

Abstract
Cyclooxygenase-2 (COX-2) has an established role in the pathogenesis of hypoxic-ischemic encephalopathy (HIE). In this study we sought to determine whether COX-2 was induced by asphyxia in newborn pigs, and whether neuronal COX-2 levels were affected by H2 treatment. Piglets were subjected to either 8 min of asphyxia or a more severe 20 min of asphyxia followed by H2 treatment (inhaling room air containing 2.1% H2 for 4 h). COX-2 immunohistochemistry was performed on brain samples from surviving piglets 24 h after asphyxia. The percentages of COX-2-immunopositive neurons were determined in cortical and subcortical areas. Only in piglets with more severe HIE, we observed significant, region-specific increases in neuronal COX-2 expression within the parietal and occipital cortices and in the CA3 hippocampal subfield. H2 treatment essentially prevented the increases in COX-2-immunopositive neurons. In the parietal cortex, the attenuation of COX-2 induction was associated with reduced 8'-hydroxy-2′-deoxyguanozine immunoreactivity and retained microglial ramifcation index, which are markers of oxidative stress and neuroinfiammation, respectively. This study demonstrates for the first time that asphyxia elevates neuronal COX-2 expression in a piglet HIE model. Neuronal COX-2 induction may play region-specific roles in brain lesion progression during HIE development, and inhibition of this response may contribute to the antioxidant/anti-infiammatory neuroprotective effects of H2 treatment.
Keywords: hypoxic-ischemic encephalopathy, COX-2, 8′-hydroxy-2′-deoxyguanozine, microglia, molecular hydrogen, neuroprotection, newborn pigs
Introduction
In neonates, brain damage is most often associated with perinatal asphyxia (PA), which occurs in approximately 1–6 per 1000 live full-term births1,2. PA may result in neonatal mortality, or depending on its severity, hypoxic-ischemic encephalopathy (HIE) may develop, which results in long-lasting neurodevelopmental motor and cognitive dysfunctions in approximately 25% of survivors3.
Cyclooxygenases (COXs) are the rate-limiting enzymes that catalyze the conversion of arachidonic acid released from membrane phospholipids by phospholipase-A2 to prostaglandin (PG)-H2, which is further converted to biologically active prostaglandins, including PGD2, PGE2, PGF2α, PGI2 and thromboxane A2 4. The two COX isozymes, COX-1 and COX-2, have similar catalytic activities but differ in their pharmacological properties and tissue distributions5. Although COX-1 and COX-2 are both constitutively expressed in the central nervous system6, COX-2 is enriched in the hippocampus and the cerebral cortex7. Furthermore, COX-2 is the dominant isoform in the newborn brain8, providing up to 80% of the total brain COX activity9. In the central nervous system, prostanoids modulate synaptic transmission and neurovascular coupling10,11. Neuronal COX-2 plays an essential role in the modulation of excitatory glutamatergic synaptic transmission and long-term synaptic plasticity10. Simultaneously, COX-2-derived prostanoids also participate in flow-metabolism coupling, for instance in the local blood flow response to whisker stimulation in the barrel cortex12. Expression of the inducible COX-2 isoform is well-known to be upregulated by various deleterious stimuli including brain trauma13,14, cerebral ischemia15,16, and proinflammatory insults17. The neuronal induction of COX-2 suggests the enzyme's participation in neuroinflammation, the pathogenesis of neurodegenerative diseases, traumatic brain injury, and ischemia-induced neuronal damage and epileptogenesis18.
COX-2 has an established role in the pathogenesis of HIE. For instance, a previous study showed that administration of COX-2 inhibitors before as well as soon after ischemic insult provided neuronal protection in a rat HIE model19. Furthermore, brain ischemia has been associated with high neuronal COX-2 levels also in the neonatal human brain in an autopsy study20. Therefore, the role of COX-2 in the mechanism of hypoxic-ischemic neuronal injury could perhaps be best studied in a translational large animal model that could also be used to establish possible neuroprotective treatments.
The newborn pig is an accepted large animal model for the human neonate, as brain size, gyrencephalic structure, neurodevelopmental state of the brain and cerebral metabolism at birth are similar in both species21. COX-2 has been quite extensively studied in the piglet model and has been identified as the major constitutive isoform in this species8. According to immunohistochemistry analysis, COX-2 expression in the brain and cerebral arteries of newborn piglets is markedly induced by global cerebral ischemia15,16 but, confoundingly, not by asphyxia16,22. In these studies, global cerebral ischemia was induced by raising the intracranial pressure above the arterial pressure for 10 min while venous blood was withdrawn to control the blood pressure and asphyxia was induced by suspending ventilation for 10 min. The translational significance of the findings concerning the possible role of neuronal COX-2 induction in neuronal damage during HIE development has thus become difficult to determine, as the clinical presentation of HIE is associated with asphyxia and not global no-flow ischemia. Furthermore, animals in these studies survived only 4–8 h, so it was also possible that the expression changes after asphyxia would have occurred at a later and thus undetected time point.
In recent years, we established and published two piglet PA/HIE models of increasing severity22,23 to assess the neuroprotective effect of inhaled molecular hydrogen (H2)23,24 that was first reported by Ohsawa et al. in an adult rat stroke model25. H2 was described as a neuroprotective agent that penetrates the blood–brain barrier and selectively reduces cytotoxic hydroxyl radicals25. H2 was also found to be effective in rodent HIE models26,27 and in various other neuropathological conditions, such as Parkinson's disease28, auditory neuropathy29 and even stress-induced learning impairment30. The mechanistic details of H2-induced neuroprotection are still unclear but likely involve inhibition of oxidative injury and inhibition of neuroinflammation31. However, we have virtually no information on the mechanism of H2-induced neuroprotection in PA/HIE, let alone in a large animal model.
Therefore, the major purpose of the present study was to determine the effect of H2 on asphyxia-induced neuronal COX-2 expression correlated with 8′-hydroxy-2′-deoxyguanozine23 (8-OHdG) and Iba-1 immunohistochemistry markers of oxidative stress and neuroinflammation, respectively, in our translational PA/HIE piglet model.
Materials and methods
To study neuronal COX-2 expression, we used brain samples of newborn male Large-White piglets (age < 24 h at beginning of experiments, body weight: 1.5–2.5 kg, n=59), which were obtained mainly from previously published studies23,32. In both studies, the anesthetized, artificially ventilated animals were divided into 3-3 experimental groups (n=7-7) including a normoxic time control group ventilated with room air, an asphyxiated group reventilated with room air for 24 h, and an asphyxiated group reventilated with room air containing 2.1% H2 for 4 h followed by room air for the remaining (20 h) survival time. The major difference between the two previously published studies23,32 was the induction method and the duration of asphyxia. Asphyxia was elicited by 8 min of trachea occlusion and suspended ventilation in the first study and by 20 min of ventilation with a hypoxic-hypercapnic gas mixture (6% O2, 20% CO2) in the second study. Our preliminary results suggested that the length of anesthesia per se in the absence of asphyxia might have affected COX-2 expression, therefore we included an additional naïve control group to study baseline neuronal COX-2 expression. Furthermore, we also used brain samples from normoxic time control animals with 4 h survival from another previous study24. In the new naïve control group, piglets (n =5) were anesthetized with sodium thiopental (45 mg/kg, ip; Sandoz, Kundl, Austria), and the brains were immediately perfused with 100 mL cold physiological saline solution through the catheterized common carotid arteries and harvested for immunohistochemical analysis. All procedures were approved by the Animal Care and Use Committee of the University of Szeged. The brain tissues were immersion-fixed with 4% paraformaldehyde at 4 °C for 2 weeks. Paraffin-embedded, 4-μm sections were produced using a microtome (Leica Microsystems, Wetzlar, Germany) and mounted on sylanized slides. Hematoxylin-eosin staining was performed to assess the extent of neuronal lesions, which was determined by cell counting in most areas. In the cerebral cortex, however, neuropathology scores were determined (0–9) as described previously23. Briefly, in each region, 20 fields of view were assessed by two independent observers. In each field of view, the neuronal lesions were described as none, scattered, grouped/laminar or panlaminar. The neuropathology score was determined by the incidence of the most severe lesion observed. If the most severe lesions represented less than 20% of the total lesions, the scores were 1, 4, or 7; if 20%–50%, 2, 6, or 8; and if more than 50%, 3, 6, or 9. Thus, higher scores represented increasingly severe neuronal damage.
COX-2 immunohistochemistry was performed using the LEICA BOND-MAX automated immunostainer (Leica Microsystems, Wetzlar, Germany). The slides were dewaxed at 72 °C, and antigen retrieval was performed at pH 6. The slides were then incubated with a rabbit monoclonal antibody against COX-2 diluted at 1:100 (clone SP21, Labvision, Fremont, California, USA) for 30 min followed by horseradish peroxidase-conjugated anti-rabbit antibody (EnVision®; Dako, Glostrup, Denmark). 3,3′-Diaminobenzidine (DAB) tetrahydrochloride solution was used for visualization, and the slides were then counterstained with hematoxylin to visualize the cell nuclei. The slides were dehydrated in an ascending alcohol series, cleared in xylene, covered with a coverslip, scanned in a slide scanner (Pannoramic MIDI, 3DHISTECH Ltd, Budapest, Hungary) and visualized on a personal computer using the Pannoramic Viewer software (3DHISTECH Ltd) at 20x magnification. Areas of interest were randomly selected, and photomicrographs were acquired from each region observed, including 10-10 images of the frontal, temporal, parietal and occipital cortex; 3-3 images of the CA1, CA3 subfields and the dentate gyrus of the hippocampus; 10-10 images of the thalamus and the basal ganglia; and 15 images of the cerebellar cortex for the study of Purkinje-cells. The cells were manually counted by two independent observers using ImageJ software (Wayne Rasband, NIH, Bethesda, Maryland, USA). Neurons were identified by their shape and size. The ratio of COX-2-immunopositive neurons relative to all neurons was determined, and the averages/cerebral areas/animals were counted and plotted on box plots using R 3.3.1 software (The R Foundation for Statistical Computing, Vienna, Austria). For statistical analysis, ANOVA on ranks followed by the Student-Newman-Keuls post hoc test was performed using SigmaPlot 12.0 (Systat Software Inc., Chicago, Illinois, USA). P<0.05 was considered statistically significant. The ratio of COX-2-immunopositive neurons was correlated with the previously determined histopathological score23, and the result was plotted on a dot plot using R 3.3.1 software.
Based on the results of our COX-2 studies, the parietal cortex was selected for further studies. Tissue microarrays from the parietal cortex were produced from the paraffin-embedded tissue blocks of the second study (including the time control, 20 min asphyxia, and 20 min asphyxia+H2 groups) using a custom-made stainless steel tissue puncher (3 mm). The microarrays were also sectioned at 4 μm, mounted on sylanized slides and processed for immunohistochemistry. To determine the extent of oxidative damage in the neurons, 8-OHdG immunohistochemistry was performed23. The slides were incubated with a mouse monoclonal primary antibody against 8-OHdG diluted at 1:200 (JaICA Inc, Fukuroi, Japan) for 20 min, followed by incubation with a horseradish peroxidase-conjugated rabbit anti-mouse secondary antibody for 15 min (n=6-6-6). Iba-1 immunohistochemistry was performed to assess microglial activation (n=7-6-6). Slides were incubated with rabbit anti-Iba-1 antibody (Wako Chemicals GmbH, Neuss, Germany) for 30 min, followed by horseradish peroxidase-conjugated anti-rabbit antibody (EnVision®; Dako, Glostrup, Denmark) for 15 min. In both cases, 3,3′-diaminobenzidine was used to visualize the immunostaining, and the slides were then counterstained with hematoxylin to visualize the cell nuclei. The slides were covered with coverslips, scanned in the slide scanner, and visualized on a personal computer using the Pannoramic Viewer software at x40 magnification. Homogeneous, strong nuclear 8-OHdG immunoreactivity indicated oxidative damage. The ratio of these nuclei to the total neuronal nuclei was determined using ImageJ software, similarly to the previously described method. The average/sample was plotted on box plots. The groups were compared with one-way ANOVA on ranks followed by the Student-Newman-Keuls post hoc test (P<0.05). Microglial activation was characterized by determining the so-called ramification index (RI)33. We applied a 0.20 mm×0.25 mm grid to the microphotographs of 3 randomly selected areas/sample at x40 magnification. The immunopositive cell bodies (CBD) in the grid were counted as well as the microglial branches (B) crossing the gridlines. From these data, the RI was defined according to the following equation: RI=B2/CBD. The RI values for each field of view were plotted on box plots, and the groups were compared with one-way ANOVA on ranks followed by Dunn's post hoc test (P=0.015). Correlations were plotted and tested using R 3.3.1 software (Pearson's correlation, P<0.05).
Results
Neuronal COX-2 expression in the time controls
Neuronal COX-2 expression in the naïve animals was similar to our previous observations and showed marked regional differences: the highest percentages of COX-2-positive neurons were observed in the frontal and parietal cortices as described previously22. However, this regional expression pattern appeared markedly changed in the 24-h time controls, as COX-2 expression was significantly reduced in all neocortical regions compared with the naïve or 4-h survival animals (Figure 1). This reduction was found to be limited to the cortex, as the ratio of COX-2-immunopositive neurons remained unchanged in the hippocampus (Figure 1).
Figure 1.
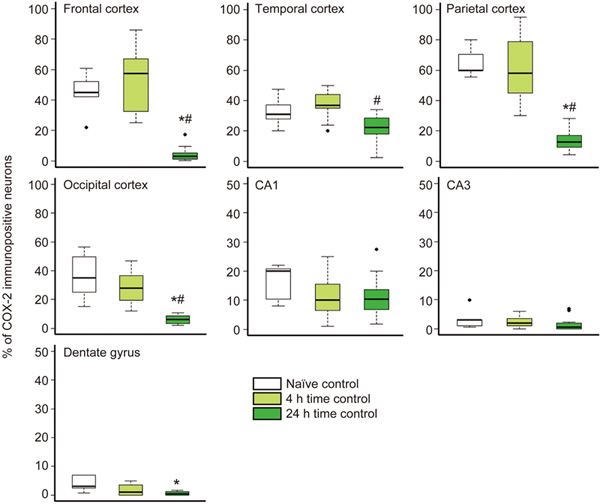
The ratio of neocortical COX-2-immunopositive neurons decreased over time under normoxic conditions in anesthetized time control animals. In the different cortical lobes, the percentages of COX-2-immunopositive cells determined in samples from naïve animals (brains harvested immediately after anesthesia, n=5) and from normoxic time controls with 4 h of survival after anesthesia (n=12) displayed similar COX-2 expression in accordance with previous data showing regional differences22. However, the percentage of COX-2-immunopositive neurons in the 24 h survival time control group (n=14) was significantly reduced compared with those of both the naïve and the 4 h time control groups in all neocortical regions. Interestingly, this reduction did not affect the CA1 and CA3 hippocampal subfields. (* P<0.05 vs naïve animals, # P<0.05 vs 4 h time control; ANOVA on ranks, Dunn's post hoc test. Bold line, box, and whiskers represent the median, 25th–75th, and 10th–90th percentiles, respectively; black dots are outliers).
Effect of asphyxia on neuronal COX-2 expression
In the brain regions obtained from the study of animals exposed to 8-min asphyxia, there was no significant alteration in neuronal COX-2 expression (Figure 2), as no difference was noted between the time control and asphyxiated groups in any of the observed regions (Figure 2). In contrast, 20-min asphyxia elicited significant increases in neuronal COX-2 immunopositivity in the parietal and occipital cortices, as well as in the hippocampal CA3 region (Figure 2). Furthermore, the asphyxia-induced elevation of COX-2-immunopositive neurons was also observed in the frontal and temporal cortices and the basal ganglia, although these changes did not reach statistical significance in these regions (Figure 2). In the H2-treated group, despite exposure to the same level of asphyxia, the ratio of COX-2-immunopositive cells was similar to that in the time control group (Figure 2). Notably, strong COX-2-immunopositive areas were only recorded in the asphyxia group (Figure 3). In contrast to the CA3 subfield, the hippocampal CA1 region and the dentate gyrus displayed low percentages of COX-2-immunopositive neurons in the time controls. In addition, the ratio of immunopositive neurons was unchanged by asphyxia, as these percentages were similar among all three groups. In a similar fashion, COX-2 was present in approximately 30% of the cerebellar Purkinje cells in all experimental groups (Figure 2).
Figure 2.
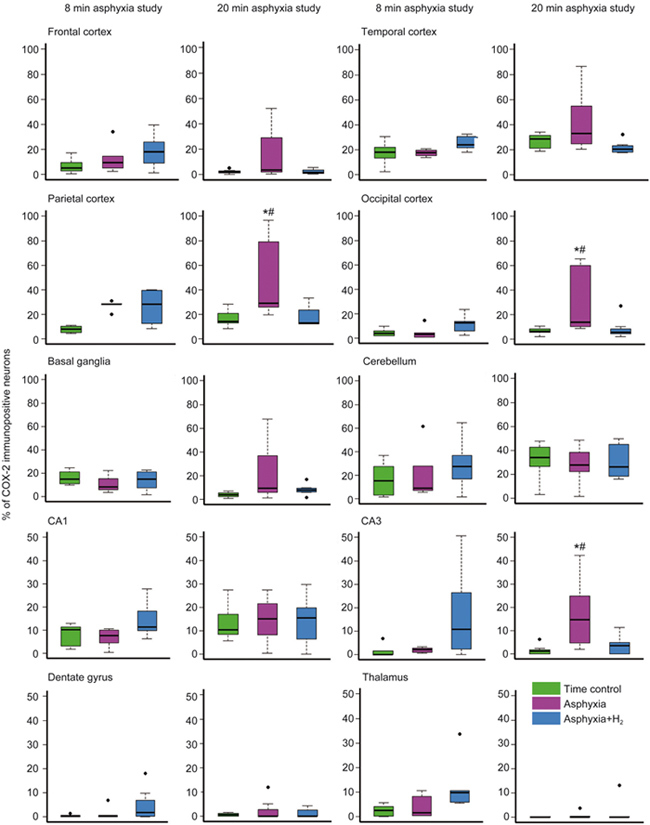
The effect of perinatal asphyxia and neuroprotective molecular H2 on the ratio of COX-2-immunopositive neurons determined after 24 h of survival. Two levels of perinatal asphyxia of different durations (8 and 20 min) were studied, and both asphyxia groups (n=7-7) were compared with the corresponding time control (n=7-7) and H2-treated asphyxia groups (n=7-7). In the first study using 8-min asphyxia, neuronal COX-2 expression was not significantly affected in either the asphyxia or the H2-treated asphyxia group. On the other hand, in the second study, 20-min asphyxia elicited significant elevations in the ratios of COX-2-immunopositive neurons in the parietal and occipital cortices and in the hippocampal CA3 region. The trends towards increased COX-2 expression were also observed in the frontal and temporal cortices and the basal ganglia, although these differences did not reach statistical significance in these regions. The hippocampal CA1 region, dentate gyrus, and thalamus showed low immunopositivity, which was unaffected by both asphyxia and H2 treatment. Approximately one-third of cerebellar Purkinje-cells were COX-2-immunopositive, and no marked differences were detected among the three groups. (* P<0.05 vs time control. # P<0.05 vs asphyxia+H2; ANOVA on ranks, Student-Newman-Keuls post hoc test. Bold line, box, and whiskers represent the median, 25th–75th, and 10th–90th percentiles, respectively; black dots are outliers).
Figure 3.
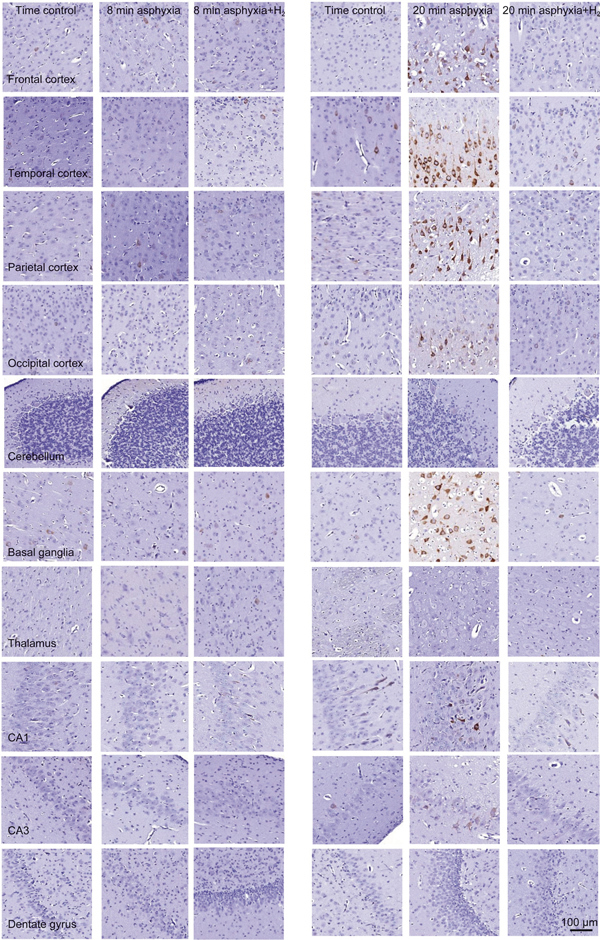
Representative photomicrographs (x20) showing cyclooxygenase-2 (COX-2)-immunopositive neurons (brown colored neurons) from the assessed brain regions in the matching time control, asphyxia and asphyxia+H2 groups of the 8-min asphyxia and the 20-min asphyxia studies. The displayed fields of view were selected from the 10 analyzed fields of view of those animals that yielded the median values of the respective groups in Figure 2. No remarkable differences were detected among the experimental groups subjected to 8 min of asphyxia. The increased abundance of COX-2-immunopositive neurons was striking in virtually all neocortical areas, in the CA3 hippocampal region and in the basal ganglia from the 20-min asphyxia group.
Assessment of correlation between neuronal COX-2 expression and neuronal damage
As neuronal COX-2 expression was affected by 20-min asphyxia exposure only, we performed all subsequent studies on these animals. We first investigated whether the ratio of COX-2-immunopositive neurons correlated with the previously determined neuropathology scores23 with higher scores reflecting more severe neuronal damage in the cortex (Figure 4). We found no correlation between the neuropathology scores and neuronal COX-2 expression (r=0.38; P<0.01); however, we clearly identified 3 expression patterns. Low neuropathology scores were always associated with low-moderate levels of neuronal COX-2 expression, and high neuropathology scores were associated either with high or with low COX-2 expression levels. Notably, low neuropathology score never coincided with high COX-2 expression levels (Figure 4).
Figure 4.
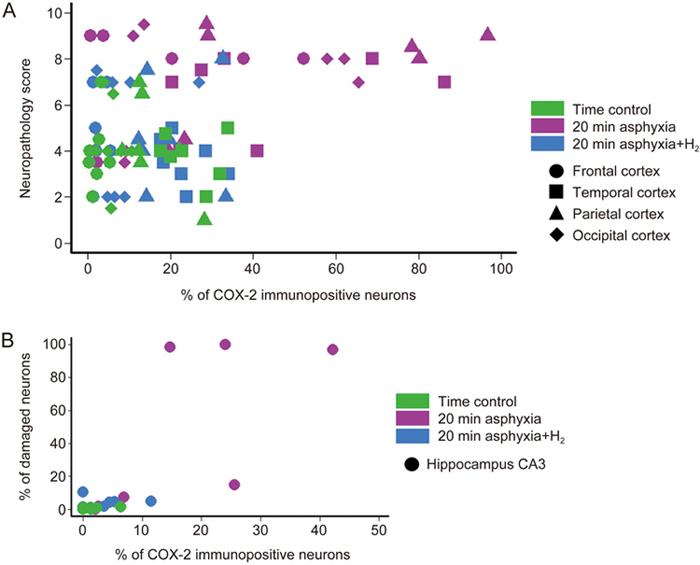
High ratios of COX-2-immunopositive neurons/total neurons are exclusively found in severely damaged cortical and hippocampal regions. (A) In the cortical regions of the normoxic time control group (n=7), the neuropathology scores were typically low and associated with low levels of COX-2-immunopositive neurons. Similar observations were made for the 20-min asphyxia+H2 group (n=7) as well. However, in the 20-min asphyxia group (n=7), severe neuronal lesions may or may not be associated with high percentages of COX-2-immunopositive neurons. Importantly, high COX-2 immunopositivity was not associated with low neuropathology scores in any animals/regions. (B) In the hippocampal CA3 subfield, the highest number COX-2-immunopositive neurons was from the 20-min asphyxia group with largest degree of neuronal damage.
Assessment of the correlation between neuronal COX-2 expression and oxidative DNA damage
In the parietal cortex, 20-min asphyxia significantly increased the ratio of 8-OHdG-immunopositive neuronal nuclei (Figure 5A) compared with both the time control and the H2-treated asphyxia group, indicating the oxidative stress induced by asphyxia and the mitigating effect of molecular H2 (Figure 5B). By assessing the correlation between the ratios of COX-2- and 8-OHdG-immunopositive neurons (Figure 5C), we found a marked tendency towards a positive correlation (r=0.71), considering data either from all groups or from the 20-min asphyxia group alone.
Figure 5.
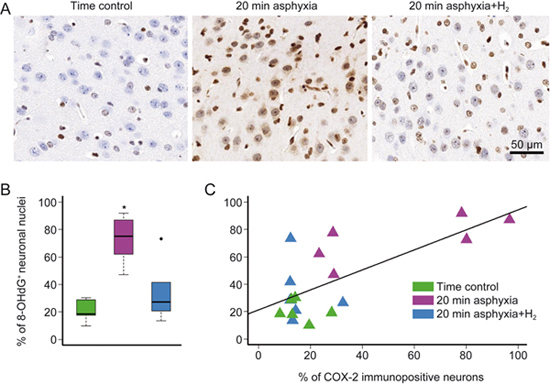
8-OHdG immunopositivity in the parietal cortex of the matching time control, 20-min asphyxia and 20-min asphyxia+H2 groups. Representative photomicrographs (A) demonstrate the oxidative damage of nuclei (×40) in the 20-min asphyxia group compared with the time control and the 20-min asphyxia+H2 groups. The ratio of 8-OHdG-immunopositive neuronal nuclei/total neuronal nuclei (B) was markedly increased in the 20-min asphyxia group compared with time control and 20-min asphyxia+H2 groups (n=6-6-6, ANOVA on ranks, Student-Newman-Keuls post hoc test, * P<0.05; bold line, box, and whiskers represent the median, 25th-75th, and 10th-90th percentiles, respectively; black dots are outliers). Correlation analysis of the 8-OHdG and COX-2 immunopositivity (C) detected a significant tendency (Pearson's method, r=0.71, P<0.05).
Assessment of the correlation between neuronal COX-2 expression and microglial activation
The Iba-1 immunohistochemistry results revealed the distribution of microglia in the parietal cortex (Figure 6A). Microglial activation is associated with reduced branching and the adoption of an amoeboid shape, and these changes were quantified by RI determination. We found that the RI was significantly lower in the group subjected to 20-min asphyxia than in the time control group (Figure 6B). However, there was no significant difference between the time controls and the H2-treated asphyxiated animals. When all data points were considered, the RI showed no correlation with neuronal COX-2 expression (r=-0.56, Figure 6C), but when data from only the 20-min asphyxia group were considered, a significant negative correlation (r=-0.75) was observed.
Figure 6.
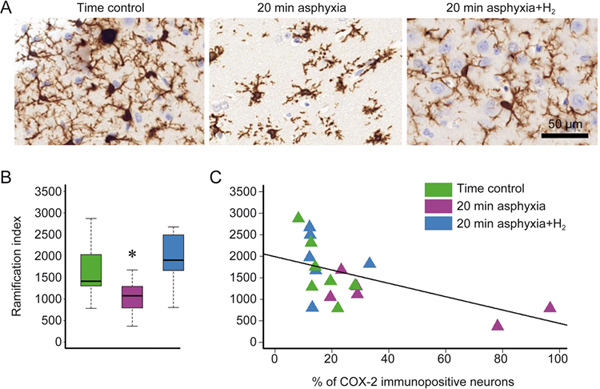
Iba-1 immunopositivity and microglial ramification index in the parietal cortex of the matching time control, 20-min asphyxia and 20-min asphyxia+H2 groups. Representative photomicrographs (A) show that while in the time control and 20-min asphyxia+H2 groups, the shape of the microglial cells becomes ramified, in the 20-min asphyxia group, the microglial cells become activated and start assuming a more amoeboid shape (×40). The ramification index (B) was evidently decreased in the 20-min asphyxia group compared with the time control group; however, the 20-min asphyxia+H2 group was more similar to the time control group (n=7-6-6, ANOVA on ranks, Dunn's post hoc test, * P=0.015). The correlation between the ramification index and neuronal COX-2 expression (% of total neurons) (C) considering all 3 groups was less pronounced (Pearson's method, r=-0.56, P<0.05); however, when regarding the 20-min asphyxia group only, a significant negative correlation was detected (r=-0.75).
Discussion
The major findings of the present study are as follows: 1) neuronal COX-2 expression in the neocortical areas is greatly reduced in 24-h anesthetized time controls; 2) 20-min, but not 8-min, asphyxia exposure elevates the number of COX-2-positive neurons both in the cortex and the hippocampal CA3 subfield but not in the assessed subcortical areas or other hippocampal structures; 3) the effect of asphyxia on neuronal COX-2 abundance is associated and correlated with oxidative stress and microglial activation as shown by enhanced 8-OHdG staining and reduced RI, respectively; and 4) post-asphyxia administration of neuroprotective molecular H2 prevents the upregulation of neuronal COX-2 expression in all sensitive brain regions with simultaneous reductions in oxidative stress and prevention of microglial activation.
This study is the first to describe the induction of neuronal COX-2 expression following asphyxia in a translational subacute piglet PA/HIE model. In our previous study23, we extensively characterized the effect of 20-min asphyxia elicited in newborn (<1 day old) piglets by ventilation with a hypoxic-hypercapnic (6% O2, 20% CO2) gas mixture on hemodynamics, blood gases, metabolites, electroencephalogram and neuropathology. The applied insult resulted in alterations that matched both human pathology and naturally occurring birth asphyxia in swine34 and corresponded to moderate to severe HIE in all of the animals. Our present results elucidated the confounding results from previous studies in which elevations in COX-2 levels were reported after 10 min of global cerebral ischemia but not 10 min of asphyxia16. Our current results suggest that this reported difference was due to the more severe hypoxic/ischemic insult elicited by global cerebral ischemia, and in the present study, the longer asphyxia duration rather than the shorter treatment elicited conditions similar to those observed with global ischemia, which resulted in the upregulation of COX-2.
In the present study, neuronal COX-2 abundance was conspicuously reduced in all neocortical areas of the 24-h time control animals compared with the values previously reported in our 4-h survival study22. This difference cannot be attributed to differences in methodology such as for COX-2 immunostaining or cell counting, as the values obtained from the naïve animals in the present study yielded virtually identical data to the previously published values. The decreased number of COX-2-expressing neurons may be in part explained by the inactivation of cortex due to anesthesia, as COX-2 expression is stimulated by neuronal activity35. The applied anesthetic/analgesic drugs could exert an inhibitory effect on COX-2 expression by interacting with the nuclear transcription factor NF-κB signaling pathway, which is a well-known transcriptional regulator of COX-236. Although morphine has been reported to have ambiguous stimulatory and inhibitory effects on NF-κB activation37,38, midazolam is unequivocally known to inhibit the NF-κB pathway39. The applied anesthetic regimen was chosen to enhance the translational potential of our animal model, as morphine/midazolam analgesia/sedation is routinely used in the management of human neonates affected by PA/HIE40. Furthermore, experimental data suggested that morphine analgesia may be an important permissive factor by allowing neuroprotective therapies such as therapeutic hypothermia to be effective; mild hypothermia failed to exhibit neuroprotection in the absence of anesthesia and analgesia in newborn pigs41. Our current results suggested that this “supportive” analgesia may have a beneficial effect on neuronal survival in part through the attenuation of COX-2 expression. Our findings also suggested a time-dependent role of COX-2-derived ROS and prostanoids in the pathomechanism of HIE development in different brain regions, as COX-2 activity-dependent neuronal injury in the early reventilation/reoxygenation phase will be most likely pronounced in regions with high baseline COX-2 expression (especially the frontoparietal neocortex). However, in the delayed secondary energy failure phase, COX-2 will likely remain a more important pathogenic factor for neuronal injury in those areas where the asphyxia-induced elevation dominates the anesthesia-induced depression of COX-2 levels.
The present data thus suggested that at least in the neocortical areas, two factors affect neuronal COX-2 abundance. Long-term anesthesia tends to decrease the enzyme levels, whereas asphyxia elevates them. Both the increase in COX-2 levels and the increase in the level of neuronal injury were variable in the piglets subjected to asphyxia, in accordance with the spectrum of human HIE severity. We found that very high percentages of COX-2-immunopositive neurons were inevitably accompanied by the most severe types of cortical neuronal damage. In some cases, however, similarly high neuropathology scores coincided with rather low COX-2 immunopositivity. These areas may perhaps represent those severely damaged areas where the hypoxia/ischemia-induced translational blockade might have prevented the expression of COX-2. Thus, the areas displaying very high neuronal COX-2 levels may represent those areas that were still able after asphyxia to translate new proteins, and the deleterious effects of COX-2 may have contributed most in these areas to the observed neuronal damage.
In the present study, we assessed the effect of a neuroprotective (2.1%) concentration of H2 on neuronal COX-2 expression. In addition to our previous studies22,23,24, this concentration was found to be neuroprotective in a number of other disease models as well25,42,43. Inhaled H2 can easily penetrate into the brain, as has been shown numerous times by the successful application of the so-called H2-clearance technique to quantitatively determine cerebral blood flow in newborn pigs44,45,46. After rapid equilibration with the blood, the applied H2 concentration likely resulted in approximately 10–20 μmol/L H2 brain levels, as determined in both rats and humans25,47. Our current results concerning the correlation of nuclear 8-OHdG immunoreactivity with COX-2 expression and the remarkable efficacy of molecular H2 inhalation to attenuate elevations in both 8-OHdG and COX-2 levels after asphyxia suggest a role for ROS in the mechanism of COX-2 expression. 8-OHdG is used as a biomarker of oxidative modifications to DNA48,49,50 and is one of the most studied catabolites. Guanine is the best electron donor that has the lowest oxidation potential among the DNA bases49,51. 8-Hydroxylation in the guanine base occurs after an attack by hydroxyl radicals under oxidative stress52. Thus, elevations in the number of 8-OHdG-positive nuclei indirectly indicate significant oxidative stress imposed by hydroxyl radicals. Importantly, molecular H2 was originally described as a selective hydroxyl radical scavenger25, and thus, the efficacy of H2 to attenuate elevations in 8-OHdG levels after asphyxia further confirms the presence of significant oxidative stress perhaps characterized by significant production of hydroxyl radicals in our present PA/HIE piglet model. The connection between this oxidative stress and the observed induction of COX-2 expression may be the activation of NF-κB, which is a transcription factor known to be induced by ROS and inhibited by antioxidants53. The general physiological functions of NF-κB include the regulation of apoptosis, cell growth, cellular stress responses and intracellular signaling54. NF-κB affects various brain functions as well as neuronal development, inflammation and neurodegeneration55. Brain injury has been shown to increase NF-κB activity56. The COX-2 gene is a neuronal target of NF-κB36, and therefore the asphyxia-induced increase in neuronal COX-2 expression is very likely mediated via NF-κB. Indeed, a very recent report in neonatal rats demonstrated reduced NF-κB activation in H2-treated rat pups, which lends experimental support to our hypothesis57. Our results with Iba-1 immunohistochemistry that was used to visualize microglia to determine the RI as an indicator of microglial activation are also in favor of the above-mentioned mechanism. We found that microglia were activated by asphyxia only but not by asphyxia followed by H2. As microglial activation has also been reported to be accompanied by NF-κB activation58, hydroxyl radicals may have activated the microglia by promoting NF-κB activity in the present study.
The efficacy of molecular H2 to prevent increases in COX-2 levels after asphyxia suggest that the ROS responsible for triggering COX-2 induction were produced during the early reventilation/reoxygenation phase, as H2 was administered only in the first four hours of survival. This is interesting, as ROS may arise at later time points as well, for instance from mitochondria during the secondary energy failure59, from COX activity itself 60, or from the activated microglia61.
Conclusion
In our translational HIE model, PA-induced increases in neuronal COX-2 expression were dependent on regional vulnerability and on the severity of asphyxia. ROS are known to trigger COX-2 expression, and in turn, COX-2 activity is also an important source of ROS after PA22,62. One of the limitations of the present study was that although it showed changes in neuronal COX-2 abundance, it could not directly demonstrate the changes in neuronal COX activity. However, the demonstrated increases in oxidative DNA damage and microglial activation may also indirectly signal enhanced COX-2 activity via the effects of ROS and neuroinflammation. Furthermore, these mechanisms indicate the development of a vicious cycle where ROS-inflicted changes (COX-2 induction, neuroinflammation triggered by microglial activation, and ROS-inflicted mitochondrial damage) lead to progressive neuronal injury. Molecular H2 appears to interrupt this vicious cycle when applied immediately during the early reoxygenation phase after asphyxia.
We conclude that H2 remains a promising neuroprotective agent to combat HIE development. H2-induced neuroprotection involves not only direct scavenging of ROS but also the inhibition of deleterious neuronal COX-2 induction in vulnerable neurons.
Author contribution
Viktória VARGA participated in animal experiments, evaluated the immunostaining results, analyzed the data and wrote the paper. Orsolya OLAH designed the study, participated in animal experiments, evaluated the immunostaining results, and analyzed the data. János NEMETH participated in animal experiments, evaluated the immunostaining results, and analyzed the data. Valéria TOTH-SZUKI participated in animal experiments and research management. Viktória KOVACS and Gábor REMZSO participated is animal experiments. Ferenc DOMOKI designed the study, participated in animal experiments, analyzed the data and wrote the paper.
Acknowledgements
This study was supported by grants from the Hungarian Brain Research Program (No KTIA_13_NAP-A-I/13) and from the EU-funded Hungarian grant EFOP-3.6.1-16-2016–00014. János NÉMETH was supported by the “Nemzeti Tehetség Program” of the “Emberi Erőforrás Támogatáskezelő” from the Hungarian Ministry of Human Capacities.
References
- 1.Thornberg E, Thiringer K, Odeback A, Milsom I. Birth asphyxia: incidence, clinical course and outcome in a Swedish population. Acta Paediatr. 1995;84:927–32. doi: 10.1111/j.1651-2227.1995.tb13794.x. [DOI] [PubMed] [Google Scholar]
- 2.Pierrat V, Haouari N, Liska A, Thomas D, Subtil D, Truffert P. Prevalence, causes, and outcome at 2 years of age of newborn encephalopathy: population based study. Arch Dis Child Fetal Neonatal Ed. 2005;90:F257–61. doi: 10.1136/adc.2003.047985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vannucci RC, Perlman JM. Interventions for perinatal hypoxic-ischemic encephalopathy. Pediatrics. 1997;100:1004–14. doi: 10.1542/peds.100.6.1004. [DOI] [PubMed] [Google Scholar]
- 4.Smith WL, Dewitt DL. Prostaglandin endoperoxide H synthases-1 and -2. Adv Immunol. 1996;62:167–215. doi: 10.1016/S0065-2776(08)60430-7. [DOI] [PubMed] [Google Scholar]
- 5.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–82. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 6.Kaufmann WE, Andreasson KI, Isakson PC, Worley PF. Cyclooxygenases and the central nervous system. Prostaglandins. 1997;54:601–24. doi: 10.1016/S0090-6980(97)00128-7. [DOI] [PubMed] [Google Scholar]
- 7.Yamagata K, Andreasson KI, Kaufmann WE, Barnes CA, Worley PF. Expression of a mitogen-inducible cyclooxygenase in brain neurons: regulation by synaptic activity and glucocorticoids. Neuron. 1993;11:371–86. doi: 10.1016/0896-6273(93)90192-T. [DOI] [PubMed] [Google Scholar]
- 8.Peri KG, Hardy P, Li DY, Varma DR, Chemtob S. Prostaglandin G/H synthase-2 is a major contributor of brain prostaglandins in the newborn. J Biol Chem. 1995;270:24615–20. doi: 10.1074/jbc.270.41.24615. [DOI] [PubMed] [Google Scholar]
- 9.Weerasinghe GR, Coon SL, Bhattacharjee AK, Harry GJ, Bosetti F. Regional protein levels of cytosolic phospholipase A2 and cyclooxygenase-2 in Rhesus monkey brain as a function of age. Brain Res Bull. 2006;69:614–21. doi: 10.1016/j.brainresbull.2006.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H, Chen C. Cyclooxygenase-2 in synaptic signaling. Curr Pharm Des. 2008;14:1443–51. doi: 10.2174/138161208784480144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hahn T, Heinzel S, Plichta MM, Reif A, Lesch KP, Fallgatter AJ. Neurovascular coupling in the human visual cortex is modulated by cyclooxygenase-1 (COX-1) gene variant. Cereb Cortex. 2011;21:1659–66. doi: 10.1093/cercor/bhq236. [DOI] [PubMed] [Google Scholar]
- 12.Lecrux C, Toussay X, Kocharyan A, Fernandes P, Neupane S, Levesque M, et al. Pyramidal neurons are "neurogenic hubs" in the neurovascular coupling response to whisker stimulation. J Neurosci. 2011;31:9836–47. doi: 10.1523/JNEUROSCI.4943-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Strauss KI, Barbe MF, Marshall RM, Raghupathi R, Mehta S, Narayan RK. Prolonged cyclooxygenase-2 induction in neurons and glia following traumatic brain injury in the rat. J Neurotrauma. 2000;17:695–711. doi: 10.1089/089771500415436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dash PK, Mach SA, Moore AN. Regional expression and role of cyclooxygenase-2 following experimental traumatic brain injury. J Neurotrauma. 2000;17:69–81. doi: 10.1089/neu.2000.17.69. [DOI] [PubMed] [Google Scholar]
- 15.Domoki F, Veltkamp R, Thrikawala N, Robins G, Bari F, Louis TM, et al. Ischemia-reperfusion rapidly increases COX-2 expression in piglet cerebral arteries. Am J Physiol. 1999;277:H1207–14. doi: 10.1152/ajpheart.1999.277.3.H1207. [DOI] [PubMed] [Google Scholar]
- 16.Dégì R, Bari F, Thrikawala N, Beasley TC, Thore C, Louis TM, et al. Effects of anoxic stress on prostaglandin H synthase isoforms in piglet brain. Dev Brain Res. 1998;107:265–76. doi: 10.1016/S0165-3806(98)00022-4. [DOI] [PubMed] [Google Scholar]
- 17.Seibert K, Zhang Y, Leahy K, Hauser S, Masferrer J, Perkins W, et al. Pharmacological and biochemical demonstration of the role of cyclooxygenase 2 in inflammation and pain. Proc Natl Acad Sci U S A. 1994;91:12013–7. doi: 10.1073/pnas.91.25.12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SH, Aid S, Bosetti F. The distinct roles of cyclooxygenase-1 and -2 in neuroinflammation: implications for translational research. Trends Pharmacol Sci. 2009;30:174–81. doi: 10.1016/j.tips.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fathali N, Ostrowski RP, Lekic T, Jadhav V, Tong W, Tang J, et al. Cyclooxygenase-2 inhibition provides lasting protection against neonatal hypoxic-ischemic brain injury. Crit Care Med. 2010;38:572–8. doi: 10.1097/CCM.0b013e3181cb1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toti P DFC, Schürfeld K, Stumpo M, Bartolommei S, Lombardi A, Petraglia E, Buonocore G. Cyclooxygenase-2 immunoreactivity in the ischemic neonatal human brain. An autopsy study. J Submicrosc Cytol Pathol. 2001;33:245–9. [PubMed] [Google Scholar]
- 21.Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early HumDev 1979; 3: 79–83. [DOI] [PubMed]
- 22.Olah O, Nemeth I, Toth-Szuki V, Bari F, Domoki F. Regional differences in the neuronal expression of cyclooxygenase-2 (COX-2) in the newborn pig brain. Acta Histochem Cytochem 2012; 45: 187–92. [DOI] [PMC free article] [PubMed]
- 23.Nemeth J, Toth-Szuki V, Varga V, Kovacs V, Remzso G, Domoki F. Molecular hydrogen affords neuroprotection in a translational piglet model of hypoxic-ischemic encephalopathy. J Physiol Pharmacol. 2016;67:677–89. [PubMed] [Google Scholar]
- 24.Domoki F, Olah O, Zimmermann A, Nemeth I, Toth-Szuki V, Hugyecz M, et al. Hydrogen is neuroprotective and preserves cerebrovascular reactivity in asphyxiated newborn pigs. Pediatr Res. 2010;68:387–92. doi: 10.1203/PDR.0b013e3181f2e81c. [DOI] [PubMed] [Google Scholar]
- 25.Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, et al. Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med. 2007;13:688–94. doi: 10.1038/nm1577. [DOI] [PubMed] [Google Scholar]
- 26.Cai J, Kang Z, Liu K, Liu W, Li R, Zhang JH, et al. Neuroprotective effects of hydrogen saline in neonatal hypoxia–ischemia rat model. Brain Res. 2009;1256:129–37. doi: 10.1016/j.brainres.2008.11.048. [DOI] [PubMed] [Google Scholar]
- 27.Cai J, Kang Z, Liu WW, Luo X, Qiang S, Zhang JH, et al. Hydrogen therapy reduces apoptosis in neonatal hypoxia–ischemia rat model. Neurosci Lett. 2008;441:167–72. doi: 10.1016/j.neulet.2008.05.077. [DOI] [PubMed] [Google Scholar]
- 28.Fu Y, Ito M, Fujita Y, Ito M, Ichihara M, Masuda A, et al. Molecular hydrogen is protective against 6-hydroxydopamine-induced nigrostriatal degeneration in a rat model of Parkinson's disease. Neurosci Lett. 2009;453:81–5. doi: 10.1016/j.neulet.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 29.Qu J, Gan YN, Xie KL, Liu WB, Wang YF, Hei RY, et al. Inhalation of hydrogen gas attenuates ouabain-induced auditory neuropathy in gerbils. Acta Pharmacol Sin. 2012;33:445–51. doi: 10.1038/aps.2011.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nagata K, Nakashima-Kamimura N, Mikami T, Ohsawa I, Ohta S. Consumption of molecular hydrogen prevents the stress-induced impairments in hippocampus-dependent learning tasks during chronic physical restraint in mice. Neuropsychopharmacology. 2008;34:501–8. doi: 10.1038/npp.2008.95. [DOI] [PubMed] [Google Scholar]
- 31.Iketani M, Ohsawa I. Molecular hydrogen as a neuroprotective agent. Curr Neuropharmacol. 2017;15:324–31. doi: 10.2174/1570159X14666160607205417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olah O, Toth-Szuki V, Temesvari P, Bari F, Domoki F. Delayed neurovascular dysfunction is alleviated by hydrogen in asphyxiated newborn pigs. Neonatology. 2013;104:79–86. doi: 10.1159/000348445. [DOI] [PubMed] [Google Scholar]
- 33.Faulkner S, Bainbridge A, Kato T, Chandrasekaran M, Kapetanakis AB, Hristova M, et al. Xenon augmented hypothermia reduces early lactate/N-acetylaspartate and cell death in perinatal asphyxia. Ann Neurol. 2011;70:133–50. doi: 10.1002/ana.22387. [DOI] [PubMed] [Google Scholar]
- 34.Randall GCB. The relationship of arterial blood pH and pCO2 to the viability of the newborn piglet. Can J Comp Med. 1971;35:141–6. [PMC free article] [PubMed] [Google Scholar]
- 35.Niwa K, Araki E, Morham SG, Ross ME, Iadecola C. Cyclooxygenase-2 contributes to functional hyperemia in whisker-barrel cortex. J Neurosci. 2000;20:763–70. doi: 10.1523/JNEUROSCI.20-02-00763.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaltschmidt B, Linker RA, Deng J, Kaltschmidt C. Cyclooxygenase-2 is a neuronal target gene of NF-kappaB. BMC Mol Biol. 2002;3:16. doi: 10.1186/1471-2199-3-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hou YN, Vlaskovska M, Cebers G, Kasakov L, Liljequist S, Terenius L. A μ-receptor opioid agonist induces AP-1 and NF-κB transcription factor activity in primary cultures of rat cortical neurons. Neurosci Lett. 1996;212:159–62. doi: 10.1016/0304-3940(96)12799-3. [DOI] [PubMed] [Google Scholar]
- 38.Cadet P, Rasmussen M, Zhu W, Tonnesen E, Mantione KJ, Stefano GB. Endogenous morphinergic signaling and tumor growth. Front Biosci. 2004;9:3176–86. doi: 10.2741/1471. [DOI] [PubMed] [Google Scholar]
- 39.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-κB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta, Gene Regul Mech. 2010;1799:775–87. doi: 10.1016/j.bbagrm.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roka A, Kelen D, Halasz J, Beko G, Azzopardi D, Szabo M. Serum S100B and neuron-specific enolase levels in normothermic and hypothermic infants after perinatal asphyxia. Acta Paediatr. 2012;101:319–23. doi: 10.1111/j.1651-2227.2011.02480.x. [DOI] [PubMed] [Google Scholar]
- 41.Tooley J, Eagle R, Satas S, Thoresen M. Significant head cooling can be achieved while maintaining normothermia in the newborn piglet. Arch Dis Child Fetal Neonatal Ed. 2005;90:F262–F6. doi: 10.1136/adc.2003.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xin Y, Liu H, Zhang P, Chang L, Xie K. Molecular hydrogen inhalation attenuates postoperative cognitive impairment in rats. NeuroReport. 2017;28:694–700. doi: 10.1097/WNR.0000000000000824. [DOI] [PubMed] [Google Scholar]
- 43.Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Maekawa Y, et al. H2 gas improves functional outcome after cardiac arrest to an extent comparable to therapeutic hypothermia in a rat model. J Am Heart Assoc 2012; 1: e003459. doi: 10.1161/JAHA.112.003459. [DOI] [PMC free article] [PubMed]
- 44.Park TS, Gidday JM, Gonzales E. Local cerebral blood flow response to locally infused 2-chloroadenosine during hypotension in piglets. Dev Brain Res. 1991;61:73–7. doi: 10.1016/0165-3806(91)90115-Y. [DOI] [PubMed] [Google Scholar]
- 45.Gidday JM, Park TS. Effect of 2-chloroadenosine on cerebrovascular reactivity to hypercapnia in newborn pig. J Cereb Blood Flow Metab. 1992;12:656–63. doi: 10.1038/jcbfm.1992.90. [DOI] [PubMed] [Google Scholar]
- 46.Ruth VJ, Park TS, Gonzales ER, Gidday JM. Adenosine and cerebrovascular hyperemia during insulin-induced hypoglycemia in newborn piglet. Am J Physiol, Heart Circ Physiol. 1993;265:H1762–H8. doi: 10.1152/ajpheart.1993.265.5.H1762. [DOI] [PubMed] [Google Scholar]
- 47.Ono H, Nishijima Y, Adachi N, Sakamoto M, Kudo Y, Kaneko K, et al. A basic study on molecular hydrogen (H2) inhalation in acute cerebral ischemia patients for safety check with physiological parameters and measurement of blood H2 level. Med Gas Res. 2012;2:21. doi: 10.1186/2045-9912-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Anjana Vaman VS, Tinu SK, Geetha CS, Lissy KK, Mohanan PV. Effect of fibrin glue on antioxidant defense mechanism, oxidative DNA damage and chromosomal aberrations. Toxicol Mech Methods. 2013;23:500–8. doi: 10.3109/15376516.2013.785625. [DOI] [PubMed] [Google Scholar]
- 49.Villaño D, Vilaplana C, Medina S, Cejuela-Anta R, Martínez-Sanz JM, Gil P, et al. Effect of elite physical exercise by triathletes on seven catabolites of DNA oxidation. Free Radic Res. 2015;49:973–83. doi: 10.3109/10715762.2015.1025388. [DOI] [PubMed] [Google Scholar]
- 50.Hintsala HR, Jokinen E, Haapasaari KM, Moza M, RistimÄKi ARI, Soini Y, et al. Nrf2/Keap1 pathway and expression of oxidative stress lesions 8-hydroxy-2′-deoxyguanosine and nitrotyrosine in melanoma. Anticancer Res. 2016;36:1497–506. [PubMed] [Google Scholar]
- 51.Nunomura A, Moreira PI, Castellani RJ, Lee HG, Zhu X, Smith MA, et al. Oxidative damage to RNA in aging and neurodegenerative disorders. Neurotox Res. 2012;22:231–48. doi: 10.1007/s12640-012-9331-x. [DOI] [PubMed] [Google Scholar]
- 52.Huang Q, Chen Z, Yang Y, Wang B, Li F, Zhan M. Content change and clinical significance of oxidation damage products 8-OHdG, 4-HNE and NTY in hemangioma tissue. Int J Clin Exp Pathol. 2016;9:8847–57. [Google Scholar]
- 53.Campo GM, Avenoso A, Campo S, D'Ascola A, Traina P, Samà D, et al. The antioxidant effect exerted by TGF-1β-stimulated hyaluronan production reduced NF-kB activation and apoptosis in human fibroblasts exposed to FeSo4 plus ascorbate. Mol Cell Biochem. 2008;311:167–77. doi: 10.1007/s11010-008-9707-7. [DOI] [PubMed] [Google Scholar]
- 54.Hoffmann A, Levchenko A, Scott ML, Baltimore D. The IκB-NF-κB signaling module: temporal control and selective gene activation. Science. 2002;298:1241–5. doi: 10.1126/science.1071914. [DOI] [PubMed] [Google Scholar]
- 55.O'Neill LAJ, Kaltschmidt C. NF-kB: a crucial transcription factor for glial and neuronal cell function. Trends Neurosci. 1997;20:252–8. doi: 10.1016/S0166-2236(96)01035-1. [DOI] [PubMed] [Google Scholar]
- 56.Salminen A, Liu PK, Hsu CY. Alteration of transcription factor binding activities in the ischemic rat-brain. Biochem Biophys Res Commun. 1995;212:939–44. doi: 10.1006/bbrc.1995.2060. [DOI] [PubMed] [Google Scholar]
- 57.Shi Y, Wang G, Li J, Yu W. Hydrogen gas attenuates sevoflurane neurotoxicity through inhibiting nuclear factor kappa-light-chain-enhancer of activated B cells signaling and proinflammatory cytokine release in neonatal rats. Neuroreport. 2017;28:1170–5. doi: 10.1097/WNR.0000000000000899. [DOI] [PubMed] [Google Scholar]
- 58.Kaltschmidt C, Kaltschmidt B, Lannes-Vieira J, Kreutzberg GW, Wekerle H, Baeuerle PA, et al. Transcription factor NF-κB is activated in microglia during experimental autoimmune encephalomyelitis. J Neuroimmunol. 1994;55:99–106. doi: 10.1016/0165-5728(94)90151-1. [DOI] [PubMed] [Google Scholar]
- 59.Indo HP, Davidson M, Yen HC, Suenaga S, Tomita K, Nishii T, et al. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion. 2007;7:106–18. doi: 10.1016/j.mito.2006.11.026. [DOI] [PubMed] [Google Scholar]
- 60.Phaniendra A, Jestadi DB, Periyasamy L. Free radicals: properties, sources, targets, and their implication in various diseases. Indian J Clin Biochem. 2015;30:11–26. doi: 10.1007/s12291-014-0446-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- 62.Domoki F, Perciaccante JV, Puskar M, Bari F, Busija DW. Cyclooxygenase-2 inhibitor NS398 preserves neuronal function after hypoxia/ischemia in piglets. Neuroreport. 2001;12:4065–8. doi: 10.1097/00001756-200112210-00041. [DOI] [PubMed] [Google Scholar]