

Abstract
The present study investigated the efficacy of the hepatoprotective drug matrine (Mtr) for its new application for hepatosteatosis and associated disorders in glucose homeostasis. The study was performed in two nutritional models of hepatosteatosis in mice with various abnormal glucose homeostasis: (1) high-fructose diet (HFru) induced hepatosteatosis and glucose intolerance from hepatic, and (2) hepatosteatosis and hyperglycemia induced by high-fat (HF) diet in combination with low doses of streptozotocin (STZ). Administration of Mtr (100 mg/kg every day in diet for 4 weeks) abolished HFru-induced hepatosteatosis and glucose intolerance. These effects were associated with the inhibition of HFru-stimulated de novo lipogenesis (DNL) without altering hepatic fatty acid oxidation. Further investigation revealed that HFru-induced endoplasmic reticulum (ER) stress was inhibited, whereas heat-shock protein 72 (an inducible chaperon protein) was increased by Mtr. In a type 2 diabetic model induced by HF-STZ, Mtr reduced hepatosteatosis and improved attenuated hyperglycemia. The hepatoprotective drug Mtr may be repurposed for the treatment of hepatosteatosis and associated disorders in glucose homeostasis. The inhibition of ER stress associated DNL and fatty acid influx appears to play an important role in these metabolic effects.
Keywords: matrine, hepatosteatosis, glucose intolerance, hyperglycemic control
Introduction
The liver plays an important role in regulating whole-body lipid metabolism and glucose homeostasis. Excess accumulation of lipids (namely hepatosteatosis), either from endogenous de novo lipogenesis (DNL) and/or influx of exogenous fatty acids (FA), can disturb glucose homeostasis, increasing the risk of type 2 diabetes (T2D) [1, 2]. Although the degree of hepatosteatosis and T2D do not necessarily tightly coupled [3], inhibition of excess hepatic DNL has been shown to ameliorate hepatosteatosis and associated glucose intolerance [4]. Shulman and colleagues have also demonstrated that correction of hepatosteatosis in patients with T2D is important for hyperglycemia control [5]. In search for a new therapeutic for the treatment of hepatic steatosis from DNL, we have taken the approach of repurposing existing drugs for these conditions [6, 7].
Matrine (Mtr) is a small molecule (MW: 248) found in Sophora and it is structurally different from the drugs currently used to treat T2D [8–10]. Mtr has been used clinically as a hepatoprotective drug for the treatment of tumors and viral hepatitis [8], where DNL is often increased [11]. A recent study from our laboratory found that Mtr is able to reduce hepatosteatosis, fasting blood glucose and glucose intolerance in high fat (HF)-fed mice [9]. However, the HF model does not exhibit the characteristics of DNL-induced hepatosteatosis and glucose intolerance because the accumulation of triglyceride (TG) in the liver is due to a direct influx of lipids into the liver from the HF diet [12, 13].
The liver is a major site of DNL production from carbohydrates [12, 14] and interestingly the inhibition of hepatic DNL reduces hepatosteatosis and hyperglycemia [4, 15]. It has been suggested that an increase in DNL is the second major source of lipid accumulation in the liver and contributes about 26% of patients with hepatosteatosis [16]. In mice, a high-fructose (HFru) diet induces hepatosteatosis as early as 1 day [17] prior to the development of glucose intolerance [12, 18, 19]. Dietary fructose is almost entirely metabolised in the liver in its first pass, and serves mainly as a substrate for DNL in both animals [13, 17, 20] and humans [19, 21–23]. HFru diets increase the expression of lipogenic transcription factors, sterol regulatory element binding protein (SREBP1c) and carbohydrate response element binding protein (ChREBP), which upregulate lipogenic genes. Upregulation of these lipogenic transcription factors can result in hepatosteatosis and glucose intolerance via promoting DNL [24, 25]. Notably, HFru-stimulated hepatic DNL via SREBP1c is dependent on the activation of the ER stress pathway [12, 26].
The primary aim of the present study was to investigate whether the hepatoprotective drug Mtr can limit the hepatosteatosis and the associated glucose intolerance that usually results from increased DNL in HFru-fed mice. If so, the second aim was to investigate whether the action of Mtr is via the ER stress pathway. Finally, we also evaluated whether Mtr assists glycemic control in a T2D mouse model generated by a combination of HF and streptozotocin (STZ) where an increased DNL via SREBP1c is also involved [27, 28].
Materials and methods
Animals and diets
C57BL/6J mice aged 10–12 weeks and weighing 21–24 g were obtained from the Animal Resource Centre Pty. Ltd. (Perth, Australia). The animals were housed in a temperature-controlled room (22 ± 1 °C) on a 12-h light/dark cycle with free access to food and water. Mice were fed ad libitum for 1 week on a normal chow diet (~70% calories from starch, ~10% calories from fat, and ~20% calories from protein; Gordon’s Specialty Stock Feeds, Yanderra, Australia). Mtr (≥ 98% by HPLC) was purchased from Sigma Aldrich. The experiments described in this manuscript were approved by the Animal Ethics Committee of RMIT University (Application ID: 1012) and conducted in compliance with the guidelines of the National Health and Medical Research Council of Australia for Animal Experimentation.
Two sets of animal experiments were performed. In the first set of experiments, mice were fed a HFru diet (35% fructose, 35% starch, ~10% fat and ~20% protein) to generate hepatosteatosis. Mice were fed for 8 weeks with or without Mtr at a dose of 100 mg/kg every day as a food additive in the last 4 weeks as described previously [9, 10]. Body weight gain and food intake were measured twice a week. For blood glucose levels, blood samples were collected from the tail tip and measured using a glucometer (AccuCheck II; Roche, New South Wales, Castle Hill, Australia) after 2 weeks of Mtr treatment. In the second set of experiments, the effects of Mtr on hepatosteatosis and hyperglycemia were examined in a T2D model induced by HF feeding in combination with low doses of STZ as previously reported [28–30]. Briefly, mice were fed a HF diet (45% calorie from lard, 20% calories from protein and 35% calories from carbohydrate) for 14 weeks to induce insulin resistance. After 8 weeks of HF feeding, mice were injected with STZ at a low dose (40 mg/kg per day, ip) for 5 consecutive days to reduce the level of plasma insulin by ~50% [28–30]. One week after the last injection of STZ, fasting blood glucose was usually increased by 50%–100% (hyperglycemia, defined as T2D). The T2D mice were then divided into 2 groups: one group receiving Mtr added in the HF diet (100 mg/kg per day) for 4 weeks (T2D-Mtr) whereas the other group was fed HF alone (T2D-Con) for the same period of time. During the period of Mtr treatment, fasting blood glucose was monitored once a week. A normal control group of mice (CH-Con) was included for the same period. At the end of both sets of experiments, mice were killed by cervical dislocation and liver tissues were collected and freeze-clamped immediately for further analysis.
Assessment of the effect on hepatosteatosis
Hepatosteatosis was assessed by measuring TG content in the liver. Mice were fasted for 5–7 h before being killed; the liver was collected and freeze-clamped immediately. As described previously [13, 28] plasma and liver TG levels were determined with a Peridochrom triglyceride GPO-PAP biochemical kit (Roche diagnostics). The method of lipid extraction from liver with chloroform/methanol has been described previously [17].
Assessment of the effect on hepatic FA oxidation
FA oxidation was assessed in fresh liver tissue ex vivo as described [12, 13]. Briefly, fresh liver samples were homogenised in an isolating medium which contained 100 mM sucrose, 50 mM Tris, 100 mM KCl, 1 mM KH2PO4 and 0.1 mM EGTA, 0.2% FA-free BSA at pH 7.0. The liver homogenate was incubated with [14 C]-palmitate and [14 C]–CO2 produced from the incubation was collected in 1 M sodium hydroxide. Palmitate oxidation rates were determined by counting the 14 C radioactivity of captured CO2 and acid-soluble metabolites and oxidation rate were expressed as nanomoles of CO2 per gram of wet weight per hour [12].
Assessment of the effects on DNL and ER stress
DNL and ER stress were assessed by immunoblotting with specific anti-bodies for the key proteins in the DNL, ER stress and heat shock protein (HSP) pathways based on our recent work [9, 10, 12, 13, 17]. Briefly, freeze-clamped liver was homogenized in ice-cold lysis buffer supplemented with fresh protease inhibitor and phosphatase inhibitor (Sigma Aldrich). The key proteins in the DNL pathway included SREBP-1 (Santa Cruz), ChREBP (Abcam), acetyl-CoA carboxylase (ACC, Upstate), fatty acid synthase (FAS, Abcam) and stearoyl-CoA desaturase 1 (SCD-1, Cell Signaling). The key proteins measured in the ER stress pathway included inositol-requiring kinase 1 (IRE1, Abcam), eukaryotic translation initiation factor 2α (eIF2α, Cell Signaling) and CHOP (Santa Cruz). The effect on the HSP pathway was assessed by heat shock protein 72 (HSP72, Abcam) based on our recent work [9, 10]. Proteins were quantified using a ChemiDoc and densitometry analysis was performed using Image Lab software (Bio-Rad Laboratories, USA).
Statistical analysis
All results are presented as means ± s.e.m. One-way analysis of variance was used to assess the statistical significance across all groups. When significant differences were found, the Tukey-Kramer multiple comparisons post hoc test was used to establish differences between groups. Differences at P < 0.05 were considered to be statistically significant and P < 0.01 were considered to be highly significant.
Results
Effects on body weight, adiposity, hepatosteatosis and glucose tolerance in HFru-fed mice
HFru feeding is a well-defined model of hepatosteatosis, visceral adiposity and glucose intolerance resulting from increased DNL in the liver [12]. As expected, HFru feeding moderately increased the mass of epididymal fat (by 40%, P < 0.01) without altering body weight or food intake (P > 0.05; Fig. 1a–c). The TG content (indicative of hepatosteatosis) was increased dramatically in the liver (by threefold) but only moderately in muscle (~35%) (both P < 0.01; Fig. 1d). As shown in Fig. 1e, HFru-fed mice also showed moderate glucose intolerance.
Fig. 1.

Effects of Mtr on body weight gain, visceral adiposity, hepatosteatosis and glucose tolerance in HFru-fed mice. Mice were fed a high-fructose (HFru) diet for 8 weeks and matrine (Mtr, 100 mg/kg per day in diet) was administered in the last 4 weeks. A glucose tolerance test (GTT at 3 g glucose/kg BW, ip) was conducted after 2 weeks of treatment with Mtr. Epididymal (Epi) fat weight and liver TG content were determined at the end of the study. (a) Body weight gain, (b) Epididymal fat weight as a percentage of body weight, (c) Caloric intake, (d) TG content in liver and muscle. (e) Glucose tolerance. **P < 0.01 vs CH; #P < 0.05, ##P < 0.01 vs HFru (n = 7–8 mice/group)
Administration of Mtr prevented the moderate body weight gain (8%–10%) in HFru-fed mice during this period of time (Fig. 1a). It corrected HFru-induced increases in epididymal fat, liver TG content and glucose intolerance (all, P < 0.01 vs untreated HFru-fed mice) to the levels similar to CH-fed normal mice (Fig. 1b–e). Although not significantly reduced, muscle TG content in Mtr-treated HFru-fed mice was no longer different from the value of CH-fed normal mice (Fig. 1d).
Effects on FA oxidation and DNL in the liver of HFru-fed mice
We first examined whether Mtr treatment may promote FA oxidation in the liver of HFru-fed mice. As shown in Fig. 2a, palmitate oxidation by the liver homogenates was not affected by the treatment with Mtr, suggesting that the reduced hepatosteatosis by Mtr is not likely to be due to an increased FA oxidation in the liver. We next examined the DNL pathway because HFru-induced hepatosteatosis is believed to result from the stimulation to this pathway in the liver [13, 17, 20]. As expected, HFru-fed mice exhibited dramatic increases in DNL proteins in the liver (Fig. 2b–e), including SREBP-1c (by 2-fold), ChREBP (by 33%) ACC (by 3-fold), FAS (by 3.4-fold) and SCD-1 (by 4-fold; all P < 0.05). Interestingly, these lipogenic proteins except for ACC were significantly reduced by the treatment with Mtr, including SREBP-1c (by 45%, P < 0.01), ChREBP (by 33%, P < 0.05), SCD-1 (by 32%, P < 0.01) and FAS (by 24%, P < 0.05). These results suggest that the reduced TG content in the liver by Mtr can be attributed to its inhibitory effect on HFru-induced increase in hepatic DNL.
Fig. 2.

Effects of Mtr on FA oxidation and DNL pathways in the liver of HFru-fed mice. FA oxidation was detected by incubating fresh liver homogenates with [14 C]–palmitate and DNL was assessed by the protein expression of palmitate in this pathway. (a) Liver lyzates from mice were immunoblotted with the mature form of SREBP-1c and ChREBP (b), SCD-1 (c), FAS (d) and ACC (e) and then quantified for statistical analysis. *P < 0.05, **P < 0.01 versus CH-Con; #P < 0.05, ##P < 0.01 versus HFru-Con, n = 7–8 mice per group
Effects on ER stress and HSP72 in the liver of HFru-fed mice
By promoting DNL in the liver, the activation of ER stress represents a key step in the pathogenesis of hepatosteatosis [12, 31, 32]. As shown in Fig. 3a, HFru-fed mice exhibited marked increases of the mature form of eIF2α (by 2-fold), CHOP (by 2.3-fold) and IRE1 (by 2-fold) along with the upregulation of the DNL pathway in the liver. Treatment with Mtr markedly reduced the protein levels of these hepatic ER markers towards the levels seen in CH fed mice. These results indicate that Mtr induced-suppression of ER markers may be associated with the improvement in lipogenesis, which could account for its beneficial effects on hepatosteatosis. Recent studies indicate that HSP72 is likely to mediate the effect of Mtr on hepatosteatosis and glucose intolerance [9, 33, 34]. As shown in Fig. 3b, there was ~50% suppression of HSP72 (P < 0.05 vs CH fed mice) in the liver of HFru-fed mice and this reduction was reversed following treatment with Mtr (P < 0.05 vs untreated HFru-fed mice).
Fig. 3.

Effects on ER stress and HSP72 in the liver of HFru-fed mice. Liver lyzates from mice were immunoblotted for (a) eIF2α, CHOP and IRE1 and (b) HSP72 and quantified for statistical analysis. *P < 0.05, **P < 0.01 versus CH-Con; #P < 0.05, ##P < 0.01 versus HFru-Con, n = 7–8 mice per group
Effects on hyperglycemia in T2D mice
To investigate the relationship of the effect on hepatosteatosis with glycemic control, we examined the metabolic effects of Mtr in T2D mice generated by a HF diet in combination with low doses of STZ [9, 27, 28]. The body weight were reduced in HF-STZ induced T2D mice (by ~15%) and treatment with Mtr had no effect on the body weight (Fig. 4a), while visceral adiposity remained unchanged in T2D-Con mice compared to CH-fed mice Mtr significantly reduced epididymal fat in T2D mice (P < 0.05, Fig. 4b). T2D-Con mice displayed typical fasting hyperglycemia and Mtr treatment significantly reduced the degree of the hyperglycemia over the period of 4 weeks (by 20%–30%, Fig. 4c). As expected, HF-STZ-induced T2D showed severe glucose intolerance but this was not attenuated by the treatment with Mtr (Fig. 4b).
Fig. 4.

Effects of Mtr on body weight, visceral adiposity, blood glucose and glucose tolerance in T2D mice. T2D was generated by a high-fat (HF) diet plus low-dose of STZ injections. After the development of hyperglycemia, Mtr (100 mg/kg per day in diet) was administered to diabetic mice for 4 weeks. Body weight at the end of the study (a). Epididymal (Epi) fat weight (b). Blood glucose levels (after 5–7 h of fasting) (c) were monitored once a week. An ipGTT (1.0 g glucose/g body weight) was performed after 2 weeks of treatment with Mtr (d). **P < 0.01 vs CH-Con; ##P < 0.01 vs T2D-Con (n = 7–8 mice/group)
Effect on TG levels in T2D mice
Recent studies indicate that hepatosteatosis can contribute to hyperglycemia and hepatic insulin resistance [12, 35]. To determine whether the reduced hepatosteatosis by resulting from Mtr treatment is associated with the control of hyperglycemia in T2D mice, we measured the TG content in the liver. As shown in Fig. 5a, b, T2D mice exhibited hypertriglyceridemia and hepatosteatosis, however Mtr significantly reduced these conditions (P < 0.05). Together, these results clearly indicate that Mtr reduces the T2D-induced hepatosteatosis that is associated with hyperglycemia and this could account for its beneficial effects on the regulation of lipid metabolism.
Fig. 5.

Effects of Mtr on TG level in the plasma and livers of T2D mice. Plasma levels of TG were measured from blood samples collected in week 2 of the treatment (a). Liver TG content was determined from freeze-clamped samples obtained at the end of the study (b). **P < 0.01 vs CH-Con; ##P < 0.01 vs T2D-Con (n = 7–8 mice/group)
Discussion
The present study investigated whether the hepatoprotective drug Mtr can treat the hepatosteatosis and associated glucose intolerance in HFru-fed mice resulting from increased DNL. Consistent with our previous studies [13, 17, 36], HFru-fed mice developed hepatosteatosis and glucose intolerance by promoting ER stress associated DNL. Treatment of these mice with Mtr ameliorated hepatosteatosis and glucose intolerance. Within the liver, Mtr decreased the protein expression of DNL enzymes concomitant with reduced ER stress. We further examined the effects of Mtr on hepatosteatosis in relation to glycemic control in T2D mice, which display a phenotype of hyperglycemia and hepatosteatosis associated with increased DNL [27, 28]. The results showed that Mtr treatment reduced hepatosteatosis and improved hyperglycemia. Collectively, these findings suggest that Mtr has the potential to be repurposed for the treatment of hepatosteatosis resulting from increased DNL and associated disorders in glucose metabolism.
Overconsumption of dietary fructose can lead to DNL and hepatosteatosis [12, 37], which in turn can result in glucose intolerance and contribute to hyperglycemia [19, 38]. Therefore, correction of hepatosteatosis is beneficial for improving glucose homeostasis in metabolic syndrome. For example, in obese patients with T2D, a reversal of hepatosteatosis can improve hepatic insulin action and glycemic control [5]. We have investigated drugs that have previously been used for the treatment of liver conditions, in order to determine whether they can be repurposed to treat hepatosteatosis [7]. One such candidate we have identified by this approach is Mtr because liver has been shown to be the major target site of Mtr [39, 40]. Indeed, our recent work has demonstrated that Mtr is able to attenuate the increased fasting blood glucose and improve glucose tolerance in insulin resistant mice induced by a HF diet [9]. The same study found that these anti-diabetic effects of Mtr appears to result from its effect in reducing hepatosteatosis without affecting HF diet induced lipid accumulation in muscle.
Mtr is clinically used for treatment of chronic liver conditions including hepatocellular carcinoma and viral hepatitis with minimal adverse effects [7–9]. Interestingly, both hepatocellular carcinoma and viral hepatitis are associated with an increase in DNL [10, 41, 42]. Indeed, our studies in 3T3L1 adipocytes have found that Mtr can reduce DNL and lipid accumulation within the cells [43]. Although our recent studies have demonstrated that Mtr is able to reduce hepatosteatosis and glucose intolerance in mice that have been fed a HF diet [9], the source of hepatosteatosis in this mouse model is from the exogenous FA due to the intake of dietary fat rather than endogenous FA from an increased DNL. Therefore, it is not clear yet whether Mtr is effective for metabolic disorders by that involve an increased hepatic DNL [12].
Several studies have demonstrated that DNL enzymes are over-expressed during the development of hepatosteatosis [24, 44]. HFru-fed mice are a well-defined animal model of DNL-induced hepatosteatosis and insulin resistance [12, 13], and DNL-induced hepatosteatosis can be observed as early as 1 day after HFru feeding [36]. Indeed, the present study showed that chronic HFru feeding resulted in hepatosteatosis (increased TG level) by promoting DNL (indicated by SREBP1c, ChREBP, acetyl-CoA carboxylase (ACC) and fatty acid synthase (FAS) and stearoyl-CoA desaturase-1 (SCD-1)) without affecting FA oxidation, as indicated by unchanged level of [14C]–palmitate. However, reduced hepatic FA oxidation and mitochondrial enzyme activity has been demonstrated to occur prior to the appearance of hepatosteatosis, it has been shown that DNL is a primary cause of the development of hepatosteatosis [12, 45]. As expected, treatment with Mtr significantly reduced steatosis in the liver (not muscle because high distribution of Mtr in the liver after and oral administration [40]) and the associated glucose intolerance in these mice. We then examined the key lipogenic enzymes and found that SREBP1c, ChREBP, SCD-1 and FAS in the liver were all reduced in HFru-fed mice treated with Mtr. These results suggest that Mtr is likely to reduce hepatosteatosis via inhibition of the DNL pathway.
As an increase in FA oxidation can also attenuate hepatosteatosis [46], we next investigated whether the reduction of hepatosteatosis caused by Mtr in HFru-fed mice results from an increase in liver FA oxidation. However, Mtr did not increase the oxidation of 14C-palmitate in the liver, indicating FA oxidation pathway was not activated in HFru-fed mice. This finding adds further support to our interpretation that Mtr reduces hepatosteatosis and glucose intolerance in HFru-fed mice by inhibiting DNL rather than by stimulating FA oxidation in the liver.
It has been shown that the ER stress pathway plays a critical role in HFru-induced DNL and hepatosteatosis [32, 36]. For example, an ob/ob mice hepatosteatosis is largely due to increased DNL as a result of hyperphagia in an ER stress-dependent manner [47]. The same study also showed that alleviation of hepatic ER stress by overexpression of GRP78 reduces hepatosteatosis and insulin resistance by inhibiting DNL. Similarly, in HFru-fed mice, inhibition of ER stress by TUDCA and Betulin suppress DNL and improve insulin signaling in the liver [13, 17, 20]. To investigate whether the inhibition of hepatic DNL by Mtr involves the ER stress pathway, we examined the major ER stress markers in response to HFru-induced DNL. Interestingly, the results showed that HFru-induced ER stress (indicated by eIF2α, CHOP and IRE1) were all inhibited by Mtr. These findings suggest that attenuation of ER stress may be a novel mode of action for the inhibitory effect of Mtr on DNL and the resultant hepatosteatosis.
In terms of the possible cellular target of Mtr, our previous work suggested that a downregulation of HSP72 may contribute to lipid accumulation in vivo [9], and in vitro [43]. We showed that Mtr is able to increase HSP72 expression and protect against lipid accumulation and glucose intolerance in the liver [9]. Consistent with this observation, the present study found that liver tissue from HFru-fed mice had significantly lower concentrations of HSP72 protein, and this reduction was prevented by Mtr treatment. HSPs have been implicated in the regulation of diverse metabolic disorders including hepatosteatosis (the major metabolic defect of non-alcoholic fatty liver disease) and insulin resistance (the major metabolic defect of T2D) [33, 48, 49]. It has been reported that an enhanced expression of HSP72 can inhibit ER stress to protect cell survival [50]. Collectively, our findings suggest that Mtr may inhibit the ER-DNL axis by up-regulating HSP72 to reduce hepatosteatosis and the associated glucose intolerance.
Hepatic DNL and hepatosteatosis also occur in transgenic diabetic mice such as db/db [24]. Therefore, in the present study we explored whether Mtr is able to reduce hepatosteatosis in a mouse model of T2D induced by HF-STZ [27, 28]. Our results showed that Mtr reduced epididymal fat and lowered hyperglycemia, indicating that Mtr may have the potential to control hyperglycemia in T2D. Because Mtr showed no effect on normal fasting blood glucose, glucose tolerance or liver TG content in chow-fed mice [9], the anti-diabetic effects of Mtr could be attributed to its effect in reducing hepatosteatosis.
In summary, we report here a potential novel application of the hepatoprotective drug Mtr for the treatment of hepatosteatosis and associated abnormal glucose homeostasis. This study is the first to evaluate the effect of Mtr on hepatosteatosis induced by the ER stress-DNL signaling pathway in HFru-fed mice. As suppression of ER stress can reduce hepatosteatosis by inhibiting DNL [16, 36], it is likely that Mtr may exert these beneficial effects by suppressing ER stress-induced increase in hepatic DNL. We speculate that the upregulation of the chaperon protein HSP72 may play a critical role in suppressing ER stress (as illustrated in Fig. 6) but this hypothesis requires validation by further studies using HSP72 knock-down animal model. Together with our recent findings in HF-fed mice [9], our results suggest that Mtr may be repurposed for the treatment of hepatosteatosis and associated disorders in glucose homeostasis including T2D.
Fig. 6.
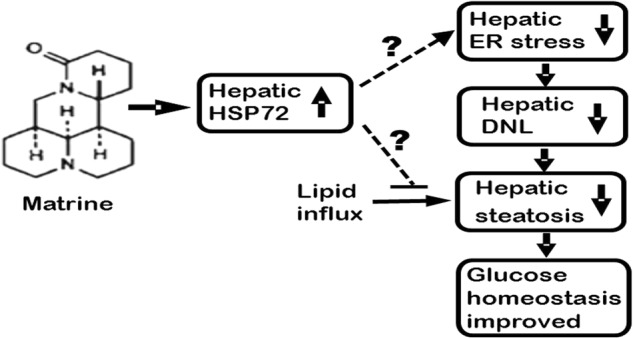
Proposed hypothesis for the therapeutic effects of Mtr for hepatosteatosis and associated disorders in glucose homeostasis
Acknowledgements
This work was supported by funding from a Program Grant (535921 allocation to JMY) and a Project Grant (535930 to JMY) of the National Health and Medical Research Council of Australia. AM is supported by Al-Baha University (Ministry of Higher Education, Saudi Arabia).
Author contribution
J.M.Y. conceived and designed the study. A.M., S.L., X.Y.Z. and J.M.Y. performed the experiments. J.X., W.T., R.V. and S.R. provided intellectual input and participated in data analysis. J.M.Y. and J.X. contributed reagents/materials/analysis tools. A.M. and J.M.Y. prepared the manuscript. R.V. and S.R. contributed to the revision of the MS.
References
- 1.Ballestri S, Zona S, Targher G, Romagnoli D, Baldelli E, Nascimbeni F, et al. Nonalcoholic fatty liver disease is associated with an almost twofold increased risk of incident type 2 diabetes and metabolic syndrome. Evidence from a systematic review and meta-analysis. J Gastroenterol Hepatol. 2016;31:936–44. doi: 10.1111/jgh.13264. [DOI] [PubMed] [Google Scholar]
- 2.Cusi K, Sanyal AJ, Zhang S, Hartman ML, Bue-Valleskey JM, Hoogwerf BJ, et al. Non-alcoholic fatty liver disease (NAFLD) prevalence and its metabolic associations in patients with type 1 diabetes and type 2 diabetes. Diabetes Obes Metab. 2017;19:1630–34. doi: 10.1111/dom.12973. [DOI] [PubMed] [Google Scholar]
- 3.Sun Z, Lazar MA. Dissociating fatty liver and diabetes. Trends Endocrinol Metab. 2013;24:4–12. doi: 10.1016/j.tem.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu M, Singh SB, Wang J, Chung CC, Salituro G, Karanam BV, et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc Natl Acad Sci U S A. 2011;108:5378–83. doi: 10.1073/pnas.1002588108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Petersen KF, Dufour S, Befroy D, Lehrke M, Hendler RE, Shulman GI. Reversal of nonalcoholic hepatic steatosis, hepatic insulin resistance, and hyperglycemia by moderate weight reduction in patients with type 2 diabetes. Diabetes. 2005;54:603–8. doi: 10.2337/diabetes.54.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu JY, Hu JH, Zhu QG, Li FQ, Wang J, Sun HJ. Effect of matrine on the expression of substance P receptor and inflammatory cytokines production in human skin keratinocytes and fibroblasts. Int Immunopharmacol. 2007;7:816–23. doi: 10.1016/j.intimp.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 7.Turner N, Zeng XY, Osborne B, Rogers S, Ye JM. Repurposing Drugs to Target the Diabetes Epidemic. Trends Pharmacol Sci. 2016;37:379–89. doi: 10.1016/j.tips.2016.01.007. [DOI] [PubMed] [Google Scholar]
- 8.Liu J, Zhu M, Shi R, Yang M. Radix Sophorae flavescentis for chronic hepatitis B: a systematic review of randomized trials. Am J Chin Med. 2003;31:337–54. doi: 10.1142/S0192415X03001107. [DOI] [PubMed] [Google Scholar]
- 9.Zeng XY, Wang H, Bai F, Zhou X, Li SP, Ren LP, et al. Identification of matrine as a promising novel drug for hepatic steatosis and glucose intolerance with HSP72 as an upstream target. Br J Pharmacol. 2015;172:4303–18. doi: 10.1111/bph.13209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Syed GH, Amako Y, Siddiqui A. Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010;21:33–40. doi: 10.1016/j.tem.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Q, Pene V, Krishnamurthy S, Cha H, Liang TJ. Hepatitis C virus infection activates an innate pathway involving IKK-alpha in lipogenesis and viral assembly. Nat Med. 2013;19:722–9. doi: 10.1038/nm.3190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ren LP, Chan SM, Zeng XY, Laybutt DR, Iseli TJ, Sun RQ, et al. Differing endoplasmic reticulum stress response to excess lipogenesis versus lipid oversupply in relation to hepatic steatosis and insulin resistance. PLoS One. 2012;7:e30816. doi: 10.1371/journal.pone.0030816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan SM, Sun RQ, Zeng XY, Choong ZH, Wang H, Watt MJ, et al. Activation of PPARalpha ameliorates hepatic insulin resistance and steatosis in high fructose-fed mice despite increased endoplasmic reticulum stress. Diabetes. 2013;62:2095–105. doi: 10.2337/db12-1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thorburn AW, Storlien LH, Jenkins AB, Khouri S, Kraegen EW. Fructose-induced in vivo insulin resistance and elevated plasma triglyceride levels in rats. Am J Clin Nutr. 1989;49:1155–63. doi: 10.1093/ajcn/49.6.1155. [DOI] [PubMed] [Google Scholar]
- 15.Iizuka K, Bruick RK, Liang G, Horton JD, Uyeda K. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101:7281–6. doi: 10.1073/pnas.0401516101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources offatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115:1343–51. doi: 10.1172/JCI23621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sun RQ, Wang H, Zeng XY, Chan SM, Li SP, Jo E, et al. IRE1 impairs insulin signaling transduction offructose-fed mice via JNK independent ofexcess lipid. Biochim Biophys Acta. 2015;1852:156–65. doi: 10.1016/j.bbadis.2014.11.017. [DOI] [PubMed] [Google Scholar]
- 18.Turner N, Kowalski GM, Leslie SJ, Risis S, Yang C, Lee-Young RS, et al. Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia. 2013;56:1638–48. doi: 10.1007/s00125-013-2913-1. [DOI] [PubMed] [Google Scholar]
- 19.Stanhope KL, Schwarz JM, Keim NL, Griffen SC, Bremer AA, Graham JL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest. 2009;119:1322–34. doi: 10.1172/JCI37385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee AH, Scapa EF, Cohen DE, Glimcher LH. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science. 2008;320:1492–6. doi: 10.1126/science.1158042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lecoultre V, Egli L, Carrel G, Theytaz F, Kreis R, Schneiter P, et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity. 2013;21:782–5. doi: 10.1002/oby.20377. [DOI] [PubMed] [Google Scholar]
- 22.Schwarz JM, Noworolski SM, Wen MJ, Dyachenko A, Prior JL, Weinberg ME, et al. Effect of a High-Fructose Weight-Maintaining Diet on Lipogenesis and Liver Fat. J Clin Endocrinol Metab. 2015;100:2434–42. doi: 10.1210/jc.2014-3678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ameer F, Scandiuzzi L, Hasnain S, Kalbacher H, Zaidi N. De novo lipogenesis in health and disease. Metabolism. 2014;63:895–902. doi: 10.1016/j.metabol.2014.04.003. [DOI] [PubMed] [Google Scholar]
- 24.Shimomura I, Bashmakov Y, Horton JD. Increased levels ofnuclear SREBP-1c associated with fatty livers in two mouse models of diabetes mellitus. J Biol Chem. 1999;274:30028–32. doi: 10.1074/jbc.274.42.30028. [DOI] [PubMed] [Google Scholar]
- 25.Dentin R, Benhamed F, Hainault I, Fauveau V, Foufelle F, Dyck JR, et al. Liver-specific inhibition of ChREBP improves hepatic steatosis and insulin resistance in ob/ob mice. Diabetes. 2006;55:2159–70. doi: 10.2337/db06-0200. [DOI] [PubMed] [Google Scholar]
- 26.Zheng Z, Zhang C, Zhang K. Role ofunfolded protein response in lipogenesis. World J Hepatol. 2010;2:203–7. doi: 10.4254/wjh.v2.i6.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mu J, Woods J, Zhou YP, Roy RS, Li Z, Zycband E, et al. Chronic inhibition of dipeptidyl peptidase-4 with a sitagliptin analog preserves pancreatic beta-cell mass and function in a rodent model of type 2 diabetes. Diabetes. 2006;55:1695–704. doi: 10.2337/db05-1602. [DOI] [PubMed] [Google Scholar]
- 28.Zeng XY, Wang YP, Cantley J, Iseli TJ, Molero JC, Hegarty BD, et al. Oleanolic acid reduces hyperglycemia beyond treatment period with Akt/FoxO1-induced suppression of hepatic gluconeogenesis in type-2 diabetic mice. PLoS One. 2012;7:e42115. doi: 10.1371/journal.pone.0042115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ye JM, Tid-Ang J, Turner N, Zeng XY, Li HY, Cooney GJ, et al. PPARdelta agonists have opposing effects on insulin resistance in high fat-fed rats and mice due to different metabolic responses in muscle. Br J Pharmacol. 2011;163:556–66. doi: 10.1111/j.1476-5381.2011.01240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ye JM, Iglesias MA, Watson DG, Ellis B, Wood L, Jensen PB, et al. PPARalpha /gamma ragaglitazar eliminates fatty liver and enhances insulin action in fat-fed rats in the absence of hepatomegaly. Am J Physiol Endocrinol Metab. 2003;284:E531–40. doi: 10.1152/ajpendo.00299.2002. [DOI] [PubMed] [Google Scholar]
- 31.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–17. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306:457–61. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 33.Chung J, Nguyen AK, Henstridge DC, Holmes AG, Chan MH, Mesa JL, et al. HSP72 protects against obesity-induced insulin resistance. Proc Natl Acad Sci U S A. 2008;105:1739–44. doi: 10.1073/pnas.0705799105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kurucz I, Morva A, Vaag A, Eriksson KF, Huang X, Groop L, et al. Decreased expression of heat shock protein 72 in skeletal muscle of patients with type 2 diabetes correlates with insulin resistance. Diabetes. 2002;51:1102–9. doi: 10.2337/diabetes.51.4.1102. [DOI] [PubMed] [Google Scholar]
- 35.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–77. doi: 10.1016/S0140-6736(10)60408-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang H, Sun RQ, Zeng XY, Zhou X, Li S, Jo E, et al. Restoration of autophagy alleviates hepatic ER stress and impaired insulin signalling transduc-tion in high fructose-fed male mice. Endocrinology. 2015;156:169–81. doi: 10.1210/en.2014-1454. [DOI] [PubMed] [Google Scholar]
- 37.Herman MA, Samuel VT. The sweet path to metabolic demise: fructose and lipid synthesis. Trends Endocrinol Metab. 2016;27:719–30. doi: 10.1016/j.tem.2016.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perry RJ, Samuel VT, Petersen KF, Shulman GI. The role of hepatic lipids in hepatic insulin resistance and type 2 diabetes. Nature. 2014;510:84–91. doi: 10.1038/nature13478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang HF, Shi LJ, Song GY, Cai ZG, Wang C, An RJ. Protective effects of matrine against progression of high-fructose diet-induced steatohepatitis by enhancing antioxidant and anti-inflammatory defences involving Nrf2 translo-cation. Food Chem Toxicol. 2013;55:70–7. doi: 10.1016/j.fct.2012.12.043. [DOI] [PubMed] [Google Scholar]
- 40.Gao G, Law FC. Physiologically based pharmacokinetics of matrine in the rat after oral administration of pure chemical and ACAPHA. Drug Metab Dispos. 2009;37:884–91. doi: 10.1124/dmd.108.023788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Milner KL, van der Poorten D, Trenell M, Jenkins AB, Xu A, Smythe G, et al. Chronic hepatitis C is associated with peripheral rather than hepatic insulin resistance. Gastroenterology. 2010;138:932–41. doi: 10.1053/j.gastro.2009.11.050. [DOI] [PubMed] [Google Scholar]
- 42.McPherson S, Jonsson JR, Barrie HD, O'Rourke P, Clouston AD, Powell EE. Investigation of the role of SREBP-1c in the pathogenesis of HCV-related steatosis. J Hepatol. 2008;49:1046–54. doi: 10.1016/j.jhep.2008.06.022. [DOI] [PubMed] [Google Scholar]
- 43.Zeng XY, Zhou X, Xu J, Chan SM, Xue CL, Molero JC, et al. Screening for the efficacy on lipid accumulation in 3T3-L1 cells is an effective tool for the identification of new anti-diabetic compounds. Biochem Pharmacol. 2012;84:830–7. doi: 10.1016/j.bcp.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 44.Postic C, Girard J. Contribution of de novo fatty acid synthesis to hepatic steatosis and insulin resistance: lessons from genetically engineered mice. J Clin Invest. 2008;118:829–38. doi: 10.1172/JCI34275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rector RS, Thyfault JP, Uptergrove GM, Morris EM, Naples SP, Borengasser SJ, et al. Mitochondrial dysfunction precedes insulin resistance and hepatic steatosis and contributes to the natural history of non-alcoholic fatty liver disease in an obese rodent model. J Hepatol. 2010;52:727–36. doi: 10.1016/j.jhep.2009.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reddy JK. Nonalcoholic steatosis and steatohepatitis. III. Peroxisomal beta-oxidation, PPAR alpha, and steatohepatitis. Am J Physiol Gastrointest Liver Physiol. 2001;281:G1333–9. doi: 10.1152/ajpgi.2001.281.6.G1333. [DOI] [PubMed] [Google Scholar]
- 47.Kammoun HL, Chabanon H, Hainault I, Luquet S, Magnan C, Koike T, et al. GRP78 expression inhibits insulin and ER stress-induced SREBP-1c activation and reduces hepatic steatosis in mice. J Clin Invest. 2009;119:1201–15. doi: 10.1172/JCI37007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gupte AA, Bomhoff GL, Swerdlow RH, Geiger PC. Heat treatment improves glucose tolerance and prevents skeletal muscle insulin resistance in rats fed a high-fat diet. Diabetes. 2009;58:567–78. doi: 10.2337/db08-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Di Naso FC, Porto RR, Fillmann HS, Maggioni L, Padoin AV, Ramos RJ, et al. Obesity depresses the anti-inflammatory HSP70 pathway, contributing to NAFLD progression. Obesity. 2015;23:120–9. doi: 10.1002/oby.20919. [DOI] [PubMed] [Google Scholar]
- 50.Gupta S, Deepti A, Deegan S, Lisbona F, Hetz C, Samali A. HSP72 protects cells from ER stress-induced apoptosis via enhancement of IRE1a-XBP1 signaling through a physical Interaction. PLoS Biol. 2010;8:e1000410. doi: 10.1371/journal.pbio.1000410. [DOI] [PMC free article] [PubMed] [Google Scholar]