

Abstract
SND p102 was first described as a transcriptional co-activator, and subsequently determined to be a co-regulator of Pim-1, STAT6 and STAT5. We previously reported that SND p102 expression was increased in high glucose-treated mesangial cells (MCs) and plays a role in the extracellular matrix (ECM) accumulation of MCs by regulating the activation of RAS. In this study, we further examined the roles of SND p102 in diabetic nephropathy (DN)-induced glomerulosclerosis. Rats were injected with STZ (50 mg/kg, ip) to induce diabetes. MCs or isolated glomeruli were cultured in normal glucose (NG, 5.5 mmol/L)- or high glucose (HG, 25 mmol/L)-containing DMEM. We found that SND p102 expression was significantly increased in the diabetic kidneys, as well as in HG-treated isolated glomeruli and MCs. In addition, HG treatment induced significant fibrotic changes in MCs evidenced by enhanced protein expression of TGF-β, fbronectin and collagen IV, and significantly increased the proliferation of MCs. We further revealed that overexpression of SND p102 significantly increased the protein expression of angiotensin II (Ang II) type 1 receptor (AT1R) in MCs by increasing its mRNA levels via directly targeting the AT1R 3′-UTR, which resulted in activation of the ERK/Smad3 signaling and subsequently promoted the up-regulation of fbronectin, collagen IV, and TGF-β in MCs, as well as the cell proliferation. These results demonstrate that SND p102 is a key regulator of AT1R-mediating ECM synthesis and cell proliferation in MCs. Thus, small molecule inhibitors of SND p102 may be a novel therapeutic strategy for DN.
Keywords: diabetic nephropathy, SND p102, isolated glomeruli, mesangial cells, extracellular matrix accumulation, cell proliferation, Ang II type 1 receptor, ERK, Smad3
Introduction
Diabetic nephropathy (DN) is one of the most severe and common chronic complications of diabetes mellitus (DM)1. The principal pathological changes associated with DN include glomerular extracellular matrix (ECM) accumulation, glomerular hypertrophy, glomerular basement membrane (GBM) thickening and glomerulosclerosis, which may devolve into chronic renal insufficiency and become the main cause of end-stage renal disease (ESRD)1,2. Multiple mechanisms, including dyslipidemia, hyperglycemia, hemodynamic abnormalities, oxidative stress and autophagy, contribute to DN initiation and progression, eventually leading to ESRD1,3.
These morphological changes in the renal glomeruli are ultimately derived from excess or inappropriate activation of the renin-angiotensin system (RAS), a well-known endocrine system involved in the regulation of blood pressure and fluid balance4. Growing evidence has revealed that there is local RAS in the kidney, which can affect cellular activity and tissue injury5, and the classical renin/angiotensin I-converting enzyme (ACE)/angiotensin II (Ang II)/Ang II type 1 receptor (AT1R) axis of RAS is a critical pathway leading to ESRD6,7. In addition, AT1R plays an important role in regulating renal fibrosis and eventual renal failure by modulating functions of renal cells and surrounding cells. Every known component of RAS is contained within mesangial cells (MCs). We have previously found that Ang II/AT1R can induce ECM accumulation and cell proliferation in rat renal MCs8. Ang II also induces transforming growth factor-β (TGF-β) up-regulation, which plays a central role in driving renal fibrosis and DN through Smad3 signaling9,10,11,12. It has been demonstrated that ACE inhibitors (ACEI) and AT1R blockers (ARB) have significant inhibitory effects on the progression of DN in clinical patients, renal fibrosis of experimental murine models and dysfunction of cultured MCs13,14,15,16.
SND p102 is a rat homologue of human staphylococcal nuclease and tudor domain-containing 1 (SND1), first described as an Epstein-Barr virus nuclear antigen 2 (EBNA 2) co-activator17,18,19. SND1, also known as Tudor staphylococcal nuclease (Tudor-SN) or p100 protein, is a multifunctional protein that regulates various intracellular processes, such as transcriptional activation, RNA interference, mRNA splicing, editing, and stability, protein ubiquitination and proteasomal degradation20,21,22. We have previously demonstrated that SND p102 is increased in MCs under hyperglycemic conditions and is potentially involved in ECM accumulation in MCs. However, the mechanism by which SND p102 participates in high glucose (HG)-induced MC activation remains unclear.
It has been previously demonstrated that Ang II can directly activate Smad3 by a TGF-β-independent AT1R/ERK signaling pathway23. In the present study, we investigated whether SND p102 overexpression or knockdown could directly affect ECM accumulation and proliferation in MCs and whether the downstream AT1R/ERK/Smad3 signaling pathway was involved in SND p102-induced cell dysfunction.
Materials and methods
Cell culture
The rat mesangial cell line HBZY-1 was purchased from the Center of Type Culture Collection (Wuhan, China) and cultured in normal glucose (NG) DMEM medium (5.5 mmol/L D-glucose) (Gibco, USA) containing 10% FBS (Gibco, USA) in an atmosphere of 5% CO2 at 37 °C. 293T cells were kindly provided by Prof Si-Feng CHEN and were cultured in NG DMEM medium containing 10% FBS. HG culture medium was generated by supplementing normal DMEM medium with additional D-glucose at a final concentration of 25 mmol/L. An osmotic control medium was generated by supplementing normal medium with 19.5 mmol/L mannitol.
Isolation and culture of rat renal glomeruli
Sprague Dawley rats (180-200 g) were obtained from Shanghai SLAC Laboratory Animal Co Ltd (Shanghai, China). All experimental procedures followed the Criteria of the Medical Laboratory Animal Administrative Committee of Shanghai and the Guide for the Care and Use of Laboratory Animals of Fudan University and were approved by the Ethics Committee for Experimental Research, Shanghai Medical College, Fudan University. Glomeruli were isolated from euthanized SD rats as described previously24. Briefly, glomeruli were separated by gentle grinding of the renal cortex fragments on a 200-mm stainless steel screen, followed by sequentially passing the filtrates through sieves with mesh sizes of 150, 100, and 75 mm. The glomeruli were then collected and cultured in NG DMEM medium with 1% FBS (Gibco, USA) and cultured under an atmosphere of 5% CO2 at 37°C. High-glucose (HG) culture medium was generated by supplementing normal DMEM medium with additional D-glucose to a final concentration of 25 mmol/L.
Animal model
Male Sprague-Dawley rats, weighing 180-210 g, were provided by the Shanghai SLAC Laboratory Animal Center. All experimental procedures followed the Criteria of the Medical Laboratory Animal Administrative Committee of Shanghai and the Guide for the Care and Use of Laboratory Animals of Fudan University and were approved by the Ethics Committee for Experimental Research, Shanghai Medical College, Fudan University. Rats were rendered diabetic by a single intraperitoneal injection of STZ (50 mg/kg) dissolved in 0.1 mol/L sodium citrate buffer (pH 4.0). Only animals with plasma glucose concentrations >16.7 mmol/L one week after STZ injection were included in the study, as described previously25. Twelve weeks after STZ injection, rats were euthanized, kidney tissues were collected, and the renal cortex was dissected and used for Western blot analyses.
Plasmids, siRNA and cell transfection
A plasmid expressing full-length SND p102, called pcDNA3.0-SND p102, and a rat AT1R 3′-untranslated region (3′-UTR)-luciferase reporter construct were obtained from TaiHe Biotechnology Co, Ltd. Rat SND p102 siRNA and AT1R siRNA were purchased from the Ribobio Company (Guangzhou, Guangdong, China). The sequences used were as follows: SND p102 (F5′--3′, R5′--3′); AT1R (F5′--3′, R5′--3′). A nonsilencing siRNA oligonucleotide (Ribobio, Guangzhou, China) was used as a negative control. Cells were transfected with either of the recombinant plasmids (1 μg)/siRNAs (50 nmol/L) or control plasmids (1 μg)/siRNA (50 nmol/L) using Lipofectamine 2000 Reagent (Invitrogen) according to manufacturer's instructions. Forty-eight hours after transfection, cells were harvested for immunoblotting analysis, real-time PCR and luciferase assays.
Real-time PCR analysis
Total RNA was extracted from kidney tissues using TRIzol® (Invitrogen), and AT1R mRNA was measured by real-time PCR, as described previously23. The housekeeping gene β-actin was used as an endogenous control to normalize the levels of AT1R.
Western blotting and antibodies
Cell lysates and renal cortex tissue homogenates were prepared as previously described26. The antibodies used were as follows: anti-SND p102, anti-collagen IV, anti-AT1R, anti-phospho-Smad3 (p-Smad3), anti-phospho-ERK (p-ERK), anti-Smad3, anti-ERK and anti-fibronectin (Abcam, Cambridge, MA); anti-TGF-β1 (Bioworld Technology); and anti-β-actin (Sigma, St. Louis, MO, USA).
Cell proliferation assay
MCs in the logarithmic growth phase were seeded in 96-well plates at a cell density of 2000 cells/well. After HG treatment or transfection with siRNAs or plasmids for 48 or 72 h, cell proliferation was measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (Roche, Mannheim, Germany) according to the manufacturer's instructions, as described previously25.
Luciferase assay of the 3′-UTR of AT1R
293T cells were seeded at 5×104 cells in 500 μL of medium per well in a 24-well plate (37 °C, 5% CO2). After 18 to 24 h of incubation, 200 ng of firefly luciferase reporter plasmid (pmirGLO Dual-Luciferase miRNA Target Expression Vector) containing the 3′-UTR of AT1R or control luciferase reporter plasmid along with 10 ng of pRL-TK reporter plasmid containing the renilla luciferase gene (Promega) were cotransfected with 200 ng of pcDNA3.0-SND p102 plasmid or pcDNA3.0 plasmid using Lipofectamine 2000 (Invitrogen). Luciferase activity was measured using the Dual-Luciferase reporter assay system (Promega) according to the manufacturer's instructions. Firefly luciferase activity was normalized to renilla luciferase activity.
Statistical analysis
Numerical data are presented as the mean±SEM from at least three independent experiments and compared by Student's t test or one-way ANOVA using SPSS 17.0 software. P<0.05 was considered statistically significant when comparing mean values.
Results
SND p102 expression is up-regulated in diabetic kidneys and HG-treated glomeruli and MCs
STZ injection-induced diabetic mice developed renal fibrosis, as indicated by increases in TGF-β, fibronectin and collagen IV protein expression compared with normal mice (Figure 1A). To determine the relevance of SND p102 expression in DN, we measured SND p102 expression in diabetic kidneys and glomeruli cultured in HG medium. SND p102 was weakly expressed in normal kidneys and glomeruli but highly expressed in diabetic kidneys and glomeruli cultured in HG medium, as demonstrated by Western blot analysis (Figure 1B).
Figure 1.
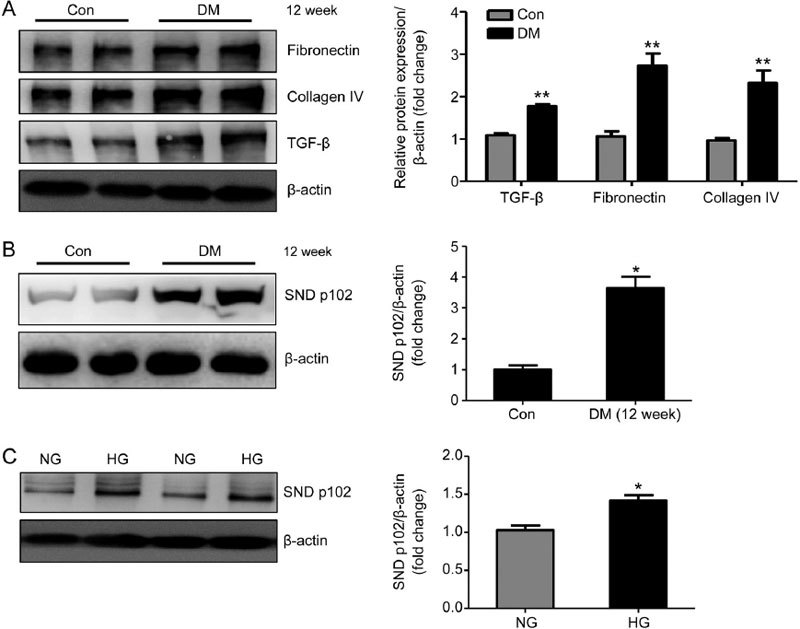
Enhanced SND p102 expression in diabetic kidneys and high glucose (HG)-treated glomeruli. Rats were injected with STZ to induce type I diabetes. Twelve weeks after injection, renal fibronectin, collagen IV, transforming growth factor-β (TGF-β) (A), and SND p102 protein levels (B) were measured by Western blot analysis. (C) Glomeruli were treated with normal glucose (NG) (5.5 mmol/L D-glucose) or high-glucose (HG) (25 mmol/L D-glucose) medium for 48 h. SND p102 protein levels were measured by Western blot analysis. Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with control or NG samples.
In cultured MCs, we found that HG significantly induced fibrotic changes characterized by enhanced TGF-β, fibronectin and collagen IV protein expression, as shown by Western blot analysis (Figure 2A). BrdU incorporation assays also showed increased proliferation of MCs under HG conditions (Figure 2B). These pathologic changes were similarly accompanied by elevated protein levels of SND p102 (Figure 2C).
Figure 2.
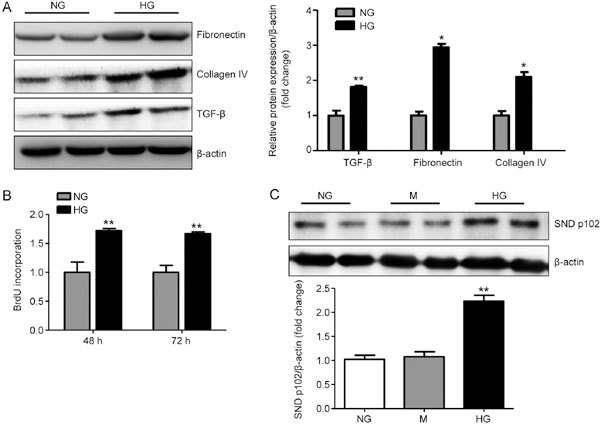
Enhanced SND p102 expression in HG-treated mesangial cells (MCs). MCs were treated with NG, HG or NG with 19.5 mmol/L mannitol (M) for 48 h or 72 h. Fibronectin, collagen IV, TGF-β (A), and SND p102 protein levels (C) were measured by Western blot analysis. Cell proliferation was measured by 5-bromo-2′-deoxyuridine (BrdU) incorporation assay (B). Data represent mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with NG samples.
SND p102 regulates ECM production and proliferation in cultured MCs
To evaluate potential regulation of ECM production and proliferation of MCs by differential SND p102 expression, the recombinant plasmid pcDNA3.0-SND p102, containing full-length SND p102 cDNA, and SND p102 siRNA were used to overexpress or silence SND p102. A significant increase in SND p102 protein levels was observed in MCs transfected with pcDNA3.0-SND p102 (Figure 3A), accompanied by enhanced TGF-β, fibronectin and collagen IV protein expression and cell proliferation (Figure 3B, C). In contrast, SND p102 siRNA treatment significantly decreased SND p102 protein levels (Figure 4A), which resulted in decreased proliferation and TGF-β, fibronectin, collagen IV protein expression (Figure 4B and C), indicating that SND p102 is involved in the regulation of ECM production and the proliferation of MCs.
Figure 3.
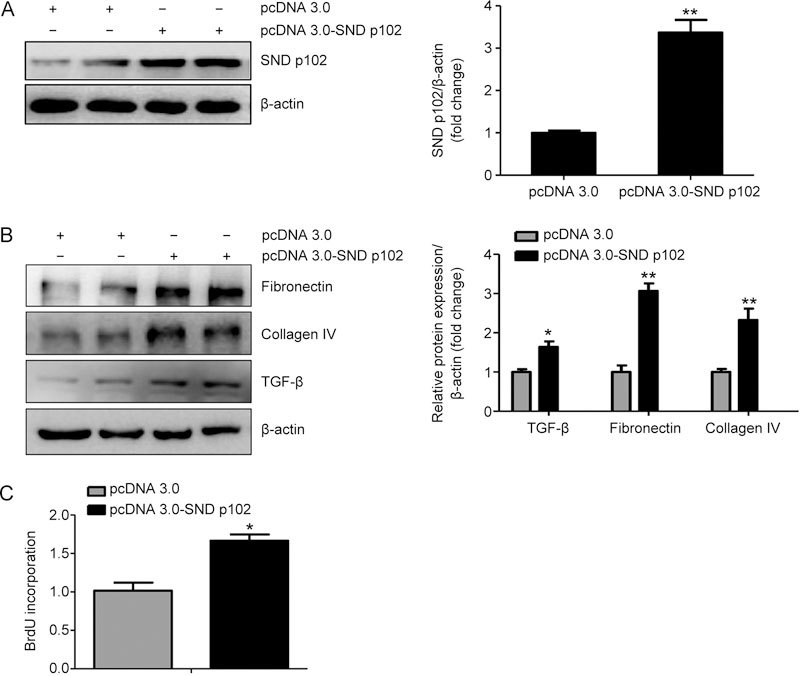
SND p102 overexpression promotes ECM production and cell proliferation in cultured MCs. MCs were transfected with pcDNA3.0-SND p102 plasmid (1 μg) or pcDNA3.0 plasmid (1 μg) for 48 h. SND p102 (A), fibronectin (B), collagen IV (B), and TGF-β (B) protein levels were measured by Western blot analysis. Cell proliferation was measured by BrdU incorporation assay (C). Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with pcDNA3.0 plasmid-transfected samples.
Figure 4.
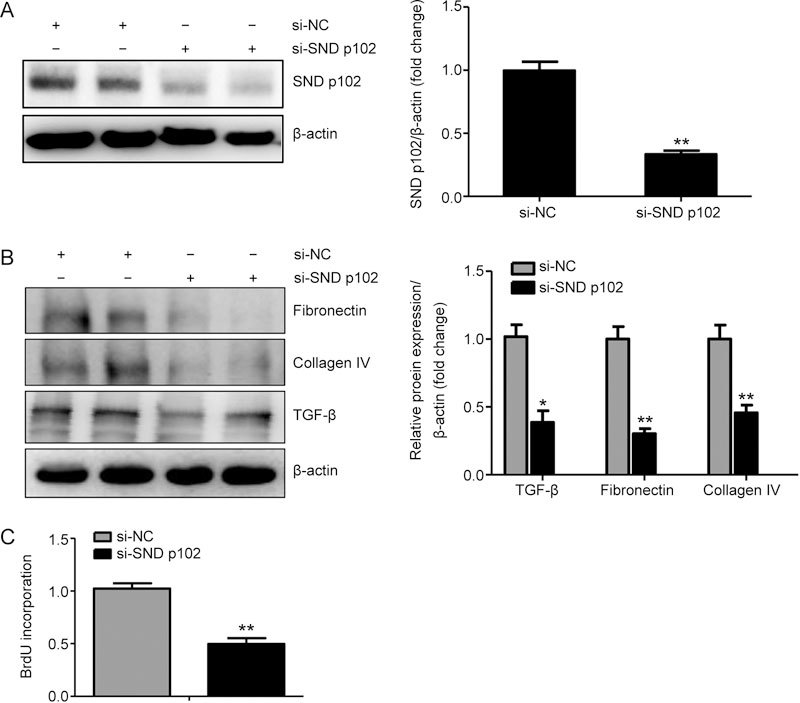
SND p102 knockdown attenuates ECM production and cell proliferation in cultured MCs. Cells were transfected with SND p102 siRNA (50 nmol/L) or control siRNA (50 nmol/L) for 48 h. SND p102 (A), fibronectin (B), collagen IV (B), and TGF-β (B) protein levels were measured by Western blot analysis. Cell proliferation was measured by BrdU incorporation assay (C). Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with control siRNA-transfected samples.
SND p102 regulates AT1R expression in cultured MCs
The excessive activation of intrarenal RAS plays an important role in the pathogenesis of diabetic nephropathy. Western blot analysis showed that compared with pcDNA3.0, pcDNA3.0-SND p102 transfection significantly up-regulated AT1R protein levels in MCs (Figure 5A). In addition, treatment with SND p102 siRNA, but not control siRNA, significantly decreased AT1R protein levels (Figure 5B).
Figure 5.
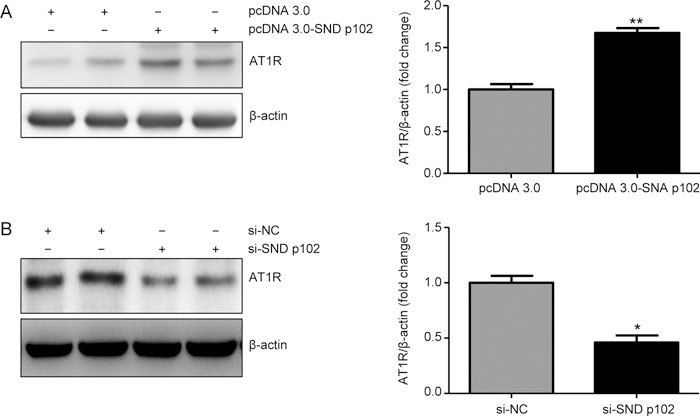
SND p102 regulates Ang II type 1 receptor (AT1R) protein levels in cultured MCs. (A) Cells were transfected with pcDNA3.0-SND p102 plasmid (1 μg) or pcDNA3.0 plasmid (1 μg) for 48 h. AT1R protein levels were measured by Western blot analysis. (B) Cells were transfected with SND p102 siRNA (50 nmol/L) or control siRNA (50 nmol/L) for 48 h. AT1R was measured by Western blot analysis. Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with pcDNA3.0 plasmid- or control siRNA-transfected samples.
Knockdown of AT1R expression blocks SND p102-induced ECM production and cell proliferation in cultured MCs
AT1R siRNA treatment remarkably down-regulated SND p102 overexpression-induced AT1R up-regulation (Figure 6A). This led to a reduction in cell proliferation and protein expression of fibrotic markers resulting from SND p102 overexpression (Figure 6A and B).
Figure 6.
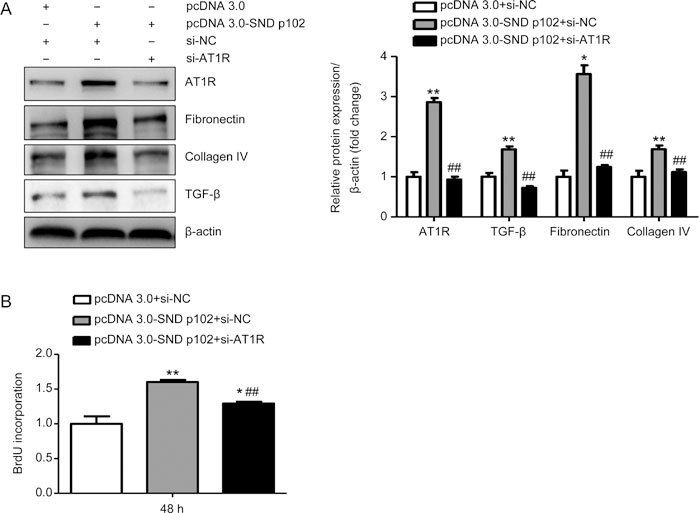
AT1R knockdown blocks SND p102-induced ECM production and cell proliferation in cultured MCs. A 50 nmol/L aliquot of AT1R siRNA or control siRNA and 1 μg of pcDNA3.0-SND p102 plasmid or pcDNA3.0 plasmid were cotransfected into cells for 48 h. AT1R, fibronectin, collagen IV, and TGF-β protein levels were measured by Western blot analysis (A). Cell proliferation was measured by BrdU incorporation assay (B). Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with pcDNA3.0 plasmid- and control siRNA-transfected samples. ## P<0.01 compared with pcDNA3.0-SND p102 plasmid- and control siRNA-transfected samples.
Knockdown of AT1R expression blocks SND p102-induced AT1R/ERK/Smad3 activation in cultured MCs
As previous studies have demonstrated that Ang II can induce rapid activation of Smad3 in TGF-β knockout tubular epithelial cells via a TGF-β-independent, AT1R/ERK-mediated protein kinase cross-signaling pathway24, we evaluated whether the AT1R/ERK/Smad3 signaling pathway was involved in SND p102-induced MC dysfunction. Our findings suggested that SND p102 overexpression activated AT1R/ERK/Smad3 signaling, as indicated by increased p-ERK and p-Smad3 (Figure 7A). Furthermore, silencing of AT1R significantly attenuated these changes in cultured MCs (Figure 7B).
Figure 7.
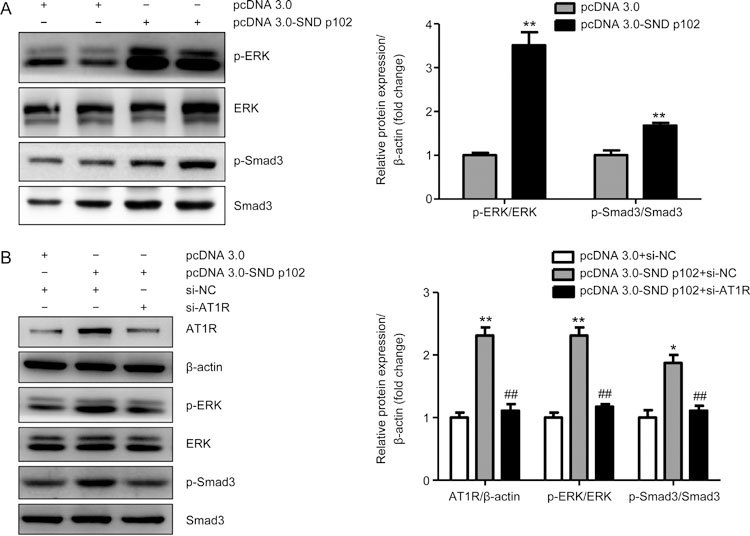
AT1R Knockdown blocks SND p102-induced AT1R/ERK/Smad3 signaling in cultured MCs. (A) Cells were transfected with pcDNA3.0-SND p102 plasmid (1 μg) or pcDNA3.0 plasmid (1 μg) for 48 h. p-ERK and p-Smad3 protein levels were measured by Western blot analysis. (B) A 50 nmol/L aliquot of AT1R siRNA or control siRNA and 1 μg of pcDNA3.0-SND p102 plasmid or pcDNA3.0 plasmid were cotransfected into cells for 48 h. AT1R, p-ERK, and p-Smad3 levels were measured by Western blot analysis. Data represent the mean±SEM of at least 3 experiments. * P<0.05, ** P<0.01 compared with pcDNA3.0 plasmid- or pcDNA3.0 plasmid and control siRNA-transfected samples. ## P<0.01 compared with pcDNA3.0-SND p102 plasmid- and control siRNA-transfected samples.
SND p102 enhances AT1R expression by increasing its mRNA levels via the AT1R 3′-UTR in cultured 293T cells
As demonstrated by real-time PCR, AT1R was highly expressed at the mRNA level in 293T cells transfected with pcDNA3.0-SND p102 plasmid compared with cells transfected with pcDNA3.0 plasmid (Figure 8A). To explore the possibility of a direct targeting mechanism of SND p102 to the AT1R 3′-UTR, we performed a luciferase assay of the 3′-UTR of AT1R. A luciferase reporter plasmid containing the 3′-UTR of AT1R or a control reporter plasmid was cotransfected separately with pcDNA3.0-SND p102 plasmid or pcDNA3.0 plasmid. Luciferase activity was measured at 48 h post-transfection. Compared with the control samples, the luciferase activity of Luc (AT1R 3′-UTR) increased after SND p102 overexpression (Figure 8B), indicating that SND p102 up-regulates AT1R expression by increasing its mRNA levels via direct interactions with the AT1R 3′-UTR.
Figure 8.
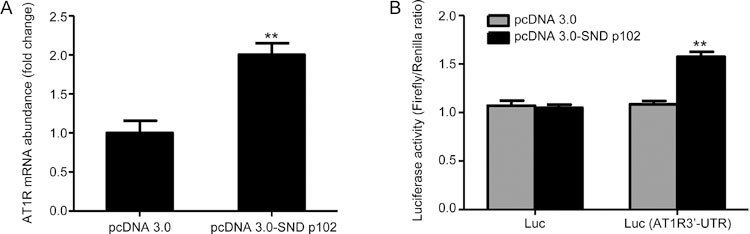
SND p102 up-regulates AT1R mRNA levels by interacting with the AT1R 3′-UTR in cultured 293T cells. Cells were transfected with pcDNA3.0-SND p102 plasmid (1 μg) or pcDNA3.0 plasmid (1 μg) for 48 h. AT1R mRNA was measured by real-time PCR analysis (A). A 200 ng aliquot of firefly luciferase reporter plasmid containing the 3′-UTR of AT1R or control reporter plasmid along with 10 ng of pRL-TK reporter plasmid containing the renilla luciferase gene were cotransfected with 200 ng of pcDNA3.0-SND p102 plasmid or pcDNA3.0 plasmid for 48 h. Firefly luciferase activity was measured and normalized to renilla luciferase activity. Data represent the mean±SEM of at least 3 experiments. ** P<0.01 compared with pcDNA3.0 plasmid-, pcDNA3.0 plasmid and control reporter plasmid- or pcDNA3.0 plasmid and AT1R 3′-UTR reporter plasmid-transfected samples.
Discussion
Glomerulopathy, characterized by thickening of the GBM and mesangial cell expansion, is the most important renal structural change associated with DN. Morphological lesions develop concomitantly in the arterioles, tubules and interstitium27, and the pathogenic processes of glomerulopathy eventually lead to glomerulosclerosis and fibrotic scar formation. Renal MCs are important interstitial cells in glomeruli that play a crucial role in the maintenance of the normal morphology and function of glomeruli28,29. Abnormalities in renal MCs will lead to functional and morphological changes in renal glomeruli.
Although the role of the ACE/Ang II/AT1R axis is well established in diabetic nephropathy, regulating functions of renal tubular epithelial cells, endothelial cells, podocytes and mesangial cells, the molecular mechanism underlying RAS dysfunction has not been clearly demonstrated30,31. In the present study, we describe a novel molecular signaling pathway between SND p102 and AT1R with subsequent activation of ERK/Smad3 signaling that contributes to ECM accumulation and proliferation of rat MCs.
SND1 (p100, SND p102) was first identified as a co-activator of Epstein-Barr virus nuclear antigen 2 (EBNA-2)17,19 and subsequently determined to be a co-regulator of Pim-132, signal transducer and activator of transcription 6 (STAT6)33, and STAT534. We have previously reported that SND p102 expression is increased in HG-treated MCs and plays a role in the ECM accumulation of MCs by regulating the activation of RAS23. However, the underlying mechanisms remain to be further clarified. In this study, we also confirmed that SND p102 expression was remarkably increased in diabetic kidneys and glomeruli and MCs under hyperglycemic conditions, which was accompanied by enhanced fibrotic changes.
A major, novel finding in this study was the facilitating effect of SND p102 on AT1R expression and subsequent activation of the ERK/Smad3 signaling pathway. Our results demonstrated that AT1R protein levels were increased in SND p102-overexpressing cells, without any addition of the natural AT1R ligand Ang II, and were decreased in SND p102-silenced cells. In addition, AT1R mRNA levels were also increased by SND p102. We also used luciferase reporter assays to further illustrate that the AT1R 3′-UTR is required for the regulation of AT1R expression by SND p102. Although it is challenging to determine whether the increased AT1R mRNA levels were due to enhanced mRNA stability or translation by luciferase assay, we have shown that SND p102 can mediate the progression of diabetic nephropathy by targeting the AT1R 3′-UTR. Furthermore, a previous study has revealed that p100 (SDN p102) can bind to the 3′-UTR of AT1R mRNA, resulting in increased AT1R expression by enhancing the stability of AT1R mRNA and translation by RNA-binding protein immunoprecipitation (RIP) assay, mRNA half-life analysis and in vitro translation, which further supports our present findings35. RNA electrophoretic mobility-shift assay (REMSA) was also used in that study to show that the AT1R 3′-UTR residues 118-120 are required for p100 binding. Thus, we propose that p100 can regulate AT1R mRNA levels by affecting the mRNA stability through a specific binding site in the AT1R 3′-UTR35.
We also found that knockdown of AT1R could block SND p102-induced activation of the downstream ERK/Smad3 signaling pathway, as well as ECM production and cell proliferation in MCs, indicating that the overexpression of AT1R led to the activation of downstream ERK/Smad3 signaling that contributed to subsequent fibrotic changes. These findings were consistent with a previous report that revealed TGF-β-independent AT1R/ERK-mediated rapid activation of Smad3.
In the present study, we provide supportive evidence to indicate that HG can activate SND p102 expression, consequently enhancing the posttranscriptional activation of AT1R in which SND p102 recognizes and binds to the 3′-UTR of AT1R. Furthermore, SND p102 promotes the activation of the AT1R/ERK/Smad3 signaling pathway, leading to ECM accumulation and proliferation of rat MCs. Our study demonstrates the role and underlying mechanism of SND p102 in regulating MC dysfunction and may suggest a novel therapeutic strategy for DN treatment.
Author contribution
Li-min LU and Wei ZHANG designed the research project; Jin-lan XU and Xin-xin GAN performed experiments; Jun NI and De-cui SHAO wrote the manuscript; Yang SHEN contributed reagents and materials; Nai-jun MIAO, Dan XU and Li ZHOU analyzed the data.
Acknowledgements
This research was financially supported by the National Natural Science Foundation of China (No 81470591 and 81670664 to Li-min LU; No 81400695 to De-cui SHAO). This work was also supported by the Science and Technology Commission of Shanghai Municipality (14DZ2260200, the project of Shanghai Key Laboratory of Kidney and Blood Purification). All authors declare that no conflicts of interest exist.
Contributor Information
Wei Zhang, Email: wzhang@shmu.edu.cn.
Li-min Lu, Email: lulimin@shmu.edu.cn.
References
- 1.Kitada M, Ogura Y, Monno I, Koya D. Regulating autophagy as a therapeutic target for diabetic nephropathy. Curr Diab Rep. 2017;17:53. doi: 10.1007/s11892-017-0879-y. [DOI] [PubMed] [Google Scholar]
- 2.Mauer M, Caramori ML, Fioretto P, Najafian B. Glomerular structural-functional relationship models of diabetic nephropathy are robust in type 1 diabetic patients. Nephrol Dial Transplant. 2015;30:918–23. doi: 10.1093/ndt/gfu279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cao Z, Cooper ME. Pathogenesis of diabetic nephropathy. J Diabetes Investig. 2011;2:243–7. doi: 10.1111/j.2040-1124.2011.00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yacoub R, Campbell KN. Inhibition of RAS in diabetic nephropathy. Int J Nephrol Renovasc Dis. 2015;8:29–40. doi: 10.2147/IJNRD.S37893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ferrao FM, Lara L S, Lowe J. Renin-angiotensin system in the kidney: What is new? World J Nephrol. 2014;3:64–76. doi: 10.5527/wjn.v3.i3.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838–43. doi: 10.1074/jbc.M200581200. [DOI] [PubMed] [Google Scholar]
- 7.Koka V, Huang XR, Chung AC, Wang W, Truong LD, Lan HY. Angiotensin II up-regulates angiotensin I-converting enzyme (ACE), but down-regulates ACE2 via the AT1-ERK/p38 MAP kinase pathway. Am J Pathol. 2008;172:1174–83. doi: 10.2353/ajpath.2008.070762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xue H, Zhou L, Yuan P, Wang Z, Ni J, Yao T, et al. Counteraction between angiotensin II and angiotensin-(1-7) via activating angiotensin type I and Mas receptor on rat renal mesangial cells. Regul Pept. 2012;177:12–20. doi: 10.1016/j.regpep.2012.04.002. [DOI] [PubMed] [Google Scholar]
- 9.Lopez-Hernandez FJ, Lopez-Novoa JM. Role of TGF-beta in chronic kidney disease: an integration of tubular, glomerular and vascular effects. Cell Tissue Res. 2012;347:141–54. doi: 10.1007/s00441-011-1275-6. [DOI] [PubMed] [Google Scholar]
- 10.Lan HY. Diverse roles of TGF-beta/Smads in renal fibrosis and inflammation. Int J Biol Sci. 2011;7:1056–67. doi: 10.7150/ijbs.7.1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meng XM, Tang PM, Li J, Lan HY. TGF-beta/Smad signaling in renal fibrosis. Front Physiol. 2015;6:82. doi: 10.3389/fphys.2015.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen HY, Huang XR, Wang W, Li JH, Heuchel RL, Chung AC, et al. The protective role of Smad7 in diabetic kidney disease: mechanism and therapeutic potential. Diabetes. 2011;60:590–601. doi: 10.2337/db10-0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mauer M, Zinman B, Gardiner R, Drummond KN, Suissa S, Donnelly SM, et al. ACE-I and ARBs in early diabetic nephropathy. J Renin Angiotensin Aldosterone Syst. 2002;3:262–9. doi: 10.3317/jraas.2002.048. [DOI] [PubMed] [Google Scholar]
- 14.Jacobsen P, Andersen S, Jensen BR, Parving HH. Additive effect of ACE inhibition and angiotensin II receptor blockade in type I diabetic patients with diabetic nephropathy. J Am Soc Nephrol. 2003;14:992–9. doi: 10.1097/01.ASN.0000054495.96193.BF. [DOI] [PubMed] [Google Scholar]
- 15.Mohamed RH, Abdel-Aziz HR, Abd EMD, Abd ET. Effect of RAS inhibition on TGF-beta, renal function and structure in experimentally induced diabetic hypertensive nephropathy rats. Biomed Pharmacother. 2013;67:209–14. doi: 10.1016/j.biopha.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Wang C, Min C, Rong X, Fu T, Huang X, Wang C. Irbesartan can improve blood lipid and the kidney function of diabetic nephropathy. Discov Med. 2015;20:67–77. [PubMed] [Google Scholar]
- 17.Tong X, Drapkin R, Yalamanchili R, Mosialos G, Kieff E. The Epstein-Barr virus nuclear protein 2 acidic domain forms a complex with a novel cellular coactivator that can interact with TFIIE. Mol Cell Biol. 1995;15:4735–44. doi: 10.1128/MCB.15.9.4735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palacios L, Ochoa B, Gomez-Lechon M J, Castell JV, Fresnedo O. Overexpression of SND p102, a rat homologue of p100 coactivator, promotes the secretion of lipoprotein phospholipids in primary hepatocytes. Biochim Biophys Acta. 2006;1761:698–708. doi: 10.1016/j.bbalip.2006.05.005. [DOI] [PubMed] [Google Scholar]
- 19.Callebaut I, Mornon JP. The human EBNA-2 coactivator p100: multidomain organization and relationship to the staphylococcal nuclease fold and to the tudor protein involved in Drosophila melanogaster development. Biochem J. 1997;321:125–32. doi: 10.1042/bj3210125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caudy AA, Ketting RF, Hammond SM, Denli AM, Bathoorn AM, Tops BB, et al. A micrococcal nuclease homologue in RNAi effector complexes. Nature. 2003;425:411–4. doi: 10.1038/nature01956. [DOI] [PubMed] [Google Scholar]
- 21.Jariwala N, Rajasekaran D, Srivastava J, Gredler R, Akiel MA, Robertson CL, et al. Role of the staphylococcal nuclease and tudor domain containing 1 in oncogenesis. Int J Oncol. 2015;46:465–73. doi: 10.3892/ijo.2014.2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rajasekaran D, Jariwala N, Mendoza RG, Robertson CL, Akiel M A, Dozmorov M, et al. Staphylococcal nuclease and tudor domain containing 1 (SND1 Protein) promotes hepatocarcinogenesis by inhibiting monoglyceride lipase (MGLL) J Biol Chem. 2016;291:10736–46. doi: 10.1074/jbc.M116.715359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877–84. doi: 10.1161/HYPERTENSIONAHA.109.136531. [DOI] [PubMed] [Google Scholar]
- 24.Wang Z, Ni J, Shao D, Liu J, Shen Y, Zhou L, et al. Elevated transcriptional co-activator p102 mediates angiotensin II type 1 receptor up-regulation and extracellular matrix overproduction in the high glucose-treated rat glomerular mesangial cells and isolated glomeruli. Eur J Pharmacol. 2013;702:208–17. doi: 10.1016/j.ejphar.2013.01.031. [DOI] [PubMed] [Google Scholar]
- 25.Kong Y L, Shen Y, Ni J, Shao D C, Miao N J, Xu J L, et al. Insulin deficiency induces rat renal mesangial cell dysfunction via activation of IGF-1/IGF-1R pathway. Acta Pharmacol Sin. 2016;37:217–27. doi: 10.1038/aps.2015.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.He M, Zhang L, Shao Y, Xue H, Zhou L, Wang XF, et al. Angiotensin II type 2 receptor mediated angiotensin II and high glucose induced decrease in renal prorenin/renin receptor expression. Mol Cell Endocrinol. 2010;315:188–94. doi: 10.1016/j.mce.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 27.Ponchiardi C, Mauer M, Najafian B. Temporal profile of diabetic nephropathy pathologic changes. Curr Diab Rep. 2013;13:592–9. doi: 10.1007/s11892-013-0395-7. [DOI] [PubMed] [Google Scholar]
- 28.Dalla VM, Saller A, Mauer M, Fioretto P. Role of mesangial expansion in the pathogenesis of diabetic nephropathy. J Nephrol. 2001;14 Suppl 4:S51–7. [PubMed] [Google Scholar]
- 29.Ellis EN, Warady BA, Wood EG, Hassanein R, Richardson WP, Lane PH, et al. Renal structural-functional relationships in early diabetes mellitus. Pediatr Nephrol. 1997;11:584–91. doi: 10.1007/s004670050342. [DOI] [PubMed] [Google Scholar]
- 30.Burns KD. Angiotensin II and its receptors in the diabetic kidney. Am J Kidney Dis. 2000;36:449–67. doi: 10.1053/ajkd.2000.16192. [DOI] [PubMed] [Google Scholar]
- 31.Kennefick TM, Anderson S. Role of angiotensin II in diabetic nephropathy. Semin Nephrol. 1997;17:441–7. [PubMed] [Google Scholar]
- 32.Leverson JD, Koskinen PJ, Orrico FC, Rainio EM, Jalkanen KJ, Dash AB, et al. Pim-1 kinase and p100 cooperate to enhance c-Myb activity. Mol Cell. 1998;2:417–25. doi: 10.1016/S1097-2765(00)80141-0. [DOI] [PubMed] [Google Scholar]
- 33.Valineva T, Yang J, Palovuori R, Silvennoinen O. The transcriptional co-activator protein p100 recruits histone acetyltransferase activity to STAT6 and mediates interaction between the CREB-binding protein and STAT6. J Biol Chem. 2005;280:14989–96. doi: 10.1074/jbc.M410465200. [DOI] [PubMed] [Google Scholar]
- 34.Paukku K, Yang J, Silvennoinen O. Tudor and nuclease-like domains containing protein p100 function as coactivators for signal transducer and activator of transcription 5. Mol Endocrinol. 2003;17:1805–14. doi: 10.1210/me.2002-0256. [DOI] [PubMed] [Google Scholar]
- 35.Paukku K, Kalkkinen N, Silvennoinen O, Kontula K K, Lehtonen JY. p100 increases AT1R expression through interaction with AT1R 3′-UTR. Nucleic Acids Res. 2008;36:4474–87. doi: 10.1093/nar/gkn411. [DOI] [PMC free article] [PubMed] [Google Scholar]