

Abstract
SMARCA2 is a critical catalytic subunit of the switch/sucrose non-fermenting (SWI/SNF) chromatin remodeling complexes. Dysregulation of SMARCA2 is associated with several diseases, including some cancers. SMARCA2 is multi-domain protein containing a bromodomain (BRD) that specifically recognizes acetylated lysine residues in histone tails, thus playing an important role in chromatin remodeling. Many potent and specific inhibitors targeting other BRDs have recently been discovered and have been widely used for cancer treatments and biological research. However, hit discovery targeting SMARCA2-BRD is particularly lacking. To date, there is a paucity of reported high-throughput screening (HTS) assays targeting the SMARCA2-BRD interface. In this study, we developed an AlphaScreen HTS system for the discovery of SMARCA2-BRD inhibitors and optimized the physicochemical conditions including pH, salt concentrations and detergent levels. Through an established AlphaScreen-based high-throughput screening assay against an in-house compound library, DCSM06 was identified as a novel SMARCA2-BRD inhibitor with an IC50 value of 39.9±3.0 μmol/L. Surface plasmon resonance demonstrated the binding between SMARCA2-BRD and DCSM06 (K d=38.6 μmol/L). A similarity-based analog search led to identification of DCSM06-05 with an IC50 value of 9.0±1.4 μmol/L. Molecular docking was performed to predict the binding mode of DCSM06-05 and to decipher the structural basis of the infiuence of chemical modifications on inhibitor potency. DCSM06-05 may be used as a starting point for further medicinal chemistry optimization and could function as a chemical tool for SMARCA2-related functional studies.
Keywords: AlphaScreen, high-throughput screening, SMARCA2, bromodomain, small molecule inhibitor
Introduction
The switch/sucrose non-fermenting (SWI/SNF) complexes are evolutionarily conserved nucleosome remodeling factors that use the energy derived from ATP hydrolysis to remodel chromatin1. SWI/SNF complexes contain the evolutionarily conserved subunit SMARCA2 (SWI/SNF related, Matrix associated, Actin dependent Regulator of Chromatin, subfamily A, member 2), which forms the catalytic core of the remodeling complexes2,3.
SMARCA2 has distinct domain structures containing various motifs, including an ATP domain, a bromodomain (BRD) and a chromodomain, that contribute specifically to chromatin-remodeling activities4,5. The BRD is a protein module of ∼110 amino acids that contains a conserved structure composed of four α helices linked by flexible loops; this domain specifically recognizes acetylated lysine residues (KAc) on protruding histone tails6,7,8. Emerging evidence demonstrates that bromodomains are involved in a plethora of biological processes, including transcriptional regulation, the formation of scaffolding proteins and signal transduction9,10,11. Recent findings indicated that dysregulation of SMARCA2 occurs in approximately 10%–20% of solid tumors, including lung cancer, prostate cancer and
gastric cancer12; thus, this target has gained considerable attention in both academia and industry13,14. Although many potent and specific inhibitors targeting other BRDs have recently been discovered and widely applied to cancer treatments and biological research15,16,17,18,19, hit discovery targeting SMARCA2-BRD is particularly lacking. To date, only compound PFI-3 has been identified as a bona fide inhibitor targeting the SMARCA2 bromodomain20. There is still an urgent need to develop inhibitors of novel chemotypes that may function as chemical probes for in vivo SMARCA2-related mechanism studies.
In the present work, we have developed an optimized AlphaScreen HTS assay for the discovery of small-molecule inhibitors targeting the SMARCA2-BRD and histone H4 interface. The high Z' factors and signal-to-background ratio at different dimethyl sulfoxide (DMSO) concentrations indicate that the assay is robust and reproducible. Based on this platform, we performed a high-throughput screen against an in-house compound library containing ca. 20 000 diverse compounds, leading to the identification of a novel SMARCA2-BRD inhibitor, DCSM06. Through similarity-based analogue searching, we discovered the more potent derivative DCSM06-05, which may provide a novel chemotype for further in vivo SMARCA2-related functional studies.
Materials and methods
Protein expression and purification
The human SMARCA2 bromodomain (1373 to 1493) DNA sequence was cloned into a pET28a vector. The fusion protein was expressed with an N-terminal 6×His-tag in Escherichia coli BL21 (DE3) cells. When the OD 600 reached 0.6–0.8 at 37 °C, cell cultures were then induced with 0.4 mmol/L isotropy-β-D-thiogalactoside (IPTG) overnight (14–16 h) at 16 °C. The cell pellets were collected and sonicated in pre-cooled lysis buffer (50 mmol/L HEPES pH 8.0, 150 mmol/L NaCl, 1 mmol/L DTT and 10 mmol/L imidazole). After centrifugation at 18 000 r/min for 30 min at 4 °C, the supernatant was loaded onto nickel affinity columns (HisTrap FF, GE Healthcare), and the recombinant protein was eluted with 350 mmol/L imidazole. Then, the protein was further purified by gel-filtration chromatography using a Superdex 200 10/300 GL column (GE Healthcare) in storage buffer containing 25 mmol/L HEPES pH 8.0, 150 mmol/L NaCl, and 1 mmol/L DTT. The protein was concentrated to 5 mg/mL and flash-frozen in liquid nitrogen for further use.
Protein thermal shift assay
Protein thermal shift assays were performed as previously described21. In each well, 5 μmol/L SMARCA2-BRD protein and 5×SYPRO Orange (Invitrogen) were mixed in a total volume of 20 μL containing 25 mmol/L buffer and 100 mmol/L NaCl. The assay plate was heated from 25 °C to 95 °C at a ramp rate of 0.05 °C/s. The changes in SYPRO Orange fluorescence intensity were monitored and collected for further analysis. The melting temperature (Tm) was calculated with the Boltzmann fitting method in the Protein Thermal Shift Software v1.2. Different buffer salts, including MES (pH 5.5–6.8), BIS-TRIS (pH 5.8–8.0), PIPES (pH 6.1–7.9) and MOPS (pH 6.5–8.0), were tested for buffer optimization. The experiments were performed in triplicate and independently repeated three times.
Peptide synthesis
The peptide abbreviated as “H4 peptide” was synthesized by Shanghai China Peptide Corporation with the sequence SGRG-K (Ac)-GG-K (Ac)-GLG-K (Ac)-GGA-K (Ac)-RHRKVGG-K-Biotin6. The peptide was purified by reverse phase preparative HPLC using a Waters C-18 reverse phase column. The molecular weight and purity of the peptide were validated by using an API150-ESI mass spectrometry system and HPLC. Fractions were lyophilized into white powder for long-term storage.
Molecular library
A ligand database containing ca. 200 000 diverse structures (each with more than 10 mg of stored compound) was extracted from the SPECS Company (SPECS_SC_10mg_Dec2016). The database was filtered by Lipinski's rule and the pan-assay interference compounds (PAINS) rule22 using Pipeline Pilot, version 7.5 (Pipeline Pilot; Accelrys Software Inc., San Diego, CA, USA). The remaining molecules were clustered into 20 000 groups according to their structural differences using the Cluster Molecules component of Pipeline Pilot, version 7.5. Then, we selected 20 000 stable and structurally representative drug-like compounds from each group to build an in-house molecular library for the assays in this study. All compounds dissolved in DMSO were stored at 4 °C for long-term storage.
AlphaScreen high-throughput screening assay
As shown in Table S1, all reagents were diluted with 1×assay buffer and allowed to equilibrate to room temperature prior to addition to plates. A total of 2.5 μL of assay buffer or compounds was pre-plated into 384-well plates (OptiPlate, PerkinElmer). Then, 2.5 μL of 200 nmol/L SMARCA2-BRD protein was transferred into the assay plate. Plates were sealed and incubated at room temperature for 20 min, and 5 μL of biotinylated peptide H4 was added to a final concentration of 100 nmol/L. Plates were sealed and incubated at room temperature for another 30 min. Then, 5 μL of nickel-chelate acceptor beads (PerkinElmer) and 5 μL of streptavidin-conjugated donor beads (PerkinElmer) were mixed and added under subdued light. Plates were sealed and incubated at room temperature for 60 min, and signals were read on a Multilabel Reader (EnVision, PerkinElmer) using a 680 nm dichroic AlphaScreen™ mirror for excitation and a 570 nm cutoff filter for emission. The compound PFI-3 was used as the positive control.
Z′ factor and S/B calculation
The Z′ factor is commonly used as an indicator of high-throughput screening assay performance and is calculated as follows:
Z′=1-3(δp+δn) /∣(μn−μp)∣
In this formula, the means and standard deviations of the positive (p) and negative (n) controls are denoted as μp, δp and μn, δn respectively23,24. DMSO and PFI-3 (40 μmol/L) are the negative and positive controls, respectively, and were included on each plate to calculate the Z′ factor. The S/B value is the ratio of the mean of the negative controls to the mean of the positive controls in the reactions treated with 40 μmol/L PFI-3.
Surface plasmon resonance (SPR)-based binding assays
The SPR binding assays were performed on a Biacore T200 instrument (GE Healthcare) at 25 °C. SMARCA2-BRD protein was covalently immobilized on a CM5 chip using a standard amine-coupling procedure in 10 mmol/L sodium acetate (pH 5.0). The chip was first equilibrated with HBS buffer (10 mmol/L HEPES pH 7.4, 150 mmol/L NaCl, and 0.1% (v/v) DMSO) overnight. The compound was serially diluted with HBS buffer and injected for 120 s to allow binding; this was followed by a 120 s dissociation step. The K d values of the compound (representing binding to SMARCA2-BRD) were determined using Biacore T200 evaluation software (GE Healthcare).
Similarity-based analogue searching
The prepared SPECS library was searched in Pipeline Pilot, version 7.5 (Accelrys Software Inc, San Diego, CA, USA) to investigate preliminary structure-activity relationships (SAR). Derivatives of interest were identified and purchased for biological activity tests.
Molecular modeling
The molecular modeling software Maestro (Schrödinger LLC 2015, USA) was selected to conduct molecular docking experiments. PFI-3 and DCSM06-05 were prepared with LigPrep and Advanced Conformational Search to generate protonation states and conformations. The crystal structure of SMARCA2-BRD bound to its inhibitor PFI-3 (from the PDB database, PDB ID: 5DKC) was chosen as the molecular docking receptor. The protein was prepared with the Protein Preparation Wizard Workflow, with a pH value of 7.4. SP and XP modes of induced-fit docking were used for the experiment. The docking center was set to the PFI-3 binding site. Other parameters were set as the default.
Results
Optimization of physicochemical conditions
Buffer conditions–including the pH value, salt concentration, and detergent percentage–were optimized in order to select the buffer in which SMARCA2-BRD is most stable and to improve the detection limit for high-throughput screening assays. Protein thermal shift assays were carried out to investigate the stability of SMARCA2-BRD under different pH conditions (5.5 to 8.0). We observed a relative increase in the Tm value when using HEPES buffer at pH 8.0 (Figure 1A and Figure S1A). Thus, HEPES buffer pH 8.0 was selected for the development of our AlphaScreen assay.
Figure 1.
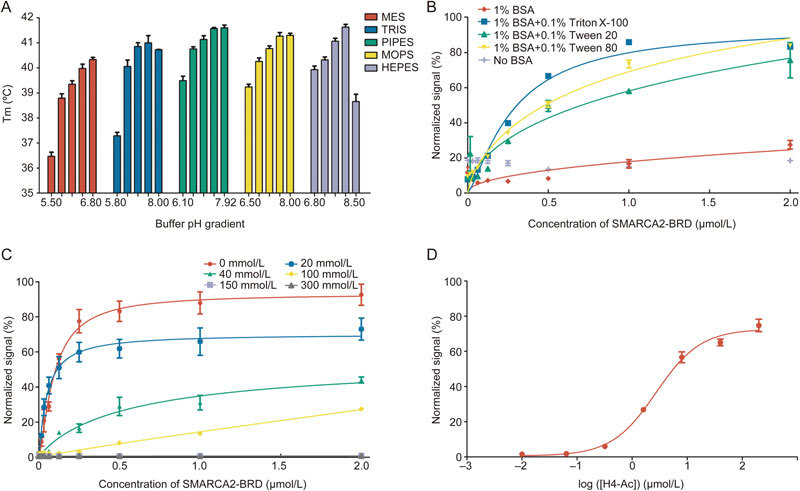
AlphaScreen assay development and optimization. (A) The thermostability of SMARCA2-BRD in different buffers. (B) AlphaScreen saturation binding curves with increasing concentrations of SMARCA2-BRD in the absence or presence of different detergents. (C) AlphaScreen saturation binding curves with different salt concentrations. (D) AlphaScreen saturation binding curves with increasing concentrations of H4 peptide in optimized buffer.
Previous studies have demonstrated that the application of additives could prevent polypeptide aggregation, thus reducing nonspecific signals and ensuring the solubility and stability of components25. Therefore, we studied different additives during our AlphaScreen assay development. The SMARCA2-BRD AlphaScreen normalized signal was plotted as a function of protein titration (from 0.01 to 2 μmol/L) in the absence or presence of different detergents (Figure 1B). Of these detergents, 1% (w/v) BSA and 0.01% (v/v) Triton X-100 proved to be most efficient at elevating the signal-background ratio and decreasing non-specific binding. Hence, both 1% (w/v) BSA and 0.1% (v/v) Triton X-100 were added to the AlphaScreen assay buffer.
Salt concentration is another important component of biological assays that must be optimized to maintain ionic strength and satisfy the specific requirements of individual proteins. Thus we studied a range of different NaCl concentrations (0 mmol/L–300 mmol/L) (Figure 1C). As the salt concentration increased, the signal-background ratio of the AlphaScreen signal decreased significantly. With these results in mind, the optimal AlphaScreen assay buffer was selected using 25 mmol/L HEPES pH 8.0, 1% (w/v) BSA, and 0.01% (v/v) Triton X-100. Replacement of HEPES pH 8.0 with more acidic buffer salts led to decreased SMARCA2-BRD binding activity, which is consistent with previous observations (Figure S1B). Under the optimized conditions described above, the relative low concentrations (200 nmol/L SMARCA2-BRD and 100 nmol/L biotinylated H4 peptide) that provided reliable AlphaScreen signals were selected for future application (Figure 1D).
Evaluation of DMSO tolerance and Z' factor
DMSO is a widely applied solvent typically used to dissolve small-molecule compounds. Thus, DMSO tolerance is one of most important factors influencing the stability of high-throughput screens. Therefore, we studied various concentrations (0%–10%) of DMSO in our assay. As illustrated in Figure 2A–2B, although increased DMSO concentrations led to lower signal-to-background ratios (S/B), at 8% DMSO, the S/B was still greater than 10-fold, and the K d values were stable. This indicated that our AlphaScreen assay platform was highly robust and qualified for inhibitor screening.
Figure 2.
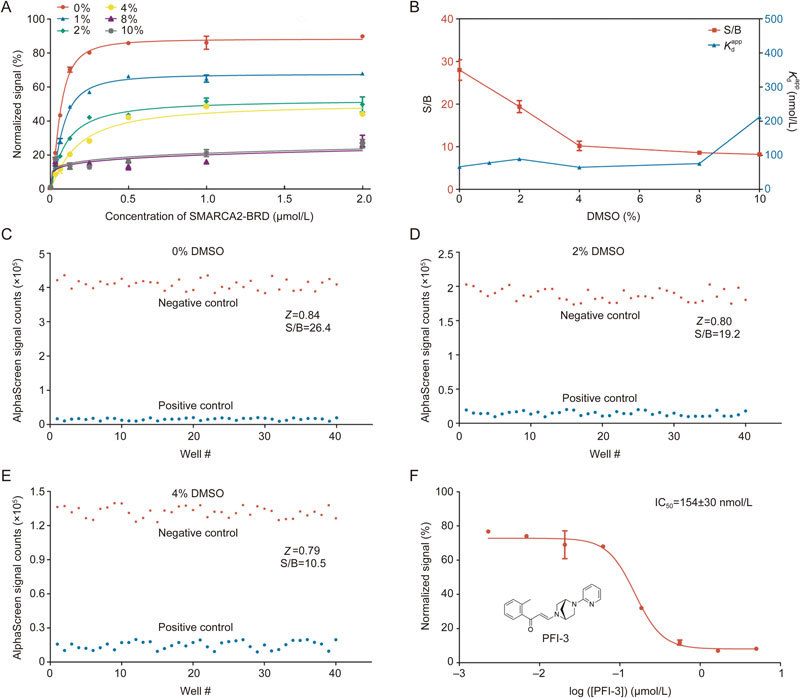
Evaluation of DMSO tolerance in the optimized AlphaScreen binding assay. (A) AlphaScreen saturation binding curves in the absence or presence of different DMSO concentrations. (B) Signal-background ratio and apparent K d values at different DMSO concentrations. (C–E) Evaluation of Z' factors for the AlphaScreen high-throughput screening assay in the absence or presence of 0%, 2%, and 4% DMSO. The AlphaScreen normalized signals from both negative and positive controls are shown in a scatter plot. (F) IC50 curve for PFI-3. All experiments were performed at least three times. The data were analyzed in GraphPad Prism 5.0.
In addition, the Z' factor was evaluated to analyze data variation and signal dynamic range in this assay26. We evaluated the effects of DMSO concentration (0%, 2% and 4%) on the Z' factor; these concentrations are commonly used in HTS assays (Figure 3C–3E). Generally, high-throughput screens with a Z' factor value greater than 0.5 are considered to be highly qualified systems27,28. The Z' factors in our AlphaScreen assays were greater than 0.7, even in the presence of 4% DMSO. These results clearly revealed that our AlphaScreen assay is robust, reproducible, and appropriate for HTS with DMSO concentrations up to 4%. The feasibility of this high-throughput method was further verified by testing the inhibitory activities of the positive compound PFI-3; the reported IC50 value for this compound was 0.154±0.030 μmol/L, which is in line with previous reports (Figure 3F)29.
Figure 3.
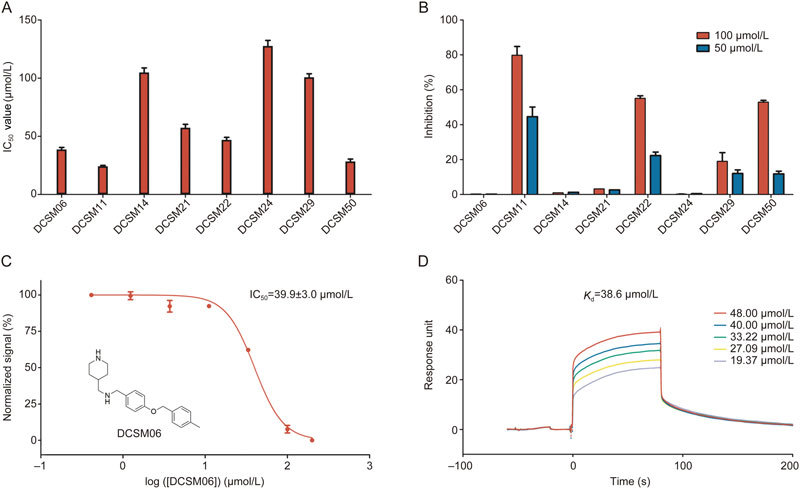
Discovery and validation of novel SMARCA2-BRD inhibitors. (A) Primary AlphaScreen high-throughput screening results. (B) AlphaScreen TruHits kit validation. (C) Inhibitory activities of DCSM06. (D) SPR assay demonstrated binding between SMARCA2-BRD and DCSM06.
High-throughput screening against an in-house compound library
We established our in-house compound library containing ca. 20 000 compounds of diverse chemical scaffolds through filtering and optimization using the Lipinski rules of five and the PAINS filter in Pipeline Pilot, version 7.5 (Pipeline Pilot; Accelrys Software Inc., San Diego, CA). We performed a primary screen at a single concentration (200 μmol/L), and then, compounds with inhibitory activity >90% were identified for further evaluation.
After a primary AlphaScreen-based high-throughput screen, we identified 8 hits with novel scaffolds, the IC50 values of which are shown in Figures 3A and S2. Because fluorescence quenching agents, light scattering agents (insoluble compounds), singlet oxygen quenching agents and biotin mimetics commonly interfere with AlphaScreen signals and result in false positives, the AlphaScreen TruHits kit was used to identify false positives in the screening process. As shown in Figure 3B, three of eight compounds, including DCSM11, DCSM22, and DCSM50, displayed greater than 50% inhibition at a 100 μmol/L concentration. Thus, these compounds were excluded from further validation steps. Among the remaining hit compounds, DCSM06 showed the greatest inhibitory activity, with an IC50 value of 39.9±3.0 μmol/L (Figure 3C).
SPR-based binding assay
Surface plasmon resonance (SPR), one of most useful biophysical methods for hit validation, was used to demonstrate the binding between DCSM06 and SMARCA2-BRD30. As shown in Figure 3D, DCSM06 directly bound to SMARCA2-BRD with an equilibrium dissociation constant (K d) of 38.6 μmol/L, which was consistent with the IC50 value of 39.9±3.0 μmol/L. These results suggest that DCSM06 binds to SMARCA2-BRD in vitro.
Similarity-based analogue searching
To further study the structure activity relationship (SAR), we used Pipeline Pilot to identify several derivatives, and we purchased these derivatives from the SPECS Company. The structures and their inhibitory activities against SMARCA2-BRD are summarized in Table 1 and Figure S2. Of these, the derivative DCSM06-05 displayed the best inhibitory activity, with an IC50 value of 9.0±1.4 μmol/L and a K d value of 22.4 μmol/L based on SPR binding assays (Figure S3, Figure 4A–4B). Thus, DCSM06-05 was chosen for further analyses.
Table 1.
Inhibitory activities of DCSM06 derivatives.
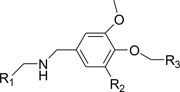
| ||||
|---|---|---|---|---|
| Compound ID | R1 | R2 | R3 | IC50 (μmol/L) |
| DCSM06-01 |
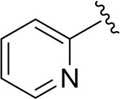
|
H |
|
9.7±1.4 |
| DCSM06-02 |
|
H |
|
13.2±2.9 |
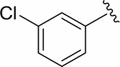
| DCSM06-03 |
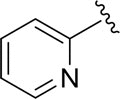
|
Cl |
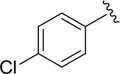
|
35.2±3.2 |
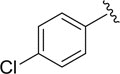
| DCSM06-04 |
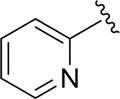
|
Cl |
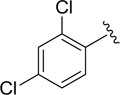
|
9.1±1.8 |
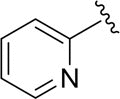
| DCSM06-05 |
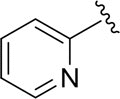
|
H |
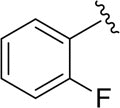
|
9.0±1.4 |
| DCSM06-06 |
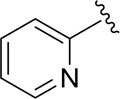
|
H |
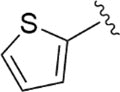
|
17.8±1.9 |
| DCSM06-07 |
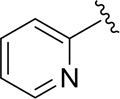
|
H |
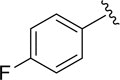
|
16.7±2.3 |
| DCSM06-08 |
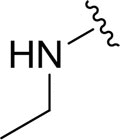
|
H |
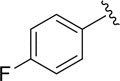
|
aN.D. |
| DCSM06-09 |
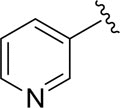
|
H |
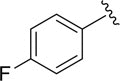
|
81.3±3.1 |
| DCSM06-10 |
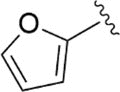
|
Cl |
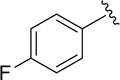
|
73.3±4.1 |
aN.D. represents not determined.
Figure 4.
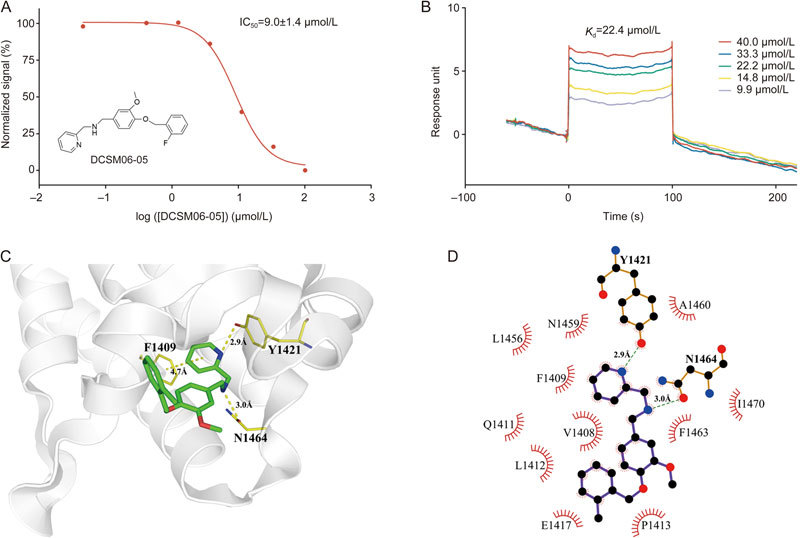
Hit validation and analysis of the mechanism of action of the derivative DCSM06-05. (A) Inhibitory activities of DCSM06-05. (B) SPR assay demonstrated binding between SMARCA2-BRD and DCSM06-05. (C) Molecular docking analysis elucidated key interactions with SMARCA2-BRD. (D) Schematic diagram showing interactions between SMARCA2-BRD and DCSM06-05. Hydrogen bonds are shown in green dashed lines, and residues involved in these interactions were illustrated in the LigPlot+ program33.
Docking simulations and SAR analysis
To reveal the binding modes and mechanisms of interaction between DCSM06 derivatives and SMARCA2-BRD, the crystal structures of DCSM06-05 and SMARCA2 (PDB ID: 5DKC) were selected for a molecular docking study using the molecular modeling software Maestro (Schrödinger LLC 2015, USA). First, we re-docked the positive control compound, PFI-3, into the SMARCA2-BRD binding pocket to validate the docking model. As illustrated in Figure S4, the model of PFI-3 docking (in yellow) successfully reproduced the real binding mode of this inhibitor (in green). After the reliability and practicability of the docking model were validated, we docked DCSM06-05 into the SMARCA2 binding pocket . The binding results suggested that DCSM06-05 displayed a similar binding mode to PFI-3. Further, similar hydrogen bonds stabilized the DCSM06-05 docking conformation, though the precise interaction details need further experimental validation. As shown in Figure 4D, DCSM06-05 forms π-π stacking interactions with F1409, which is a universal feature of inhibitors binding to this site in this model. The 2-pyridine (R1) group contributes significantly to improvements in inhibitory activity, and any chemical modifications to this group may reduce the potency of inhibition. This demonstrates that a hydrogen bond interaction between the nitrogen of the inhibitor's pyridine group and F1409 is preferred to ensure enhanced binding affinities in this series of inhibitors. Moreover, the benzene ring docks into a shallow hydrophobic pocket formed by P1413 and I1470, and methoxy substitution can create a hydrogen bond with F1409 in a similar manner as PFI-3. Additionally, slight differences in halogen substitutions in the R3 region do not appear to have a major influence on the inhibitory activity of these compounds. Collectively, the docking results suggest that DCSM06-05 can bind within the H4 peptide-binding site of SMARCA2-BRD in a manner quite similar to the binding of PFI-3. These results help us to decipher the structural basis of the improved potency of DCSM06-05.
Discussion
Bromodomains specifically recognize acetylated lysine residues on histones and play a pivotal role in downstream gene transcription, DNA repair and chromatin remodeling. The therapeutic targeting of bromodomains has recently generated great interest in industry and academia31,32. In sharp contrast to the rapid discovery and development of drugs targeting BET family proteins, very little investigation into non-BET family bromodomains has occurred. PFI-3 was the only potent chemical probe to be discovered as a SMARCA2-BRD interactor, limiting further biological studies. There is still an urgent need to develop potent SMARCA2-BRD inhibitors of novel chemotypes.
In this study, we developed a robust and reproducible AlphaScreen-based HTS assay targeting SMARCA2-BRD. The high Z' factors (>0.7) at different DMSO concentrations demonstrated the robustness and reliability of this AlphaScreen HTS assay. Based on this platform, we conducted a pilot screen against an in-house compound library, leading to the identification of DCSM06, which has an IC50 value of 39.9±3.0 μmol/L. Through 2D similarity-based analogue searching, DCSM06-05 was identified as a more potent inhibitor targeting SMARCA2-BRD with an IC50 value of 9.0±1.4 μmol/L. Further molecular docking analyses provided a detailed mechanism of action for DCSM06-05 and provided a preliminary structure-activity relationship that could be exploited for further modification.
Considering all these data together, this robust AlphaScreen-based HTS assay may be a reliable platform that could be used to speed up hit discovery targeting SMARCA2-BRD and other non-BET family proteins. Further, DCSM06-05 may be a starting point for further medicinal chemistry optimization and could be used as an efficient chemical probe to uncover the elusive role of SMARCA2 in physiological and pathological processes.
Author contribution
Cheng LUO, Hui-fang CHAI and Kai-xian CHEN designed the research; Wen-chao LU, Tian LU and Jie HAN performed the biological experiments; Jun-chi HU and Hao JIANG performed computational analysis; Hong DING Yuan-yuan ZHANG, Li-yan YUE and Shi-jie CHEN analyzed and interpreted the data. Tian LU, Jun-chi HU and Wen-chao LU wrote the manuscript; Cheng LUO, Hua-liang JIANG, and Kai-xian CHEN reviewed the manuscript. All authors read and approved the manuscript.
Electronic supplementary material
Supplementary Table and Figures
Acknowledgements
The authors disclose receipt of the following financial support for the research and/or authorship of this article: the Ministry of Science and Technology of China (2017YFB0202600 to Shi-jie CHEN and 2015CB910304 to Yuan-yuan ZHANG); the National Natural Science Foundation of China (21472208 and 81625022 to Cheng LUO and 81430084 to Kai-xian CHEN). We are extremely grateful to the National Centre for Protein Science Shanghai (Shanghai Science Research Center, Protein Expression and Purification system) for their instrumental support and technical assistance.
Contributor Information
Kai-xian Chen, Email: kxchen@simm.ac.cn.
Hui-fang Chai, Email: saieho@126.com.
Cheng Luo, Email: cluo@simm.ac.cn.
Electronic supplementary material
Supplementary Information is available at the website of Acta Pharmacologica Sinica.10.1038/aps.2017.188
References
- 1.Hirschhorn JN, Brown SA, Clark CD, Winston F. Evidence that Snf2/Swi2 and Snf5 activate transcription in yeast by altering chromatin structure. Gene Dev. 1992;6:2288–98. doi: 10.1101/gad.6.12a.2288. [DOI] [PubMed] [Google Scholar]
- 2.Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, et al. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34:1136–44. doi: 10.1128/MCB.01372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhou J, Zhang M, Fang H, El-Mounayri O, Rodenberg JM, Imbalzano AN, et al. The SWI/SNF chromatin remodeling complex regulates myocardin-induced smooth muscle-specific gene expression. Arterioscler Thromb Vasc Biol. 2009;29:921–8. doi: 10.1161/ATVBAHA.109.187229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13:373–80. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Z, Wang F, Du C, Guo H, Ma L, Liu X, et al. BRM/SMARCA2 promotes the proliferation and chemoresistance of pancreatic cancer cells by targeting JAK2/STAT3 signaling. Cancer Lett. 2017;402:213–24. doi: 10.1016/j.canlet.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 6.Philpott M, Yang J, Tumber T, Fedorov O, Uttarkar S, Filippakopoulos P, et al. Bromodomain-peptide displacement assays for interactome mapping and inhibitor discovery. Mol Biosyst. 2011;7:2899–908. doi: 10.1039/c1mb05099k. [DOI] [PubMed] [Google Scholar]
- 7.Chandrasekaran R, Thompson M. Polybromo-1-bromodomains bind histone H3 at specific acetyl-lysine positions. Biochem Biophys Res Commun. 2007;355:661–6. doi: 10.1016/j.bbrc.2007.01.193. [DOI] [PubMed] [Google Scholar]
- 8.Sun Z, Zhang H, Chen Z, Xie Y, Jiang H, Chen L, et al. Discovery of novel BRD4 inhibitors by high-throughput screening, crystallography, and cell-based assays. Bioorg Med Chem Lett. 2017;27:2003–9. doi: 10.1016/j.bmcl.2017.03.012. [DOI] [PubMed] [Google Scholar]
- 9.Muchardt C, Reyes JC, Bourachot B, Leguoy E, Yaniv M. The HBRM and BRG-1 proteins, components of the human SNF/SWI complex, are phosphorylated and excluded from the condensed chromosomes during mitosis. EMBO J. 1996;15:3394–402. doi: 10.1002/j.1460-2075.1996.tb00705.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanchez R, Zhou MM. The role of human bromodomains in chromatin biology and gene transcription. Curr Opin Drug Discov Devel. 2009;12:659–65. [PMC free article] [PubMed] [Google Scholar]
- 11.Marmorstein R, Berger SL. Structure and function of bromodomains in chromatin-regulating complexes. Gene. 2001;272:1–9. doi: 10.1016/S0378-1119(01)00519-4. [DOI] [PubMed] [Google Scholar]
- 12.Biegel JA, Busse TM, Weissman BE. SWI/SNF chromatin remodeling complexes and cancer. Am J Med Genet C Semin Med Genet. 2014;166C:350–66. doi: 10.1002/ajmg.c.31410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gui Y, Guo G, Huang Y, Hu X, Tang A, Gao S, et al. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat Genet. 2011;43:875–8. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hohmann AF, Vakoc CR. A rationale to target the SWI/SNF complex for cancer therapy. Trends Genet. 2014;30:356–63. doi: 10.1016/j.tig.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Delmore JE, Issa GC, Lemieux ME, Rahl PB, Shi J, Jacobs HM, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146:904–17. doi: 10.1016/j.cell.2011.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perez-Salvia M, Esteller M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics. 2017;12:323–39. doi: 10.1080/15592294.2016.1265710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith SG, Zhou MM. The bromodomain: a new target in emerging epigenetic medicine. ACS Chem Biol. 2016;11:598–608. doi: 10.1021/acschembio.5b00831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jung M, Gelato KA, Fernandez-Montalvan A, Siegel S, Haendler B. Targeting BET bromodomains for cancer treatment. Epigenomics. 2015;7:487–501. doi: 10.2217/epi.14.91. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez R, Meslamani J, Zhou MM. The bromodomain: from epigenome reader to druggable target. Biochim Biophys Acta. 2014;1839:676–85. doi: 10.1016/j.bbagrm.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gerstenberger BS, Trzupek JD, Tallant C, Fedorov O, Filippakopoulos P, Brennan PE, et al. Identification of a chemical probe for family VIII bromodomains through optimization of a fragment hit. J Med Chem. 2016;59:4800–11. doi: 10.1021/acs.jmedchem.6b00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang WY, Lu WC, Jiang H, Lv ZB, Xie YQ, Lian FL, et al. Discovery of alkyl bis (oxy) dibenzimidamide derivatives as novel protein arginine methyltransferase 1 (PRMT1) inhibitors. Chem Biol Drug Des 2017; 1–11. [DOI] [PubMed]
- 22.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–40. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 23.Wen Y, Xu L, Chen FL, Gao J, Li JY, Hu LH, et al. Discovery of a novel inhibitor of NAD(P)(+)-dependent malic enzyme (ME2) by high-throughput screening. Acta Pharmacol Sin. 2014;35:674–84. doi: 10.1038/aps.2013.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang TT, Huang ZT, Dai Y, Chen XP, Zhu P, Du GH. High-throughput fluorescence polarization method for identifying ligands of LOX-1. Acta Pharmacol Sin. 2006;27:447–52. doi: 10.1111/j.1745-7254.2006.00281.x. [DOI] [PubMed] [Google Scholar]
- 25.Zhu MR, Du DH, Hu JC, Li LC, Liu JQ, Ding H, et al. Development of a high-throughput fluorescence polarization assay for the discovery of EZH2-EED interaction inhibitors. Acta Pharmacol Sin. 2018;39:302–10. doi: 10.1038/aps.2017.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang JH, Chung TDY, Oldenburg KR. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 27.Shockley KR. Quantitative high-throughput screening data analysis: challenges and recent advances. Drug Discov Today. 2015;20:296–300. doi: 10.1016/j.drudis.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sui Y, Wu Z. Alternative statistical parameter for high-throughput screening assay quality assessment. J Biomol Screen. 2007;12:229–34. doi: 10.1177/1087057106296498. [DOI] [PubMed] [Google Scholar]
- 29.Vangamudi B, Paul TA, Shah PK, Kost-Alimova M, Nottebaum L, Shi X, et al. The SMARCA2/4 ATPase domain surpasses the bromodomain as a drug target in SWI/SNF-Mutant cancers: insights from cDNA rescue and PFI-3 inhibitor studies. Cancer Res. 2015;75:3865–78. doi: 10.1158/0008-5472.CAN-14-3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Myszka DG. Kinetic analysis of macromolecular interactions using surface plasmon resonance biosensors. Curr Opin Biotech. 1997;8:50–7. doi: 10.1016/S0958-1669(97)80157-7. [DOI] [PubMed] [Google Scholar]
- 31.Harner MJ, Chauder BA, Phan J, Fesik SW. Fragment-based screening of the bromodomain of ATAD2. J Med Chem. 2014;57:9687–92. doi: 10.1021/jm501035j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Picaud S, Fedorov O, Thanasopoulou A, Leonards K, Jones K, Meier J, et al. Generation of a selective small molecule inhibitor of the CBP/p300 bromodomain for leukemia therapy. Cancer Res. 2015;75:5106–19. doi: 10.1158/0008-5472.CAN-15-0236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laskowski RA, Swindells MB. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J Chem Inf Model. 2011;51:2778–86. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table and Figures