCT8 cells
Introduction
Colorectal cancer (CRC), one of the most common malignant tumors of the digestive tract1, was the third most common cancer and third leading cause of cancer death in the United States in 20142. Surgery and combination therapy including surgery, chemotherapy and radiotherapy are important treatments for CRC, and chemotherapy plays a vital role in the treatment of CRC. However, the prognosis of CRC patients is poor, with only a 65% five-year relative survival rate in 20133. Multidrug resistance (MDR), an important contributor to the insensitivity of chemotherapy, is crucial in cancer recurrence and metastasis, and leads to poor patient prognosis4. MDR is perceived to be the primary reason for chemotherapy resistance and is caused by different structures and molecular mechanisms, including overexpression of the ATP-binding cassette (ABC) transporter, DNA damage and repair, mutation of drug targets, activation of survival pathways and autophagy, evasion of apoptosis, epithelial changes and the presence of cancer stem cells5,6,7. Hence, novel and effective chemotherapy strategies, including seeking the mechanisms behind MDR and enhancing the effect of chemotherapeutic drugs, are urgently needed for the treatment of CRC.
Escin, a triterpene saponin isolated from horse chestnut seed, has been applied in the treatment of encephaledema in clinical practice, a condition caused by swelling that results from trauma or surgery and chronic venous insufficiency8,9. Interestingly, escin has been shown to have an antineoplastic effect by the induction of cell proliferation inhibition and apoptosis in cholangiocarcinoma cells10, rat embryo fibroblast cells11, cervical carcinoma and breast cancer cells12, lung adenocarcinoma and glioma cells13, pancreatic cancer cells14 and renal cancer cells15. Recently, escin was found to induce apoptosis of patient-derived glioblastoma-initiating cells (GIC) and inhibit the stem identity of GIC16. Escin inhibits the formation of nicotine-derived nitrosamine ketone (NNK)-induced lung tumors by regulating RhoA/Rock signaling17. Furthermore, escin has a synergistic effect on cancer cells, similar to the synergistic anti-cancer interaction of escin and gemcitabine in pancreatic cancer that inhibits the activity of NF-κB18, and a synergistic inhibitory effect with 5-FU in SMMC-7721 cells that arrests the cell cycle and induces apoptosis-related proteins19. However, the mechanisms of escin in inhibiting tumor proliferation and its synergistic antitumor effect with chemotherapeutic agents requires further exploration.
The adaptor protein p62, known as SQSTM1, was first identified as an autophagy adaptor. However, increasing evidence has shown that p62 plays a crucial role in controlling cell survival and tumorigenesis20. High expression of p62 has been detected in many cancers, such as gastrointestinal cancer21, breast tumors22,23 and lung adenocarcinoma24. Additionally, p62 promotes the proliferation of CRC cells in the absence of autophagy activity and functions as a potent prognostic factor in colorectal tumor patients25. Furthermore, p62 has been demonstrated to induce cisplatin resistance in human ovarian cancer cells by binding ubiquitinated proteins for transport to autophagic degradation26. These results suggest the importance of p62 in cancer initiation and progression, in which it not only operates as a signaling adaptor and substrate in autophagy but also has an important function in DNA damage and repair27.
Our results demonstrate that escin concomitantly induces DNA damage and upregulation of p62 in CRC cells. However, the effects of p62 on escin-induced DNA damage and cell apoptosis are unknown. We verified that p62 plays a protective role in escin-induced DNA damage and cell apoptosis. p62 knockdown increased escin-induced DNA damage and cell apoptosis in vitro and the antitumor effect of escin in vivo. Increasing DNA damage contributed to escin-induced apoptosis via p62 regulation of the ATM/γH2AX pathway in CRC cells.
Materials and methods
Cell culture and drug treatment
Human CRC cell lines (HCT116 and HCT8) were obtained from Shanghai Chinese Academy Sciences (Shanghai, China). The cells were cultured with Dulbecco's Modified Eagle's Medium (DMEM; GIBCO, C11965500BT, New York, USA) supplemented with 10% fetal bovine serum (FBS; GIBCO; 10270-106 ) at 37 °C in a 5% CO2 humidified atmosphere. Escin was purchased from Shandong Luye Pharma LTD (Yantai, Shandong, China) and dissolved in normal saline. KU-55933 (ATM Kinase Inhibitor) was purchased from Selleck (S1092, Shanghai, China) and dissolved in dimethylsulfoxide (DMSO, DT0163, Shanghai, China). N-Acetyl-L-cysteine (NAC) was purchased from Sangon Biotech (A601127, Shanghai, China) and dissolved in DMSO.
Measurement of cell viability and proliferation
The short-term effects of escin, p62 knockdown or p62 overexpression on cell growth were detected by CCK8 (CK04-3000T, Dojindo, Japan). Cells were seeded at a density of 3000 cells/well in 96-well plates. After a series of treatments were applied to the cells, 10 μL of CCK8 was added to each well, and then, the cells were incubated at 37 °C for 2 h. The absorbance was measured at 450 nm with a microculture plate reader (Bie Tek). The percentage of growth inhibition was estimated by the absorbance. The long-term effects of escin on cell growth were assessed with a clone formation assay. Cells were cultured at a density of 200 cells per well in 6-well plates for 36 h. Then, different concentrations of escin (5, 10 and 20 μg/mL) were added. Cells were cultured for approximately 14 d until the cells grew visible colonies. The medium was discarded, and the colonies were stained with crystal violet (C0121, Beyotime) for 15 min at room temperature. After carefully washing with double distilled water, the colonies were counted and photographed using an iPhone.
Immunofluorescence
Cells were plated on cover glass in 24-well plates. After a series of treatments, cells were washed with phosphate buffered saline (PBS) 3 times and fixed with precooled methanol for 20 min, followed by permeabilization with 0.2% Triton-X 100 in PBS for 15 min at 4°C. Then, the cells were blocked with 5% horse serum in PBS for 1 h at room temperature. The cells were incubated with primary antibody overnight at 4°C. After washing the cells with 0.05% Triton-X 100 in PBS for 10 min×3 times, they were incubated with 488-conjugated donkey anti-rabbit IgG (151000; Jackson ImmunoResearch Laboratories) for 30 min at room temperature. After 10 min of three washes with 0.05% Triton-X 100 in PBS, the cells were incubated with DAPI for 3 min; then, the cells were dehydrated in increasing grades of ethanol and cover-slipped with Fluoromount Aqueous Mounting Medium (Sigma, F4680; Saint Louis, MO, USA). The slides were analyzed with a laser scanning confocal unit (Zeiss LSM 710, Carl Zeiss, Jena, Germany)
RT-PCR
Total RNA was extracted from cells by TRIzol reagent (TaKaRa) according to the instructions and then reverse-transcribed into cDNA with a TransScript First-Stand cDNA Synthesis Kit (TaKaRa). Real-time quantitative PCR was performed using SYBR Green Mastermix (TaKaRa) on a 7500 real-time PCR system (Applied Biosystems). The primer pairs were as follows: for p62 (forward primer: 5′--3′, reverse primer: 5′--3′); for β-actin (forward primer: 5′--3′, reverse primer: 5′--3′).
siRNA design and transfection
To inhibit the expression of p62, p62 siRNA1/2 (1#, 5′-; 2#, 5′-)28 and scramble siRNA (5′-) were obtained from GenePharma Co (Shanghai, China). Cells were plated in 6-well plates (500 000 cells/well) or 96-well plates (3000 cells/well) and were transfected with scramble siRNA or p62 siRNAs using Lipofectamine RNAiMAX (Invitrogen, USA) that was diluted in Opti-MEM Reduced Serum Medium (GIBCO, 31985070). The final concentrations of scramble siRNA and p62 siRNAs were 60 nmol/L. After transfection with different siRNAs, cells were cultured for 36 h. After treatment with escin for 12 h, cell viability was tested by the CCK8 assay and the expression of different proteins was detected using Western blotting.
Plasmid construction and transfection
The FLAG-p62 overexpression plasmid was kindly provided by Prof Hai-gang REN29 (Paycho-disease and College of Pharmaceutical Sciences, Soochow University, Suzhou, China). Cells were plated in 6-well plates (700 000 cells/well) or 96-well plates (3000 cell/well) and transfected with the FLAG-NC or FLAG-p62 plasmids using Lipofectamine 3000 (Invitrogen, USA) that was diluted in Opti-MEM Reduced Serum Medium (GIBCO, 31985070) for 36 h. The final concentration of the FLAG-p62 and FLAG-NC plasmids was 3 μg/mL. After transfection with different plasmids, cells were cultured for 36 h. After treatment with escin for 12 h, cell viability was tested by the CCK8 assay and the expression of different proteins was detected by Western blotting.
Western blot analysis and antibodies
Cell samples were lysed in a cell lysis solution that contained protease inhibitors (Roche, 04693159001) and phosphorylase inhibitors (Roche, 04906845001). The concentrations of cell proteins were assessed with a BCA Protein Assay Kit (TaKaRa, T9300A). Equal amounts of proteins were separated on Tris-glycine SDS-polyacrylamide gels and transferred onto an NC membrane. Membranes were blocked in 5% non-fat milk for 2 h at room temperature and then incubated with primary antibodies against p-53BP1 (1:200, OmnimAbs, OM212889), p-ATM (1:1000, Abcam, ab81292), ATM (1:1000, Abcam, ab199726), p62 (1:2000, Sigma, P0067), PARP (1:1000, Cell Signaling Technology, #9532), γH2AX (1:1000, Cell Signaling Technology, #9718), cleaved-caspase 9 (1:1000, Cell Signaling Technology, #9505), or β-actin (1:10000, Sigma, A5441) in 1% BSA overnight at 4 °C. After washing in TBST 3 times, membranes were incubated with the appropriate secondary antibodies (1:10000, Odyssey LI-COR, anti-rabbit, 92632213, anti-mouse, 92632212) at room temperature for 2 h. Immunoreactivity was imaged using an Odyssey Infrared Imager (Li-COR Biosciences, USA) and quantified using Image J software (W S Rasband, Image J, NIH).
Flow cytometry detection of apoptosis and ROS
Cells were plated in 6-well plates. After a series of treatments, cells were trypsinized and harvested. Then, cells were resuspended in 300 μL of binding buffer containing 5 μL of propidium iodide (PI) and 5 μL of annexin V-FITC (Biouniquer, BU-AP0103) at room temperature for 15 min. The samples were analyzed by a FACScan flow cytometer (FACScan, Becton Dickson).
Cells were seeded onto 6-well plates and incubated for 24 h. Afterwards, different concentrations of escin were added for 12 h. Then, the ROS levels were determined by incubating cells with cell culture medium containing 10 mmol/L H2-DCFDA for 30 min at 37°C. The cells were washed twice in PBS, trypsinized, and resuspended in PBS. The ROS content was measured with FACS (FACScan, Becton Dickinson).
Tumor growth analysis in vivo
To acquire HCT116 cells with a stable p62 knockdown, cells were transfected with p62-shRNA (5′--3′; GenePharma, Shanghai, China), and the expression of green fluorescent protein (GFP) was detected by fluorescence microscopy 36 h after transfection. Then, cells were selected in medium with 4 μg/mL puromycin for 2 weeks. The expression of p62 was detected via Western blotting, and the cells were cultured for expansion. All experimental procedures were conducted according to the Regulations of Experimental Animal Administration issued by the Animal Committee of Soochow University. Cells (3 000 000 in 250 μL of PBS) were injected subcutaneously into the right iliac fossa of 6-week-old female athymic nude mice (The Animal Center of Soochow University). After approximately 6 weeks, visible tumors had developed and escin (2 mg/kg) was intraperitoneally injected every day for 2 weeks. Mice were sacrificed and the tumor tissues were removed. The weight of the tumors was calculated. Tumor tissues were used for Western blotting to evaluate the expression of p62.
Statistical analysis
All data were analyzed with at least three independent experiments and are presented as the mean±SD. GraphPad Prism 6 (GraphPad Software, Inc, La Jolla, CA, USA) was used for statistical analysis by using one-way ANOVA analysis.
Results
Escin inhibited proliferation of CRC cells
To verify the short-term effect of escin-induced growth inhibition of human CRC HCT116 and HCT8 cells, cells were treated with different concentrations of escin (0–100 μg/mL) for 12, 24 or 48 h, and cell viability was measured by the CCK8 assay. As shown in Figure 1A–B, escin inhibited the proliferation of two CRC cell lines in a time- and concentration-dependent manner. A long-term proliferation inhibition effect was observed with a colony formation assay, which indicated that escin significantly reduced the number of colonies (Figure 1C–F). These results suggest that escin inhibits CRC cell proliferation.
Figure 1.
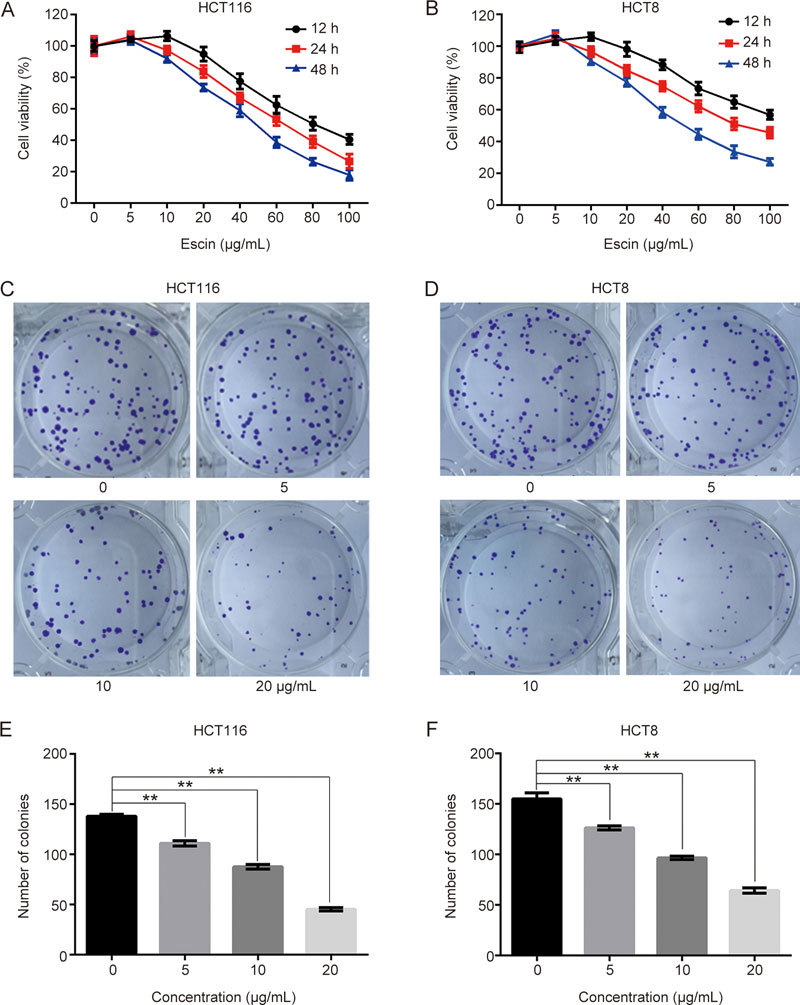
Escin inhibited proliferation in HCT 116 and HCT-8 cells. (A–B) Cells were treated with a series of concentrations of escin (0–100 μg/mL) for 12, 24 or 48 h, and cell viability was measured by the CCK8 assay. (C–D) A total of 200 cells were seeded in 6-well plates and cultivated for 12–14 d. Colony formation was detected. (E–F) Quantitative analysis of the colony numbers of HCT116 and HCT8 cells after escin treatment. The values are the mean±SD from three independent experiments. ** P<0.01 vs the control group.
Escin induced DNA damage and p62 upregulation
Because escin induces reactive oxygen species (ROS) generation15 (Figure 2A) and ROS activates DNA damage responses30, we speculated that escin probably induced DNA damage. To verify this assumption, we examined the effect of escin on inducing DNA damage responses. As shown in Figure 2B–D, expression of γH2AX, which is a sensitive indicator of the DNA damage response and can indicate variations in DNA damage levels, and p-ATM were increased in a concentration- and time-dependent manner, while p-53BP1 was upregulated in a concentration-dependent manner. Escin-induced DNA damage was further confirmed by immunofluorescence. Consistent with the Western blotting results, escin robustly elevated the levels of γH2AX in nucleus (Figure 2E). p62 regulates DNA repair and tumorigenesis30. Next, we investigated whether p62 participated in the DNA damage response in escin treated cells. The concentration-course study showed that p62 was elevated at concentrations of 20 and 40 μg/mL escin, but returned to the control level at a concentration of 80 μg/mL escin. Similarly, an increase of p62 was observed to occur in a time-dependent manner in the time-course study (Figure 2F). Furthermore, escin-induced p62 upregulation occurred at the transcriptional level (Figure 2G) and was eliminated by NAC (Figure 2H). These results indicate that escin induced DNA damage and upregulation of p62 at the same time and, moreover, that the upregulation of p62 was partly attributable to ROS.
Figure 2.
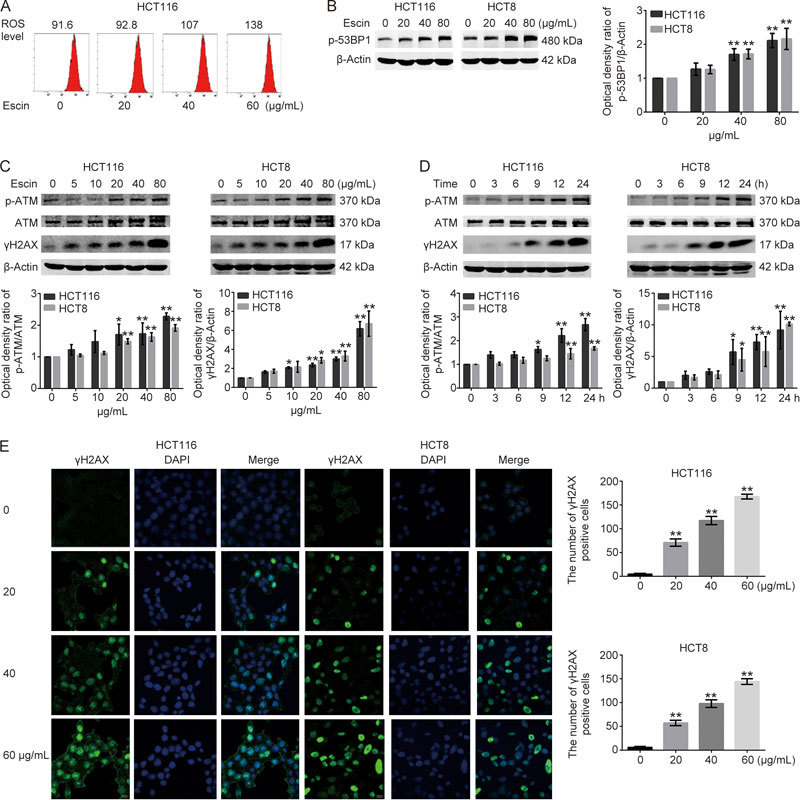
Escin induced DNA damage and upregulation of p62. Cells were treated with different concentrations of escin for 12 h or treated with 60 μg/mL escin for 3, 6, 9, 12 and 24 h. (A) Escin induced ROS generation. HCT16 cells were treated with different concentrations of escin, and the level of ROS was determined by FACS. (B, C, D) The protein levels of p-53BP1, p-ATM, ATM, γH2AX, and β-actin were detected by Western blotting. Right and down: quantitative analysis of the optical density ratio of p-53BP1, p-ATM, γH2AX compared with the loading control (β-actin) in HCT116 and HCT8. (E) Distribution of γH2AX in HCT116 and HCT8 cells treated as described above that were analyzed with confocal microscopy. γH2AX is stained red, and the nucleus is stained blue. Scale bar=10 μm. Right: the number of γH2AX positive cells determined by automated fluorescent object counting was plotted for the control, n=250. ns P>0.05, * P<0.05, ** P<0.01 vs the control group.
Figure 2 (F, G).
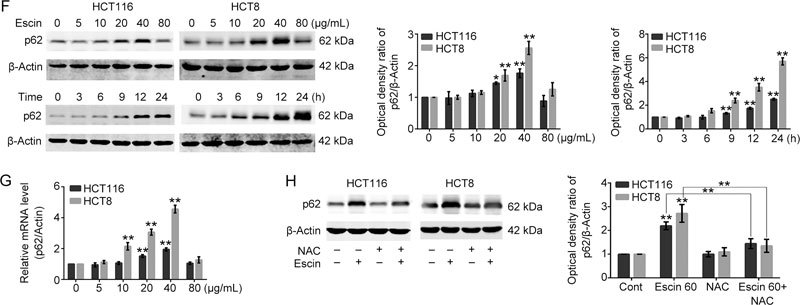
Expression of escin-induced p62 detected by real-time RT-PCR. (H) Cells were pretreated with NAC (ROS scavenger, 5 mmol/L) for 4 h and then treated with 60 μg/mL escin for 12 h. p62 was analyzed by Western blotting. Right: quantitative analysis of the optical density ratio of p62 compared with the loading control (β-actin) in HCT116 and HCT8. The values are the mean±SD from three independent experiments. ns P>0.05, * P<0.05, ** P<0.01 vs the control group.
p62 protected DNA from escin-induced DNA damage
As p62 plays an important role in DNA damage and repair and because we concurrently found that escin induces DNA damage and upregulation of p62, the next step was to illustrate the functions of p62 in escin-induced DNA damage responses. To assess the effect of p62 in downregulation, two different p62 siRNAs were used to mediate p62 suppression in HCT116 and HCT8 cells. Scramble siRNA was used as the negative control. Cells were transfected with scramble siRNA, p62 siRNA1 or p62 siRNA2 at a concentration of 60 nmol/L for 36 h. The results showed that p62 siRNA1 and siRNA2 apparently downregulated the expression of p62 compared with the negative control in two cell lines, as was detected by Western blotting. There was an 85% and 81% silencing efficiency in HCT116 and 95% and 93% silencing efficiency in HCT8, respectively (Figure 3A–B). DNA damage related proteins after p62 knockdown with or without treatment of escin were detected by Western blotting. As shown in Figure 3C–F, knockdown of p62 alone had no effect on p-ATM and γH2AX, whereas knockdown of p62 by escin treatment significantly upregulated the levels of p-ATM and γH2AX, suggesting a protective role of p62 in escin-induced DNA damage.
Figure 3.
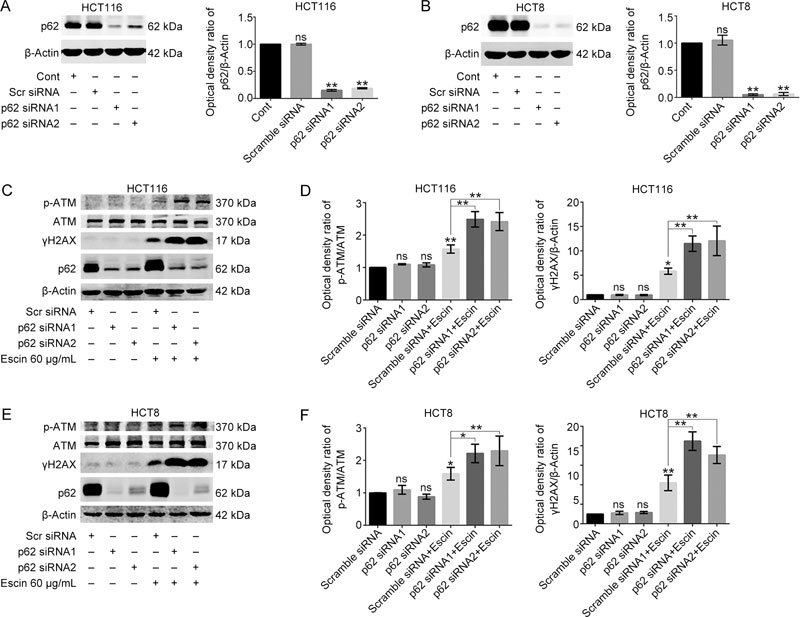
p62 protected DNA from escin-induced DNA damage. (A–B) Knockdown efficiency of p62 in HCT116 and HCT8 cells. Cells were transiently transfected with scramble siRNA and p62 siRNA1/2 for 36 h. The expression level of p62 was tested by Western blotting. Quantitative analysis of the optical density ratio of p62 compared with β-actin is shown. (C–F) Expression of DNA damage related proteins after p62 knockdown with escin treatment. Cells were treated with 60 μg/mL escin for 12 h after scramble siRNA and p62 siRNA1/2 transfection for 36 h. The protein levels of p-ATM, ATM, γH2AX, p62 and β-actin were detected by Western blotting (C and E). Quantitative analysis of the optical density ratio of p-ATM and γH2AX compared with β-actin is shown (D and F).
To further confirm the protective role of p62 in escin-induced DNA damage, p62 was overexpressed after transfection with the p62 plasmid. Cells were transfected with the FLAG-NC or FLAG-p62 plasmid at a concentration of 3 μg/mL for 36 h. P62 was successfully overexpressed in two cell lines, as shown by Western blotting, with a fourteen times overexpression efficiency in HCT116 and two-fold overexpression efficiency in HCT8 (Figure 3G–H). Overexpression of p62 alone had no significant effect on p-ATM and γH2AX. Conversely, overexpression of p62 dramatically downregulated the levels of p-ATM and γH2AX in two cell lines with escin treatment (Figure 3I–J). These findings indicate that p62 has a significantly protective function in escin-reduced DNA damage.
Figure 3 (G–H).
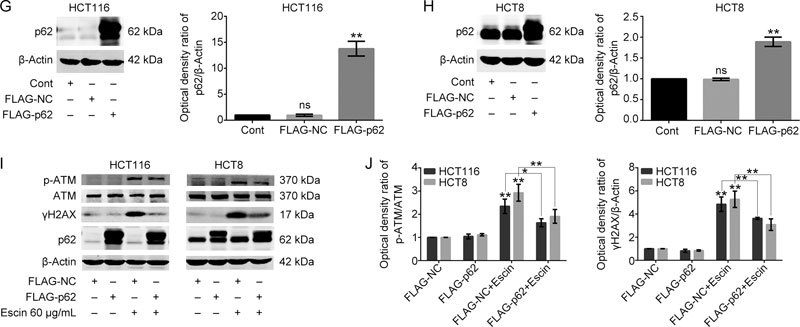
Overexpression efficiency of p62 in HCT116 and HCT8 cells. Cells were transfected with FLAG-p62 plasmids for 36 h. Analysis of p62 expression by Western blotting. Quantitative analysis of the optical density ratio of p62 compared with β-actin is shown. (I–J) Expression of DNA damage related proteins after p62 overexpression with escin treatment. Cells were treated with 60 μg/mL escin for 12 h after p62 overexpression plasmid transfection for 36 h. The protein levels of p-ATM, ATM, γH2AX, p62 and β-actin were detected by Western blotting (I). Quantitative analysis of the optical density ratio of p-ATM and γH2AX compared with β-actin is shown (J). The values are the mean±SD from three independent experiments. ns P>0.05, * P<0.05, ** P<0.01 vs the corresponding group.
Escin induced cell apoptosis
Severe DNA damage may lead to apoptosis. Escin induced cell DNA damage, which may have resulted in apoptosis. We next examined the effect of escin on apoptosis via flow cytometry analysis (FACS). The results demonstrated that escin induced apoptosis in a concentration-dependent manner in two cell lines (Figure 4A–B). We also detected the expression of apoptosis related proteins by Western blotting. The expression levels of cleaved-poly (ADP-ribose) polymerase (c-PARP) and cleaved-caspase 9 (c-Casp 9) were strikingly increased after cells were treated with high concentrations of escin or were exposed for to escin at a 60 μg/mL concentration for a long time (Figure 4C–F). All of these results suggest that escin induced cell apoptosis in a concentration- and time-dependent manner.
Figure 4.
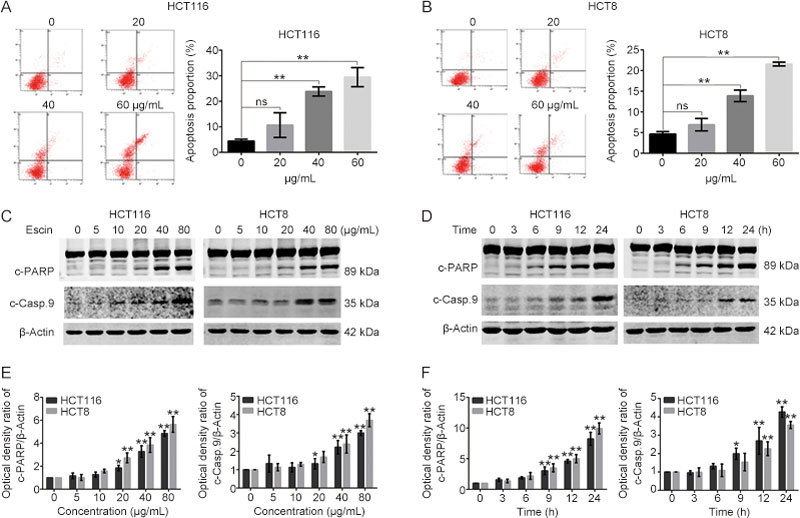
Escin induced apoptosis. (A–B) Escin induced apoptosis. Cells were treated with escin for 12 h, and apoptosis was analyzed by FACS. Quantitative analysis of apoptosis is shown. (C–F) Escin induced upregulation of apoptosis related proteins. Cells were incubated with different concentrations of escin for 12 h or treated with 60 μg/mL of escin for 3, 6, 9, 12 and 24 h. Apoptosis related proteins (cleaved-PARP and cleaved-caspase 9) were detected by Western blotting (C and D). Quantitative analysis of the optical density ratio of cleaved-PARP and cleaved-caspase 9 compared with β-actin is shown (E–F). The values are the mean±SD from three independent experiments. ns P>0.05, * P<0.05, ** P<0.01 vs the control group.
Downregulation of p62 enhanced escin-induced cell apoptosis
Escin induced p62 upregulation and induced cell apoptosis at the same time. To verify any role of p62 in escin-induced apoptosis, we performed a cell growth assay and apoptosis-related experiments to examine cell viability and apoptosis. Cell viability measured by CCK8 showed that knockdown of p62 alone had no effect on cell viability. However, cell viability was markedly decreased in cells with the p62 knockdown that were treated with escin compared to cells that were scramble siRNA transfected and received an escin treatment, indicating a protective role of p62 in cell survival (Figure 5A–B). Furthermore, p62 knockdown and escin treatment greatly increased the rate of HCT116 apoptosis compared with those treated with scramble siRNA transfection and escin (Figure 5C–D). Consistent with the results of FACS, Western blotting showed that the levels of cleaved-PARP and cleaved-caspase 9 were higher in p62 knockdown and escin-treated cells than in scramble siRNA transfection and escin-treated cells (Figure 5E–H). The enhancement of apoptosis in cells with p62 knockdown and escin treatment may be attributed to the augmentation of DNA damage.
Figure 5.
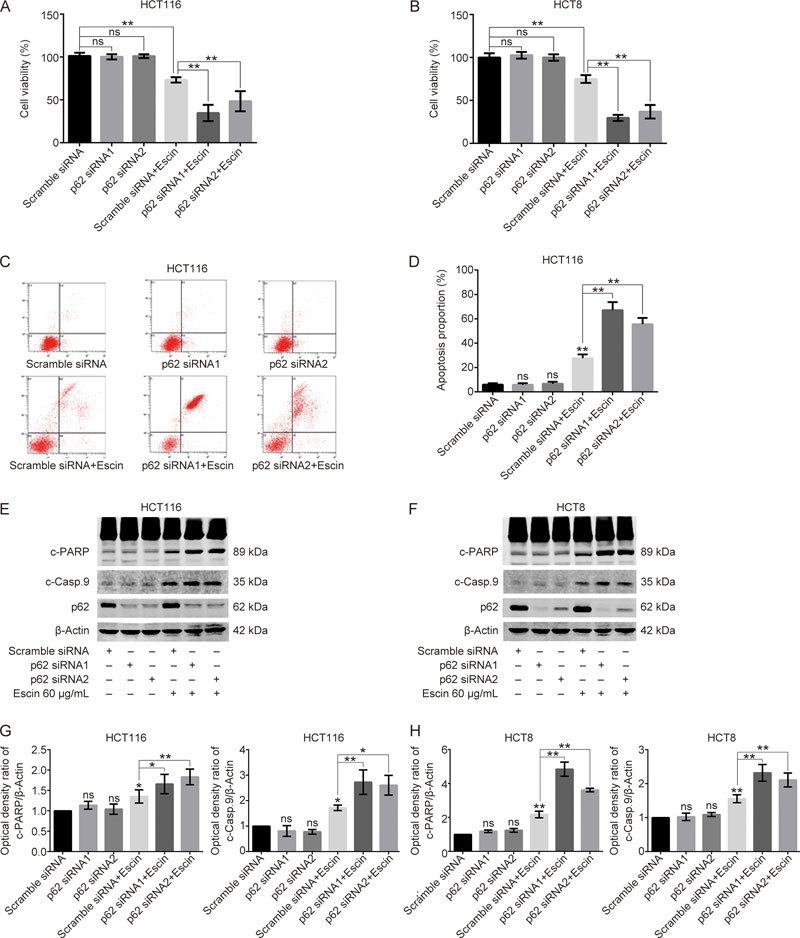
Downregulation of p62 enhanced escin-induced cell apoptosis. (A–B) The effect of escin on cell viability after p62 knockdown. Cells were transiently transfected with scramble siRNA or p62 siRNA1/2 for 36 h and then treated with 60 μg/mL escin for 12 h. Cell viability was assessed by CCK8 assay. (C–D) Analysis of apoptosis with FACS in HCT116 cells. Cells were treated as described above, and apoptosis was analyzed with FACS. Quantitative analysis of apoptosis after p62 knockdown with or without treatment of escin is shown (D). (E–H) Expression of apoptosis related proteins. Cells were treated as described above, and apoptosis-related proteins were detected with Western blotting (E and F). Quantitative analysis of the optical density ratio of cleaved-PARP and cleaved-caspase 9 compared with β-actin is shown (G and H). ns P>0.05, * P<0.05, ** P<0.01 vs the corresponding group.
Overexpression of p62 attenuated escin-induced cell apoptosis
To further confirm the anti-apoptosis effect of p62, p62 was overexpressed to detect cell viability and the expression of apoptosis-related proteins. Escin apparently inhibited cell viability, while p62 overexpression rescued the inhibition in two cell lines (Figure 6A). Furthermore, p62 overexpression with escin treatment dramatically decreased HCT116 apoptosis compared with those treated with escin alone (Figure 6B). Consistent with the cell viability and FACS results, p62 overexpression remarkably suppressed the expression of cleaved-PARP and cleaved-caspase 9 (Figure 6C–D). p62 not only protected DNA from DNA damage but also antagonized cell apoptosis. These results indicate that p62 obviously reduced escin-induced cell viability inhibition and apoptosis, which is consistent with previous reports of p62 promoting tumorigenesis31 and multidrug resistance26.
Figure 6.
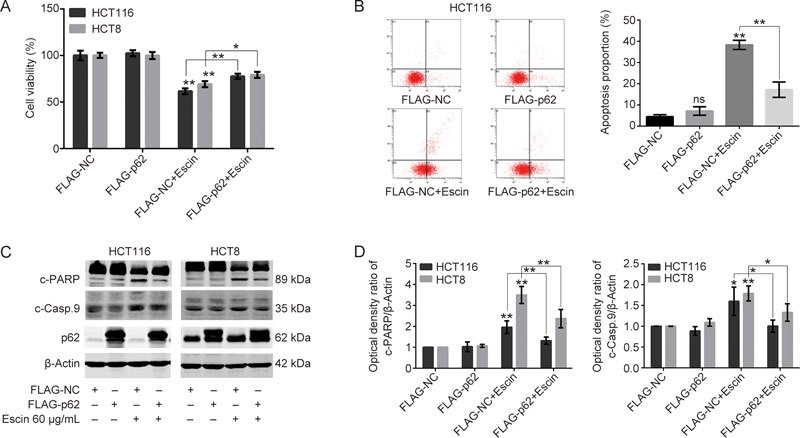
Overexpression of p62 attenuated escin-induced cell apoptosis. (A) The effects of escin on cell viability after p62 overexpression. Cells were transiently transfected with FLAG-NC and FLAG-p62 for 36 h and then treated with 60 μg/mL escin for 12 h. Cell viability was assessed by the CCK8 assay. (B) Analysis of apoptosis with FACS in HCT116 cells. Cells were treated as described above, and apoptosis was analyzed with FACS. Right: quantitative analysis of apoptosis after p62 overexpression with or without treatment of escin is shown. (C–D) Expression of apoptosis related proteins. Cells were treated as described above, and apoptosis-related proteins were detected by Western blotting (C). Quantitative analysis of the optical density ratio of cleaved-PARP and cleaved-caspase 9 compared with β-actin is shown (D). ns P>0.05, * P<0.05, ** P<0.01 vs the corresponding group.
Downregulation of p62 enhanced the antitumor effect of escin in vivo
To assess the function of p62 in the escin antitumor effect in vivo, xenografts of HCT116 cells transfected with vector or p62 shRNA were examined. HCT116 cells with a stably expressed vector or p62 shRNA were successfully established (Figure 7A–B). The cells were injected subcutaneously into the right iliac fossa of 6-week-old female athymic nude mice. These mice were separated into four groups after visible tumor development. The tumor tissues were removed, and the weight of the tumors was calculated after escin (2 mg/kg) was intraperitoneally injected into mice every day for 2 weeks. As shown in Figure 7C–D, the p62 knockdown group showed a slight inhibition of tumor growth as measured by weight in comparison with the vector group, which indicated that p62 enhanced tumor proliferation. In addition, the vector with escin treatment group inhibited the growth of tumors. Furthermore, the p62 knockdown with escin treatment group achieved a more pronounced inhibition of cell growth than the vector with escin treatment group, which indicated that p62 knockdown enhanced the antitumor effect of escin in vivo. Expression of p62 in xenograft tumor tissues was detected to confirm the downregulation of p62 (Figure 7E).
Figure 7.
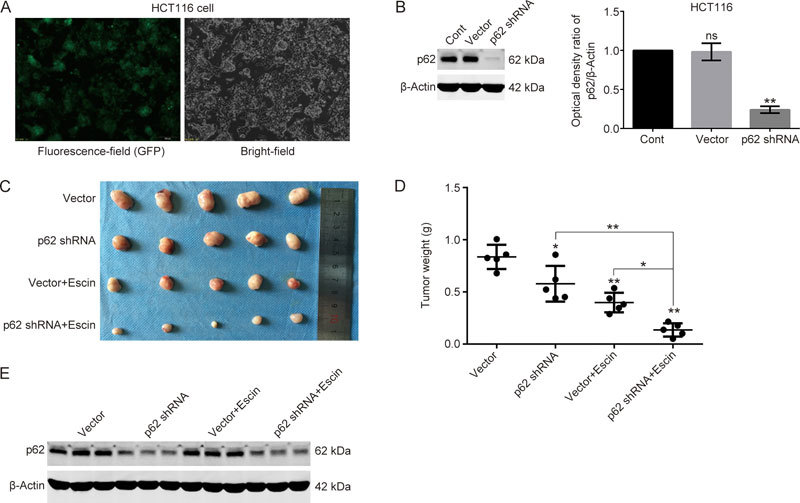
Downregulation of p62 enhanced the antitumor effect of escin in vivo. (A) The transfection efficiency of p62 shRNA in HCT116 cells. Cells were transfected with vector and GFP-p62-shRNA. GFP-green fluorescence was verified with fluorescence microscopy after cells were selected in medium with 4 μg/mL puromycin for 2 weeks. (B) Expression and knockdown efficiency of p62 in control, vector transfection and p62 shRNA transfection cells. (C) Comparison of tumor sizes from various groups (n=5). (D) Comparison of tumor weights from various groups. (E) Expression of p62 in xenograft tumor tissues by Western blotting. ns P>0.05, * P<0.05, ** P<0.01 vs the corresponding group.
p62 regulation of the ATM/γH2AX pathway was involved in escin-induced DNA damage and apoptosis
To verify the potential molecular mechanisms by which p62 reduced escin-induced DNA damage and apoptosis, an ATM specific inhibitor, KU55933, was used to illustrate the function of the ATM/γH2AX pathway in this process. Scramble siRNA, p62 siRNA1 and p62 siRNA2 were transfected for 36 h, and then, KU55933 was applied for 2 h before escin treatment for 12 h. As shown in Figure 8A–B, p62 knockdown with escin treatment increased the expression of p-ATM, cleaved-PARP, cleaved-caspase 9 and γH2AX. When all cells were incubated with escin, p62 knockdown with KU55933 significantly increased the levels of cell apoptosis proteins and decreased the level of γH2AX in comparison with p62 knockdown individually, as shown in Figure 8A–B. There was upregulation of apoptosis-related proteins cleaved-PARP and cleaved-caspase 9, and downregulation of DNA damage-related protein γH2AX in the two cell lines. Similarly, the flow cytometry results of the HCT116 cells showed the same outcomes for apoptosis (Figure 8C–D). These results suggest that inhibition of DNA repair, which was induced by KU55933 and p62 knockdown, increased escin-induced apoptosis. The protective functions of p62 in escin-induced DNA damage attenuated apoptosis by regulating the ATM/γH2AX pathway.
Figure 8A–B.
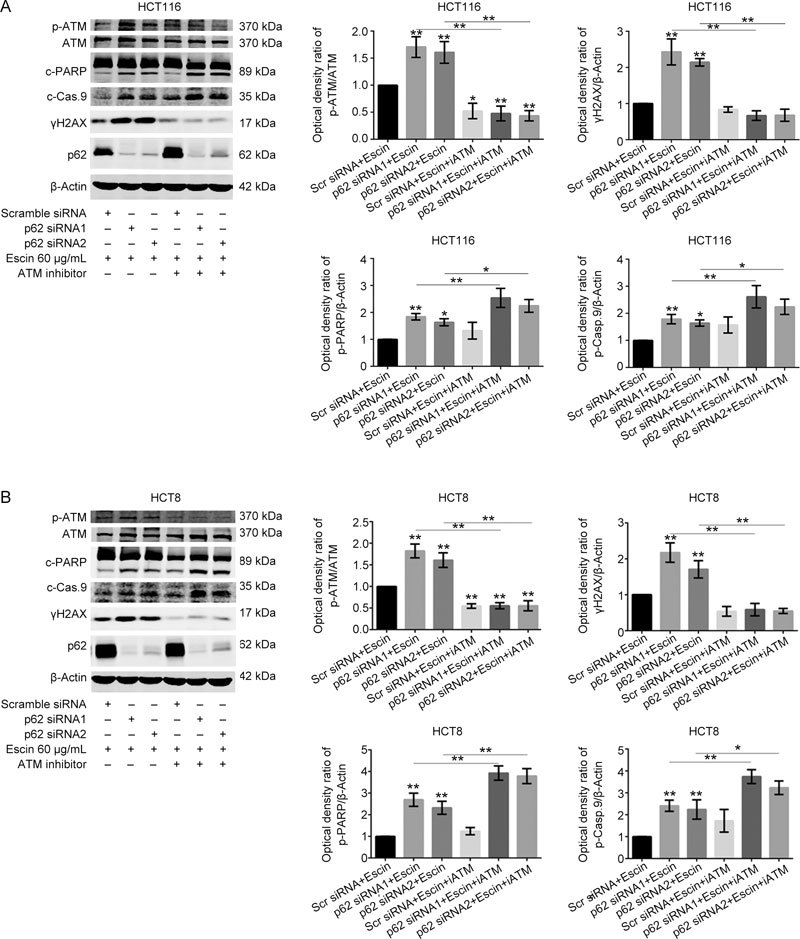
p62 regulation of the ATM/γH2AX pathway was involved in escin-induced DNA damage and apoptosis. (A–B) p62 knockdown combined with the ATM inhibitor KU55933 increased DNA damage and cell apoptosis. Cells were transfected with scramble siRNA or p62 siRNA1/2, and after 36 h, cells were treated with 10 μmol/L KU55933 for 2 h before escin treatment for 12 h. Protein levels of p-ATM, ATM, cleaved-PARP, cleaved-caspase 9, γH2AX, p62 and β-actin were detected by Western blotting. Down: quantitative analysis of the optical density ratio of p-ATM, γH2AX, cleaved-PARP and cleaved-caspase 9 compared with β-actin are shown. * P<0.05, ** P<0.01.
Figure 8C–D.
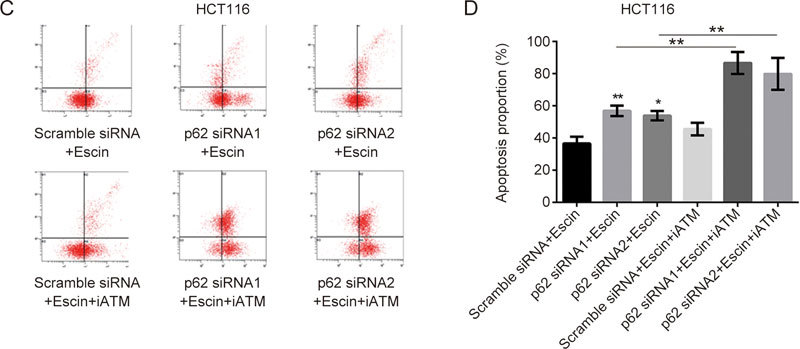
p62 regulation of the ATM/γH2AX pathway was involved in escin-induced DNA damage and apoptosis. (C) Analysis of apoptosis with FACS in HCT116 cells. Cells were treated as described above, and apoptosis was analyzed with FACS. Quantitative analysis of apoptosis after p62 knockdown and escin with or without KU55933 is shown (D). ns P>0.05, * P<0.05, ** P<0.01 vs the corresponding group.
In summary, the results of the ATM inhibitor experiment suggested that the degree of DNA damage induced by p62 knockdown was involved in escin-induced apoptosis. P62 modulated escin-induced apoptosis by monitoring DNA damage and repair as well as by regulating the ATM/γH2AX pathway.
Escin activated autophagy and inhibition of autophagy augmented escin-induced apoptosis
Escin elevated the level of ROS in Figure 2a, which is a key upstream signaling to activate autophagy. We next examined the level of cellular autophagy in response to escin. The results showed that escin markedly increased the level of LC3-II in a concentration-dependent manner in two cell lines (Figure 9A). Next, we investigated the role of autophagy in escin-induced apoptosis. Atg5 siRNA was used to suppress autophagy in HCT116 and HCT8 cells. As shown in Figure 9B, Atg5 siRNA markedly down-regulated the expression of Atg5 compared with the negative control in two cell lines. Furthermore, inhibition of autophagy with escin treatment for 12 h significantly increased the protein levels of cleaved-PARP, cleaved-caspase 9 and γH2AX (Figure 9C). These results suggested that a protective role of autophagy in escin-induced apoptosis and DNA damage. Inhibition of autophagy activity enhanced the antitumor effect of escin.
Figure 9.
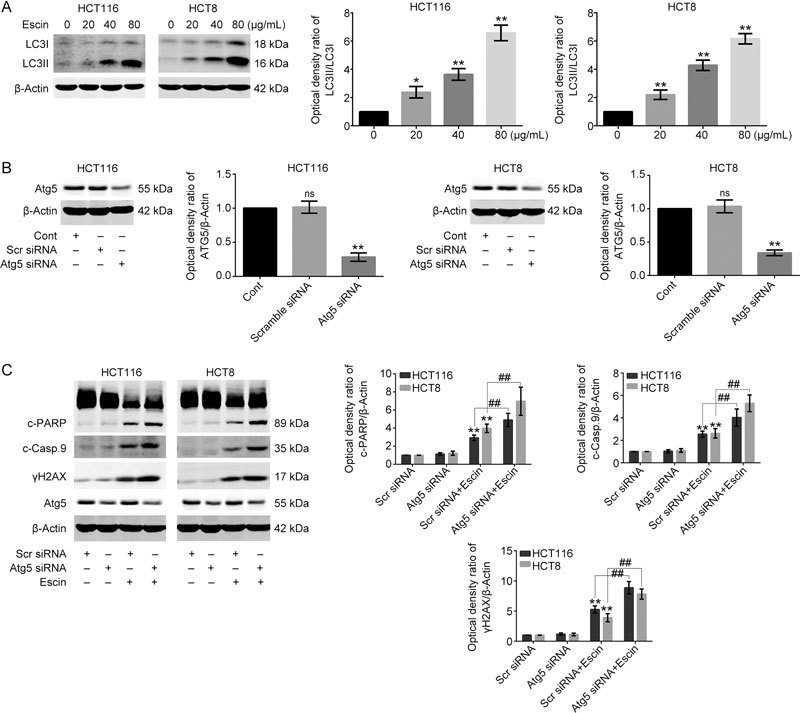
Escin activated autophagy and inhibition of autophagy augmented escin-induced apoptosis. (A) Escin activated autophagy. HCT116 and HCT8 cells were treated with different concentrations of escin for 12 h, the level of autophagy was detected by Western blot. Right: the quantitative analysis of the optical density ratio of LC3 compared with the loading control (β-actin) in HCT116 and HCT8. (B) Knockdown efficiency of Atg5 in HCT116 and HCT8 cells. Cells were transiently transfected with scramble siRNA and Atg5 siRNA for 36 h. The level of Atg5 was tested by Western blot. Right: the quantitative analysis of the optical density ratio of Atg5 compared with β-actin is shown. (C) Expression of γH2AX and apoptosis related proteins after Atg5 knockdown with escin treatment. Cells were treated with 60 μg/mL escin for 12 h after scramble siRNA and Atg5 siRNA transfection for 36 h. The protein levels of γH2AX, cleaved-PARP, cleaved-caspase 9 and β-actin were detected by Western blot. Right: the quantitative analysis of the optical density ratio of γH2AX, cleaved-PARP, cleaved-caspase 9 compared with β-actin is shown. ** P<0.01. ## P<0.01.
Discussion
Chemotherapy is one of the most important treatments for human CRC. However, chemo-resistance, including multidrug resistance (MDR) and insensitivity to clinical antitumor drugs, contributes to the poor prognosis of cancer32,33. Escin has been reported to inhibit the growth of many cancer cell lines13,34,35. Consistent with inhibition of tumor proliferation34 and induction of cell cycle arrest15, the same inhibitory action of escin was found in vitro (Figure 1) and in vivo (Figure 7). Escin is an inducer of apoptosis36,37 and operates by inhibiting activation of the JAK/STAT signaling pathway38. Escin also has an anti-tumor effect that operates by inhibiting NF-κB phosphorylation and nuclear translocation, which affects the expression of its downstream genes and proteins39. However, whether the mechanisms of escin induction of apoptosis involved DNA damage and repair are still unclear.
Severe DNA damage activates ATM or ATR, followed closely by phosphorylation of various downstream signaling pathway proteins, such as CHK1, CHK2 and p5340,41. Activated ATM phosphorylates γH2AX42,43, which forms a focus that recruits DNA repair proteins around the damage site42. In addition, inhibition of ATM phosphorylation increases cellular sensitivity to PARP inhibitors in various cell lines44,45,46. These studies showed the relationship between DNA damage and apoptosis.
In this study, escin was found to induce DNA damage, as shown by the upregulation of p-ATM and γH2AX, and increase of p62. Furthermore, expression analysis of p-ATM, γH2AX and p62 suggested that escin-induced DNA damage and upregulation of p62 occurred in a concentration- and time-dependent manner (Figure 2). Severe DNA damage was detected after p62 knockdown along with escin treatment (Figure 3A–F). Compared with escin treatment, we found that severe DNA damage could be rescued in p62 overexpression and escin treated cells (Figure 3G–J), suggesting that p62 regulates escin-induced DNA damage and has a protective effect in this model. In addition, there were no changes in DNA damage and repair proteins in both the p62 knockdown and overexpression models.
Recent studies have demonstrated that p62 freely shuttles from the cytoplasm to the nucleus and that nuclear-localized p62 increases the sensitivity of tumor cells to radiation27. In autophagy-defective cells, accumulated p62 directly binds to and inhibits nuclear RNF168, an E3 ligase essential for histone H2A ubiquitination and DNA damage responses, which results in the inability of DNA repair proteins, such as BRCA1, RAP80 and Rad51, to recruit to the sites of DNA double-strand breaks27. In the absence of autophagy, p62 inhibits DNA repair and histone ubiquitinated degradation. However, there is a contradiction in autophagy integrated cells. Surprisingly, we found that escin apparently increases the autophagy levels in HCT116 and HCT-8 cells (Figure 9A), simultaneously upregulating the expression of p62, which may indicate that RNF168 leads to ubiquitination of DNA repair proteins to recruit them to the sites of DNA damage to repair the damage. In addition, escin apparently increases the levels of cellular ROS and autophagy (Figure 2A, Figure 9A). ROS, which induces autophagy and is also eliminated by autophagy, may be involved in the functions of p62 in escin-induced DNA damage and repair. The detailed mechanisms as to how p62 induces DNA damage repair in escin-treated cells requires further study.
PARP1 is one of the proteins that links DNA damage and apoptosis47. Apoptosis is induced by inhibition of PARP1 and EGFR47. We speculated that severe DNA damage, which was induced by p62 knockdown, was related to escin-induced apoptosis. Escin induced apoptosis (Figure 4) and p62 knockdown augmented escin-induced apoptosis (Figure 5), and there was an antitumor effect of escin in vivo (Figure 7). However, p62 overexpression partly rescued escin-induced cell viability inhibition and cell apoptosis (Figure 6). P62 increased escin-induced apoptosis by augmenting escin-induced DNA damage. The mechanisms involved in this regulation were further investigated. Escin-induced apoptosis was increased when p-ATM was inhibited by KU55933 (Figure 8), suggesting that activation of the ATM pathway is important in causing apoptosis. Our results showed that escin activated ROS levels (Figure 2A), however, there is no evidence to expound the relationship between escin-induced autophagy and apoptosis. Autophagy is a protective factor against DNA damage, ROS and endoplasmic reticulum stress. In response to escin-induced oxidative stress, intracellular autophagy level was increased by escin (Figure 9A). Furthermore, knockdown of Atg5 with escin treatment increased escin-induced apoptosis and DNA damage compared with escin treatment alone (Figure 9B, C). These results suggested that escin-induced autophagy played a protective role in escin-induced apoptosis and DNA damage.
In conclusion, this study demonstrated that the signaling adaptor p62 reduced escin-induced apoptosis in CRC cells via p62-mediated DNA damage and the ATM/γH2AX pathway. p62-mediated DNA damage may be a potential target for the antitumor effect of escin.
Author contribution
All of the listed authors contributed to the idea generation, design, and completion of this study. Zhong WANG and Qiang CHEN contributed equally to the idea generation, experimental work and manuscript preparation; Bin LI, Jia-ming XIE, Xiao-dong YANG, Kui ZHAO, Yong WU Zhen-yu YE, and Zheng-rong CHEN contributed to the experimental work and manuscript preparation; Chun-gen XING and Zheng-hong QIN guided the idea generation, experimental work and manuscript preparation. All of the authors reviewed the manuscript.
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (No 81672970), the Natural Science Foundation of Jiangsu Province (No BK20160338), and supported by Suzhou Key Medical Center (No LCZX201505), the Projects of Suzhou Technology Bureau (No SS201753, SYS201552, SZS201618), the Second Affiliated Hospital of Soochow University Preponderant Clinic Discipline Group Project Funding, the Project of Invigorating Health Care through Science, Technology and Education (Jiangsu Provincial Medical Youth Talent, QNRC2016249) and Graduate Student Scientific Research Innovation Projects of Jiangsu Province (No KYCX17_1999), National Natural Science Youth Foundation of China (No 81602613).
Contributor Information
Zheng-hong Qin, Email: qinzhenhong@suda.edu.cn.
Chun-gen Xing, Email: xingcgchn@163.com.
References
- 1.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Siegel R, Desantis C, Jemal A. Colorectal cancer statistics, 2014. CA Cancer J Clin. 2014;64:104–17. doi: 10.3322/caac.21220. [DOI] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Fedewa SA, Ahnen DJ, Meester RGS, Barzi A, et al. Colorectal cancer statistics, 2017. CA Cancer J Clin. 2017;67:177–93. doi: 10.3322/caac.21395. [DOI] [PubMed] [Google Scholar]
- 4.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–26. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 5.Fletcher JI, Williams RT, Henderson MJ, Norris MD, Haber M. ABC transporters as mediators of drug resistance and contributors to cancer cell biology. Drug Resist Updat. 2016;26:1–9. doi: 10.1016/j.drup.2016.03.001. [DOI] [PubMed] [Google Scholar]
- 6.Fojo T, Bates S. Strategies for reversing drug resistance. Oncogene. 2003;22:7512–23. doi: 10.1038/sj.onc.1206951. [DOI] [PubMed] [Google Scholar]
- 7.Wu Q, Yang Z, Nie Y, Shi Y, Fan D. Multi-drug resistance in cancer chemotherapeutics: mechanisms and lab approaches. Cancer Lett. 2014;347:159–66. doi: 10.1016/j.canlet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 8.Diehm C, Trampisch HJ, Lange S, Schmidt C. Comparison of leg compression stocking and oral horse-chestnut seed extract therapy in patients with chronic venous insufficiency. Lancet. 1996;347:292–4. doi: 10.1016/S0140-6736(96)90467-5. [DOI] [PubMed] [Google Scholar]
- 9.Pittler MH, Ernst E. Horse chestnut seed extract for chronic venous insufficiency. Cochrane Database Syst Rev. 2012;11:CD003230. doi: 10.1002/14651858.CD003230.pub2. [DOI] [PubMed] [Google Scholar]
- 10.Shen DY, Kang JH, Song W, Zhang WQ, Li WG, Zhao Y, et al. Apoptosis of human cholangiocarcinoma cell lines induced by beta-escin through mitochondrial caspase-dependent pathway. Phytother Res. 2011;25:1519–26. doi: 10.1002/ptr.3435. [DOI] [PubMed] [Google Scholar]
- 11.Guney G, Kutlu HM, Iscan A. The apoptotic effects of escin in the H-Ras transformed 5RP7 cell line. Phytother Res. 2013;27:900–5. doi: 10.1002/ptr.4804. [DOI] [PubMed] [Google Scholar]
- 12.Mojzisova G, Kello M, Pilatova M, Tomeckova V, Vaskova J, Vasko L, et al. Antiproliferative effect of beta-escin–an in vitro study. Acta Biochim Pol. 2016;63:79–87. doi: 10.18388/abp.2015_1013. [DOI] [PubMed] [Google Scholar]
- 13.Ciftci GA, Iscan A, Kutlu M. Escin reduces cell proliferation and induces apoptosis on glioma and lung adenocarcinoma cell lines. Cytotechnology. 2015;67:893–904. doi: 10.1007/s10616-015-9877-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rimmon A, Vexler A, Berkovich L, Earon G, Ron I, Lev-Ari S. Escin chemosensitizes human pancreatic cancer cells and inhibits the nuclear factor-kappab signaling pathway. Biochem Res Int. 2013;2013:251752. doi: 10.1155/2013/251752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yuan SY, Cheng CL, Wang SS, Ho HC, Chiu KY, Chen CS, et al. Escin induces apoptosis in human renal cancer cells through G2/M arrest and reactive oxygen species-modulated mitochondrial pathways. Oncol Rep. 2017;37:1002–10. doi: 10.3892/or.2017.5348. [DOI] [PubMed] [Google Scholar]
- 16.Harford-Wright E, Bidere N, Gavard J. Beta-escin selectively targets the glioblastoma-initiating cell population and reduces cell viability. Oncotarget. 2016;7:66865–79. doi: 10.18632/oncotarget.11784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Patlolla JM, Qian L, Biddick L, Zhang Y, Desai D, Amin S, et al. Beta-Escin inhibits NNK-induced lung adenocarcinoma and ALDH1A1 and RhoA/Rock expression in A/J mice and growth of H460 human lung cancer cells. Cancer Prev Res (Phila) 2013;6:1140–9. doi: 10.1158/1940-6207.CAPR-13-0216. [DOI] [PubMed] [Google Scholar]
- 18.Wang YW, Wang SJ, Zhou YN, Pan SH, Sun B. Escin augments the efficacy of gemcitabine through down-regulation of nuclear factor-kappaB and nuclear factor-kappaB-regulated gene products in pancreatic cancer both in vitro and in vivo. J Cancer Res Clin Oncol. 2012;138:785–97. doi: 10.1007/s00432-012-1152-z. [DOI] [PubMed] [Google Scholar]
- 19.Ming ZJ, Hu Y, Qiu YH, Cao L, Zhang XG. Synergistic effects of beta-aescin and 5-fluorouracil in human hepatocellular carcinoma SMMC-7721 cells. Phytomedicine. 2010;17:575–80. doi: 10.1016/j.phymed.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 20.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137:1001–4. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ren F, Shu G, Liu G, Liu D, Zhou J, Yuan L, et al. Knockdown of p62/sequestosome 1 attenuates autophagy and inhibits colorectal cancer cell growth. Mol Cell Biochem. 2014;385:95–102. doi: 10.1007/s11010-013-1818-0. [DOI] [PubMed] [Google Scholar]
- 22.Thompson HG, Harris JW, Wold BJ, Lin F, Brody JP. p62 overexpression in breast tumors and regulation by prostate-derived Ets factor in breast cancer cells. Oncogene. 2003;22:2322–33. doi: 10.1038/sj.onc.1206325. [DOI] [PubMed] [Google Scholar]
- 23.Rolland P, Madjd Z, Durrant L, Ellis IO, Layfield R, Spendlove I. The ubiquitin-binding protein p62 is expressed in breast cancers showing features of aggressive disease. Endocr Relat Cancer. 2007;14:73–80. doi: 10.1677/erc.1.01312. [DOI] [PubMed] [Google Scholar]
- 24.Inoue D, Suzuki T, Mitsuishi Y, Miki Y, Suzuki S, Sugawara S, et al. Accumulation of p62/SQSTM1 is associated with poor prognosis in patients with lung adenocarcinoma. Cancer Sci. 2012;103:760–6. doi: 10.1111/j.1349-7006.2012.02216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakayama S, Karasawa H, Suzuki T, Yabuuchi S, Takagi K, Aizawa T, et al. p62/sequestosome 1 in human colorectal carcinoma as a potent prognostic predictor associated with cell proliferation. Cancer Med. 2017;6:1264–74. doi: 10.1002/cam4.1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu H, Su J, Xu Y, Kang J, Li H, Zhang L, et al. p62/SQSTM1 involved in cisplatin resistance in human ovarian cancer cells by clearing ubiquitinated proteins. Eur J Cancer. 2011;47:1585–94. doi: 10.1016/j.ejca.2011.01.019. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y, Zhang N, Zhang L, Li R, Fu W, Ma K, et al. Autophagy regulates chromatin ubiquitination in dna damage response through elimination of SQSTM1/p62. Mol Cell. 2016;63:34–48. doi: 10.1016/j.molcel.2016.05.027. [DOI] [PubMed] [Google Scholar]
- 28.Xu LZ, Li SS, Zhou W, Kang ZJ, Zhang QX, Kamran M, et al. p62/SQSTM1 enhances breast cancer stem-like properties by stabilizing MYC mRNA. Oncogene. 2017;36:304–17. doi: 10.1038/onc.2016.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo D, Ying Z, Wang H, Chen D, Gao F, Ren H, et al. Regulation of autophagic flux by CHIP. Neurosci Bull. 2015;31:469–79. doi: 10.1007/s12264-015-1543-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mathew R, Karp CM, Beaudoin B, Vuong N, Chen G, Chen HY, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137:1062–75. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Duran A, Linares JF, Galvez AS, Wikenheiser K, Flores JM, Diaz-Meco MT, et al. The signaling adaptor p62 is an important NF-kappaB mediator in tumorigenesis. Cancer Cell. 2008;13:343–54. doi: 10.1016/j.ccr.2008.02.001. [DOI] [PubMed] [Google Scholar]
- 32.Lage H. An overview of cancer multidrug resistance: a still unsolved problem. Cell Mol Life Sci. 2008;65:3145–67. doi: 10.1007/s00018-008-8111-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ren F, Shen J, Shi H, Hornicek FJ, Kan Q, Duan Z. Novel mechanisms and approaches to overcome multidrug resistance in the treatment of ovarian cancer. Biochim Biophys Acta. 2016;1866:266–75. doi: 10.1016/j.bbcan.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 34.Patlolla JM, Raju J, Swamy MV, Rao CV. Beta-escin inhibits colonic aberrant crypt foci formation in rats and regulates the cell cycle growth by inducing p21(waf1/cip1) in colon cancer cells. Mol Cancer Ther. 2006;5:1459–66. doi: 10.1158/1535-7163.MCT-05-0495. [DOI] [PubMed] [Google Scholar]
- 35.Huang GL, Shen DY, Cai CF, Zhang QY, Ren HY, Chen QX. Beta-escin reverses multidrug resistance through inhibition of the GSK3beta/beta-catenin pathway in cholangiocarcinoma. World J Gastroenterol. 2015;21:1148–57. doi: 10.3748/wjg.v21.i4.1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Niu YP, Li LD, Wu LM. Beta-aescin: a potent natural inhibitor of proliferation and inducer of apoptosis in human chronic myeloid leukemia K562 cells in vitro. Leuk Lymphoma. 2008;49:1384–91. doi: 10.1080/10428190802090151. [DOI] [PubMed] [Google Scholar]
- 37.Zhang Z, Gao J, Cai X, Zhao Y, Wang Y, Lu W, et al. Escin sodium induces apoptosis of human acute leukemia Jurkat T cells. Phytother Res. 2011;25:1747–55. doi: 10.1002/ptr.3457. [DOI] [PubMed] [Google Scholar]
- 38.Tan SM, Li F, Rajendran P, Kumar AP, Hui KM, Sethi G. Identification of beta-escin as a novel inhibitor of signal transducer and activator of transcription 3/Janus-activated kinase 2 signaling pathway that suppresses proliferation and induces apoptosis in human hepatocellular carcinoma cells. J Pharmacol Exp Ther. 2010;334:285–93. doi: 10.1124/jpet.110.165498. [DOI] [PubMed] [Google Scholar]
- 39.Harikumar KB, Sung B, Pandey MK, Guha S, Krishnan S, Aggarwal BB. Escin, a pentacyclic triterpene, chemosensitizes human tumor cells through inhibition of nuclear factor-kappaB signaling pathway. Mol Pharmacol. 2010;77:818–27. doi: 10.1124/mol.109.062760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med. 2006;12:440–50. doi: 10.1016/j.molmed.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 41.Chipuk JE, Green DR. Dissecting p53-dependent apoptosis. Cell Death Differ. 2006;13:994–1002. doi: 10.1038/sj.cdd.4401908. [DOI] [PubMed] [Google Scholar]
- 42.Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–67. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Burma S, Chen BP, Murphy M, Kurimasa A, Chen DJ. ATM phosphory-lates histone H2AX in response to DNA double-strand breaks. J Biol Chem. 2001;276:42462–7. doi: 10.1074/jbc.C100466200. [DOI] [PubMed] [Google Scholar]
- 44.Gilardini Montani MS, Prodosmo A, Stagni V, Merli D, Monteonofrio L, Gatti V, et al. ATM-depletion in breast cancer cells confers sensitivity to PARP inhibition. J Exp Clin Cancer Res. 2013;32:95. doi: 10.1186/1756-9966-32-95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–87. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 46.Williamson CT, Muzik H, Turhan AG, Zamo A, O'Connor MJ, Bebb DG, et al. ATM deficiency sensitizes mantle cell lymphoma cells to poly(ADP-ribose) polymerase-1 inhibitors. Mol Cancer Ther. 2010;9:347–57. doi: 10.1158/1535-7163.MCT-09-0872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–32. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]