

Abstract
The immune checkpoint molecules are emerged in the evolution to protect the host from self-attacks by activated T cells. However, cancer cells, as a strategy to survive and expand, can hijack these molecules and mechanisms to suppress T cell-mediated immune responses. Therefore, an idea of blocking the checkpoint molecules to enhance the anti-tumor activities of the host immune system has been developed and applied to the cancer therapy after discovery of the inhibitory T cell co-receptor, cytotoxic T-lymphocyte associated protein 4 (CTLA-4), and further enhanced on the identification of PD-1 and its ligands. Since 2010, several checkpoint inhibitors have been approved by FDA and many more are in clinical trials. In the treatment of advanced cancers, these inhibitors significantly increased response rates and survival benefits. However, accompanied with the striking results, immune-related adverse events (irAEs) that broadly occurred in many organs were observed and reported, some of which were fatal. Herein, we first review the recent progressions in the research of the immune checkpoint molecules and the application of their blocking antibodies in cancer treatment, and then discuss the cardiac toxicity induced by the therapy and the strategy to monitor, manage this adverse event when it occurs.
Keywords: cancer immunotherapy, immune checkpoint inhibitor, CTLA-4, PD-1, PD-L1, cardiac toxicity, myocarditis
Introduction
Cancer comprises a group of diseases in which cells divide uncontrollably, without following the normal process of cellular growth, proliferation and differentiation. Cancer cells undergo multiple mutations and express different antigens known as tumor-specific antigens (TSA). They also upregulate the expression of non-mutated molecules to abnormally high levels, referred to as tumor-associated antigens (TAA) [1]. Both TSA and TAA can be detected by the host immune system-activating downstream pathways that can eliminate cancer cells. Cancer cells are able to evade immune surveillance through overexpressing checkpoint proteins that prevent immune cells from killing them [2]. This process allows for them to survive and persist in the host [3]. To overcome this survival mechanism adopted by cancer cells, immunotherapy has emerged as a method to allow for the immune system to activate, recognize and attack neoplastic cells. Compared to the traditional surgical, chemotherapeutic or radiotherapeutic approaches, immunotherapy exhibits a more favorable toxicity profile while treating metastatic solid tumors systemically and provides clinical benefit with long-term disease control [4].
Immune-checkpoint inhibition is currently one of the most promising types of immunotherapy employed in cancer management. It has revolutionized the treatment of various malignancies including melanoma, non-small-cell lung cancer, renal cell carcinoma, Hodgkin’s lymphoma, bladder cancer, head and neck cancer, gastric cancer, liver cancer and microsatellite instability high or DNA mismatch repair-deficient colorectal cancer and solid tumors by improving the response rates and overall prognosis of cancer patients. Immune checkpoints are comprised of multiple inhibitory pathways that involve the interactions of co-receptors and ligands expressed on the surfaces of T cells and antigen-presenting cells. Once a T cell recognizes and binds to its cognate antigen through the T cell receptor (TCR), the interaction exerts a co-stimulatory or inhibitory downstream signaling to either suppress or activate the T cell. These co-stimulatory and inhibitory interactions also allow for the maintenance of self-tolerance under normal physiological conditions, preventing autoimmunity and tissue damage upon pathogenic insults [5]. In light of the immune tolerance that occurs within the tumor microenvironment, pharmaceutical companies have devoted significant efforts to develop drugs that block the immune checkpoints while activating the host’s immune system against cancer. Indeed, since the approval of ipilimumab—a monoclonal antibody against cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) by the Food and Drug Administration (FDA) in 2011—the era of immunotherapy has emerged rapidly (a full list of FDA-approved checkpoint inhibitors is shown in Table 1). Following ipilimumab, two anti-programmed cell death-1 (PD-1) antibodies (nivolumab and pembrolizumab) and three anti-programmed cell death ligand (PD-L1) antibodies (atezolizumab, durvalumab, and avelumab) have also been developed and subsequently approved by the FDA for treatment of various metastatic solid tumors [6–9]. With the rapid emergence and use of the checkpoint inhibitors, a wide spectrum of immune-related adverse events (irAEs) has been documented [10–13]. Of all the irAEs, cardiovascular toxicity, although rare but potentially deadly, has not been well recognized or reported [11]. In this review, we will elaborate on the mechanisms and clinical applications of immune checkpoint inhibitors, focusing on CTLA-4 and PD-1/PD-L1 inhibitors and their associated autoimmune cardiotoxicity. We will discuss the preclinical modeling and clinical investigation of immunotherapy-induced cardiac adverse effects, the prophylactic strategies, and the potential treatments for checkpoint inhibitor-induced cardiotoxicity.
Table 1.
Summary of FDA-approved checkpoint inhibitors
| Name | Target | Trade name | Company | First approval year |
|---|---|---|---|---|
| Ipilimumab | CTLA-4 | Yervoy | Bristol-Myers Squibb Co. | 2011 |
| Pembrolizumab | PD-1 | Keytruda | Merck & Co., Inc. | 2014 |
| Nivolumab | PD-1 | Opdivo | Bristol-Myers Squibb Co. | 2014 |
| Atezolizumab | PD-L1 | Tecentriq | Genentech, Inc. | 2016 |
| Avelumab | PD-L1 | Bavencio | EMD Serono, Inc. | 2017 |
| Durvalumab | PD-L1 | Imfinizi | AstraZeneca UK Limited | 2017 |
Immune checkpoint therapy
Role of co-stimulatory and co-inhibitory molecules
The immune system performs the surveillance and clearance of transformed malignant cells. T lymphocytes, as a major component of the human immune system, play an essential role in both processes [14]. Once the T cell receptor recognizes a tumor antigen presented by major histocompatibility complex I or II molecules expressed on antigen-presenting cells (APCs) or tumor cells, co-stimulatory and co-inhibitory interactions take place on the cell surface (summarized in Table 2), resulting in either the activation or inhibition of T cells [5, 15]. CD28 is an example of a well-studied stimulatory T cell co-receptor, which can bind to CD80 (also known as B7-1) or CD86 (also known as B7-2) expressed on ACPs. This interaction subsequently decreases the threshold required for the full activation of T cells (Figs. 1a, b). Furthermore, CD28 also mediates co-stimulatory signaling that contributes to cytokine production and enhances T cell proliferation and differentiation [16].
Table 2.
Summary of T cell co-receptors, their ligands, and functions
| Co-receptor | Ligand (alias) | Stimulatory or inhibitory |
|---|---|---|
| CD28 | CD80 (B7-1), CD86 (B7-2) | Stimulatory |
| ICOS (CD278) | ICOSL (B7-H2) | Stimulatory |
| 4-1BB (CD137) | 4-1BBL | Stimulatory |
| OX40 (TNFSF4, CD134) | OX40L | Stimulatory |
| CD27 | CD70 | Stimulatory |
| GITR (glucocorticoid-induced TNFR-related protein) | GITR ligand | Stimulatory |
| DR3 (death receptor 3) | TL1A (TNF-like ligand 1A) | Stimulatory |
| HVEM (herpesvirus entry mediator) | LIGHT | Stimulatory |
| LIGHT | HVEM | Stimulatory |
| CTLA-4 | CD80, CD86 | Inhibitory |
| PD-1 | PD-L1, PD-L2 | Inhibitory |
| BTLA (B and T-lymphocyte attenuator) | HVEM | Inhibitory |
| CD160 | HVEM | Inhibitory |
| KIR (killer-cell immunoglobulin-like receptors) | HLA class I | Inhibitory |
| LAG3 (lymphocyte-activation gene 3) | HLA class II | Inhibitory |
| TIM3 (T-cell immunoglobulin and mucin-domain containing-3) | Galectin-9 | Inhibitory |
| TIGIT (T cell immunoreceptor with Ig and ITIM domains) | CD112, CD113, CD155 | Inhibitory |
| Adenosine A2a receptor | Adenosine | Inhibitory |
| Unknown | B7-H3 | Inhibitory |
| Unknown | B7-H4 | Inhibitory |
Fig. 1.
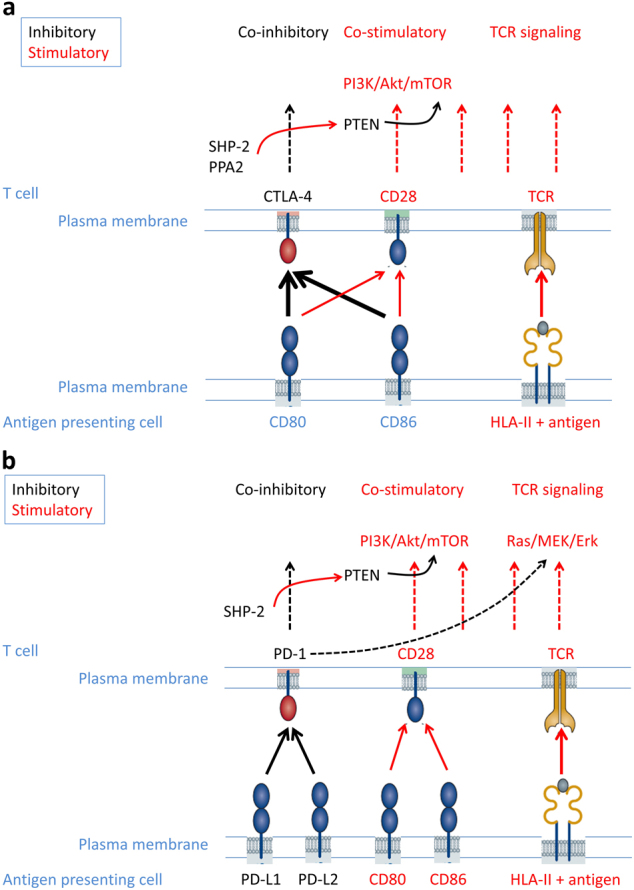
Schematic diagram of the molecular actions of CTLA-4 and PD-1 on inhibiting T cell function. TCR binds to the antigen peptide presented by HLA class I and class II molecules to initiate cellular signaling. CD80 and CD86 expressed on antigen-presenting cells interact with CD28 on T cells, inducing the activation of the PI3K-Akt-mTOR pathway, which co-operates with the TCR signaling to activate the downstream Ras/MEK/Erk pathway for the full activation of T cells. a Once TCR signaling is initiated, intracellular CTLA-4 translocates to the cell surface and competes with CD28 for binding to CD80 and CD86; phosphorylated CTLA-4 also recruits and activates phosphatases PP2A and SHP-2, which dephosphorylates PTEN and subsequently inactivates PI3K/Akt/mTOR signaling. b TCR signaling induces the upregulation of PD-1. The interaction of PD-1 with its ligands leads to the phosphorylation of PD-1, which recruits and activates SHP-2. Similar to the scenario for CTLA-4, PTEN is activated, and PI3K/Akt/mTOR signaling is inhibited. In addition, the activation of PD-1 impairs the activity of the Ras/MEK/Erk pathway
Inhibitory co-receptor CTLA-4
CTLA-4 is a co-inhibitory molecule that can suppress T cell activity upon binding of CTLA-4 to B7 expressed on the surface of APCs. The regulation of T cell inhibition is critical in that it balances T cell activation to prevent auto-immunity [9, 16–18]. Therefore, CTLA-4 (also known as CD152) represents a relevant checkpoint to target in clinical practice given its role in overcoming tumor induced immunosuppression. The CTLA-4 molecule is structurally related to CD28 with approximately 30% sequence homology but has a greater affinity for binding to the ligands B7-1 and B7-2 [19]. CTLA-4 is originally located in the intracellular compartment, but after binding with CD28 and B7, it is upregulated and translocates to the surface of T lymphocytes and subsequently competes with CD28 for binding of B7-1 and B7-2 [9, 16, 20], thereby dampening the synergetic CD28 co-stimulatory effect on TCR activation (Fig. 1a) [9, 16, 20–22]. In addition to the physical competition, the engagement of CTLA-4 with ligands leads to the activation of downstream phosphatases SHP-2 and PP2A, which dephosphorylate kinases, including FYN, LCK and ZAP-70 as well as the members of the RAS pathway, therefore reducing IL-2 production and preventing the progression of the cell cycle in activated T cells (Fig. 1a) [16]. CTLA-4 is consistently expressed in Treg cells, both on the cell surface and intracellularly [16, 23, 24]. The specific depletion of CTLA-4 in Treg cells results in spontaneous lympho-proliferation and autoimmune diseases [16, 24]. These findings indicate that the molecule may also negatively regulate an immune response via upregulation of Treg cell activities. Overall, CLTA-4 plays a key role in immune suppression through both the inhibition of conventional effector T cells and promotion of regulatory T cells.
Clinical applications of CTLA-4 blocking antibody
Allison et al. explored in preclinical models the effects of CTLA-4 blockade. They used mouse models that were injected with murine colon carcinoma cells and showed that administration of anti-CTLA-4 antibody dramatically restrained the tumor cell growth compared with the control group [25]. Although the authors did not investigate the mechanism by which CTLA-4 blockade induced tumor regression, the results provided evidence supporting their hypothesis that releasing the suppression of inhibitory co-receptors on T cell activation and T cell-mediated immune responses can greatly enhance tumor rejection by the host immune system. In a novel syngeneic murine prostate cancer model, Allison’s lab further confirmed the effectiveness of CTLA-4 blockade-mediated cancer cell rejection [26]. In combination with a granulocyte/macrophage colony-stimulating factor (GM-CSF)-expressing tumor cell vaccine, the blocking anti-CTLA-4 antibody successfully evoked CD8+ T cell-dependent immune responses to induce tumor regression in mice infused with highly tumorigenic, poorly immunogenic murine melanoma cells, while either the tumor vaccine or the CTLA-4 blocking antibody alone exhibited no or little effect [27].
These encouraging preclinical findings led to the design of clinical trials to test two fully humanized CTLA-4 blocking antibodies, tremelimumab and ipilimumab. In a phase III clinical trial, tremelimumab failed to show survival benefit in treating patients with naive, unresectable stage IIIc or IV melanoma compared to the standard-of-care chemotherapy (temozolamide or dacarbazine). On the other hand, ipilimumab tested in two phase III, double blinded, randomized clinical trials showed improved response rates, overall survival, progression-free survival and duration of response in patients with treated or untreated advanced melanoma. Up to 60% of patients in the ipilimumab group had more than 24 months of clinical response. Overall, 11% of patients in the ipilimumab group also exhibited objective clinical responses compared to 2% in the gp100 (melanoma vaccine) group. The average survival time for patients in the ipilimumab group was 3.5 months; 20% of patients experienced long-term survival (2 year median survival was 18% in the ipilimumab group vs 9% in the dacarbazine group) [28–30]. In 2011, the FDA approved ipilimumab for treating late-stage melanoma, and the drug is currently under study for its use in treating non-melanoma cancers, with or without other conventional or immuno-therapies. [5, 9].
Inhibitory PD-1/PD-L1 signaling
In 1992, Tasuku and colleagues at Tokyo University identified PD-1 in the search of genes upregulated during programmed cell death using subtractive hybridization [31]. Although the molecule was named “programmed cell death-1”, it was later discovered that the main function of PD-1 was not directly related to apoptosis, but rather, the main function was as a negative regulator of T cell activation and T cell-mediated immune responses. These experiments demonstrated that compared to wild-type controls, the PD-1 deficient mice were more susceptible to developing T cell-mediated autoimmune diseases such as arthritis, glomerulonephritis and autoimmune myocarditis. [17].
PD-1 protein is composed of 288 amino acid residues, forming four domains [32–35]. Unlike CTLA-4, whose expression is mainly restricted to T cells, PD-1 is more broadly expressed on many cell types, including activated CD4+ and CD8+ T cells, B cells, monocytes, natural killer cells and dendritic cells (DCs) [36–38]. Specifically, PD-1 expression on T cells can be induced by cytokine (IL-2, IL-7, IL-15 and IL-21) receptors that share a common γ chain (CD132), as well as by different T cell regulators, including NFATc1, Foxo1, Notch and IRF9 [39–43].
The primary effect of PD-1 signaling is to inhibit TCR activation through both direct inhibition of TCR signaling and indirect interference of co-stimulatory receptor-mediated signaling cascades. Upon engaging its ligand, PD-1 recruits and activates a tyrosine phosphatase, SHP-2, which dephosphorylates TCR CD3ζ and Zap-70, preventing the initiation of TCR signaling and transduction to the downstream effects [44]. Meanwhile, active SHP-2 also releases the inhibitory phosphorylation on PTEN (phosphatase and tensin homolog), which leads to the activation of PTEN’s lipid phosphatase function and consequently inhibits CD28-mediated PI3K-Akt-mTOR (mechanistic target of rapamycin) signaling cascades, as well as impairs the activity of the Ras/MEK/Erk signaling pathway through dephosphorylation of the kinases (Fig. 1b) [45, 46]. Similar to CTLA-4, PD-1 is also highly expressed in Treg cells. By increasing the expression of Foxp3 (forkhead box P3), a master transcription factor of Treg cell population, PD-1 promotes the growth and function of these regulatory T cells [47].
To this day, two ligands of PD-1 have been identified and characterized, PD-L1 (also known as B7-H1, CD274) and PD-L2 (programmed cell death ligand 2, also known as B7-DC, CD273). PD-L1 was first discovered in 1999 by Chen and colleagues using an expressed-sequence tag database search based on the homology sequence of CD80 and CD86 [48]. 1 year later, the molecule was identified as the binding ligand and function partner of PD-1 [2]. 2 years after the discovery of PD-L1, two laboratories independently identified the second PD-1 ligand and named it PD-L2. PD-L1 and PD-L2 have 38% homology at the amino acid sequence level [49], but they exhibit distinctly different cellular distribution profiles. PD-L1 is expressed on many types of immune cells, including T cells, B cells, monocytes, macrophages and DCs, as well as some non-hematopoietic cell populations. However, PD-L2 is mainly expressed on DCs, although it is upregulated on monocytes and macrophages once they are activated [37]. Notably, PD-L1 is often overexpressed on various cancer cells as a mechanism for triggering PD-1 signaling that suppresses the anti-tumor activities of tumor-infiltrating T lymphocytes [50].
Clinical applications of PD-1 and its ligand PD-L1
The mechanism employed by cancer cells to evade tumor suppression via PD-1 signaling and downstream T cell inhibition led to the development of PD-1/PD-L1 inhibitors. These monoclonal antibodies disrupt PD-1 signaling, reduce its downstream inhibitory effects on TCR, and ultimately enhance the T cell-mediated tumor cell elimination [50]. Since 2010, two PD-1 monoclonal antibodies (mAbs) and three PD-L1 mAbs have been approved by the FDA to treat malignant tumors. The clinical trial results showed that anti-PD-1/PD-L1 therapies have induced regression and improved survival in several solid organ cancers and hematological malignancies [6, 50, 51]. Nivolumab is the first anti-PD-1 antibody that showed significant clinical benefits with a consistent objective response rate (ORR) of 30–40% in patients with melanoma (NCT00730639, NCT01721772 and NCT01844505) and up to 87% in those with relapsed or refractory Hodgkin’s lymphoma [52–54]. In addition, compared to patients receiving chemotherapy, nivolumab demonstrated an overall extended survival rate in patients with advanced (stage IIIB and IV) squamous-cell non-small-cell lung cancer (NSCLC) who had progressed. First-line chemotherapy patients who received nivolumab lived 3.2 months longer than those treated with docetaxel. At 1-year follow-up, the overall survival rate was 42% (95% CI, 34 to 50) in the nivolumab group vs. 24% (95% CI, 17 to 31) in the docetaxel group. The median progression-free survival with nivolumab was 3.5 months vs. 2.8 months with docetaxel [55]. Pembrolizumab is another PD-1 inhibitor similar to nivolumab in efficacy and safety for the treatment of melanoma and NSCLC. Compared with ipilimumab, however, perbrolizumab exhibits a higher efficacy and a far better survival rate, which suggests different mechanisms underlying the PD-1 and CTLA-4 signaling pathways and their respective immune system regulations. To date, pembrolizumab has been FDA approved for the treatment of many metastatic solid tumors, including advanced melanoma, NSCLC with PD-L1 expression greater than 50% (a first line monotherapy), or metastatic non-squamous NSCLC (as combination therapy with carboplatin and pemetrexed), refractory classical Hodgkin lymphoma, bladder cancer, recurrent or metastatic head and neck cancer that progressed during or after platinum-based therapy, gastric cancer and microsatellite instability high-grade tumors or with deficient mismatch repair mechanisms that have progressed on prior treatments [6, 50, 51].
PD-L1-blocking mAbs can eliminate the interactions of PD-1 and PD-L1 as well, therefore attenuating the suppressive effect mediated by PD-1. The clinical trial on anti-PD-L1 mAb used to treat patients with metastatic urothelial bladder cancer supported this hypothesis. The trial results showed significant induction of cancer regression in the anti-PD-L1 cohort. Patients with PD-L1-positive tumors treated with PD-L1 inhibitor had a 43% ORR vs. 11% in those with PD-L1 negative tumors [56]. In an expanded clinical trial conducted on patients with multiple cancer types being treated with anti-PD-L1 mAb, the best anti-tumor responses observed were in those with tumors expressing high levels of PD-L1. This finding suggests that the level of PD-L1 expression may be a useful screening and prognostic tool when initiating anti-PD-L1 inhibitor therapy [57].
The adverse effects induced by the checkpoint inhibitor immunotherapy
Immune checkpoints play a critical role in limiting excess T cell activation and immune responses; therefore, the inhibition of these co-receptors often breaks the balance between immunity and tolerance. Immune checkpoint-blocking mAbs have been associated with a variety of irAEs, including colitis, dermatitis, nephritis, endocrinopathies, hepatitis, pneumonitis and myocarditis [9, 11, 12] (summarized in the reference article [58]). The irAEs are graded according to the Common Terminology Criteria for Adverse Events. In patients receiving ipilimumab, 64–80% developed irAEs, and of those, 23% were Grade 3/4. Up to 79% patients treated with pembrolizumab had irAEs, with 13% being Grade 3/4. When ipilimumab and nivolumab were combined, the incidence of irAEs reached 96% with 55% being Grade 3/4. [29, 59–61]. Although most irAEs were mild, transient and reversible, the ipilimumab and nivolumab combination treatment was discontinued in approximately 40% of patients [11]. Furthermore, some adverse events can be associated with severe consequences. For example, cardiac toxicities such as myocarditis can be fatal.
Preclinical evidence of autoimmune myocarditis
In a CTLA-4-deficient mouse model in which CTLA-4 expression was specifically disrupted in the resident cells in lymph node and spleen, the accumulation of activated T lymphocytes was observed in the heart, liver, lung, bone marrow and pancreas tissues. Electron microscopic examination of the myocardium from these mice revealed the presence of fibroblast proliferation, neutrophils, macrophages and few lymphocytes in ill-defined edematous areas, which eventually led to myocardial infarctions. Myocardial failure was thought to be the cause of early death of these mice at the 3rd and 4th weeks [18].
Similarly, PD-1-deficient mice started to die at the 5th week [17]. The study showed that the depletion of PD-1 resulted in dilated cardiomyopathy with severely impaired contraction and congestive heart failure followed by sudden death. The hearts collected from knockout mice exhibited diffuse deposition of immunoglobulin G (IgG) on the surface of cardiomyocytes, and all PD-1 knockout mice had circulating high-titer IgG autoantibodies against an autoantigen expressed on the cell surface of cardiomyocytes. Interestingly, the defect was not observed in PD-1 and Rag-2 (recombination-activating gene-2) double knockout mice in which only a few T and B cells were present due to loss of Rag-2 mediated TCR and B cell receptor gene rearrangement. This finding indicated the contribution of self-reactive T cell-mediated B cell autoantibody production. Tarrio et al. determined that myocardial damage was induced by PD-1-CD4+ T cells and PD-1-CD8+ T cells and concluded that both subsets of T cells required PD-1 to maintain their tolerance on self-components on the myocardium [62]. Meanwhile, PD-L1, a ligand of PD-1, is expressed in both human [48] and murine heart [2]. The Lichtman group used cytotoxic T cells to induce myocarditis. They found that the genetic deletion of both PD-1 ligands, PD-L1 and PD-L2, as well as treatment with PD-L1 inhibitor, caused a transient myocarditis to progress to a lethal disease, which confirms the pivotal role of PD-1 signaling in protecting the myocardium from self-reactive T lymphocytes [63]. In another investigation published in the same year, a Japanese group utilized PD-L1 and PD-L2 blocking antibodies to study the role of these two PD-1 ligands in the development of murine acute myocarditis caused by viral infection. They found that PD-L1, but not PD-L2, played a pivotal role in suppressing myocardial inflammation during infection, as demonstrated by the increased expression of IFNγ, FasL, CD40L, perforin and viral genomes in myocardial tissue in the presence of PD-L1 blocking antibody [64]. Furthermore, PD-L1 deficiency in Murphy Roths Large mice (genetically predisposed to autoimmunity) caused lethal autoimmune myocarditis, with infiltrating macrophages and T cells predominantly detected throughout the heart along with heart-specific autoantibodies [65].
The incidences of cardiotoxicity have been reported in patients receiving checkpoint inhibitor therapy, including several fatal cases. In patients treated with ipilimumab and combination of ipilimumab and nivolumab, myocardial fibrosis, left ventricular dysfunction, Takotsubo cardiomyopathy and late-onset pericarditis were reported [10, 66–69]. There was one fatal case of myocardial infarction in a patient with NSCLC who received pembrolizumab [70]. In another clinical trial with ipilimumab plus nivolumab, two cases of fulminant myocarditis and myositis were described [71]. Both were melanoma patients who had a history of hypertension without any other cardiac risk factors. Histological analysis of myocardial biopsy demonstrated the infiltration of CD4+ T and CD8+ T cells and macrophages in the myocardium, cardiac sinus and atrioventricular nodes. Generally, patients who received combined immune checkpoint inhibitors (anti-CLTA-4 plus anti-PD-1 or anti-PD-L1) were more susceptible to developing severe myocarditis than those getting nivolumab immunotherapy [71]. Myocarditis was also noted to be an early cardiotoxicity diagnosed on average 17 days after the initial treatment [71].
To manage myocarditis associated with immune checkpoint inhibitor therapy, cardiac monitoring with serum troponin I and electrocardiogram early after treatment initiation is recommended [11, 13]. When a diagnosis of myocarditis is suspected, immunotherapy should be discontinued temporarily while starting corticosteroids. Grade 3 or 4 myocarditis requires permanent discontinuation of the immunotherapy, and systemic oral or IV steroids should be started at 1 to 2 mg/kg/day for 3 days followed by a gradual taper over at least 1 month to avoid the recurrence or worsening of irAEs. The steroid doses can be adjusted based upon the severity or grade of the adverse effect. Endomyocardial biopsy may be considered before the therapy is permanently discontinued [13, 72]. Of note, extra caution may be required in using checkpoint inhibitors in patients with systemic autoimmune diseases, as they may develop subclinical myocarditis without obvious signs and symptoms [11]. In addition, the development of cardiac protective agents that can be co-administered with checkpoint inhibitors will be critical to preventing cardiac irAEs.
Conclusion
The ultimate goal in battling cancer is to achieve the specific recognition and effective elimination of malignant cells with minimum/tolerable side effects. The immune checkpoint inhibitor therapy has emerged as a milestone in the cancer treatment in recent years. It inhibits the suppression on T cell activation and anti-tumor activities and thereby effectively increases the response rate, induces cancer regression and improves patient survival. However, a high incidence of adverse events is associated with this therapy, ranging from mild to severe cases. Cardiotoxicity related to immunotherapy use is one of the rare but life-threatening irAEs that deserves special attention. Better understanding of the mechanism, early recognition and diagnosis and prompt treatment are critical in managing cancer patients with cardiotoxicity related to immunotherapy.
Acknowledgements
The Zhu laboratory is supported by grants from the National Institute of Health (HL124122 and AR067766). The He laboratory is also supported by the National Institute of Health (grant 2K12CA133250).
Competing interests
The authors declare to have no competing financial interests.
Contributor Information
Ning Zou, Email: sunnyning116@163.com.
Kai He, Email: kai.he@osumc.edu.
Hua Zhu, Email: Hua.Zhu@osumc.edu.
References
- 1.Neville AM, Mackay AM, Westwood J, Turberville C, Laurence DJ. Human tumour-associated and tumour-specific antigens: some concepts in relation to clinical oncology. J Clin Pathol Suppl. 1975;6:102–12. doi: 10.1136/jcp.s1-6.1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science. 2011;331:1565–70. doi: 10.1126/science.1203486. [DOI] [PubMed] [Google Scholar]
- 4.Dimberu PM, Leonhardt RM. Cancer immunotherapy takes a multi-faceted approach to kick the immune system into gear. Yale J Biol Med. 2011;84:371–80. [PMC free article] [PubMed] [Google Scholar]
- 5.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252–64. doi: 10.1038/nrc3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen L, Han X. Anti-PD-1/PD-L1 therapy of human cancer: past, present, and future. J Clin Invest. 2015;125:3384–91. doi: 10.1172/JCI80011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Callahan MK, Postow MA, Wolchok JD. Targeting T cell co-receptors for cancer therapy. Immunity. 2016;44:1069–78. doi: 10.1016/j.immuni.2016.04.023. [DOI] [PubMed] [Google Scholar]
- 8.Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348:56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- 9.Buchbinder E, Hodi FS. Cytotoxic T lymphocyte antigen-4 and immune checkpoint blockade. J Clin Invest. 2015;125:3377–83. doi: 10.1172/JCI80012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, et al. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J Immunother Cancer. 2016;4:50. doi: 10.1186/s40425-016-0152-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Varricchi G, Galdiero MR, Marone G, Criscuolo G, Triassi M, Bonaduce D, et al. Cardiotoxicity of immune checkpoint inhibitors. ESMO Open. 2017;2:e000247. doi: 10.1136/esmoopen-2017-000247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yshii LM, Hohlfeld R, Liblau RS. Inflammatory CNS disease caused by immune checkpoint inhibitors: status and perspectives. Nat Rev Neurol. 2017;13:755–63. doi: 10.1038/nrneurol.2017.144. [DOI] [PubMed] [Google Scholar]
- 13.Wang DY, Okoye GD, Neilan TG, Johnson DB, Moslehi JJ. Cardiovascular toxicities associated with cancer immunotherapies. Curr Cardiol Rep. 2017;19:21. doi: 10.1007/s11886-017-0835-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. doi: 10.1146/annurev-immunol-031210-101324. [DOI] [PubMed] [Google Scholar]
- 15.Mahoney KM, Rennert PD, Freeman GJ. Combination cancer immunotherapy and new immunomodulatory targets. Nat Rev Drug Discov. 2015;14:561–84. doi: 10.1038/nrd4591. [DOI] [PubMed] [Google Scholar]
- 16.Alegre ML, Frauwirth KA, Thompson CB. T-cell regulation by CD28 and CTLA-4. Nat Rev Immunol. 2001;1:220–8. doi: 10.1038/35105024. [DOI] [PubMed] [Google Scholar]
- 17.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 18.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 19.Ling V, Wu PW, Finnerty HF, Sharpe AH, Gray GS, Collins M. Complete sequence determination of the mouse and human CTLA4 gene loci: cross-species DNA sequence similarity beyond exon borders. Genomics. 1999;60:341–55. doi: 10.1006/geno.1999.5930. [DOI] [PubMed] [Google Scholar]
- 20.Walker LS, Sansom DM. Confusing signals: recent progress in CTLA-4 biology. Trends Immunol. 2015;36:63–70. doi: 10.1016/j.it.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schneider H, Rudd CE. Diverse mechanisms regulate the surface expression of immunotherapeutic target ctla-4. Front Immunol. 2014;5:619. doi: 10.3389/fimmu.2014.00619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Egen JG, Allison JP. Cytotoxic T lymphocyte antigen-4 accumulation in the immunological synapse is regulated by TCR signal strength. Immunity. 2002;16:23–35. doi: 10.1016/S1074-7613(01)00259-X. [DOI] [PubMed] [Google Scholar]
- 23.Tai X, Van Laethem F, Pobezinsky L, Guinter T, Sharrow SO, Adams A, et al. Basis of CTLA-4 function in regulatory and conventional CD4( + ) T cells. Blood. 2012;119:5155–63. doi: 10.1182/blood-2011-11-388918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. CTLA-4 control over Foxp3 + regulatory T cell function. Science. 2008;322:271–5. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 25.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–6. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- 26.Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci USA. 1997;94:8099–103. doi: 10.1073/pnas.94.15.8099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190:355–66. doi: 10.1084/jem.190.3.355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ribas A, Kefford R, Marshall MA, Punt CJ, Haanen JB, Marmol M, et al. Phase III randomized clinical trial comparing tremelimumab with standard-of-care chemotherapy in patients with advanced melanoma. J Clin Oncol. 2013;31:616–22. doi: 10.1200/JCO.2012.44.6112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–23. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maio M, Grob JJ, Aamdal S, Bondarenko I, Robert C, Thomas L, et al. Five-year survival rates for treatment-naive patients with advanced melanoma who received ipilimumab plus dacarbazine in a phase III trial. J Clin Oncol. 2015;33:1191–6. doi: 10.1200/JCO.2014.56.6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. EMBO J. 1992;11:3887–95. doi: 10.1002/j.1460-2075.1992.tb05481.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang X, Schwartz JC, Guo X, Bhatia S, Cao E, Lorenz M, et al. Structural and functional analysis of the costimulatory receptor programmed death-1. Immunity. 2004;20:337–47. doi: 10.1016/S1074-7613(04)00051-2. [DOI] [PubMed] [Google Scholar]
- 33.Neel BG, Gu H, Pao L. The Shp’ing news: SH2 domain-containing tyrosine phosphatases in cell signaling. Trends Biochem Sci. 2003;28:284–93. doi: 10.1016/S0968-0004(03)00091-4. [DOI] [PubMed] [Google Scholar]
- 34.Long EO. Regulation of immune responses through inhibitory receptors. Annu Rev Immunol. 1999;17:875–904. doi: 10.1146/annurev.immunol.17.1.875. [DOI] [PubMed] [Google Scholar]
- 35.Sidorenko SP, Clark EA. The dual-function CD150 receptor subfamily: the viral attraction. Nat Immunol. 2003;4:19–24. doi: 10.1038/ni0103-19. [DOI] [PubMed] [Google Scholar]
- 36.Agata Y, Kawasaki A, Nishimura H, Ishida Y, Tsubata T, Yagita H, et al. Expression of the PD-1 antigen on the surface of stimulated mouse T and B lymphocytes. Int Immunol. 1996;8:765–72. doi: 10.1093/intimm/8.5.765. [DOI] [PubMed] [Google Scholar]
- 37.Yamazaki T, Akiba H, Iwai H, Matsuda H, Aoki M, Tanno Y, et al. Expression of programmed death 1 ligands by murine T cells and APC. J Immunol. 2002;169:5538–45. doi: 10.4049/jimmunol.169.10.5538. [DOI] [PubMed] [Google Scholar]
- 38.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Terawaki S, Chikuma S, Shibayama S, Hayashi T, Yoshida T, Okazaki T, et al. IFN-alpha directly promotes programmed cell death-1 transcription and limits the duration of T cell-mediated immunity. J Immunol. 2011;186:2772–9. doi: 10.4049/jimmunol.1003208. [DOI] [PubMed] [Google Scholar]
- 40.Oestreich KJ, Yoon H, Ahmed R, Boss JM. NFATc1 regulates PD-1 expression upon T cell activation. J Immunol. 2008;181:4832–9. doi: 10.4049/jimmunol.181.7.4832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Staron MM, Gray SM, Marshall HD, Parish IA, Chen JH, Perry CJ, et al. The transcription factor FoxO1 sustains expression of the inhibitory receptor PD-1 and survival of antiviral CD8( + ) T cells during chronic infection. Immunity. 2014;41:802–14. doi: 10.1016/j.immuni.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathieu M, Cotta-Grand N, Daudelin JF, Thebault P, Labrecque N. Notch signaling regulates PD-1 expression during CD8( + ) T-cell activation. Immunol Cell Biol. 2013;91:82–88. doi: 10.1038/icb.2012.53. [DOI] [PubMed] [Google Scholar]
- 43.Kao C, Oestreich KJ, Paley MA, Crawford A, Angelosanto JM, Ali MA, et al. Transcription factor T-bet represses expression of the inhibitory receptor PD-1 and sustains virus-specific CD8 + T cell responses during chronic infection. Nat Immunol. 2011;12:663–71. doi: 10.1038/ni.2046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta. FEBS Lett. 2004;574:37–41. doi: 10.1016/j.febslet.2004.07.083. [DOI] [PubMed] [Google Scholar]
- 45.Parry RV, Chemnitz JM, Frauwirth KA, Lanfranco AR, Braunstein I, Kobayashi SV, et al. CTLA-4 and PD-1 receptors inhibit T-cell activation by distinct mechanisms. Mol Cell Biol. 2005;25:9543–53. doi: 10.1128/MCB.25.21.9543-9553.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patsoukis N, Brown J, Petkova V, Liu F, Li L, Boussiotis VA. Selective effects of PD-1 on Akt and Ras pathways regulate molecular components of the cell cycle and inhibit T cell proliferation. Sci Signal. 2012;5:ra46. doi: 10.1126/scisignal.2002796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK, et al. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009;206:3015–29. doi: 10.1084/jem.20090847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med. 1999;5:1365–9. doi: 10.1038/70932. [DOI] [PubMed] [Google Scholar]
- 49.Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand for PD-1 and inhibits T cell activation. Nat Immunol. 2001;2:261–8. doi: 10.1038/85330. [DOI] [PubMed] [Google Scholar]
- 50.Chinai JM, Janakiram M, Chen F, Chen W, Kaplan M, Zang X. New immunotherapies targeting the PD-1 pathway. Trends Pharmacol Sci. 2015;36:587–95. doi: 10.1016/j.tips.2015.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bardhan K, Anagnostou T, Boussiotis VA. The PD1:PD-L1/2 pathway from discovery to clinical implementation. Front Immunol. 2016;7:550. doi: 10.3389/fimmu.2016.00550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol. 2014;32:1020–30. doi: 10.1200/JCO.2013.53.0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Robert C, Long GV, Brady B, Dutriaux C, Maio M, Mortier L, et al. Nivolumab in previously untreated melanoma without BRAF mutation. N Engl J Med. 2015;372:320–30. doi: 10.1056/NEJMoa1412082. [DOI] [PubMed] [Google Scholar]
- 54.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med. 2015;372:311–9. doi: 10.1056/NEJMoa1411087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non-small-cell lung cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515:558–62. doi: 10.1038/nature13904. [DOI] [PubMed] [Google Scholar]
- 57.Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–7. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Postow MA, Sidlow R, Hellmann MD. Immune-related adverse events associated with immune checkpoint blockade. N Engl J Med. 2018;378:158–68. doi: 10.1056/NEJMra1703481. [DOI] [PubMed] [Google Scholar]
- 59.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369:134–44. doi: 10.1056/NEJMoa1305133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Graziani G, Tentori L, Navarra P. Ipilimumab: a novel immunostimulatory monoclonal antibody for the treatment of cancer. Pharmacol Res. 2012;65:9–22. doi: 10.1016/j.phrs.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 62.Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J Immunol. 2012;188:4876–84. doi: 10.4049/jimmunol.1200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8 + T-cell mediated injury in the heart. Circulation. 2007;116:2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360. [DOI] [PubMed] [Google Scholar]
- 64.Seko Y, Yagita H, Okumura K, Azuma M, Nagai R. Roles of programmed death-1 (PD-1)/PD-1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res. 2007;75:158–67. doi: 10.1016/j.cardiores.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 65.Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol. 2008;181:2513–21. doi: 10.4049/jimmunol.181.4.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Voskens CJ, Goldinger SM, Loquai C, Robert C, Kaehler KC, Berking C, et al. The price of tumor control: an analysis of rare side effects of anti-CTLA-4 therapy in metastatic melanoma from the ipilimumab network. PLoS One. 2013;8:e53745. doi: 10.1371/journal.pone.0053745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Roth ME, Muluneh B, Jensen BC, Madamanchi C, Lee CB. Left ventricular dysfunction after treatment with ipilimumab for metastatic melanoma. Am J Ther. 2016;23:e1925–e1928. doi: 10.1097/MJT.0000000000000430. [DOI] [PubMed] [Google Scholar]
- 68.Yun S, Vincelette ND, Mansour I, Hariri D, Motamed S. Late onset ipilimumab-induced pericarditis and pericardial effusion: a rare but life threatening complication. Case Rep Oncol Med. 2015;2015:794842. doi: 10.1155/2015/794842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Geisler BP, Raad RA, Esaian D, Sharon E, Schwartz DR. Apical ballooning and cardiomyopathy in a melanoma patient treated with ipilimumab: a case of takotsubo-like syndrome. J Immunother Cancer. 2015;3:4. doi: 10.1186/s40425-015-0048-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Herbst RS, Baas P, Kim DW, Felip E, Perez-Gracia JL, Han JY, et al. Pembrolizumab versus docetaxel for previously treated, PD-L1-positive, advanced non-small-cell lung cancer (KEYNOTE-010): a randomised controlled trial. Lancet. 2016;387:1540–50. doi: 10.1016/S0140-6736(15)01281-7. [DOI] [PubMed] [Google Scholar]
- 71.Johnson DB, Balko JM, Compton ML, Chalkias S, Gorham J, Xu Y, et al. Fulminant myocarditis with combination immune checkpoint blockade. N Engl J Med. 2016;375:1749–55. doi: 10.1056/NEJMoa1609214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Champiat S, Lambotte O, Barreau E, Belkhir R, Berdelou A, Carbonnel F, et al. Management of immune checkpoint blockade dysimmune toxicities: a collaborative position paper. Ann Oncol. 2016;27:559–74. doi: 10.1093/annonc/mdv623. [DOI] [PubMed] [Google Scholar]