

Abstract
Xuezhikang capsule (XZK) is a traditional Chinese medicine that contains lovastatin (Lv) for hyperlipidemia treatment, although it has fewer side effects than Lv. However, the pharmacokinetic mechanisms contributing to its distinct efficacy and low side effects are unclear. Mice were fed a high-fat diet (HFD) for 6 weeks to induce hyperlipidemia. We first conducted the pharmacokinetic studies in HFD mice following oral administration of Lv (10 mg/kg, i.g.) and found that HFD remarkably decreased the active form of Lv (the lovastatin acid, LvA) exposure in the circulation system, especially in the targeting organ liver, with a declined conversion from Lv to LvA, whereas the Lv (responsible for myotoxicity) exposure in muscle markedly increased. Then we compared the pharmacokinetic profiles of Lv in HFD mice after the oral administration of XZK (1200 mg/kg, i.g.) or an equivalent dose of Lv (10 mg/kg, i.g.). A higher exposure of LvA and lower exposure of Lv were observed after XZK administration, suggesting a pharmacokinetic interaction of some ingredients in XZK. Further studies revealed that HFD promoted the inflammation and inhibited carboxylesterase (CES) activities in the intestine and the liver, thus contributing to the lower transformation of Lv into LvA. In contrast, XZK inhibited the inflammation and upregulated CES in the intestine and the liver. Finally, we evaluated the effects of monacolins and phytosterols, the fractional extracts of isoflavones, on inflammatory LS174T or HepG2 cells, which showed that isoflavones inhibited inflammation, upregulated CES, and markedly enhanced the conversion of Lv into LvA. For the first time, we provide evidence that isoflavones and Lv in XZK act in concert to enhance the efficacy and reduce the side effects of Lv.
Keywords: Xuezhikang capsules, lovastatin, lovastatin acid, hyperlipidemia, inflammation, carboxylesterase, isoflavones, pharmacokinetics
Introduction
Cardiovascular disease (CVD) remains the primary cause of morbidity and mortality globally, and hyperlipidemia is generally recognized as a major risk factor in CVD [1, 2]. Red yeast rice (RYR) refers to a rice product fermented by a specific strain of red yeast called Monascus purpureus. RYR has been shown to be efficacious in the improvement of blood circulation and serum cholesterol levels, and currently serves as a dietary supplement [3–10]. In addition to lovastatin (Lv), which acts as the major bioactive component [11–13], several other ingredients such as natural statins, natural pigments, unsaturated fatty acids, plant sterols, amino acids, isoflavones, alkaloids, and trace elements are contained in RYR [13–17]. Remarkably, the cholesterol-lowering efficacy of RYR, which contains ~2–7.5 mg of Lv, is equivalent to that of a 20 mg Lv monomer [18–21].
As an extract of RYR, Xuezhikang capsules (XZK) have been widely used in Chinese medicine for the treatment of CVD for more than two thousand years. At present, XZK has been officially recommended for dyslipidemia treatment [22]. Clinical observations and research studies have both demonstrated that when the clinical dosage of the Lv monomer is four times the content of Lv found in XZK, its capacity for lowering serum total cholesterol (TC), triglyceride (TG), and low-density lipoprotein cholesterol (LDL-C) levels is equal to that of Lv in XZK. Moreover, XZK not only greatly improves the serum high-density lipoprotein cholesterol (HDL-C) levels, but also rarely causes the severe side effects of the statin monomers observed in clinical applications including liver damage, myopathy, and even rhabdomyolysis [23, 24]. Based on a comparison of clinical efficacy and safety, we propose that the interaction between other components in XZK and Lv explains how XZK exhibits a higher efficiency and a lower toxicity than the Lv monomer. Our previous studies demonstrated that non-statins in XZK showed a synergistic enhancement of Lv [25]. Nevertheless, the pharmacokinetic mechanism for enhancing the efficacy and reducing the toxicity of XZK capsules has not been reported.
Carboxylesterase (CES), which was originally discovered in the 1930s, is involved in the hydrolysis process of numerous drugs of various structures and determines the pharmacokinetic behaviors of most drugs containing ester and amide bonds, thus affecting the therapeutic efficacies and toxicities. The mammalian CES is a multigene family whose genetic products are located in the endoplasmic reticulum of multiple tissues. Stronger CES activity exists in some tissues including intestine and liver tissues [26]. In addition, CES also participates in the metabolism of various endogenous substances such as short-chain and long-chain acylglycerol, long-chain acylcarnitine, and long-chain acyl-COA [27–29]. It has been demonstrated that diverse CES family members are expressed in the tissues and organs of multiple mammals [30]. For example, the human CES family 1 (CES1) is mainly expressed in human livers, and CES family 2 (CES2) is mainly expressed in the intestinal tissues. Accordingly, Ces1c and Ces1d, belonging to the mouse Ces family 1 (Ces1), are highly expressed in the livers of mice, and Ces2a, Ces2c, and Ces2e, belonging to Ces family 2 (Ces2), are highly expressed in the intestines of mice [31].
It has been generally accepted that it is of extreme vital clinical significance to investigate the influence of CES on the pharmacokinetics, pharmacodynamics, and drug toxicity. Because it serves as a substrate for CES, Lv was researched as a pro-drug for the treatment of hyperlipidemia [32]. Absorbed through the intestinal tract, Lv is hydrolyzed by esterases such as CES into its loop-opening active metabolite lovastatin acid (LvA) [32], which has lipid-lowering effects. It has been reported that the myotoxicity of statins is closely associated with the exposure of Lv in muscles, and this toxicity is attributed to statins in lactone forms [33, 34]. Obviously, CES plays a well-defined role in the efficacy and toxicity of Lv.
Fewer studies focusing on the influence of hyperlipidemia or a high-fat diet (HFD) on CES activity exist, and most of these studies focus only on blood and adipose tissues. For example, an HFD did not alter most mRNA expression levels of most lipase/esterases in the adipose tissue [35]. Importantly, the CES1 mRNA expression levels in the adipose tissues were positively associated with the body-mass index and the TG levels [36]. An HFD appeared to induce a slight upregulation of CES in the plasma [37]. In this case, it is noteworthy that patients with hyperlipidemia tended to be accompanied by an inflammatory response [38–40]. Numerous reports have described that the significantly increased secretion of various cytokines under pathological circumstances such as inflammation and infection is accompanied by the significantly decreased hydrolytic metabolism of multiple drugs. Richardson TA demonstrated that the expression levels and the activities of various Cytochrome P450s (CYPs) were downregulated in lipopolysaccharide (LPS)-treated mice [41]. Wait found that serum CES levels in mice were decreased after LPS treatment [42]. Data from Xiong further revealed that inflammation regulated CES via the nuclear pregnane X receptor (PXR) [43]. There is every reason to believe that hyperlipidemia or HFD-associated inflammation is involved in the lower efficacy and higher toxicity of Lv, which might be improved by XZK, probably via the regulation of CES (which mediates the conversion from Lv to LvA).
Based on the above research background, the present study was designed to compare the pharmacokinetic behaviors of Lv and LvA in controls or HFD mice after the oral administration of Lv or XZK (which contained an equal amount of Lv), and to investigate the effects of XZK on inflammation and CES in liver and intestine tissues, which mediates the conversion from Lv to LvA, validating the underlying pharmacokinetic mechanism and identifying active ingredients from the four extracted fractions in the inflammatory cells. Our aim is to provide a scientific basis for the high efficacy and low toxicity of XZK and determine which components confer pharmacokinetic advantages to XZK.
Materials and methods
Chemicals and reagents
XZK capsules were provided by the WBL Peking University Biotech Co. (Beijing, China). Every capsule contains 300 mg of XZK powder with 2.5 mg of Lv. As we described previously [25], four fractions from the primary extract of XZK (with a yield of 6.7%) including isoflavones (F1), monacolins (F2), sterols (F3), and other components (F4) were obtained, with yields of 34%, 14%, 44%, and 8% relative to the primary extract, respectively, according to HPLC identification (Shimadzu, Kyoto, Japan). Lv capsules were purchased from the Yangtze River Pharmaceutical Group Co., Ltd. (Nanjing, China). Lv or LvA reference (IS) were obtained from the China National Institute for Drug and Biological Products (Beijing, China). Monoclonal primary antibodies against CES1, CES2, and PXR were purchased from Abcam (Cambridge, UK). The polyclonal primary antibody against GAPDH and horseradish peroxidase-conjugated anti-mouse/rabbit IgG secondary antibodies were purchased from Bioworld Technology, Inc. (Minnesota, USA). A SYBR Prime Script RT-PCR Kit was purchased from Takara Bio Inc. (Otsu, Shiga, Japan).
Diets, animals, and pharmacokinetic assays
Diets were purchased from TROPHIC Animal Feed Hightech Co., Ltd. (Nantong, China), as described previously [25]. Six-week-old specific-pathogen-free (SPF) male C57BL/6 mice were purchased from the Animal Center of Nanjing Medical University. All animal experiments were approved by the Animal Ethics Committee of China Pharmaceutical University (Nanjing, China). Mice were housed at 25 °C under a 12-h light–dark cycle. Mice were fed with a HFD for 6 weeks before the pharmacokinetics experiments. A total of 108 (31 ± 2 g) male hyperlipidemia mice were randomly divided into three groups (n = 36): Lv administration (10 mg/kg, i.g.) in control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in HFD mice (HFD + Lv), and XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in HFD mice (HFD + XZK). Mice were treated according to their group assignment for the following 3 days. Food was removed 12 h before the end of the experiment. Post drug administration, blood was collected at 0 min, 30 min, 45 min, 1 h, 1.5 h, 2 h, 3 h, 4 h, and 5 h via the retro-orbital venous sinus into the heparinized tubes (containing 2 mM PMSF and 1% acetic acid to inactivate esterase) and immediately separated into plasma. It was then stored at −80 °C before analysis. For every time point, 4 mice were used for blood sampling. Biological samples including liver, intestine, and muscle were immediately collected at 0 h, 1 h, 2 h, 4 h, and 5 h and stored at –80 °C until analysis.
Sample preparation
Blood was obtained from the retinal vein and was centrifuged at 5000 × g for 10 min at 4 °C. A total of 500 μL of 50% acetonitrile solution (containing 2 mM PMSF and 1% acetic acid) was added to ~0.1 g of liver, intestine, or muscle (normalized to the actual weight). It was then homogenized and broken up with an ultrasonic unit (3 s/time, a total of six times) to obtain tissue homogenate. Next, 150 μL of acetonitrile, including simvastatin (10 ng/mL) and tolbutamide (10 ng/mL), was added into the plasma sample (50 μL), tissue homogenate (50 μL), and cell suspension (50 μL), and the area was vortex mixed for 3 min. After centrifugation for 10 min at 15,000 × g twice, the supernatant was collected for LC–MS/MS analysis.
Cell culture
Human hepatoma G2 HepG2 cells and human colon cancer LS174T cells were purchased from the American Type Culture Collection (ATCC, Rockville, MD). These cells were grown in DMEM medium (high glucose of 4.5 g/L, Gibco-Invitrogen, USA) supplemented with 10% (v/v) fetal bovine serum, 100 U/mL penicillin, and streptomycin (Invitrogen, Carlsbad, CA, USA) in an atmosphere of 5% CO2 at 37 °C.
Lv and LvA accumulation in HepG2 and LS174T cells
HepG2 and LS174T cells were cultured in 24-well plates evenly (5 × 103 cells/well). After reaching 70% confluence, 1 μg/mL LPS (HepG2) or 10 ng/mL TNF-ɑ (LS174T) was administered to induce inflammation for 24 h. The cells were rinsed three times with Hank’s balanced salt solution (Hank’s, 37 °C, pH 7.4) and then treated with 1 mL of Hank’s liquid containing Lv (10 µM), XZK (480 μg/mL, an equivalent dose to 10 μM Lv), or Lv (10 µM) plus F1 (10.9 μg/mL). After incubation at 37 °C for 2 h, the retention was stopped by rinsing the cells three times with Hank’s solution (4 °C). Cells were lysed in 1 mL of deionized water by applying three freeze-thaw cycles, and 100 µL of the cell suspension was collected for the protein concentration measurement with a BCA protein assay kit (Beyotime, Jiangsu, China). The rest was preserved at –80 °C until LC–MS/MS analysis.
Inflammatory factors, cytotoxicity, and TC assays
Inflammatory factors (TNF-α, IL-1β, and IL-6) in HepG2 and LS174T cells were measured by PCR analysis. The respective cell viabilities of HepG2 and LS174T were evaluated by 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays. Cells were cultured in a 96-well plate (2 × 103 cells/well). Control cells were incubated with a medium containing dimethyl sulfoxide (DMSO) as the vehicle. HepG2 or LS174T cells were incubated with various concentrations of irinotecan (0.3, 3, and 30 μM) or clopidogrel (1, 10, and 100 μM). After 24 h of treatment, the cells were incubated with 20 µL (5 mg/mL) of MTT for 4 h at 37 °C. The medium was removed, and the formazan crystals produced by viable cells were dissolved in 150 µL of DMSO for 15 min at 37 °C. The absorbance was detected by a microplate reader Powerwave 200 (Bio-Tek Instruments, Winooski, VT, USA) at a wavelength of 570/695 nm. The cholesterol-lowering efficacy of Lv (ranging from 20 to 80 μM, over 24 h) in HepG2 cells was measured according to a total cholesterol assay kit (Jiancheng, Nanjing, China).
LC–MS/MS analysis
Briefly, 10 μL of the supernatant was injected into an AB SCIEX API-4000 tandem mass spectrometer (Foster City, CA, USA) equipped with electrospray ionization (ESI), LC-20AD pumps, a SIL-20A autosampler, and a CTO-20A oven (Shimadzu, Kyoto, Japan). Liquid chromatographic separations were conducted on a Thermo BDS Hypersil C18 (100 × 2.1 mm, 2.4 μm, Thermo Scientific, Waltham, MA), with the column temperature maintained at 40 °C. A binary gradient consisting of solvent A (0.02% ammonia water with 1 mM ammonium acetate) and solvent B (acetonitrile) was employed for the elution of Lv, LvA, simvastatin, and tolbutamide. The flow rate was kept at 0.2 mL/min by the following gradient program: 0–2.5 min, 30–85% (B); 2.5–3.0 min, 85–30% (B); and 3.0–5.0 min, 30% (B). The retention times were as follows: tR (Lv)≈3.11 min; tR (simvastatin)≈3.30 min; tR (LvA)≈2.68 min; and tR (tolbutamide)≈2.09 min.
The parameters of the ESI source were set as follows: curtain gas, 30 Arb; gas 1, 65 Arb and gas 2, 70 Arb; and temperature/TEM, 450 °C. Five minutes of analysis time was divided into two periods, and period 1 (0–3 min) was operated in the multiple reaction monitoring (MRM) mode under the negative mode: ion spray voltage/IS, −4500 V. MRM was performed at m/z values of 421.00 → 319.00 for LvA and 269.00 → 170.00 for tolbutamide. Period 2 (3–5 min) was operated in the MRM mode under the positive mode: ion spray voltage/IS, 5000 V. MRM was performed at m/z values of 405.00 → 199.00 for Lv and m/z values of 419.00 → 285.00 for simvastatin. Methodology validation data are presented in Tables S1-S3.
Real-time PCR analyses
Total RNA was extracted from the liver, intestine, and cells using a TRIzol Plus RNA Purification Kit (Invitrogen, Life Technologies, USA), according to the manufacturer’s instructions. The resulting RNA was reverse-transcribed into cDNA using a PrimeScript™ 1st Reagent (Takara, Kyoto, Japan). The mRNA levels were determined by quantitative reverse transcription (qRT)-PCR using the SYBR Premix Ex TaqTM (TaKaRa Bio Inc., Japan) in a Bio-Rad real-time PCR machine. The sequences of the primers used for this study are listed in Table S4. The results were normalized to GAPDH expression.
Western blotting analysis
Total proteins from tissues and cells were extracted according to a previously described method [25]. The total proteins were electrophoresed by 8% SDS-PAGE and then transferred to a 0.22-μm polyvinylidene difluoride membrane (Bio-Rad, Hercules, CA, USA). After being blocked in 5% (w/v) nonfat milk, membranes were incubated with anti-CES1, anti-CES2, anti-PXR, or anti-GAPDH antibody overnight at 4 °C. After the wash step, the blots were incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG for 1 h at 37 °C. After washing in TBS-T, signals were detected using an ECL (enhanced chemiluminescence) kit (Thermo Fisher Scientific, Waltham, MA, USA) and were captured with a ChemiDoc XRS + System (Bio-Rad, Hercules, CA, USA). Densitometry was quantified using the Image Lab statistical software (Bio-Rad Laboratories, Hercules, CA, USA). The CES1, CES2, and PXR abundance values were normalized to GAPDH.
The total activity of carboxylate esterase
Samples of the tissues and the cells were homogenized in three volumes of ice-cold 1.15% (w/v) potassium chloride solution and were centrifuged at 10,000 × g for 10 min, and the supernatant was centrifuged at 105,000 × g for 60 min. The obtained microsomal pellet was suspended in a 67.5 mM sodium/potassium phosphate buffer (pH 7.4). Microsomal proteins were quantified by a BCA protein assay kit (Beyotime, China), with bovine serum albumin (Wako Pure Chemical Ind., Osaka, Japan) as the standard. Microsome samples were analyzed for the para-nitrophenyl acetate (PNPA) hydrolysis. A sample cuvette (1 mL) contained 20 μg of protein in 100 mM potassium phosphate buffer (pH 7.4) and 1 mM substrate at room temperature. After initiation with PNPA (10 μL of 100 mM stock in acetonitrile), the hydrolytic rate was recorded as an increase in the absorbance at 400 nm. The extinction coefficient (E400) was determined to be 13 mM/cm. Several controls were conducted including incubation without proteins, and then, non-enzymatic hydrolysis was subtracted. Each sample was conducted in triplicate and averaged. The values were expressed in nmol/mg/min.
Statistical analysis
Pharmacokinetic parameters were calculated using the WinNonlin 6.3 software (Pharsight, USA). Conversions of the unit of AUC from h*ng/mL to h*nmol/mL (plasma) or from h*ng/mg to h*nmol/mg (tissues) were calculated, obtaining the conversion ratio = AUC (LvA)/ (AUC (LvA) + AUC (Lv))*100%. Data were expressed as the mean ± standard deviation (SD). Statistical analyses were conducted by the two-tailed Student’s t-test and one-way ANOVA. When the probability value was less than 0.05 (P < 0.05), the differences were considered to be statistically significant.
Results
Validation of the hyperlipidemia mouse model
As shown in Table 1-1, the body and liver weights of C57BL/6 mice fed with HFD were significantly higher than those of the control mice (P < 0.01). Serum TC and TG, as well as LDL-C and HDL-C were increased in HFD mice for 10 weeks successively (Tables 1 and 2). In particular, the serum TC and LDL-C levels were significantly elevated by 1.86 and 2.29 times, respectively (P < 0.01), compared to those of the control group. These data confirmed that a hyperlipidemia mouse model was successfully established.
Table 1-1.
Anthropometric data of the control or HFD mice
| Body weight (g) | Body weight gain (g/10 weeks) | Food intake (g/day) | Liver weight (g) | |
|---|---|---|---|---|
| Control | 26.18 ± 1.15 | 6.11 ± 0.88 | 2.97 ± 0.34 | 0.91 ± 0.11 |
| HFD | 31.28 ± 1.47** | 10.99 ± 1.34** | 3.02 ± 0.61 | 1.52 ± 0.18** |
Values are means ± SD, n = 6
*P < 0.05, **P < 0.01 vs. Control
Table 1-2.
Serum levels of TC, TG, LDL-C, and HDL-C in the control or HFD mice
| Serum TC (mmol/L) | Serum TG (mmol/L) | Serum LDL-C (mmol/L) | Serum HDL-C (mmol/L) | |
|---|---|---|---|---|
| Control | 3.02 ± 0.65 | 1.75 ± 0.41 | 1.12 ± 0.24 | 2.13 ± 0.54 |
| HFD | 5.63 ± 0.83** | 2.23 ± 0.34 | 2.56 ± 0.46** | 2.83 ± 0.76 |
Values are means ± SD, n = 6
*P < 0.05, **P < 0.01 vs Control
HFD decreased the conversion of Lv into LvA
The mean concentration–time curves (in the plasma and tissues) after the oral administration of Lv (10 mg/kg) in the HFD mice or control mice are presented in Fig. 1. The mean pharmacokinetic parameters of Lv and LvA in the mice were analyzed by a non-compartmental model with Winnonlin 6.3 software and are summarized in Tables 2–5.
Fig. 1.

Pharmacokinetic profiles of Lv and LvA in the control and HFD mice. Concentration–time curves of Lv and LvA in plasma (a, e) and tissues including liver (b, f), intestine (c, g), and muscle (d, h) are presented. The experimental groups were as follows: Lv administration (10 mg/kg, i.g.) in the control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in the HFD mice (HFD + Lv), and XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in the HFD mice (HFD + XZK). The values are expressed as the mean ± SD of four mice for each time point in each group
Table 2.
Mean pharmacokinetic parameters of Lv and LvA in plasma
| Lv | LvA | |||||
|---|---|---|---|---|---|---|
| Parameters | Control+Lv | HFD+Lv | HFD+XZK | Control+Lv | HFD+Lv | HFD+XZK |
| Tmax (h) | 2.00 | 2.00 | 2.00 | 1.50 | 1.50 | 1.50 |
| Cmax (ng/mL) | 81.33 | 111.23 | 154.33 | 473.33 | 173.33 | 839.00 |
| AUC0−t (h*ng/mL) | 130.90 | 173.11 | 269.20 | 778.24 | 325.74 | 1366.58 |
| AUC0−∞ (h*ng/mL) | 132.57 | 175.80 | 278.96 | 779.68 | 330.13 | 1376.21 |
| t1/2 (h) | 0.58 | 0.62 | 0.75 | 0.43 | 0.71 | 0.57 |
| Conversion ratio (Lv to LvA, %) | 84.9 | 64.3 | 82.5 | |||
The experimental groups were as follows: Lv administration (10 mg/kg, i.g.) in the control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in the HFD mice (HFD + Lv), XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in the HFD mice (HFD + XZK). n = 6
Tmax time to peak concentration, Cmax peak concentration, AUC0–t area under the concentration–time curve from zero to the time of last measurable concentration, AUC0−∞ area under the concentration–time curve from zero to infinity, t1/2 apparent elimination half-time
Table 5.
Mean pharmacokinetic parameters of Lv and LvA in muscle
| Lv | LvA | |||||
|---|---|---|---|---|---|---|
| Parameters | Control+Lv | HFD+Lv | HFD+XZK | Control+Lv | HFD+Lv | HFD+XZK |
| Tmax (h) | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 |
| Cmax (ng/mg) | 0.33 | 0.70 | 0.41 | 0.55 | 0.36 | 0.70 |
| AUC0−t (h*ng/mg) | 0.76 | 1.71 | 0.81 | 1.29 | 1.13 | 1.68 |
| AUC0−∞ (h*ng/mg) | 0.79 | 1.82 | 0.84 | 1.32 | 1.32 | 1.74 |
| t1/2 (h) | 0.80 | 0.96 | 0.75 | 0.74 | 1.48 | 0.79 |
| Conversion ratio (Lv to LvA, %) | 61.6 | 41.0 | 66.6 | |||
The experimental groups were as follows: Lv administration (10 mg/kg, i.g.) in the control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in the HFD mice (HFD + Lv), XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in the HFD mice (HFD + XZK). n = 6
Tmax time to peak concentration, Cmax peak concentration, AUC0–t area under the concentration–time curve from zero to the time of last measurable concentration, AUC0−∞ area under the concentration–time curve from zero to infinity, t1/2 apparent elimination half-time
After the oral administration of Lv monomer in the control mice, the plasma levels of Lv and its ring-opening metabolite LvA reached maximum concentrations (peak concentration, Cmax) of 81.33 and 473.33 ng/mL after 2 h and 1.5 h (time to peak concentration, Tmax), respectively. Additionally, the t1/2 (the elimination half-life) values of Lv and LvA were 0.58 and 0.43 h and the AUC0−∞ values were 132.57 and 779.68 h*ng/mL, respectively. Compared to those in the control mice, the Cmax and AUC0−∞ values of LvA were decreased by 63% and 58%, and t1/2 was prolonged by 1.64 times after the oral administration of Lv monomer in the HFD mice. In contrast, the pharmacokinetic parameters of Lv remained approximately the same (Table 2).
Given that the intestinal tract was the main absorption site, Lv was exposed mostly in the intestine, instead of the other tissues. After the oral administration of the Lv monomer, the Cmax, AUC0−∞, and t1/2 values of LvA in the intestine were decreased by 41%, 58%, and 41% in HFD mice, respectively, compared to those in the control mice, although the pharmacokinetic parameters of Lv showed little change (Table 3).
Table 3.
Mean pharmacokinetic parameters of Lv and LvA in intestine
| Lv | LvA | |||||
|---|---|---|---|---|---|---|
| Parameters | Control+Lv | HFD+Lv | HFD+XZK | Control+Lv | HFD+Lv | HFD+XZK |
| Tmax (h) | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 |
| Cmax (ng/mg) | 12.47 | 16.47 | 23.98 | 4.41 | 2.60 | 5.67 |
| AUC0−t (h*ng/mg) | 29.45 | 40.45 | 47.23 | 13.66 | 6.47 | 18.25 |
| AUC0−∞ (h*ng/mg) | 29.50 | 40.50 | 47.25 | 16.47 | 6.86 | 25.21 |
| t1/2 (h) | 0.44 | 0.42 | 0.35 | 1.64 | 0.98 | 2.21 |
| Conversion ratio (Lv to LvA, %) | 34.8 | 13.9 | 33.8 | |||
The experimental groups were as follows: Lv administration (10 mg/kg, i.g.) in the control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in the HFD mice (HFD + Lv), XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in the HFD mice (HFD + XZK). n = 6
Tmax time to peak concentration, Cmax peak concentration, AUC0–t, area under the concentration–time curve from zero to the time of last measurable concentration, AUC0−∞ area under the concentration–time curve from zero to infinity, t1/2 apparent elimination half-time
In contrast to the intestine, only 4.78 and 6.18 h*ng/mg Lv were exposed in the liver of both the control and the HFD mice after Lv administration, whereas a higher exposure of LvA was observed. Compared to those of the control mice, the Cmax and AUC0−∞ values of LvA in the livers of the HFD mice were decreased by 52% and 41% (Table 4).
Table 4.
Mean pharmacokinetic parameters of Lv and LvA in liver
| Lv | LvA | |||||
|---|---|---|---|---|---|---|
| Parameters | Control+Lv | HFD+Lv | HFD+XZK | Control+Lv | HFD+Lv | HFD+XZK |
| Tmax (h) | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 | 2.00 |
| Cmax (ng/mg) | 2.53 | 3.53 | 4.65 | 10.26 | 4.92 | 16.26 |
| AUC0−t (h*ng/mg) | 4.73 | 6.13 | 9.33 | 24.41 | 14.24 | 37.11 |
| AUC0−∞ (h*ng/mg) | 4.78 | 6.18 | 9.35 | 24.60 | 14.58 | 37.28 |
| t1/2 (h) | 0.56 | 0.51 | 0.40 | 0.58 | 0.80 | 0.52 |
| Conversion ratio (Lv to LvA, %) | 83.1 | 69.3 | 79.2 | |||
The experimental groups were as follows: Lv administration (10 mg/kg, i.g.) in the control mice (Control + Lv), Lv administration (10 mg/kg, i.g.) in the HFD mice (HFD + Lv), XZK administration (1200 mg/kg, i.g., an equivalent dose to 10 mg/kg of Lv) in the HFD mice (HFD + XZK). n = 6
Tmax time to peak concentration, Cmax peak concentration, AUC0–t area under the concentration–time curve from zero to the time of last measurable concentration, AUC0−∞ area under the concentration–time curve from zero to infinity, t1/2 apparent elimination half-time
Obviously, lower contents of Lv and LvA were exposed in the muscles, compared to other tissues. Compared to those of the control mice, the Cmax and AUC0−∞ of Lv in the muscles of HFD mice were increased by 2.08 and 2.31 times, respectively, while the Cmax of LvA was decreased by 34% (Table 5).
According to the above parameters, the conversion ratios from Lv to LvA in the plasma and the major tissues were calculated in each group, as shown in Tables 2–5. After Lv treatment, the conversion ratios in the plasmas, livers, intestines, and muscles of the HFD mice were decreased by 24%, 17%, 60%, and 33%, compared to the control mice, respectively. These results revealed that HFD significantly increased the Lv exposure in the muscle and decreased the LvA exposure in the plasma and tissues of mice.
The conversion of Lv into LvA was increased in HFD mice treated with XZK
Furthermore, we investigated the pharmacokinetic profiles of Lv and LvA after the oral administration of XZK (1200 mg/kg) or an equivalent dose of Lv (10 mg/kg) in the HFD mice (Fig. 1 and Tables 2–5).
It was shown that the plasma Lv and LvA concentrations at each sampling point were higher in the XZK-treated mice than in the Lv-treated mice, especially for LvA (Fig. 1a, e). Specifically, in the XZK group, the AUC0−∞ value of Lv increased to 278.96 h*ng/mL (a 1.6-fold increase), and the Cmax and AUC0−∞ values of LvA increased to 839.00 ng/mL (a 4.8-fold increase) and 1376.21 h*ng/mL (a 4.2-fold increase), respectively, compared to the Lv group. However, there was no significant difference in either the Tmax or t1/2 values of Lv and LvA between the two groups (Table 2).
In HFD mice, compared with Lv administration, the Cmax of Lv in the intestine increased from 16.47 to 23.98 ng/mg (a 1.46-fold increase) after XZK administration. In addition, the pharmacokinetic parameters of LvA in the intestine were altered more notably by XZK. For example, Cmax and AUC0−∞ increased greatly (2.18- and 3.68-fold increases, respectively). It is interesting to note that the t1/2 of LvA was elevated by 2.26 times (Table 3).
Moreover, the effects of XZK on the pharmacokinetic parameters of LvA in the liver resembled those in the intestine. For example, XZK could significantly increase the Cmax, AUC0−∞, and t1/2 (3.3-, 2.6-, and 1.5-fold increases, respectively) (Table 4).
In muscle tissues, XZK could increase the Cmax of LvA by 1.9 times, compared to Lv administration. It is noteworthy that the Cmax and AUC0−∞ values of Lv were decreased by XZK, accounting for 60% and 50% of those in the Lv group, respectively (Table 5).
In HFD mice, the conversion ratios in the plasma, liver, intestine, and muscle of the XZK group were increased by 1.28, 1.14, 2.42, and 1.62 times, respectively, compared to the Lv group (Tables 2–5). Hence, these results suggested that XZK could effectively reverse the pharmacokinetic alterations of Lv and LvA induced by an HFD.
XZK increased the expression levels and activities of CES that was inhibited by a HFD
It has been generally accepted that the loop-opening conversion from Lv to LvA is regulated by CES. Therefore, to preliminarily explore the mechanism by which HFD or XZK affected the conversion ratios, we investigated expression levels and activities of CES in the intestine and the liver.
Compared to the control mice, the mRNA levels of Ces2a, Ces2c, and Ces2e (expressed predominantly in mouse intestinal tissues), as well as Ces1c and Ces1d (expressed predominantly in mouse liver tissues), were significantly downregulated in HFD mice (P < 0.01) (Fig. 2a–e). Consistent results were also achieved by Western blot analysis and enzymatic activity assay (Fig. 2f–i). In the HFD mice, compared with Lv administration, expressions and activities of CES in the liver and the intestine were significantly upregulated after XZK administration (P < 0.01) (Fig. 2).
Fig. 2.

Expression levels and activities of CES in the intestine and the liver. Expression levels of Ces2 (a–d) and the total esterase activity (e) in the intestine; expression levels of Ces1 (f–h) and the total esterase activity (i) in the liver in each experimental group. The values are expressed as mean ± SD, n = 4. *P < 0.05, **P < 0.01 vs. Control; #P < 0.05, ##P < 0.01 vs. HFD + Lv
However, XZK and Lv administration had no significant impact on the expression levels and activities of CES in the control mice (data not shown), indicating that there was no direct regulatory effect of XZK on CES.
XZK downregulated the enhanced inflammatory reaction induced by a HFD
The CES level was clearly consistent with the pharmacokinetic data, indicating that a HFD can modulate the pharmacokinetic behaviors of Lv and LvA through the suppression of CES after the oral administration of Lv. Nevertheless, XZK could elevate the hydrolytic activity of CES and enhance the conversion from Lv to LvA, consequently blocking the alteration of the pharmacokinetic characteristics induced by a HFD.
Clinical observation and accumulating evidence have suggested that obesity or hyperlipidemia is often accompanied by inflammation [38–40], which has been reported to regulate CES via PXR in vitro and in vivo [31, 43]. We proposed that the regulation of HFD on CES was based on an inflammatory pathway. For this reason, we investigated the levels of inflammatory factors in the tissues of the control or the HFD mice. This investigation implied that the tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6) levels in the intestines and livers of the HFD mice were significantly higher than those of the control mice (P < 0.01) (except for IL-6 in the liver, P > 0.05) (Fig. 3a–c, e–g), indicating that the pharmacokinetic profiles were altered by a HFD with aggravating inflammation. In contrast, in HFD mice, XZK could not only improve the pharmacokinetic behaviors of Lv and LvA, compared to Lv alone, but also inhibit the inflammation and the upregulation of PXR in the liver and the intestine (Fig. 3d, h), in accordance with the reported anti-inflammatory effects of XZK [44]. Considering that XZK had no influence on CES in the control mice, these findings suggested that the modulation of inflammation in the liver and the intestine offered XZK a target to improve the pharmacokinetic behaviors of Lv and LvA.
Fig. 3.

Expression levels of the inflammatory factors and PXR in the intestine and the liver. mRNA levels of TNF-α (a), IL-1β (b), and IL-6 (c), and mRNA and protein expressions of PXR (d) in the intestine; mRNA levels of TNF-α (e), IL-1β (f), and IL-6 (g), and mRNA and protein expressions of PXR (h) in the liver in each experimental group. The values are expressed as mean ± SD, n = 4. *P < 0.05, **P < 0.01 vs. Control; #P < 0.05, ##P < 0.01 vs. HFD + Lv
XZK and its fraction 1, F1, repressed inflammation in LS174T and HepG2 cells
Based on the above results, it was evident that the non-statin components of XZK had some pharmacokinetic interactions with the substrate drugs for CES through anti-inflammatory effects in HFD mice. We therefore obtained the preliminary fractions of a variety of polar regions from XZK via a phytochemistry method and explored what exactly allowed the non-statins to work in vitro.
First, we induced LS174T (intestinal cancer cells) and HepG2 cells (liver cancer cells) with TNF-α or LPS to induce inflammation in cells. This caused the inflammatory mediators including TNF-α, IL-1β and IL-6 to be extremely upregulated (P < 0.01), indicating the successful activation of the inflammatory response (Fig. 4). Hence, we investigated the influence of XZK and its fractions under the conditions of inflammation. Lv exhibited a small pro-inflammatory effect, which was identical to that reported in current studies [45]. However, XZK and its fraction 1, F1, could suppress the expression of inflammatory factors, compared to the TNF-α and LPS groups (Fig. 4). These results revealed that unlike F2, F3, and F4, F1 showed a strong anti-inflammatory effect, which was similar to that of XZK.
Fig. 4.

Effects of XZK and its fractions on inflammatory factors in LS174T or HepG2 cells. mRNA levels of TNF-α (a), IL-1β (b), and IL-6 (c) in LS174T cells; mRNA levels of TNF-α (d), IL-1β (e), and IL-6 (f) in HepG2 cells after the administration of Lv (10 μM), XZK (480 μg/mL, an equivalent dose to 10 μM Lv), F1 (10.9 μg/mL), F2 (4.5 μg/mL), F3 (14.2 μg/mL), or F4 (2.6 μg/mL) for 24 h under 24 h of pre-treatment of TNF-α (10 ng/mL), or LPS (1 μg/mL). All the experiments were conducted in triplicate, and data with error bars are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01 vs. Control; #P < 0.05, ##P < 0.01 vs. TNF-α or LPS treatment alone
XZK and F1 upregulated the expressions and activities of CES in inflammatory LS174T or HepG2 cells
Furthermore, the effects of XZK, especially F1, on PXR and CES were explored under inflammatory conditions. As shown in Fig. 5, the mRNA and protein expression levels of PXR and CES were downregulated in LS174T or HepG2 cells after TNF-α or LPS treatment (P < 0.05). Likewise, the enzymatic determination showed that the activities of CES in the cytoplasm were significantly inhibited (P < 0.05), while XZK or F1 could effectively reverse the decline of CES by upregulating PXR.
Fig. 5.

Effects of XZK and its fraction 1, F1, on PXR and CES in inflammatory LS174T or HepG2 cells. mRNA and protein expressions of PXR (a) and Ces2 (b), and the total esterase activity (c) in LS174T cells; mRNA and protein expressions of PXR (d) and Ces1 (e), and the total esterase activity (f) in HepG2 cells after the administration of Lv (10 μM), XZK (480 μg/mL, an equivalent dose to 10 μM Lv), F1 (10.9 μg/mL), or F1 (10.9 μg/mL) plus Lv (10 μM) for 24 h under 24 h of pre-treatment of TNF-α (10 ng/mL) or LPS (1 μg/mL). All experiments were conducted in triplicate, and data with error bars are presented as mean ± SD (n = 3). *P < 0.05, **P < 0.01 vs. Control; #P < 0.05, ##P < 0.01 vs. TNF-α or LPS treatment alone; $P < 0.05, $$P < 0.01 vs. TNF-α or LPS + Lv
XZK and F1 increased the intracellular accumulation of LvA and the lipid-lowering effects of Lv
The cellular pharmacokinetics of Lv and LvA were further investigated. Under an inflammatory response induced by TNF-α or LPS, the cellular accumulation of LvA was distinctly decreased and the conversion ratios from Lv to LvA were decreased in the LS174T (from 35.4 to 8.5%) or HepG2 (from 43.4 to 9.4%) cells. Compared to Lv, XZK or F1 could appreciably decrease the accumulation of Lv, but greatly increase the accumulation of LvA and the conversion ratio (Fig. 6a–f).The TC level was determined to evaluate the effects of F1 on the efficacy of Lv (0–80 μM). As shown in Fig. 6g, inflammation induced by LPS obviously attenuated the lipid-lowering efficacy of Lv, which could be reversed by the co-administration of F1.
Fig. 6.

Effects of XZK and its fraction 1, F1, on the intracellular accumulation and efficacy or toxicity of the substrates for CES in inflammatory LS174T or HepG2 cells. Cellular accumulations of Lv (a, d) and LvA (b, e), and the conversion ratios (c, f) in LS174T cells or HepG2 cells after administration for 2 h were presented. The experimental groups were as follows: control + Lv (10 μM), TNF-α, or LPS + Lv (10 μM), TNF-α or LPS + XZK (480 μg/mL, an equivalent dose to 10 μM of Lv), TNF-α or LPS + Lv (10 μM) + F1 (isoflavones, 10.9 μg/mL). TNF-α (10 ng/mL) or LPS (1 μg/mL) pre-treated the cells for 24 h. *P < 0.05, **P < 0.01 vs. Control + Lv; #P < 0.05, ##P < 0.01 vs. TNF-α or LPS + Lv; Cholesterol-lowering efficacy of Lv (g, ranging from 20 to 80 μM, 24 h), as well as cytotoxicity of irinotecan (h, at 0.3, 3.0, 30 μM, 24 h) and clopidogrel (i, at 1, 10, 100 μM, 24 h) with or without the co-administration of F1 (isoflavones, 10.9 μg/mL) under 24 h LPS pre-treatment (1 μg/mL) in HepG2 cells were presented. *P < 0.05, **P < 0.01 vs. Control; #P < 0.05, ##P < 0.01 vs. LPS treatment alone. All experiments were conducted in triplicate, and data with error bars are presented as mean ± SD
The validation of interactions between F1 and other typical substrates for CES
It was demonstrated that anti-inflammatory F1 affected the pharmacokinetics and pharmacodynamics of Lv by upregulating the hydrolytic activity of CES. We then applied two other typical substrate drugs to eventually ascertain these regulatory mechanisms.
As the two typical substrate drugs irinotecan and clopidogrel are both open-loop metabolized via CES. As a non-toxic pro-drug, irinotecan is metabolized to cytotoxic active metabolites by CES, whereas clopidogrel, a toxic pro-drug, is metabolized to non-toxic metabolites by CES. It was shown that LPS could significantly inhibit the cytotoxicity of irinotecan, and this can be reversed by the co-administration of F1. However, LPS and F1 appeared to have opposite effects on the cytotoxicity of clopidogrel (Fig. 6h, i).
Discussion
The preclinical pharmacokinetic studies of most drugs are conducted in healthy animals, and their properties in healthy animals can indeed be used to make critical decisions supporting the efficacy and safety of the drugs [46]. However, the presence of a pathological status could seriously affect drug absorption, distribution, metabolism, and excretion (ADME), which are directly related to both the drug efficacy and the severity of the side effects [47–49]. Therefore, for clinical application, it is more important to study the pharmacokinetic process of drug disposition using animal models corresponding to human diseases, rather than healthy animal models. In the present study, we established a hyperlipidemia mouse model through a long-term HFD, and this model was used in the pharmacokinetic study.
Generally, CES participates in the hydrolysis processes of drugs containing ester, amide, or thioester bonds. It is thus often involved in the activation of precursor drugs or the production of toxic metabolites. Therefore, it is important to include the influence of CES on the pharmacokinetics, pharmacodynamics, and drug toxicity in clinical research. In recent years, the morbidity of metabolic diseases has continued to grow, and hyperlipidemia is one common type of metabolic disease. To date, only a handful of studies have been conducted regarding the relationship between metabolic syndrome and CES, and these have mainly focused on the CES-mediated hydrolysis of the endogenous lipids [35–37]. Even so, there are some reports about the influence of a HFD on the hydrolysis of drugs that contain ester bonds and that are catalyzed by CES. In addition, the accumulating studies suggest that various degrees of inflammatory response have been observed in hyperlipidemia patients or experimental animals [38–40] accompanied by the alteration of enzyme activities under pathological circumstances. For example, the hydrolytic capacities of multiple drugs were decreased in patients with hepatitis and liver cirrhosis, the hydrolysis of the antihypertensive drug cilazapril was decreased by 45% in patients with hepatitis [50]. Similarly, the hydrolysis of the angiotensin-converting enzyme inhibitor perindopril was decreased by 50% in patients with liver cirrhosis [51]
In the present study, we successfully established a mouse hyperlipidemia model using a HFD, and explored the effects of the HFD model on CES in vivo. As shown in Fig. 2, the mRNA levels of Ces1c and Ces1d (the two main liver subtypes) of the HFD mice were downregulated to 76% and 63% of those of the control mice (P < 0.01). More significant downregulation was observed in the mRNA levels of Ces2a, Ces2c, and Ces2e (the three main subtypes in the intestine) of the HFD mice, accounting for 52%, 63%, and 42% of the levels in the control mice (P < 0.01). It is worth noting that a HFD had only a small influence on the CES levels in the plasma of the mice (data not shown), which was consistent with the reported research [37]. At the same time, we found that the levels of inflammatory factors TNF-α, IL-1β, and IL-6 in the liver and the intestine of the HFD mice were significantly higher than those in the control mice, in accordance with the clinical outcomes [38–40]. It was suggested that a HFD could inhibit the expression and activity of CES of mice, which was probably caused by the severe inflammation accompanying the HFD. Hence, we established an inflammatory human liver and intestinal cell model by TNF-α or LPS treatment and validated the effects of inflammation on CES. Our results confirmed that the inflammation model could distinctly decrease the mRNA and protein expression levels of CES, which are believed to be regulated by the upstream regulatory factor PXR (P < 0.05).
Lv is absorbed through the intestinal tract and is hydrolyzed by CES to its active metabolite LvA, which exerts a lipid-lowering effect in liver tissues. However, several reports have suggested that the myotoxicity induced by statins is related to their exposure in muscle tissues. It is not difficult to infer that the condition of CES activity directly influences the pharmacokinetic process of Lv, further influencing its efficacy and toxicity. The above assumptions were confirmed by the comparison of the pharmacokinetic behaviors of Lv in the control and the HFD mice. Due to the downregulation of CES, the conversions from Lv to LvA in plasma, liver, intestine, and muscle were significantly decreased in the HFD mice by 24%, 17%, 60%, and 33%, respectively, compared to the control mice. Furthermore, the inhibited conversion resulted in a lower exposure of the active metabolite LvA in the liver (the lipid-lowering target). Importantly, the Cmax and the AUC0−∞ values of LvA were 48% and 59% of those in the control mice, indicating a lower efficacy. In contrast, Lv exposure in the muscle (the toxic site) of the HFD mice was increased, and the Cmax and AUC0−∞ values of Lv were 2.08 and 2.31 times those of the control mice, consequently indicating a higher toxicity.
XZK is refined by the fermentation of specialized RYR and has been widely applied in the treatment of dyslipidemia. Conclusive clinical evidence has been presented demonstrating that XZK, which contains Lv as an active component, shows a considerably higher efficacy and a lower toxicity, compared to Lv treatment alone [23, 24]. Thus, we predicted that non-statin components of XZK interacted with Lv, likely targeting CES, resulted in a high efficacy and a low toxicity. Therefore, we compared the pharmacokinetic behaviors of Lv after the oral administration of XZK (1200 mg/kg) and an equivalent dosage of Lv (10 mg/kg) in HFD mice. We found that compared to Lv administration alone, XZK could inhibit HFD-associated inflammation, upregulate PXR expression (Fig. 3, P < 0.01), and increase the expression levels and activities of CES (Fig. 2, P < 0.01) in the liver and the intestine tissues. This led to subsequent increases in the conversion ratios from Lv to LvA by 1.28, 1.14, 2.42, and 1.62 times, respectively, in the plasma, liver, intestine, and muscle tissues of the HFD mice (Tables 2–5). It was suggested that XZK could reverse the pharmacokinetic behaviors of Lv and LvA, which were altered by the HFD, particularly in the liver and the muscle tissues. In summary, some kind of non-statin in XZK improved the pharmacokinetic behaviors of the substrates by inhibiting inflammation via CES regulation, providing a pharmacokinetic explanation for its superior lipid-lowering effect. Interestingly, we also observed that XZK had hardly any effects on the expression levels or activities of CES in the control mice (data not shown).
In recent years, it has been reported that interactions among components in extracts produce the pharmacokinetic and the pharmacodynamic alterations in the main bioactive compounds [52–54]. Likewise, in addition to Lv, XZK also contains several other ingredients including natural statins, natural pigments, unsaturated fatty acids, plant sterols, amino acids, isoflavones, alkaloids, and trace elements. To explore the definite ingredients in XZK that appear to have a pharmacokinetic interaction with Lv, we obtained four fractions primarily containing F1, F2, F3, and F4, extracted from XZK via a phytochemistry method. Studies on the inflammatory cell model in vitro showed that the effects of F1 resembled those of XZK, exhibiting a strong anti-inflammatory activity (Fig. 4, P < 0.05). Additionally, F1 reversed the downregulation of CES in the intestinal (LS174T) and the liver (HepG2) cells, induced by inflammation (Fig. 5). We thus used the typical substrate drugs (Lv, clopidogrel, and irinotecan) and demonstrated that F1 effectively affected the pharmacokinetic behaviors, as well as the effectiveness and toxicity of the substrate drugs through the identified mechanisms. In fact, the main components of isoflavones contained in XZK include soybean glycosides, flavin, and genistein.
Furthermore, we found that the AUC values of Lv and LvA in the plasma and the major tissues of the XZK group were increased, compared to that of the Lv monomer (an equivalent dose) group. We believed that there was probably a DDI (drug–drug interaction) between Lv and a non-statin in XZK at transporters or metabolic enzymes. For example, a series of flavonoids were substrates for some efflux transporters such as P-glycoprotein (P-gp), which were expressed in the intestine. Lv and LvA were also reported to be substrates for P-gp, and flavonoids could increase the exposures of Lv and LvA probably through the competition for P-gp [55]. At the same time, our studies showed that XZK could induce CES so that the AUC values of the metabolites in the plasma and the tissues were further increased. Previous reports suggested that the distribution of statins in muscle was mainly affected by influx and efflux transporters, which are specifically expressed in the muscle [33]. Thus, the decreased AUC of Lv in muscle may result from the transporters being affected by the non-statins in XZK. In summary, XZK contains a wide variety of components, and it is important to further search for the actual active component and identify the underlying mechanisms that specifically alter the pharmacokinetic profiles of Lv and LvA.
Conclusion
In the HFD mice, the secretion of inflammatory factors in the liver and the intestine were significantly upregulated and the expression levels and functions of PXR and CES were downregulated, leading to a decline in the conversion ratio from Lv to LvA. Consequently, Cmax levels and exposure of LvA in the livers (the lipid-lowering target) of the HFD mice were significantly lower than those of the control mice, whereas Cmax and Lv exposure in the muscle (toxicity site) were significantly higher. This suggested that inflammation accompanied by a HFD could weaken the lipid-lowering efficacy and enhance the myotoxicity of Lv by affecting the pharmacokinetic behaviors. However, F1 (isoflavones) contained in XZK could significantly upregulate the expression levels and activities of CES by inhibiting inflammation, thus contributing to a higher conversion ratio from Lv to LvA and thereby improving the pharmacokinetic behaviors of Lv and LvA (especially in the liver and the muscle). This would allow for improved therapeutic effects and lower toxicity levels. To summarize, our present study not only illustrated the pharmacokinetic mechanism underlying the influence of a HFD on the pharmacokinetic profiles of Lv, but also supplied an effective approach to study this mechanism (Fig. 7).
Fig. 7.
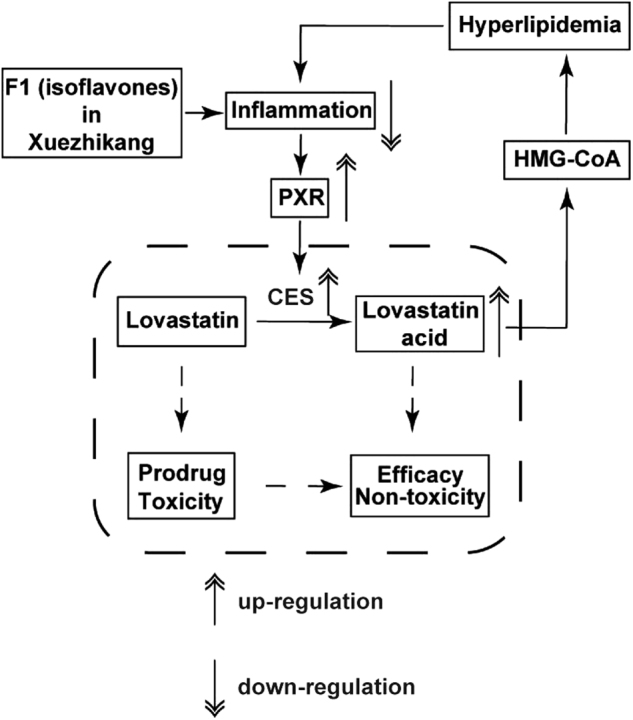
A schematic diagram of the pharmacokinetic mechanisms of F1 in XZK contributed to an improved efficacy and lower toxicity
Electronic supplementary material
Acknowledgements
The study was financially supported by the National Natural Science Foundation of China (81373481), the Jiangsu Provincial Promotion Foundation for the Key Lab of Drug Metabolism and Pharmacokinetics (Grant No. BM2012012), and the Key Technology Projects of China “Creation of New Drugs” (No. 2015ZX09501001).
Author contributions
DF contributed to the design and performed most of the experiments; CG analyzed the data and drafted the manuscript; GW and JA contributed to the development of the project and experimental design; ZT, JS, YX, LY, and CY assisted with the experiments; JA reviewed the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Ji-ye Aa, Email: jiyea@cpu.edu.cn.
Guang-ji Wang, Email: guangjiwang@hotmail.com.
Electronic supplementary material
The online version of this article (10.1038/s41401-018-0039-1) contains supplementary material, which is available to authorized users.
References
- 1.Chen ZY, Ma KY, Liang Y, Peng C, Zuo Y. Role and classification of cholesterol-lowering functional foods. J Funct Foods. 2011;3:61–9. doi: 10.1016/j.jff.2011.02.003. [DOI] [Google Scholar]
- 2.Gropper SS, Smith LJ, Groff LJ. Advanced nutrition and human metabolism. 7th ed. Ohio (USA): Peter Marshall, Wadsworth; 2005.
- 3.Ma J, Li Y, Ye Q, Li J, Hua Y, Ju D, et al. Constituents of red yeast rice, a traditional Chinese food and medicine. J Agric Food Chem. 2000;48:5220–5. doi: 10.1021/jf000338c. [DOI] [PubMed] [Google Scholar]
- 4.Wei W, Li C, Wang Y, Su H, Zhu J, Kritchevsky D. Hypolipidemic and anti-atherogenic effects of long-term Cholestin (Monascus purpureus-fermented rice, red yeast rice) in cholesterol fed rabbits. J Nutr Biochem. 2003;14:314–8. doi: 10.1016/S0955-2863(03)00051-2. [DOI] [PubMed] [Google Scholar]
- 5.Lee CL, Tsai TY, Wang JJ, Pan TM. In vivo hypolipidemic effects and safety of low dosage Monascus powder in a hamster model of hyperlipidemia. Appl Microbiol Biotechnol. 2006;70:533–40. doi: 10.1007/s00253-005-0137-0. [DOI] [PubMed] [Google Scholar]
- 6.Lin CC, Li TC, Lai MM. Efficacy and safety of Monascus purpureus Went rice in subjects with hyperlipidemia. Eur J Endocrinol. 2005;153:679–86. doi: 10.1530/eje.1.02012. [DOI] [PubMed] [Google Scholar]
- 7.Heber D, Yip I, Ashley JM, Elashoff DA, Elashoff RM, Go VL. Cholesterol-lowering effects of a proprietary Chinese red-yeast-rice dietary supplement. Am J Clin Nutr. 1999;69:231–6. doi: 10.1093/ajcn/69.2.231. [DOI] [PubMed] [Google Scholar]
- 8.Li C, Zhu Y, Wang Y, Zhu JS, Chang J, Kritchevsky D, et al. Monascus purpureus-fermented rice (red yeast rice): a natural food product that lowers blood cholesterol in animal models of hypercholesterolemia. Nutr Res. 1998;18:71–81. doi: 10.1016/S0271-5317(97)00201-7. [DOI] [Google Scholar]
- 9.Wang JJ, Pan TM, Shieh MJ, Hsu CC. Effect of red mold rice supplements on serum and meat cholesterol levels of broilers chicken. Appl Microbiol Biotechnol. 2006;71:812–8. doi: 10.1007/s00253-005-0222-4. [DOI] [PubMed] [Google Scholar]
- 10.Liu J, Zhang J, Shi Y, Grimsgaard S, Alraek T, Fønnebø V. Chinese red yeast rice (Monascus purpureus) for primary hyperlipidemia: a meta-analysis of randomized controlled trials. Chin Med. 2006;1:4. doi: 10.1186/1749-8546-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Grieco A, Miele L, Pompili M, Biolato M, Vecchio FM, Grattagliano I, et al. Acute hepatitis caused by a natural lipid-lowering product: when “alternative” medicine is no “alternative” at all. J Hepatol. 2009;50:1273–7. doi: 10.1016/j.jhep.2009.02.021. [DOI] [PubMed] [Google Scholar]
- 12.Klimek M, Wang S, Ogunkanmi A. Safety and efficacy of red yeast rice (Monascus purpureus) as an alternative therapy for hyperlipidemia. P T. 2009;34:313–27. [PMC free article] [PubMed] [Google Scholar]
- 13.Heber D, Lembertas A, Lu QY, Bowerman S, Go VL. An analysis of nine proprietary Chinese red yeast rice dietary supplements: implications of variability in chemical profile and contents. J Altern Complement Med. 2001;7:133–9. doi: 10.1089/107555301750164181. [DOI] [PubMed] [Google Scholar]
- 14.Manzoni M, Rollini M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl Microbiol Biotechnol. 2002;58:555–64. doi: 10.1007/s00253-002-0932-9. [DOI] [PubMed] [Google Scholar]
- 15.Trimarco B, Benvenuti C, Rozza F, Cimmino CS, Giudice R, Crispo S. Clinical evidence of efficacy of red yeast rice and berberine in a large controlled study versus diet. Med J Nutr Metab. 2011;4:133–9. doi: 10.1007/s12349-010-0043-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shang Q, Liu Z, Chen K, Xu H, Liu J. A systematic review of Xuezhikang, an extract from red yeast rice, for coronary heart disease complicated by dyslipidemia. Evid Based Complement Altern Med. 2012;2012:636547. doi: 10.1155/2012/636547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma KY, Zhang ZS, Zhao SX, Chang Q, Wong YM, Yeung SY, et al. Red yeast rice increases excretion of bile acids in Hamsters. Biomed Environ Sci. 2009;22:269–77. doi: 10.1016/S0895-3988(09)60056-8. [DOI] [PubMed] [Google Scholar]
- 18.Becker DJ, Gordon RY, Halbert SC, French B, Morris PB, Rader DJ. Red yeast rice for dyslipidemia in statin-intolerant patients: a randomized trial. Ann Intern Med. 2009;150:830–9. doi: 10.7326/0003-4819-150-12-200906160-00006. [DOI] [PubMed] [Google Scholar]
- 19.Bartley GE, Yokoyama W, Young SA, Anderson WH, Hung SC, Albers DR, et al. Hypocholesterolemic effects of hydroxypropyl methylcellulose are mediated by altered gene expression in hepatic bile and cholesterol pathways of male hamsters. J Nutr. 2010;140:1255–60. doi: 10.3945/jn.109.118349. [DOI] [PubMed] [Google Scholar]
- 20.Gordon RY, Cooperman T, Obermeyer W, Becker DJ. Marked variability of monacolin levels in commercial red yeast rice products: buyer beware! Arch Intern Med. 2010;170:1722–7. doi: 10.1001/archinternmed.2010.382. [DOI] [PubMed] [Google Scholar]
- 21.Kidambi S, Patel S. Cholesterol and non-cholesterol sterol transporters: ABCG5, ABCG8 and NPC1L1: a review. Xenobiotica. 2008;38:1119–39. doi: 10.1080/00498250802007930. [DOI] [PubMed] [Google Scholar]
- 22.Liu ZL, Liu JP, Zhang AL, Wu Q, Ruan Y, Lewith G, et al. Chinese herbal medicines for hypercholesterolemia. Cochrane Database Syst Rev. 2011;7:CD008305. doi: 10.1002/14651858.CD008305.pub2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang SR. Effects of Xuezhikang vs lovastatin in treating hyperlipidemia. Chin J New Drugs Clin Remedies. 2000;19:249–51. [Google Scholar]
- 24.Liu LJ, Ma SR, Liu SC. The pharmacological effects and clinical evaluation of Xuezhikang. China. Pharmacy. 2003;14:184–6. [Google Scholar]
- 25.Feng D, Sun JG, Sun RB, Ou-Yang BC, Yao L, Aa JY, et al. Isoflavones and phytosterols contained in Xuezhikang capsules modulate cholesterol homeostasis in high-fat diet mice. Acta Pharmacol Sin. 2015;36:1462–72. doi: 10.1038/aps.2015.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Satoh T, Hosokawa M. The mammalian carboxylesterases: from molecules to functions. Annu Rev Pharmacol Toxicol. 1998;38:257–88. doi: 10.1146/annurev.pharmtox.38.1.257. [DOI] [PubMed] [Google Scholar]
- 27.Furihata T, Hosokawa M, Nakata F, Satoh T, Chiba K. Purification, molecular cloning, and functional expression of inducible liver acylcarnitine hydrolase in C57BL/6 mouse, belonging to the carboxylesterase multigene family. Arch Biochem Biophys. 2003;416:101–9. doi: 10.1016/S0003-9861(03)00286-8. [DOI] [PubMed] [Google Scholar]
- 28.Furihata T, Hosokawa M, Koyano N, Nakamura T, Satoh T, Chiba K. Identification of di-(2-ethylhexyl) phthalate-induced carboxylesterase 1 in C57BL/6 mouse liver microsomes: purification, cDNA cloning, and baculovirus-mediated expression. Drug Metab Dispos. 2004;32:1170–7. doi: 10.1124/dmd.104.000620. [DOI] [PubMed] [Google Scholar]
- 29.Hosokawa M, Suzuki K, Takahashi D, Mori M, Satoh T, Chiba K. Purification, molecular cloning, and functional expression of dog liver microsomal acyl-CoA hydrolase: a member of the carboxylesterase multigene family. Arch Biochem Biophys. 2001;389:245–53. doi: 10.1006/abbi.2001.2346. [DOI] [PubMed] [Google Scholar]
- 30.Heymann E. Hydrolysis of carboxylic esters and amides. In: Jacoby WB, editor. Metabolic basis of detoxication; metabolism of functional groups. New York: Elsevier; 1982. pp. p229–45. [Google Scholar]
- 31.Jones RD, Taylor AM, Tong EY, Repa JJ. Carboxylesterases are uniquely expressed among tissues and regulated by nuclear hormone receptors in the mouse. Drug Metab Dispos. 2013;41:40–9. doi: 10.1124/dmd.112.048397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Halpin RA, Ulm EH, Till AE, Kari PH, Vyas KP, Hunninghake DB, et al. Biotransformation of lovastatin. V. Species differences in in vivo metabolite profiles of mouse, rat, dog, and human. Drug Metab Dispos. 1993;21:1003–11. [PubMed] [Google Scholar]
- 33.Knauer MJ, Urquhart BL, Meyer zu Schwabedissen HE, Schwarz UI, Lemke CJ, Leake BF, et al. Human skeletal muscle drug transporters determine local exposure and toxicity of statins. Circ Res. 2010;106:297–306. doi: 10.1161/CIRCRESAHA.109.203596. [DOI] [PubMed] [Google Scholar]
- 34.Skottheim IB, Gedde-Dahl A, Hejazifar S, Hoel K, Asberg A. Statin induced myotoxicity: the lactone forms are more potent than the acid forms in human skeletal muscle cells in vitro. Eur J Pharm Sci. 2008;33:317–25. doi: 10.1016/j.ejps.2007.12.009. [DOI] [PubMed] [Google Scholar]
- 35.Shen W, Patel S, Yu Z, Jue D, Kraemer FB. Effects of rosiglitazone and high fat diet on lipase/esterase expression in adipose tissue. Biochim Biophys Acta. 2007;1771:177–84. doi: 10.1016/j.bbalip.2006.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedrichsen M, Poulsen P, Wojtaszewski J, Hansen PR, Vaag A, Rasmussen HB. Carboxylesterase 1 gene duplication and mRNA expression in adipose tissue are linked to obesity and metabolic function. PLoS ONE. 2013;8:e56861. doi: 10.1371/journal.pone.0056861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pati S, Krishna S, Lee JH, Ross MK, de La Serre CB, Harn DA, Jr, et al. Effects of high-fat diet and age on the blood lipidome and circulating endocannabinoids of female C57BL/6 mice. Biochim Biophys Acta. 2018;1863:26–39. doi: 10.1016/j.bbalip.2017.09.011. [DOI] [PubMed] [Google Scholar]
- 38.Steven S, Dib M, Hausding M, Kashani F, Oelze M, Kröller-Schön S, et al. CD40L controls obesity-associated vascular inflammation, oxidative stress and endothelial dysfunction in mice-translational aspects for man. Cardiovasc Res. 2017;121:10552–61. doi: 10.1093/cvr/cvx197. [DOI] [PubMed] [Google Scholar]
- 39.Jung TW, Kim HC, Abd El-Aty AM, Jeong JH. Protectin DX ameliorates palmitate- or high-fat diet-induced insulin resistance and inflammation through an AMPK-PPARα-dependent pathway in mice. Sci Rep. 2017;7:1397. doi: 10.1038/s41598-017-01603-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mohammadi M, Gozashti MH, Aghadavood M, Mehdizadeh MR, Hayatbakhsh MM. Clinical significance of serum IL-6 and TNF-α levels in patients with metabolic syndrome. Rep Biochem Mol Biol. 2017;6:74–9. [PMC free article] [PubMed] [Google Scholar]
- 41.Richardson TA, Morgan ET. Hepatic cytochrome P450 gene regulation during endotoxin-induced inflammation in nuclear receptor knockout mice. J Pharmacol Exp Ther. 2005;314:703–9. doi: 10.1124/jpet.105.085456. [DOI] [PubMed] [Google Scholar]
- 42.Wait R, Chiesa GC, Parolini C, Miller I, Begum S, Brambilla D, et al. Reference maps of mouse serum acute-phase proteins: changes with LPS-induced inflammation and apolipoprotein A-I and A-II transgenes. Proteomics. 2005;5:4245–53. doi: 10.1002/pmic.200401292. [DOI] [PubMed] [Google Scholar]
- 43.Xiong J, Shang W, Wu L, Chen R, Liu W, Ning R, et al. Glucose dominates the regulation of carboxylesterases induced by lipopolysaccharide or interleukin-6 in primary mouse hepatocytes. Life Sci. 2014;112:41–8. doi: 10.1016/j.lfs.2014.07.019. [DOI] [PubMed] [Google Scholar]
- 44.Li JJ, Hu SS, Fang CH, Hui RT, Miao LF, Yang YJ, et al. Effects of Xuezhikang, an extract of cholestin, on lipid profile and C-reactive protein: a short-term time course study in patients with stable angina. Clin Chim Acta. 2005;352:217–24. doi: 10.1016/j.cccn.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 45.Zanin V, Marcuzzi A, Kleiner G, Piscianz E, Monasta L, Zacchigna S, et al. Lovastatin dose-dependently potentiates the pro-inflammatory activity of lipopolysaccharide both in vitro and in vivo. J Cardiovasc Transl Res. 2013;6:981–8. doi: 10.1007/s12265-013-9506-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhao S, Zhang Y, Ju P, Gu L, Zhuang R, Zhao L, et al. Determination of 6258-70, a new semi-synthetic taxane, in rat plasma and tissues: application to the pharmacokinetics and tissue distribution study. J Pharm Anal. 2016;6:219–25. doi: 10.1016/j.jpha.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu XF, Bi K, Chen X. Physiologically based pharmacokinetic model of docetaxel and interspecies scaling: comparison of simple injection with folate receptor-targeting amphiphilic copolymer-modified liposomes. Xenobiotica. 2016;46:1093–104. doi: 10.3109/00498254.2016.1155128. [DOI] [PubMed] [Google Scholar]
- 48.Wang Q, Chao J, Xiao Z, Zhu X, Yan S, Wang H, et al. Insight into the pharmacokinetic behavior of tanshinone IIA in the treatment of Crohn’s disease: comparative data for tanshinone IIA and its two glucuronidated metabolites in normal and recurrent colitis models after oral administration. Xenobiotica. 2017;47:66–76. doi: 10.3109/00498254.2016.1160158. [DOI] [PubMed] [Google Scholar]
- 49.Wang Y, Zhao M, Ye H, Shao Y, Yu Y, Wang M, et al. Comparative pharmacokinetic study of the main components of Cortex Fraxini after oral administration in normal and hyperuricemic rats. Biomed Chromatogr. 2017;31:3934. doi: 10.1002/bmc.3934. [DOI] [PubMed] [Google Scholar]
- 50.Gross V, Treher E, Haag K, Neis W, Wiegand U, Schölmerich J. Angiotensin-converting enzyme (ACE)-inhibition in cirrhosis: pharmacokinetics and dynamics of the ACE-inhibitor cilazapril (Ro 31-2848) J Hepatol. 1993;17:40–7. doi: 10.1016/S0168-8278(05)80519-7. [DOI] [PubMed] [Google Scholar]
- 51.Thiollet M, Funck-Brentano C, Grange JD, Midavaine M, Resplandy G, Jaillon P. The pharmacokinetics of perindopril in patients with liver cirrhosis. Br J Clin Pharmacol. 1992;33:326–8. doi: 10.1111/j.1365-2125.1992.tb04045.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hao W, Zhu ZY, Zhang GQ, Zhao L, Zhang H, Zhu D, et al. Comparative pharmacokinetic study of paeoniflorin after oral administration of pure paeoniflorin, extract of cortex Moutan and Shuang-Dan prescription to rats. J Ethnopharmacol. 2009;125:444–9. doi: 10.1016/j.jep.2009.07.019. [DOI] [PubMed] [Google Scholar]
- 53.Petiwala SM, Li G, Ramaiya A, Kumar A, Gill RK, Saksena S, et al. Pharmacokinetic characterization of mangosteen (Garcinia mangostana) fruit extract standardized to α-mangostin in C57BL/6 mice. Nutr Res. 2014;34:336–45. doi: 10.1016/j.nutres.2014.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang M, Yang X, An J, Xiao W, Wang Z, Huang W, et al. Comparative pharmacokinetic profiles of tectorigenin in rat plasma by UPLC-MS/MS after oral administration of Iris tectorum maxim extract and pure tectoridin. J Pharm Biomed Anal. 2015;114:34–41. doi: 10.1016/j.jpba.2015.05.005. [DOI] [PubMed] [Google Scholar]
- 55.Gertz M, Davis JD, Harrison A, Houston JB, Galetin A. Grapefruit juice-drug interaction studies as a method to assess the extent of intestinal availability: utility and limitations. Curr Drug Metab. 2008;9:785–95. doi: 10.2174/138920008786049276. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.