

Abstract
The serum and glucocorticoid-regulated kinase (SGK) family has been implicated in the regulation of many cellular processes downstream of the PI3K pathway. It plays a crucial role in PI3K-mediated tumorigenesis, making it a potential therapeutic target for cancer. SGK family consists of three isoforms (SGK1, SGK2, and SGK3), which have high sequence homology in the kinase domain and similar substrate specificity with the AKT family. In order to identify novel compounds capable of inhibiting SGK3 activity, a high-throughput screening campaign against 50,400 small molecules was conducted using a fluorescence-based kinase assay that has a Z' factor above 0.5. It identified 15 hits (including nitrogen-containing aromatic, flavone, hydrazone, and naphthalene derivatives) with IC50 values in the low micromolar to sub-micromolar range. Four compounds with a similar scaffold (i.e., a hydrazone core) were selected for structural modification and 18 derivatives were synthesized. Molecular modeling was then used to investigate the structure-activity relationship (SAR) and potential protein–ligand interactions. As a result, a series of SGK inhibitors that are active against both SGK1 and SGK3 were developed and important functional groups that control their inhibitory activity identified.
Keywords: serum and glucocorticoid-regulated kinase, SGK3, high-throughput screening, inhibitors, molecular modeling
Introduction
The serum and glucocorticoid-regulated kinase (SGK) family consists of serine/threonine kinases that act as effectors downstream of the phosphatidylinositol 3-kinase (PI3K) pathway [1]. SGKs are involved in the regulation of many cellular components including ion channels, enzymes, transporters and transcription factors, thereby affecting a wide range of biological processes including cell proliferation, survival, stress, and transportation of ions and nutrients [2].
This kinase family has three highly homologous isoforms: SGK1, SGK2, and SGK3 encoded by the SGK1, SGK2, and SGK3 genes, respectively [3, 4]. SGKs have three domains, an N-terminal variable domain, a catalytic domain and a C-terminal hydrophobic domain [2, 5, 6]. The catalytic domain and C-terminal hydrophobic domain can also be collectively referred to as the kinase domain. SGK shares a similar domain structure and has high sequence identity in the kinase domain with the AKT family of proteins; therefore, they also have similar substrate specificities with AKT [2, 3, 7]. The SGK family is regulated by phosphorylation downstream of the PI3K pathway and has two phosphorylation motifs [8]: a threonine residue in the activation loop of the catalytic domain (T320 in SGK3), and a serine residue in the hydrophobic motif of the C-terminal domain (S486 in SGK3) [2, 3]. SGK is phosphorylated at the threonine residue by PDK1, and mTORC2 is proposed to be responsible for phosphorylating the serine residue [1]. Phosphorylation at both sites is required for full activation of SGK [2].
The role of the PI3K/AKT signaling pathway in cancer development has been well known for many decades, however in recent years, studies have shown that a subset of tumors is dependent on PI3K and PDK1 but not AKT signaling [9–11]. Among these downstream effectors, SGK isoforms are proposed to be important for tumorigenesis, especially given the structural and substrate similarity between SGK and AKT [2]. In a screen to identify PIK3CA mutant tumors with minimal activation of AKT, SGK3 was identified as a PDK1 substrate that mediated increased cell viability [12]. It was shown that forced expression of SGK3 can increase cell growth and anchorage independent growth in hepatocellular carcinoma [13]. SGK3 is also somehow linked with estrogen-dependent breast cancer [14] as well as androgen-mediated prostate cancer [15]. In addition, SGK1 is also implicated in many cancers [16–18], and is involved in cell growth and survival regulation [19–21]. SGK activation is thus proposed to be a mechanism of resistance to AKT inhibitors, as breast cancer cell lines with high expression levels of SGK1 were resistant to AKT inhibition [17]. Altogether, these demonstrate a role of the SGK family in PI3K-mediated tumorigenesis, making it a potential therapeutic target for cancer.
Given the role of SGK in cancer signaling, a number of SGK1 inhibitors have been discovered (Fig. 1). The crystal structures of 1 (PDB: 3HDM) and 2 (PDB: 3HDN) in complex with SGK1 showed that the azaindole nitrogen of these two inhibitors interact with the linker forming donor-acceptor interactions with Ile 179 and Asp 177 [22]. In both 1 and 2, the carboxylic acid group makes an electrostatic interaction with the catalytic Lys 127. This interaction is important, as analogs lacking the carboxylic group had significantly reduced potency. GSK650394, similar in structure to 1 and 2 was discovered and characterized by Sherck et al. [23]. It is not selective between the SGK isoforms, but it is selective for SGK1 over AKT, and is able to inhibit androgen-mediated prostate cancer cell (LNCaP) growth.
Fig. 1.

Structures of published SGK inhibitors
Compounds with a different scaffold were also reported by Merck [24]. The para-phenol group in 3 is proposed to interact with the linker, and the carbonyl to interact with Lys 127 [25]. Halland et al. [25] identified 7 via virtual screening, which was proposed to form hydrogen bond interactions with Ile 179 and Asp 177 using its aminoindazole scaffold. Further optimization of 7 led to the discovery of 14g and 14n that were approximately 50-fold and 75-fold selective for SGK1 over SGK3, respectively, but not selective for SGK1 over SGK2. EMD638683, another SGK1 inhibitor similar to those unmasked by Merck, was able to lower blood pressure in mice with hyperglycemia and salt excess [26]. It is not selective between the SGK isoforms, but selective for SGK over other kinases including AKT.
An in silico study by Ortuso et al. [27] identified SI113, which is active against SGK1. It orients the OH group towards the linker, donating a hydrogen bond to Asp 177, accepting a hydrogen bond from Ile 179 and making π-stacking interaction with the lysine side chain. It was able to decrease the growth of RKO colon carcinoma, MCF-7 breast carcinoma, and A-172 glioblastoma cells [28].
To date, the number of SGK inhibitors identified is limited, and none are selective for the SGK3 isoform. Therefore, we conducted a high-throughput screening (HTS) campaign against the SGK3 isoform in an attempt to identify selective inhibitors and/or novel scaffolds that can be optimized to produce potent inhibitors.
Materials and methods
Reagents
LANCE Ultra Europium-anti-phospho-40S ribosomal protein S6 (Ser235/236) and LANCE Ultra ULight™-phospho-40S ribosomal protein S6 (pSer235/236) peptide were purchased from PerkinElmer (Boston, MA, USA). MgCl2, EGTA, EDTA, Tris, and Tween-20 were bought from Sinopharm Chemical Reagent Co., Ltd. (Shanghai, China). Dimethyl sulfoxide (DMSO) and dithiothreitol (DTT) were obtained from Sigma-Aldrich (St. Louis, MO, USA). The SGK3 recombinant human protein used for the HTS campaign was procured from Life Technologies (Carlsbad, CA, USA), and the assay plates (ProxiPlate-384 F Plus) were acquired from PerkinElmer. Staurosporine was purchased from Selleck Chemicals (Houston, TX, USA). HTRF KinEASE STK S3 kit was the product of Cisbio (Codelet, France). SGK1, SGK3, and AKT1 used for concentration-response studies of modified compounds were bought from Carna Bioscience (Kobe, Japan). Cell lines were obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). CCK-8 solution was procured from Dojindo Laboratories (Kumamoto, Japan). Cell Lytic M was acquired from Sigma-Aldrich. BCA kit was purchased from Beyotime (Shanghai, China). Denaturing buffer was bought from New Cell and Molecular Biotech Co, Ltd. (Suzhou, China). Nitrocellulose membranes (0.45 μm) were obtained from Merck Millipore (Darmstadt, Germany). Antibodies were procured from Cell signaling Technology (Beverly, MA, USA).
Kinase assay
Two protocols were used for the kinase assay. Protocol A was used for the HTS and concentration-response studies of the hit compounds. Compounds (1 μL/well) dissolved in DMSO were diluted in 20 μL/well kinase buffer (50 mmol/L Tris, pH 7.5, 1 mmol/L EGTA, 10 mmol/L MgCl2, 2 mmol/L DTT, 0.01% Tween-20), and 2 μL/well of the diluent was added to the detector plate. The wells were incubated with SGK3 protein (0.25 ng/well), ULight-rpS6 peptide (50 nmol/L) and ATP (10 μmol/L) for 1 h at room temperature with a total reaction volume of 10 μL/well. Eu-anti-phospho-rpS6 (2 nmol/L) in 12 mmol/L EDTA (10 μL) was added into the plate and incubated for another hour at room temperature. The fluorescence at 665 and 620 nm was read using the EnVision Plate Reader (PerkinElmer), and the experiment was carried out in duplicates. The fluorescence signal was calculated as 665 nm/620 nm.
Protocol B was used for concentration-response studies of the modified compounds. Compounds (1 μL/well) dissolved in DMSO were diluted in 40 μL/well kinase buffer (provided by HTRF KinEASE STK S3 kit and supplemented with 10 mmol/L MgCl2 and 1 mmol/L DTT), and 4 μL/well of the diluent was added to the detector plate. The wells were incubated with SGK1 (1.2 ng/well), SGK3 (0.08 ng/well), or AKT1 (0.12 ng/well), STK substrate 3-biotin (1 μmol/L), and ATP (10 μmol/L) for 30 min at room temperature with a total reaction volume of 10 μL/well. STK antibody-cryptate (5 μL) and 5 μL streptavidin-XL665 (62.5 nmol/L) in detection buffer were added to the plate and incubated for another hour at room temperature. The fluorescence at 665 and 620 nm was read using the EnVision Plate Reader (PerkinElmer), and the experiment was carried out in duplicates. The fluorescence signal was calculated as 665 nm/620 nm.
HTS campaign
The compound library used for the screening of SGK3 inhibitors consisted of 50,400 pure synthetic and natural compounds. Samples dissolved in 100% DMSO were applied to the primary screening, with an average final concentration of 20 μmol/L for each compound. In each 384-well assay plate, 24 wells were used as positive controls (2 μmol/L staurosporine), 24 wells as negative controls (no compounds), and 16 wells as blank controls (no protein or compounds). Compounds showing greater than 50% inhibition relative to negative controls were considered as “hits”.
Concentration-response study
Concentration-response study was performed essentially the same as described above, except that test compounds were hand-picked (average stock concentration: 10 mmol/L) and diluted (1:5) seven times to give a total of eight concentrations. The final compound concentration ranged from 100 μmol/L to 1.28 nmol/L. Each compound was tested in duplicates and the concentration-response curves and IC50 values were generated with GraphPad Prism 5 (GraphPad, San Diego, CA, USA) using log (inhibitor) vs. response equation in nonlinear regression analysis.
Cell culture and maintenance
LNCaP prostate cancer cells were maintained in RPMI1640 medium (ATCC modification) containing 10% fetal bovine serum and 100 units/mL penicillin/streptomycin. T47D breast cancer cells were maintained in RPMI1640 medium supplemented with 10% fetal bovine serum and 100 units/mL penicillin/streptomycin. The cells were incubated at 37 °C in a humidified atmosphere containing 5% CO2.
Cell viability assessment
LNCaP and T47D cells were seeded at 50,000 cells/well in 96-well plates. The plate was incubated for 42 h in a humidified incubator at 37 °C and 5% CO2. Compounds dissolved in DMSO at a concentration between 25 μmol/L and 0.39 μmol/L were added, with a final DMSO concentration of 0.5%. DMSO without inhibitor was used as the negative control. The plate was then incubated for 24 h at 37 °C, and 10 μL of CCK-8 solution were added to each well. The plate was further incubated for 4 h at 37 °C and absorbance was measured at 450 nm using the EnSpire Multimode Plate Reader (PerkinElmer). The data were then graphed and analyzed with GraphPad Prism 5.
Western blot analysis
Western blot analysis was carried out as previously described with minor modifications [29]. T47D cells were seeded in 12-well tissue culture plates at 4.2 × 105 cells/well, and after a day they were serum starved overnight. The cells were treated with DMSO only, 10 μmol/L or 20 μmol/L of test compounds for 60 min with a final DMSO concentration of 0.5%, and then stimulated with 100 nmol/L of insulin. They were lysed with Cell Lytic M containing protease inhibitor and phosphatase inhibitor cocktail on ice. The concentration of protein was determined using the BCA kit according to the manufacturer’s instructions. The samples were incubated with denaturing buffer at 37 °C for 10 min. Protein samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (6 and 8% gel) and transferred onto nitrocellulose (0.45 μm) membranes. The membranes were incubated with 5% (w/v) non-fat dried milk dissolved in TBS-T (1 mol/L Tris-HCl, 0.15 mol/L NaCl, 0.05% (v/v) Tween-20, pH 7.4) for 1 h at room temperature. The membranes were then washed three times, 10 min each and incubated with primary antibody (1:1000) overnight at 4 °C in 5% w/v BSA dissolved in TBS-T. The membranes were again washed three times, 10 min each, and then incubated with anti-rabbit IgG HRP-linked antibody (1:5000) in 5% (w/v) non-fat dried milk dissolved in TBS-T for 1 h at room temperature. Following three-time wash (10 min each), they were reacted with SuperSignal® West Dura substrate in a ChemiDocTM MP Imaging System (ThermoFisher Scientific, Waltham, MA, USA).
Molecular modeling
Homology modeling of SGK3
The amino-acid sequence of human SGK3 was downloaded from the NCBI database (accession code: Q96BR1), and sequence similarity search was performed using the NCBI BLAST server. Three crystal structures of the human SGK1 kinase domain (PDB: 2R5T, 3HDM and 3HDN) were selected as the template to construct SGK3. Sequence alignment of SGK1 and SGK3 was carried out using Discovery Studio [30]. Twenty models were generated, and the model with the lowest Discrete Optimized Protein Energy score was submitted to 200 steps of energy minimization with steepest descent. The quality of the model was evaluated using the Profiles-3D protocol in Discovery Studio, and a Ramachandran plot was also generated.
Ligand and protein preparation
All ligands were created with the two-dimensional Sketcher module in Maestro 10.1. They were prepared using the LigPrep option of Schrodinger [31]. Ligand minimization was done using the MMFFs force field, and possible ionization states at pH 7.0 ± 2.0 were generated using Epik. All possible stereoisomers were generated and a maximum of 32 conformers were obtained. The human SGK1 kinase domain (3HDM) was prepared first using Discovery Studio to clean the protein and add missing side chains and residues using CHARMm minimization, with all parameters kept as default [30]. The protein was then prepared using the protein preparation wizard tool implemented in Maestro 10.1 [31]. Water molecules were deleted and protein ionization states were generated at pH 7.0 ± 3.0, and minimized with the OPLS_2005 force field with all parameters kept as default.
Molecular docking
Two docking methods were employed in this study. The first used GLIDE implemented in Maestro 10.1 (Schrodinger) [32]. The receptor grid was defined as a 20 Å× 20 Å× 20 Å box centroid of the crystalized ligand. Hydrogen bond constraints were set at the backbone amide nitrogen of Ile 179 and carbonyl of Asp 177 in SGK1, and the backbone amide nitrogen of Val 243 and carbonyl of Asp 241 in SGK3. Docking was performed with the XP (Extra Precision) protocol and 20 poses at most were kept per ligand. All other settings were kept as default.
The second method employed was the Induced Fit Docking (IFD) protocol using the OPLS_2005 force field in Maestro 10.1 (Schrodinger) [33]. The docking cavity for SGK1 was centered on the ligand in the crystal structure, and the docking cavity for SGK3 was centered on the ligand of 1 docked into SGK3 using GLIDE. The size of the box was generated automatically. Different combinations of hydrogen bond constraints were used in the docking, with constraints set at the backbone amide nitrogen and carbonyl of Ile 179, the backbone carbonyl of Asp 177 and the side chain of Lys 127 in SGK1. Constraints were set at the backbone amide nitrogen of Val 243 and the carbonyl of Asp 241 in SGK3. Docking was carried out using SP (Standard Precision). Residues within 10 Å of ligand poses were refined, and structures within 30 kcal/mol of the best structure were re-docked. All other settings were kept as default.
Data analysis
Z' factor was evaluated using 48 positive control wells, 48 negative control wells and 32 blank control wells before the HTS campaign:
MPC means the mean value of positive control wells. SDPC means the standard deviation of positive control wells. MNC means the mean value of negative control wells. SDNC means the standard deviation of negative control wells.
Inhibition (%) was calculated using the following equation:
ST means the signal of each compound–test well. SB means the mean signal of blank control wells. SPC means the mean signal of positive control wells.
Results
Assay validation
Firstly, protein titration was performed to determine the optimal concentration of SGK3 for the HTS assay, and the EC80 (0.25 ng/well) was selected (Fig. 2a). An incubation time course was evaluated, the signal started to plateau after 1 h as shown in Fig. 2b, therefore 1 h was selected as the incubation time. ATP titration was performed to determine the optimal ATP concentration (Fig. 2c), and the Km of ATP was 15.88 μmol/L. DMSO is the most commonly employed solvent to dissolve organic compounds in HTS, and DMSO did not affect the assay performance at concentrations up to 2.5% (Fig. 2d). Under these optimized conditions, the IC50 value of the inhibitor control, staurosporine, was 0.14 μmol/L, comparable to those reported in the literature [34–36] (Fig. 2e). The final concentrations of ULight-rpS6 peptide and Eu-anti-phospho-rpS6 were 50 nmol/L and 2 nmol/L, respectively, according to the manufacturer’s instructions.
Fig. 2.

Optimization for the HTS assay. a Kinase activities at protein concentrations ranging from 100 ng/mL to 12.5 ng/mL were measured (mean ± SEM, n = 3). b Kinase activities at different incubation times (0 min to 120 min) were measured (mean ± SEM, n = 3). c ATP titrations from 1 mmol/L to 12.8 nmol/L (mean ± SEM, n = 3). d DMSO tolerance test, with DMSO concentrations ranging from 0.5 to 5% (mean ± SEM, n = 3). e Concentration-response curve of staurosporine on SGK3 activity was generated with optimal conditions, from which IC50 value was calculated (mean ± SEM, n = 3)
Assay performance
Both signals evoked by SGK3 alone and in the presence of an inhibitor control (2 μmol/L staurosporine) were studied (Fig. 3). The coefficient of variation (CV) value was 13.9%. The Z' factor for the assay was 0.56 with an S/B ratio of 14.0. These characteristics indicate that the system is of high quality and well-suited to HTS [37].
Fig. 3.
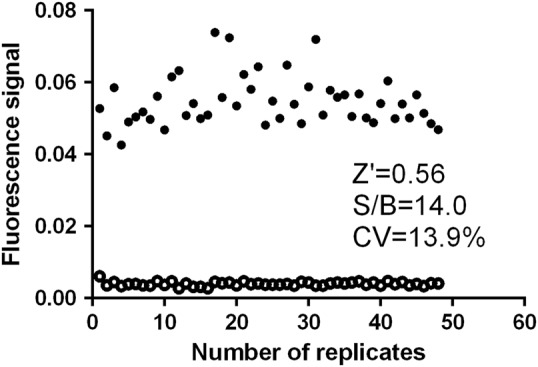
Z′ factor of the SGK3 assay was determined at optimized conditions. Forty-eight replicates of signal (1% DMSO, black circle) and background (2 μmol/L staurosporine, empty circle) were investigated
Identification of potential SGK3 inhibitors
The results of the primary screening of SGK3 inhibitors are presented in Fig. 4. 50,400 compounds were screened for potential inhibition on SGK3, and 203 hits (0.40%) that showed inhibitory activity higher than 50% were selected for confirmation. In the secondary screening, 51 compounds demonstrated consistent inhibition compared with the primary screening and showed concentration-dependency at the two concentrations used (20 μmol/L and 4 μmol/L). Among them, 15 synthetic compounds, namely, nitrogen-containing aromatic, flavone, hydrazone, and naphthalene derivatives, displayed IC50 values below 25 μmol/L (Table 1). One scaffold containing a hydrazone core of these hits was assessed as the starting point for medicinal chemistry efforts.
Fig. 4.
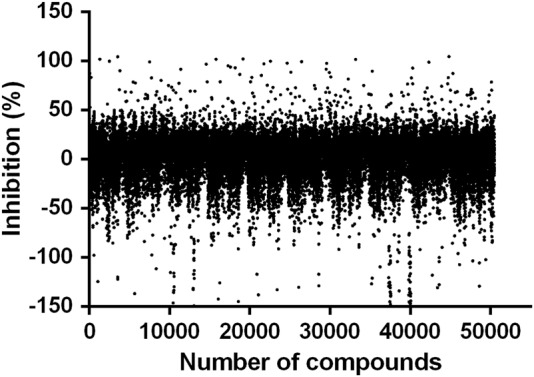
HTS campaign against SGK3 of 50,400 small-molecule compounds
Table 1.
High-throughput screening campaign against SGK3 identified 15 hits with IC50 values below 25 μmol/L
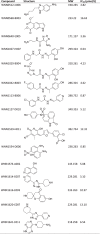
|
Structure modification
Compounds WNN1027-H007, WNN1029-B004, WNN1042-B003, and WNN1113-B006 were selected for further studies due to their potency and the consistent activity across the different analogs. Based on the hydrazone core structure, we introduced different substituent groups into the aromatic ring in an attempt to obtain SGK3 inhibitors with improved potency. The synthesis of target compounds (18a–r) is illustrated in Scheme 1. 4-(N-acetylamino) acetophenone (6a) was prepared from commercially available 4-aminoacetophenone (5) and acetic anhydride at room temperature. In the presence of sulfuric and nitric acids, 1-(4-N-acetylamino-3-nitrophenyl) ethanone (8a) and 3-nitro-4-methoxyacetophenone (8b) were synthesized via the nitration of 6a and 6b, respectively. Then, 8a was deacetylated in 6 N HCl to produce 3-nitro-4-aminoacephenone (9), and reduction of 8b with SnCl2·2H2O gave 1-(3-amino-4-methoxyphenyl) ethanone (10). Compounds 10 and 5 were treated with 2,4-difluorobenzene-1-sulfonyl chloride to provide intermediates 11 and 12. Moreover, pyrrole (13) was reacted with fuming nitric acid to form 3-nitropyrrole (15). Compounds 15, 19, and 22a–c were subsequently condensed with methyl or ethyl bromoacetate in the presence of potassium carbonate (K2CO3) to produce intermediates 16, 20a, and 23a–c. These five compounds and the commercially available 20b were then treated with hydrazine hydrate to obtain 17, 21a–b and 24a–c, which were further condensed with various aromatic aldehydes or ketones as well as 9, 10, 11, and 12, using acetic acid as a catalyst in EtOH, to provide eighteen target compounds (18a–r; see Supplementary Information).
Scheme 1.

Reagents and conditions: (a) Ac2O, then H2O, room temperature (rt); (b) H2SO4, HNO3, rt; (c) 6 N HCl, reflux; (d) 2,4-difluorobenzene-1-sulfonyl chloride, pyridine, rt, 1 h; (e) SnCl2·2H2O, DMF, rt, 24 h; (f) Ac2O, fuming HNO3, –30 °C; (g) BrCH2CO2Me or BrCH2CO2Et, K2CO3, DMF, rt; (h) H2NNH2·H2O, EtOH, 90 °C; (i) various ketones or aldehydes, EtOH, HAc, rt, or reflux
IC50 values of the modified compounds are shown in Table 2. Compound 18d has an IC50 of 1.12 μmol/L against SGK3, and when the OH group was replaced with a pyridine in 18f, potency of the compound decreased by approximately 3-fold. A similar IC50 value for 18d and 18e suggests that addition of a methyl group to 18d has minimal effects on the potency. In addition, the presence of a second OH group in the ortho-position as in 18j did not alter the potency compared with 18e. The results imply that the OH group in the para-position is critical for inhibitory activity: when it was replaced by a methyl (18g) or removed completely (18i), activities of the compounds were greatly compromised. Moreover, 18h was inactive despite having an OH group in the ortho-position, further highlighting the importance of the para-OH group.
Table 2.
IC50 values of potential inhibitors of SGK3, SGK1, and AKT

|
When the NH group of 18d was replaced by S (18b), potency decreased by approximately 20-fold, indicating that the NH group is important. This is further supported by 18c, where potency was lost when the NH group was removed, despite the compound having two OH groups. Substituting NO2 and Cl groups off the benzene as observed with 18a, 18k, and 18l did not have significant effects on potency. The data also show that these compounds in general are not selective for SGK3 over SGK1, but are selective for SGK over AKT (Table 2).
Molecular modeling
The hits were active against both SGK1 and SGK3; hence they were first docked into the SGK1 crystal structure (PDB: 3HDM) using GLIDE. Based on the crystal structure, 1 makes two hydrogen bond interactions with Asp 177 and Ile 179 at the linker (Fig. 5), therefore, hydrogen bond constraints were set at the backbone carbonyl of Asp 177 and amide of Ile 179, although only one constraint needs to be satisfied. The results demonstrate that the OH group of 18d was predicted to make a hydrogen bond donor interaction with the backbone carbonyl of Asp 177 and an acceptor interaction with the amide nitrogen of Ile 179 (Fig. 6a). Compound 18f has a pyridine instead of an OH group at the same position, and this compound is predicted to only make a hydrogen bond interaction with Ile 179 (Fig. 6b).
Fig. 5.

Crystal structure of the human SGK1 kinase domain in complex with 1 (PDB: 3HDM)
Fig. 6.

Compounds docked into the SGK1 active site. a Predicted binding mode of 18d using GLIDE with hydrogen bond constraints set at D177 and I179. b Predicted binding mode of 18f docked using GLIDE with hydrogen bond constraints set at D177 and I179. c Predicted binding mode of 18d using IFD with hydrogen bond constraints added at D177 and I179. d Predicted binding mode of 18f using IFD with hydrogen bond constraints added at D177 and I179. e Predicted binding mode of 18j using IFD with hydrogen bond constraints added at K127, D177, and I179. f Predicted binding mode of 18d using IFD with hydrogen bond constraints added at K127, D177, and I179
To give more flexibility to the active site during docking, the Induced Fit Docking (IFD) protocol within Maestro 10.1 was used. Hydrogen bond constraints were initially added at the backbone amide nitrogen of Ile 179 and the backbone carbonyl of Asp 177. The higher scoring binding modes of 18d oriented the OH group towards the linker, making a donor hydrogen bond with Asp 177 and an acceptor hydrogen bond with Ile 179, similar to the binding mode observed with GLIDE (Fig. 6c). The model also suggests that the carbonyl group can interact with the catalytic lysine. When the OH group was replaced by a pyridine (18f), the carbonyl group instead of the pyridine was preferentially oriented towards the linker (Fig. 6d).
The docked models suggest that the ligands may also interact with the catalytic lysine, therefore, additional hydrogen bond constraints were added at the side chain of Lys 127. The higher scoring poses continue to orient the OH group towards the linker; however, a second type of binding mode was also identified. In this binding mode the carbonyl group interacts with the linker, potentially making three hydrogen bond interactions with Asp 177 and Ile 179, and the OH group was predicted to interact with the catalytic lysine. Docking results of 18j and 18d suggest that the OH group in the para-position may be responsible for interacting with the lysine side chain (Fig. 6e, f). The limitation of this second type of binding mode is that it is nearly always scored lower than those that oriented the OH group towards the linker.
Both types of binding modes identified above are possible. Therefore, in order to investigate which binding mode is more likely, we designed two compounds 18m and 18n. Compound 18m has a sulfonamide group that can potentially make an electrostatic interaction with the catalytic lysine (Fig. 7a), and this has been demonstrated in other kinases [38]. Compound 18n, on the other hand, is incapable of making electrostatic interactions with the catalytic lysine. The results show that both compounds did not have very high potency, with <50% inhibition at 100 μmol/L. Docking results suggest that the sulfonamide in the para-position may be better suited than the meta-position (Fig. 7b). Therefore 18q and its analogs 18o, 18p, and 18r were synthesized and tested, and were shown to be inactive. Altogether, these suggest that the first binding mode shown in Fig. 6a–c may be more likely than binding mode two.
Fig. 7.

Compounds docked into the SGK1 active site using IFD with hydrogen bond constraints added at D177 and I179. a Predicted binding mode of 18m. b Predicted binding mode of 18q
3D structure of the SGK3 model
Currently, no crystal structure of the kinase domain of SGK3 is available in the Protein Databank (PDB), therefore, homology modeling was used to generate a model of the SGK3 kinase domain. Sequence alignment indicated that the sequence identity in the kinase domain is approximately 74% between SGK1 and SGK3. Three crystal structures of SGK1 are available from PDB, two in complex with inhibitors (PDB: 3HDM and 3HDN) and one in complex with AMP-PNP, an ATP mimetic (PDB: 2R5T). These structures were selected as templates to construct the SGK3 model using Discovery Studio. The Ramachandran plot showed that 97.7% of the residues in the SGK3 model (Fig. 8a) were either in the most favorable regions or in the additionally allowed regions, and only seven residues were located in the disallowed region (Fig. 8b). This implies that the overall conformations of the main chains and side chains are reasonable. Verification of the model using the Profiles-3D protocol showed that the verified score was 101.03 (expected high score was 135.595 and the expected low score was 61.0176). This protocol assesses the compatibility of the 3D structure of the protein model with the sequence of residues it contains. The verified score is higher than the expected low score and closer to the expected high score indicating that the model is of good quality.
Fig. 8.

Homology model of the SGK3 kinase domain. a Homology model of SGK3 kinase domain in cartoon representation. The N-terminal lobe is colored in cyan and the C-terminal lobe is colored in magenta. b Ramachandran plot calculated for the SGK3 kinase domain using Discovery Studio
The hits were also docked into the SGK3 model built by homology modeling, and the results were similar to those observed in SGK1, with the OH groups of the compounds preferentially oriented towards the linker (Fig. 9). Interestingly, binding mode 2 where the carbonyl is predicted to interact with the linker and the OH group to interact with the catalytic lysine was not observed in SGK3.
Fig. 9.

Predicted binding modes of compounds in SGK3. a Compound 18d docked into SGK3 using GLIDE, with hydrogen bond constraints added at D241 and V243. b Compound 18a docked into SGK3 using the IFD protocol, with hydrogen bond constraints added at D241 and V243
Effects of compounds in vitro
The hit compounds were tested in LNCaP and T47D cells for their effects on cell viability at a concentration between 0.39 μmol/L and 25 μmol/L using the CCK-8 assay. The results show that following 24 h of exposure, 18b, 18d, 18e, 18 f, and 18k had minimal effects on cell growth and viability (data not shown). The inhibitors were also investigated for effects downstream of SGK3, and western blot analysis was conducted in T47D cells. The cells were inhibited for 60 min at 10 and 20 μmol/L of a given test compound, and then stimulated with 100 nmol/L of insulin. It was observed that phosphorylation of SGK3, NDRG1, and TSC2 were unaffected (data not shown). These cell-based experimental data indicate that the inhibitory activity of the compounds under investigation may not be strong enough for this inhibitory activity to be translated from biochemical assays to cellular assays. Cells are more complex than protein, and compounds must cross the lipid membrane to be active within the cell, which may, therefore, reduce the concentration of compounds at the target site and contribute to the lack of inhibition in LNCaP and T47D cells.
Discussion
SGK is a family of serine/threonine kinases involved in the regulation of many cellular processes including cell growth and survival downstream of PI3K, and is considered a second AKT in cell signaling [1, 2, 8]. Through the identification of tumors that are independent on AKT but dependent on PDK1 signaling, the SGK family of proteins has become a new therapeutic target for anticancer drug discovery [9–11]. SGK and AKT have >50% sequence identity in the kinase domain, and hence share similar substrate specificity [2, 3]. Similar to AKT, SGK is also implicated in many cancers including breast and prostate cancer [13–18]. To date, only a limited number of inhibitors for the SGK family have been identified. Therefore, there is an urgent need for the discovery and development of new SGK inhibitors.
In this study, we present a HTS campaign in attempt to identify new compounds that are active against the SGK family of proteins. To apply the fluorescence-based assay to HTS, many factors were considered and optimized to maximize the S/B ratio. As shown in Fig. 2, the signal was affected by protein amount, incubation time, and ATP concentration. In addition, DMSO tolerance was also examined because it may limit the maximum concentration in which a compound could be tested. The results show that at concentrations of up to 2.5%, DMSO did not affect the assay performance. In addition to the S/B ratio, the Z' factor and CV from the validation experiments demonstrated that this assay is well-suited for HTS [37].
Our HTS campaign identified fifteen hit compounds that had IC50 values in the low micromolar to sub-micromolar range. Of these, four compounds were selected for structural modification because they have the same hydrazone core, which may be the key scaffold to display bioactivity. These compounds are also structurally similar to those discovered by Merck [24]. This indicates that our HTS method is capable of identifying hits that resemble those known to be active against SGKs. We used a combination of SAR studies and molecular modeling to identify functional groups that may be important for inhibiting SGK activity. Our modified compounds were active against both SGK3 and SGK1, indicating that they are non-selective between the isoforms. Due to the high sequence homology (74% between SGK1 and SGK3) of the SGK kinase domain, selectivity may be difficult to achieve by designing ATP-competitive inhibitors binding to the active site. Based on the currently available data, majority of the known SGK inhibitors do not appear to be selective between the isoforms [22–27], with the exception of 14g and 14n identified by Halland et al. [25] that were approximately 50-fold and 75-fold more selective for SGK1 over SGK3, respectively, but were not selective for SGK1 over SGK2.
Initial docking studies in SGK1 with hydrogen bond constraints set at the backbone amide nitrogen of Ile 179 and the carbonyl of Asp 177 (PDB: 3HDM) oriented the OH group towards the linker. Docking with the IFD protocol using hydrogen bond constraints at Ile 179 and Asp 177 also preferentially oriented the OH group towards the linker, agreeing with the binding mode suggested by Halland et al. [25] of a similar compound. In addition, we have also identified a second binding mode where the carbonyl group was oriented towards the linker, and the OH group was oriented towards the catalytic lysine (Fig. 6e, f). However, compounds designed based on this binding mode have low potency, indicating that the first predicted binding mode in agreement with those identified by Halland et al. [25] might be more likely. Furthermore, docking of the compounds into our model of SGK3 demonstrate that all the compounds were oriented in the first binding mode.
In summary, we have developed and validated a fluorescence-based assay to directly measure the kinase activity of SGK3. We used this assay to screen compounds in a 384-well plate format and identified hit compounds that were then modified and characterized in biochemical and cellular assays, resulting in the discovery of low micromolar inhibitors of SGK. SAR and molecular modeling studies were then carried out to better understand ligand–protein interactions that control the potency.
Acknowledgements
We would like to thank Prof. Wei Fu for providing the facilities and advice for the molecular modeling, and Ji Wu for technical support during the HTS. This work was partially supported by grants from the Ministry of Science and Technology of China (2014DFG32200), Shanghai Science and Technology Development Fund (15DZ2291600) and the Thousand Talents Program in China. The funders had no role in study design, data collection and analysis, decision to publish, or manuscript preparation.
Author contributions
PRS, YC, and M-WW designed research; GQG, KW, XCD, YZ, W-JL, CMB, RB, and YiC performed experiments; GQG, KW, XCD, JUF, W-JL, CMB, YiC, DHY, PRS, YC, and M-WW analyzed data; GQG, XCD, KW, and M-WW wrote the paper.
Competing interests
The authors declare no competing interests.
Contributor Information
Ying Chen, Email: yingchen71@fudan.edu.cn.
Ming-Wei Wang, Email: mwwang@simm.ac.cn.
References
- 1.Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. AKT-independent PI3-K signaling in cancer-emerging role for SGK3. Cancer Manag Res. 2013;5:281–92. doi: 10.2147/CMAR.S35178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bruhn MA, Pearson RB, Hannan RD, Sheppard KE. Second AKT: the rise of SGK in cancer signalling. Growth Factors. 2010;28:394–408. doi: 10.3109/08977194.2010.518616. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi T, Cohen P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem J. 1999;339:319–28. doi: 10.1042/bj3390319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kobayashi T, Deak M, Morrice N, Cohen P. Characterization of the structure and regulation of two novel isoforms of serum- and glucocorticoid-induced protein kinase. Biochem J. 1999;344:189–97. doi: 10.1042/bj3440189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xing Y, Liu D, Zhang R, Joachimiak A, Songyang Z, Xu W. Structural basis of membrane targeting by the Phox homology domain of cytokine-independent survival kinase (CISK-PX) J Biol Chem. 2004;279:30662–9. doi: 10.1074/jbc.M404107200. [DOI] [PubMed] [Google Scholar]
- 6.Zhao B, Lehr R, Smallwood AM, Ho TF, Maley K, Randall T, et al. Crystal structure of the kinase domain of serum and glucocorticoid-regulated kinase 1 in complex with AMP-PNP. Protein Sci. 2007;16:2761–9. doi: 10.1110/ps.073161707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tessier M, Woodgett JR. Serum and glucocorticoid-regulated protein kinases: variations on a theme. J Cell Biochem. 2006;98:1391–407. doi: 10.1002/jcb.20894. [DOI] [PubMed] [Google Scholar]
- 8.Moniz LS, Vanhaesebroeck B. AKT-ing out: SGK kinases come to the fore. Biochem J. 2013;452:e11–3. doi: 10.1042/BJ20130617. [DOI] [PubMed] [Google Scholar]
- 9.Gagliardi PA, di Blasio L, Orso F, Seano G, Sessa R, Taverna D, et al. 3-Phosphoinositide-dependent kinase 1 controls breast tumor growth in a kinase-dependent but Akt-independent manner. Neoplasia. 2012;14:719–31. doi: 10.1593/neo.12856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res. 2008;68:6084–91. doi: 10.1158/0008-5472.CAN-07-6854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang W, Haines BB, Efferson C, Zhu J, Ware C, Kunii K, et al. Evidence of mTOR activation by an AKT-independent mechanism provides support for the combined treatment of PTEN-deficient prostate tumors with mTOR and AKT inhibitors. Transl Oncol. 2012;5:422–9. doi: 10.1593/tlo.12241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasudevan KM, Barbie DA, Davies MA, Rabinovsky R, McNear CJ, Kim JJ, et al. AKT-independent signaling downstream of oncogenic PIK3CA mutations in human cancer. Cancer Cell. 2009;16:21–32. doi: 10.1016/j.ccr.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu M, Chen L, Chan TH, Wang J, Li Y, Li Y, et al. Serum and glucocorticoid kinase 3 at 8q13.1 promotes cell proliferation and survival in hepatocellular carcinoma. Hepatology. 2012;55:1754–65. doi: 10.1002/hep.25584. [DOI] [PubMed] [Google Scholar]
- 14.Wang Y, Zhou D, Phung S, Masri S, Smith D, Chen S. SGK3 is an estrogen-inducible kinase promoting estrogen-mediated survival of breast cancer cells. Mol Endocrinol. 2011;25:72–82. doi: 10.1210/me.2010-0294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Zhou D, Chen S. SGK3 is an androgen-inducible kinase promoting prostate cancer cell proliferation through activation of p70 S6 kinase and up-regulation of cyclin D1. Mol Endocrinol. 2014;28:935–48. doi: 10.1210/me.2013-1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abbruzzese C, Mattarocci S, Pizzuti L, Mileo AM, Visca P, Antoniani B, et al. Determination of SGK1 mRNA in non-small cell lung cancer samples underlines high expression in squamous cell carcinomas. J Exp Clin Cancer Res. 2012;31:4. doi: 10.1186/1756-9966-31-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sommer EM, Dry H, Cross D, Guichard S, Davies BR, Alessi DR. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem J. 2013;452:499–508. doi: 10.1042/BJ20130342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Amato R, Scumaci D, D’Antona L, Iuliano R, Menniti M, Di Sanzo M, et al. Sgk1 enhances RANBP1 transcript levels and decreases taxol sensitivity in RKO colon carcinoma cells. Oncogene. 2013;32:4572–8. doi: 10.1038/onc.2012.470. [DOI] [PubMed] [Google Scholar]
- 19.Leong ML, Maiyar AC, Kim B, O’Keeffe BA, Firestone GL. Expression of the serum- and glucocorticoid-inducible protein kinase, Sgk, is a cell survival response to multiple types of environmental stress stimuli in mammary epithelial cells. J Biol Chem. 2003;278:5871–82. doi: 10.1074/jbc.M211649200. [DOI] [PubMed] [Google Scholar]
- 20.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J Biol Chem. 2001;276:16649–54. doi: 10.1074/jbc.M010842200. [DOI] [PubMed] [Google Scholar]
- 21.Wu W, Chaudhuri S, Brickley DR, Pang D, Karrison T, Conzen SD. Microarray analysis reveals glucocorticoid-regulated survival genes that are associated with inhibition of apoptosis in breast epithelial cells. Cancer Res. 2004;64:1757–64. doi: 10.1158/0008-5472.CAN-03-2546. [DOI] [PubMed] [Google Scholar]
- 22.Hammond M, Washburn DG, Hoang HT, Manns S, Frazee JS, Nakamura H, et al. Design and synthesis of orally bioavailable serum and glucocorticoid-regulated kinase 1 (SGK1) inhibitors. Bioorg Med Chem Lett. 2009;19:4441–5. doi: 10.1016/j.bmcl.2009.05.051. [DOI] [PubMed] [Google Scholar]
- 23.Sherk AB, Frigo DE, Schnackenberg CG, Bray JD, Laping NJ, Trizna W, et al. Development of a small-molecule serum- and glucocorticoid-regulated kinase-1 antagonist and its evaluation as a prostate cancer therapeutic. Cancer Res. 2008;68:7475–83. doi: 10.1158/0008-5472.CAN-08-1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gericke R, Beier N, et al. Acylhydrazone derivatives and their use in the inhibition, regulation and/or modulation of the signal transduction of kinases. Germany. WO2005/037773 [P]. 28th April, 2005; Dorsch D, Burgdorf LT, Gericke R, et al. 3-Aminoindazoles. Germany. WO2005/123688 [P]. 29th December, 2005; Gericke R, Dorsch D, Mederski W, et al. Acyl hydrazides as kinase inhibitors, in particular for SGK. Germany. WO2006/105850 [P]. 12th October, 2006; Mederski W, Gericke R, Dorsch D, et al. Indazole squaric acid derivatives as CHK1-, CHK2- and SGK- inhibitors. Germany. WO2006/105865 [P]. 12th October, 2006.
- 25.Halland N, Schmidt F, Weiss T, Saas J, Li Z, Czech J, et al. Discovery of N-[4-(1H-pyrazolo[3,4-b]pyrazin-6-yl)-phenyl]-sulfonamides as highly active and selective SGK1 inhibitors. ACS Med Chem Lett. 2015;6:73–8. doi: 10.1021/ml5003376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ackermann TF, Boini KM, Beier N, Scholz W, Fuchß T, Lang F. EMD638683, a novel SGK inhibitor with antihypertensive potency. Cell Physiol Biochem. 2011;28:137–46. doi: 10.1159/000331722. [DOI] [PubMed] [Google Scholar]
- 27.Ortuso F, Amato R, Artese A, D’Antona L, Costa G, Talarico C, et al. In silico identification and biological evaluation of novel selective serum/glucocorticoid-inducible kinase 1 inhibitors based on the pyrazolo-pyrimidine scaffold. J Chem Inf Model. 2014;54:1828–32. doi: 10.1021/ci500235f. [DOI] [PubMed] [Google Scholar]
- 28.D’Antona L, Amato R, Talarico C, Ortuso F, Menniti M, Dattilo V, et al. SI113, a specific inhibitor of the Sgk1 kinase activity that counteracts cancer cell proliferation. Cell Physiol Biochem. 2015;35:2006–18. doi: 10.1159/000374008. [DOI] [PubMed] [Google Scholar]
- 29.Ge Y, Yang D, Dai A, Zhou C, Zhu Y, Wang MW. The putative signal peptide of glucagon-like peptide-1 receptor is not required for receptor synthesis but promotes receptor expression. Biosci Rep. 2014;34:e00152. doi: 10.1042/BSR20140120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Discovery Studio Modeling Environment, Release 16.1.0. (San Diego: Accelrys Software Inc; 2016).
- 31.Sastry GM, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27:221–34. doi: 10.1007/s10822-013-9644-8. [DOI] [PubMed] [Google Scholar]
- 32.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 33.Sherman W, Day T, Jacobson MP, Friesner RA, Farid R. Novel procedure for modeling ligand/receptor induced fit effects. J Med Chem. 2006;49:534–53. doi: 10.1021/jm050540c. [DOI] [PubMed] [Google Scholar]
- 34.Zegzouti H, Vidugiriene J, Vidugiris G, Goueli S. SGK1 kinase enzyme system product information, <http://www.promega.com.cn/resources/protocols/product-information-sheets/n/sgk1-kinase-enzyme-system-protocol/>. 2018.
- 35.Reaction Biology Corporation. SGK2, <http://www.reactionbiology.com/webapps/site/KinasePDFs/SGK2.pdf>.2006.
- 36.Reaction Biology Corporation. SGK1, <http://www.reactionbiology.com/webapps/site/KinasePDFs/SGK1.pdf>. 2010.
- 37.Zhang J, Chung T, Oldenburg K. A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]
- 38.Knight SD, Adams ND, Burgess JL, Chaudhari AM, Darcy MG, Donatelli CA, et al. Discovery of GSK2126458, a highly potent inhibitor of PI3K and the mammalian target of rapamycin. ACS Med Chem Lett. 2010;1:39–43. doi: 10.1021/ml900028r. [DOI] [PMC free article] [PubMed] [Google Scholar]