

Abstract
Cyanidin is polyphenolic pigment found in plants. We have previously demonstrated that cyanidin protects nerve cells against Aβ25-35-induced toxicity by decreasing oxidative stress and attenuating apoptosis mediated by both the mitochondrial apoptotic pathway and the ER stress pathway. To further elucidate the molecular mechanisms underlying the neuroprotective effects of cyanidin, we investigated the effects of cyanidin on neuroinflammation mediated by the TLR4/NOX4 pathway in Aβ25-35-treated human neuroblastoma cell line (SK-N-SH). SK-N-SH cells were exposed to Aβ25-35 (10 μmol/L) for 24 h. Pretreatment with cyanidin (20 μmol/L) or NAC (20 μmol/L) strongly inhibited the NF-κB signaling pathway in the cells evidenced by suppressing the degradation of IκBα, translocation of the p65 subunit of NF-κB from the cytoplasm to the nucleus, and thereby reducing the expression of iNOS protein and the production of NO. Furthermore, pretreatment with cyanidin greatly promoted the translocation of the Nrf2 protein from the cytoplasm to the nucleus; upregulating cytoprotective enzymes, including HO-1, NQO-1 and GCLC; and increased the activity of SOD enzymes. Pretreatment with cyanidin also decreased the expression of TLR4, directly improved intracellular ROS levels and regulated the activity of inflammation-related downstream pathways including NO production and SOD activity through TLR4/NOX4 signaling. These results demonstrate that TLR4 is a primary receptor in SK-N-SH cells, by which Aβ25-35 triggers neuroinflammation, and cyanidin attenuates Aβ-induced inflammation and ROS production mediated by the TLR4/NOX4 pathway, suggesting that inhibition of TLR4 by cyanidin could be beneficial in preventing neuronal cell death in the process of Alzheimer's disease.
Keywords: Alzheimer′s disease, amyloid-β, cyanidin, neuroinflammation, nuclear factor-κB, Nrf2, TLR4, oxidative stress, SK-N-SH cells
Introduction
Neuroinflammation is a key process in Alzheimer's disease (AD), which suggests that Aβ acts as a major trigger of neuronal and glial pro-inflammatory activation. Following activation, both neurons and glial cells produce pro-inflammatory molecules including cytokines and chemokines1,2. Toll-like receptor 4 (TLR4) has been implicated in the pathogenesis of AD because of its association with Aβ-triggered inflammatory activation, which results in the activation of NF-κB and production of pro-inflammatory cytokines and/or anti-inflammatory cytokines3,4,5,6. TLRs activate a common signaling pathway which culminates in the activation of NF-κB transcription factors and the PI3K/Akt pathway3,6,7,8,9. It has been suggested that Aβ binds to Toll-like receptor 4 (TLR4), causing augmented intracellular oxidative stress and the release of inflammatory factors10,11,12. In addition, Aβ has been shown to specifically activate the ROS-producing enzyme NADPH oxidase (NOX) and to induce neuronal cell death13 The PI3K/Akt pathway has been shown to control a variety of cellular processes, including cell survival and proliferation. PI3K can function as either a positive or a negative regulator of TLR signaling8,9. Previous studies have demonstrated that Aβ induces inflammation by regulation of the TLR4/NOX4/PI3K/AKT/NF-κB signaling pathway14,15,16. On the other hand, the nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway is vital in the brain's defense against oxidative insults through its upregulation of antioxidants17. Under oxidative conditions, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it binds to antioxidant response elements (AREs) and stimulates the transcription of phase II genes such as heme oxygenase (HO-1), glutamate-cysteine ligase catalytic subunit (GCLC), quinone oxidoreductase 1 (NQO-1), and superoxide dismutase (SOD) to protect the cell from oxidative stress17,18. All of the above demonstrate that Aβ is a neurotoxin and that it causes damage to nerve cells, primarily by inducing cell stress, before leading to cellular dysfunction and death. Hence, it is necessary to develop effective therapeutic strategies for AD that focus on reversing neurotoxicity by attenuating inflammation.
Cyanidin is polyphenolic pigment found in plants. It has a characteristic red to purple color and confers a wide range of pharmacological benefits, including as a potent antioxidant and a metal-chelating agent, in addition to its anti-inflammatory, antiviral, and anticarcinogenic properties19,20,21. Our previous studies have demonstrated that cyanidin protects nerve cells against Aβ25-35-induced toxicity by decreasing oxidative stress and attenuating apoptosis mediated by both the mitochondrial apoptotic pathway and the ER stress pathway22,23. In the interest of better understanding the protective role of cyanidin in AD prevention, it is important to investigate the mechanisms whereby cyanidin causes these effects. Therefore, it is of special interest to investigate the protective effect of cyanidin against Aβ25-35-induced neuroinflammation mediated by the TLR4/NOX4 pathway.
Materials and methods
Materials
Aβ25-35, cyanidin-3-glucoside (cyanidin) and N-acetylcysteine (NAC) were obtained from Sigma (St Louis, MO, USA). PrestoBlue™ was purchased from Invitrogen (Carlsbad, CA, USA). H2DCFDA was purchased from Calbiochem, USA. Minimum essential medium (MEM), fetal bovine serum (FBS), penicillin, and streptomycin were purchased from Gibco BRL (Gaithersburg, MD, USA). The following antibodies were used for the Western blot analysis: anti-p65, anti-mouse IgG peroxidase-conjugated secondary antibody, anti-rabbit IgG peroxidase-conjugated secondary antibody (Millipore, Bedford, MA, USA), anti-β-actin, anti-lamin B, anti-p-Akt, anti-Akt, anti-IκBα (Cell Signaling Technology, MA, USA), anti-TLR4, anti-Nrf2, anti-NQO-1, anti-GCLC (Abcam, Cambridge, UK), anti-HO-1, and anti-iNOS (Calbiochem, MA, USA). A superoxide dismutase assay kit was purchased from Cayman Chemical Company (Cayman, MI, USA). LY294002 (PI3K inhibitor) was obtained from Abcam (Cambridge, UK). SH-5 (Akt inhibitor), TAK-242 (TLR4 inhibitor), ochratoxin A and GKT137831 (NOX4 inhibitor) were obtained from Sigma (St. Louis, MO, USA).
Cell culture and treatment
The human neuroblastoma cell line (SK-N-SH) was a gift from Dr Damratsamon SURANGKUL (Faculty of Medical Science, Naresuan University, Thailand) (ATCC, Manassas, VA, USA). SK-N-SH cells were grown to confluence in 60-mm culture plates (Nunc, Denmark) with minimum essential medium (MEM) containing 10% heat-inactivated fetal bovine serum (FBS) and 1% penicillin–streptomycin (100 units/mL penicillin and 100 μg/mL streptomycin) (GIBCO-BRL, Gaithersburg, MD, USA) and incubated at 37 °C in a humidified 5% CO2 atmosphere. NAC is a strong antioxidant, anti-inflammatory and free radical scavenger in several neurodegenerative disorder models; thus, it was used as a positive control in this study24,25. To investigate the neuroprotective effect of cyanidin, we divided the cells into 4 groups for the experiments: non-treated control, Aβ25-35 (10 μmol/L), Aβ25-35 plus cyanidin (20 μmol/L) and Aβ25-35 plus NAC (20 μmol/L). Experiments were executed for 24 h after the cells were seeded. Cyanidin or NAC was added for 2 h prior to the treatment with Aβ25-35, and then the cells were co-incubated with Aβ25-35 and cyanidin for another 24 h.
Western blot analysis
For analysis of protein levels by Western blotting, the cells were cultured at a density of 5×105 cells/mL in a 60-mm culture dish at 37 °C. The cells were pretreated with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h in the presence of 10 μmol/L Aβ25-35 for 24 h. Our previous study found that cyanidin (20 μmol/L) and NAC (20 μmol/L) significantly reduced apoptosis caused by Aβ25-35 in SK-N-SH cells23. Thus, cyanidin (20 μmol/L) and NAC (20 μmol/L) were used in the present study. The cells were lysed in a lysis buffer containing 1% NP-40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 40 mmol/L β-glycerophosphate, 50 mmol/L sodium fluoride, 2 mmol/L sodium orthovanadate, and 1×protease inhibitors at 4 °C by vigorous shaking for 15 min. The lysates were centrifuged at 4 °C at 13 000 r/min for 20 min, and the supernatants were stored at −80 °C. By contrast, the cytoplasmic proteins were prepared as follows: cells were added into ice-cold hypotonic lysis buffer containing 10 mmol/L HEPES (pH 7.9), 1.5 mmol/L magnesium chloride, 10 mmol/L potassium chloride, 0.5 mmol/L phenylmethylsulfonyl fluoride, 0.5 mmol/L dithiothreitol, and 1×protease inhibitors at 4 °C for 15 min. The homogenates were centrifuged at 13 000 r/min for 30 s and stored at −80 °C. The nuclear pellets were resuspended in ice-cold hypotonic extraction buffer containing 10 mmol/L HEPES (pH 7.9), 0.42 mol/L sodium chloride, 1.5 mmol/L magnesium chloride, 10 mmol/L potassium chloride, 0.5 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L dithiothreitol, and 1× protease inhibitors at 4 °C for 40 min. The homogenates were centrifuged at 13 000 r/min at 4 °C for 10 min, and the supernatants were stored at −80 °C; thereafter, the protein concentrations were measured by the Bradford protein assay (BioRad, USA). The proteins in each of the fractions (50 μg) were electrophoresed in a 10%–15% SDS polyacrylamide gel and then transferred to a PVDF membrane (Immobilon-P, Millipore, Bedford, MA, USA) at 400 mA for 35 min. The blots were blocked for 3 h at room temperature in fresh blocking buffer (containing 5% skim milk in 0.1% Tween 20 in Tris-buffered saline, pH 7.4). The membranes were incubated with primary antibody (anti-TLR4 [1:1000], anti-Akt [1:1000], anti-p-Akt [1:1000], anti-p65 [1:1000], anti-IκBα [1:500], anti-iNOS [1:1000], anti-Nrf2 [1:1000], anti-HO-1 [1:1000], anti-NQO-1 [1:1000], and anti-GCLC [1:1000]) at 4 °C overnight, then probed with horseradish peroxidase-conjugated secondary antibodies (Millipore, MA, USA) for 2 h at room temperature. The blots were incubated with ECL substrate solution for 5 min. The chemiluminescent bands were detected using blue X-ray films, and the optical density of each band was analyzed by using the software ImageJ (National Institutes of Health, Bethesda, MD, USA).
Measurement of nitric oxide (NO)
The most commonly used indirect method relies on the Griess reagent; this method is based on measurement of NO2-, which is the stable nitrogen oxide formed following NO decomposition in aqueous solution in vitro. The cells were cultured at a density of 2×104 cells/well in 96-well plates at 37 °C overnight. The cells were pretreated with cyanidin or NAC for 2 h and were then treated with 10 μmol/L Aβ25-35 for 24 h. After the treatment, the cells were added to the Griess reagent (a mixture of 1% sulfanilamide in 5% phosphoric acid and 0.1% N-1-naphthylethylenediamine dihydrochloride) and further incubated at 37 °C for 10 min. The formation of nitrite was measured at 540 nm using a microplate reader (BioTek Instruments, Inc, USA).
Measurement of superoxide dismutase (SOD) activity
The specific activity of SOD in each sample was determined using an SOD assay kit in accordance with the manufacturer's instructions (BioAssay, MI, USA). The cells were cultured at a density of 5×105 cells/mL in a 60-mm culture dish at 37 °C overnight. The cells were pretreated with cyanidin or NAC for 2 h and treated with 10 μmol/L Aβ25-35 for 24 h. After the treatment, the cells were lysed and the supernatant was collected for an assay to determine the SOD activity. The enzyme activity was measured at 420 nm using a microplate reader (BioTek Instruments, Inc, USA).
Measurement of reactive oxygen species (ROS)
Nonpolar H2 DCFDA was allowed to penetrate the cell membrane, and it was de-esterified by intracellular esterase to 2′,7′-dichlorofluorescin (DCFH). In the presence of ROS, DCFH was oxidized to fluorescent 2′,7′-dichlorofluorescein (DCF). The cells were cultured at a density of 2×104 cells/well in 96-well plates at 37 °C overnight and exposed to cyanidin or NAC for 2 h, after which they were treated with 10 μmol/L Aβ25-35 for 24 h. Prior to the detection of ROS, the medium was removed and 20 μmol/L of H2DCFDA was added to each of the wells, followed by incubation at 37 °C for 2 h. The fluorescence was measured using a fluorescence microplate reader (DTX800, Beckman Coulter, Austria) at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Statistical analysis
All the values are presented as the mean±SEM. The statistical significance was determined by one-way analysis of variance (ANOVA) followed by a post hoc Dunnett's test to compare the means of individual groups. The difference was considered significant when P<0.05, as P values <0.05 are considered statistically significant.
Results
Cyanidin inhibits Aβ-induced NF-κB pathway activation and NO production
Previous studies have reported that Aβ-induces neuroinflammation. Thus, to explore the mechanism underlying the ability of cyanidin to prevent Aβ-induced neuroinflammation mediated by NF-κB pathway activation, we investigated the expression levels of proteins related to the NF-κB pathway (including the expressions of the total p65, cytosolic p65, nuclear p65, IκBα, and iNOS proteins) by Western blotting analysis. The results showed that Aβ25-35 significantly increased the total protein expression of p65 and increased the translocation of p65 from the cytosol into the nucleus (Figure 1A), which is closely related to the finding that the degradation of IκBα significantly decreased (Figure 1B). At the same time, the expression of iNOS was also found to have increased compared with that of the control group (Figure 1C). Upon pretreatment with cyanidin or NAC before Aβ25-35 treatment, the expression levels of these proteins related to the NF-κB pathway were significantly reversed compared with the levels in the group treated with Aβ25-35 alone (Figure 1A–C). Next, we investigated the level of nitrite by using the Griess test. Treatment with Aβ25-35 resulted in a significant increase in the nitrite production of the treated group compared with that of the control group (Figure 1D). Upon pretreatment with cyanidin or NAC before Aβ25-35 treatment, the level of NO production was observed to decrease significantly compared with that of the group treated with Aβ25-35 alone (Figure 1D). These results indicate that cyanidin attenuates Aβ-induced NF-κB pathway activation by inhibiting proteins related to the NF-κB pathway and thereby suppressing the NO production.
Figure 1.
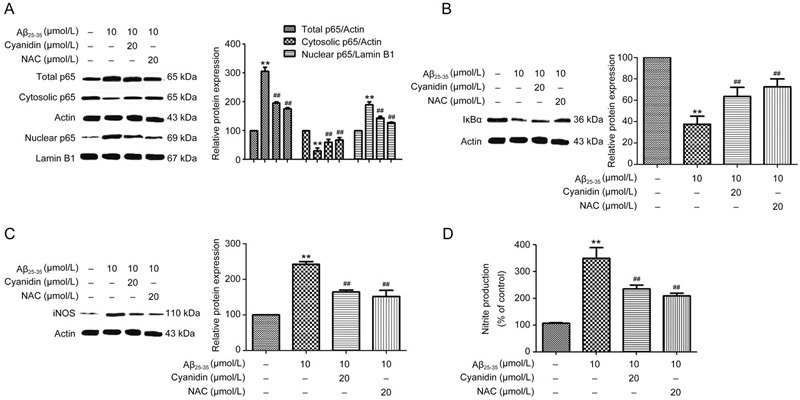
Cyanidin attenuates Aβ-induced NF-κB pathway activation and NO production in SK-N-SH cells. The SK-N-SH cells were pretreated with 20 μmol/L of cyanidin or 20 μmol/L of NAC for 2 h prior to treatment with 10 μmol/L Aβ25-35 for 24 h. The expression levels of proteins related to the NF-κB pathway were analyzed by Western blot. Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of p65 (A); IκBα (B) and iNOS (C); cytosolic proteins were normalized to actin, the nuclear proteins were normalized to lamin B1. The Griess reaction assay was performed to determine the level of nitrite production (D). The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. ## P<0.01 vs the Aβ25-35-treated group.
Cyanidin inhibits Aβ-reduced Nrf2 pathway activation and SOD activity in SK-N-SH cells
We next examined whether cyanidin activates antioxidant-pathway-mediated Nrf2 pathway activation. The Nrf2 pathway, the antagonist mechanism of NF-κB, provides defense against oxidative insults through its upregulation of antioxidants. The expression levels of proteins related to the Nrf2 pathway, including total Nrf2, cytosolic Nrf2, nuclear Nrf2, HO-1, NQO-1, and GCLC proteins, were analyzed by Western blot. Compared with the control group, the Aβ25-35-treated group showed decreased total protein expression of Nrf2 and decreased Nrf2 translocation from the cytosol into the nucleus (Figure 2A). We further examined the expression of antioxidant enzyme genes including HO-1, NQO-1, and GCLC. As shown in Figure 2B, Aβ25-35 significantly decreased the protein expression levels of HO-1, NQO-1, and GCLC compared with the levels in the control group. Upon pretreatment with cyanidin or NAC followed by treatment with Aβ25-35, the expression levels of these proteins related to the Nrf2 pathway were observed to have significantly reversed compared with those in the group treated with Aβ25-35 alone (Figure 2A–2B). Furthermore, we investigated SOD activity by using a SOD activity assay. Treatment with Aβ25-35 was found to result in a significant decrease in SOD activity compared with that of the control group (Figure 2C). Upon pretreatment with cyanidin or NAC, it was found that there was a significant increase in SOD activity compared with that of the group treated with Aβ25-35 alone (Figure 2C). Pretreatment with cyanidin or NAC decreased NO production. To confirm that the effect of cyanidin against Aβ-induced toxicity is due to the activation of the Nrf2 signaling pathway, we applied the Nrf2 inhibitor ochratoxin A (OTA). Our results demonstrated that inhibition of Nrf2 significantly decreased NO levels compared with Aβ alone group. Similar results were observed with cyanidin and NAC treatment (Figure 2D). These results suggest that the suppression of NO production by cyanidin or NAC may be due to enhancement of the Nrf2 pathway. These results indicate that cyanidin enhances Nrf2 pathway activation by activating proteins related to the Nrf2 pathway and stimulating SOD activity in cells treated with Aβ25-35.
Figure 2.
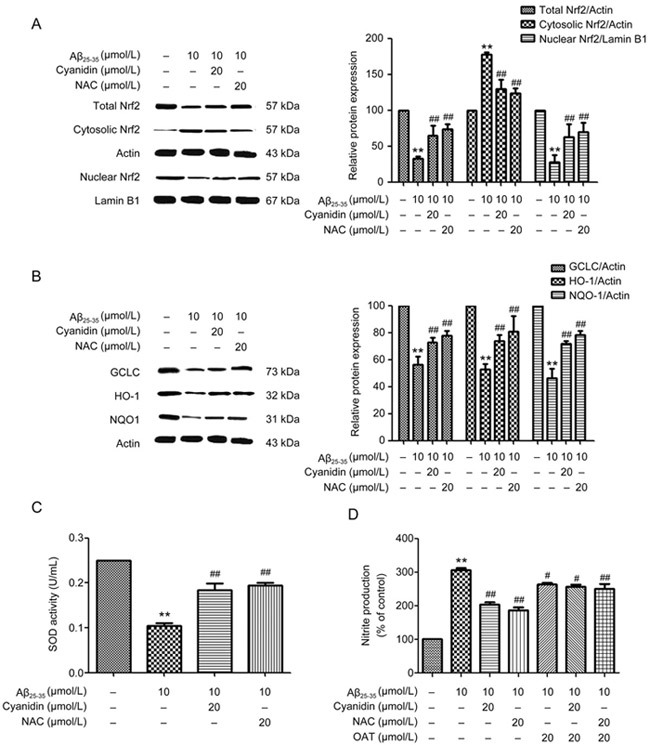
Cyanidin attenuates Aβ-induced reduction of Nrf2 pathway activation and SOD activity in SK-N-SH cells. The SK-N-SH cells were pretreated with 20 μmol/L of cyanidin or 20 μmol/L of NAC for 2 h prior to treatment with 10 μmol/L Aβ25-35 for 24 h. The expression levels of proteins related to the Nrf2 pathway were analyzed by Western blot. Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of Nrf2 (A); the expression of the antioxidant enzyme genes including HO-1, NQO-1, and GCLC (B); actin to normalize the cytosolic proteins; and lamin B1 to normalize the nuclear proteins. A SOD activity assay was carried out to determine the level of SOD activity (C). The SK-N-SH cells were pretreated with 20 μmol/L OTA for 1 h; preincubation was carried out with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h, and the cells were exposed to Aβ25-35 (10 μmol/L) for another 24 h. The Griess reaction assay was performed to determine the level of nitrite production (D). The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. ## P<0.01 vs the Aβ25-35-treated group.
Cyanidin inhibits Aβ25-35-induced PI3K/Akt pathway activation
The PI3K/Akt pathway is one of the major pathways involved in progressive inflammation; it promotes NF-κB pathway activity and inhibits the cellular defense system, especially the Nrf2 pathway, in neurons activated by Aβ25-35. To clarify the protective effect of cyanidin on Aβ-induced neuronal inflammation by determining whether it acts directly against the PI3K/Akt pathway, we applied LY294002 (PI3K inhibitor) and SH-5 (Akt inhibitor) in this experiment. As shown in Figure 3A, treatment with Aβ25-35 increased the expression of p-Akt compared with the level in the control group, whereas pretreatment with cyanidin was found to decrease the expression of p-Akt below the level associated with Aβ25-35 treatment alone. Moreover, treatment with LY294002 and cyanidin showed a similar pattern to the cyanidin pretreatment (Figure 3A). This is clearly indicative of the fact that cyanidin protects against Aβ25-35 regarding the expression of p-Akt through a mechanism that does not involve PI3K activity. Next, we investigated whether cyanidin inhibited Aβ-induced NF-κB activation and suppressed the Nrf2 via mechanism mediated by the PI3K/Akt pathway. The results showed that co-administration of LY294002 or SH5 with cyanidin before treatment with Aβ25-35 produced similar effects to cyanidin on the expression of proteins related to both the NF-κB and the Nrf2 pathways (Figure 3B,Figure 3C,Figure 3D, Figure 3E,Figure 3F,Figure 3G,Figure 3H,Figure 3I,Figure 3J,Figure 3K). These data suggest that the NF-κB and Nrf2 pathways are not involved with the PI3K/Akt pathway. In addition, it can be concluded that the inhibition of inflammation by cyanidin was also not through this pathway.
Figure 3A-D.
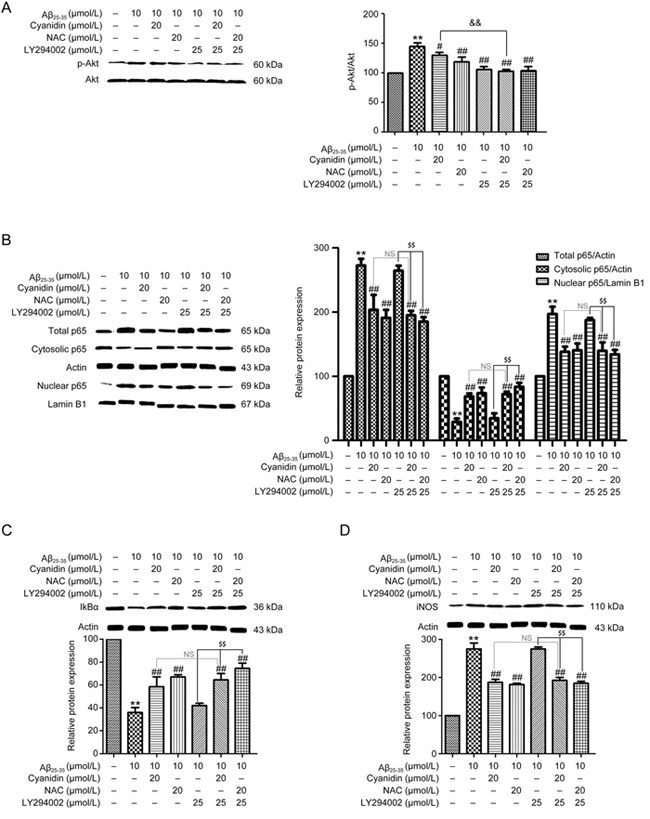
Cyanidin protects against Aβ-induced PI3K/Akt pathway activation but the effect has no relation to inflammation in SK-N-SH cells. SK-N-SH cells were pretreated with or without 25 μmol/L LY294002 for 1 h. Then, the cells were pretreated with 20 μmol/L cyanidin or 20 μmol/L NAC for 2 h prior to treatment with 10 μmol/L Aβ25-35 for 24 h. Western blotting analysis was performed to measure the protein expression levels of p-Akt and Akt, and protein loading was normalized to Akt (A). The SK-N-SH cells were pretreated with 25 μmol/L LY294002 for 1 h; preincubation was carried out with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h, and the cells were exposed to Aβ25-35 (10 μmol/L) for another 24 h. Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of p65 (B), IκBα (C), iNOS (D). The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. # P<0.05, ## P<0.01 vs the Aβ25-35-treated group. $$ P<0.01 vs the Aβ25-35-plus-inhibitor-treated group.
Figure 3E-G.
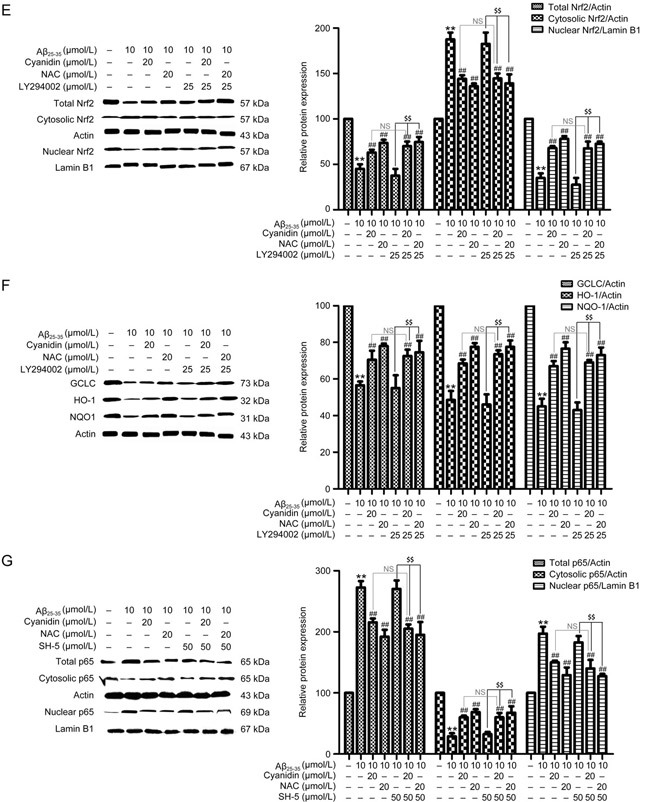
Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of Nrf2 (E), HO-1, NQO-1, and GCLC (F). The SK-N-SH cells were pretreated with 50 μmol/L SH-5 for 1 h; preincubation was carried out with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h, and the cells were exposed to Aβ25-35 (10 μmol/L) for another 24 h. Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of p65 (G). The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. # P<0.05, ## P<0.01 vs the Aβ25-35-treated group. $$ P<0.01 vs the Aβ25-35-plus-inhibitor-treated group.
Figure 3H-K.
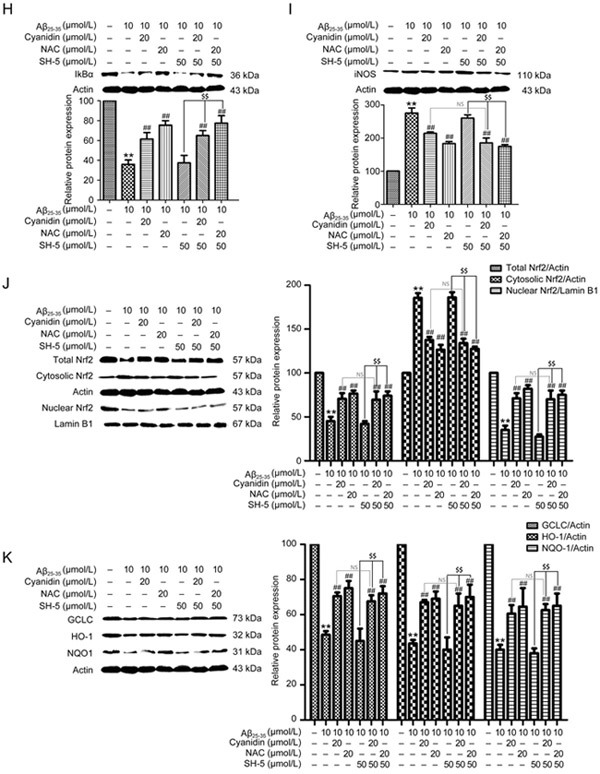
Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of IκBα (H), iNOS (I), Nrf2 (J), HO-1, NQO-1, and GCLC (K); the cytosolic proteins were normalized to actin, and the nuclear proteins were normalized to lamin B1. The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. # P<0.05, ## P<0.01 vs the Aβ25-35-treated group. $$ P<0.01 vs the Aβ25-35-plus-inhibitor-treated group.
Cyanidin inhibits Aβ-induced TLR4 signaling
We further investigated the role of cyanidin in Aβ-induced inflammation to determine whether it was mediated by the TLR4 signaling pathway. As shown in Figure 4A, treatment with Aβ25-35 was found to significantly increase the expression of TLR4 compared with that of the control group. Pretreatment with cyanidin or NAC significantly decreased the expression of TLR4 compared with Aβ25-35 treatment alone. To confirm whether cyanidin ameliorates Aβ25-35-induced inflammation in association with TLR4 signaling, we applied TAK-242, a TLR4 inhibitor. The results showed that cotreatment with TAK-242 and cyanidin or NAC ameliorated Aβ25-35-induced inflammation by significantly decreasing the translocation of NF-κB into the nucleus (Figure 4B) and that it decreased the protein expression of IκBα (Figure 4C) and iNOS (Figure 4D). In addition, the translocation of Nrf2 protein into the nucleus and the expression of proteins related to the Nrf2 pathway were also observed to have significantly increased, as in the cyanidin-treated group (Figure 4E, 4F). These data suggest that cyanidin protects against Aβ25-35-induced inflammation in SK-N-SH cells, that this protection is partly mediated by TLR4, and that it results in suppression of the NF-κB signaling pathway and activation of the Nrf2 signaling pathway.
Figure 4A-D.

Cyanidin inhibits Aβ-induced TLR4 signaling and is related to inflammation in SK-N-SH cells. SK-N-SH cells were pretreated with 20 μmol/L of cyanidin or 20 μmol/L of NAC for 2 h before being stimulated with 10 μmol/L Aβ25-35 for 24 h. The expression of TLR4 was detected by antibodies against TLR4 and then analyzed by Western blot (A). The SK-N-SH cells were pretreated with 100 nmol/L TAK-242 for 1 h; the cells were preincubated with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h and exposed to Aβ25-35 (10 μmol/L) for another 24 h. Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of p65 (B), IκBα (C), iNOS (D). The data are expressed as the mean±SEM (n=3). * P<0.05, ** P<0.01 vs the control. # P<0.05, ## P<0.01 vsthe Aβ25-35-treated group. && P<0.01 vs the Aβ25-35-plus-cyanidin-treated group.
Figure 4E-F.
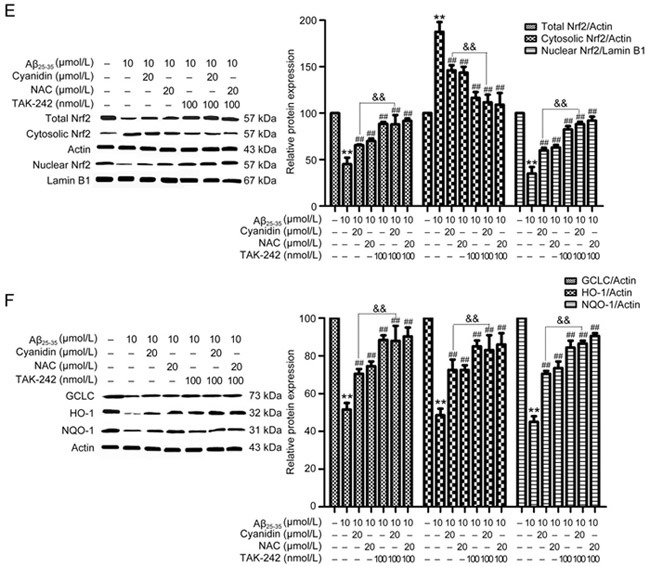
Western blotting analysis was performed to measure the total, cytosolic, and nuclear protein levels of Nrf2 (E), HO-1, NQO-1, and GCLC (F); the cytosolic proteins were normalized to actin, and the nuclear proteins were normalized to lamin B1. The data are expressed as the mean±SEM (n=3). * P<0.05, ** P<0.01 vs the control. # P<0.05, ## P<0.01 vsthe Aβ25-35-treated group .&& P<0.01 vs the Aβ25-35-plus-cyanidin-treated group.
Cyanidin protects against Aβ-induced oxidative stress and inflam-mation through the TLR/NOX4 signaling pathway
To elucidate the effect of cyanidin in protecting against Aβ-induced oxidative stress in terms of whether it acts directly against TLR/NOX4 signaling, we used the TLR4 inhibitor TAK-242 and the NOX inhibitor GKT137831 to confirm the involvement of the TLR/NOX4 signaling pathway. As shown in Figure 5A, pretreatment with TAK-242 alone or TAK-242 prior to cyanidin treatment caused the levels of ROS and NO production to decrease significantly, whereas SOD activity was found to be significantly increased compared with its activity in cells treated with Aβ25-35 alone (Figure 5A, 5B, 5C). At the same time, pretreatment with GKT137831 alone or GKT137831 prior to cyanidin treatment caused the levels of ROS and NO production to decrease significantly, whereas SOD activity was found to be significantly increased compared with its activity under cyanidin treatment alone (Figure 5A, 5B, 5C). These data suggest that cyanidin attenuates Aβ25-35-induced oxidative stress mediated by the TLR/NOX4 signaling pathway.
Figure 5.
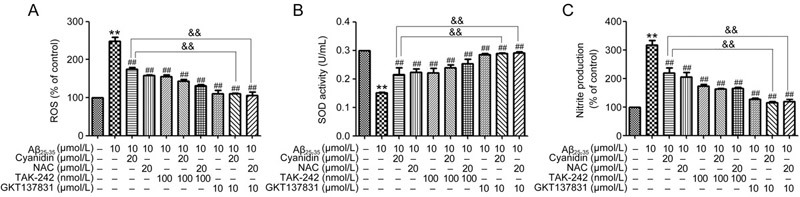
Cyanidin protects against Aβ-induced oxidation and inflammation through the TLR4/NOX4 signaling pathway. SK-N-SH cells were pretreated with 100 nmol/L TAK-242 or GKT137831 for 1 h; the cells were preincubated with cyanidin (20 μmol/L) or NAC (20 μmol/L) for 2 h and exposed to Aβ25–35 (10 μmol/L) for another 24 h. A DCF assay was carried out to determine the level of ROS (A). The Griess reaction assay was performed to determine the level of nitrite production (B). An SOD activity assay was conducted to determine the level of SOD activity (C). The data are expressed as the mean±SEM (n=3). ** P<0.01 vs the control. ## P<0.01 vs the Aβ25-35-treated group. && P<0.01 vs the Aβ25-35-plus-cyanidin-treated group.
Discussion
Aβ induces inflammation via stimulation of glial cells and neurons to produce pro-inflammatory cytokines such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α)26,27. In AD brains, increased NF-κB activity has been reported. Upon activation, the IκBα protein, which is the inhibitory subunit of the NF-κB complex, undergoes phosphorylation and degradation processes that enable the translocation of the NF-κB p65 protein from the cytosol into the nucleus and generates several cytokines including NO26,27,28,29. Previous studies have demonstrated that the anti-inflammatory activity of cyanidin-3-O-glucoside from fruits or vegetables occurs partially through suppression of NO production and TNF-α levels in several cell types including J774 cells and human microvascular endothelial cells30,31. Therefore, suppressing NF-κB signaling in AD could prevent amplification of the inflammatory cascade and neurodegeneration. Our results demonstrated that cyanidin decreased the expression of total p65 which indicated that p65 translocation from the cytosol into the nucleus, which is closely related to the degradation of IκBα, was decreased, whereas the expression of iNOS and the production of NO decreased. The major findings indicate that cyanidin inhibits NF-κB activation in terms of both activity and expression through downregulation of the p65 protein, which, in turn, decreases the expression of iNOS and the production of NO. Moreover, the Nrf2 pathway is important for cellular defense against oxidative stress17,18. Nrf2-Keap1 dissociation is triggered, with consequent translocation of Nrf2 to the nucleus, where the heterodimer formed with Maf binds to the ARE sequence in the promoter regions of genes involved in phase II detoxification and antioxidant defense including HO-1, NQO-1, GCLC, and SOD17,18. Previous studies have reported that cyanidin modulates the redox mechanism via the Nrf2 pathway, which plays an important role in the expression of antioxidant enzymes32,33,34. Our results confirm that cyanidin increases the translocation of Nrf2 into the nucleus; additionally, the activation of the phase II enzymes shows that cyanidin upregulated the production of HO-1, NQO-1, GCLC, and SOD. Nrf2 activation can have tremendous effects on the antioxidant capacity of a cell because it is a transcription factor for many genes coding for antioxidant enzymes17,18. Cyanidin can both scavenge free radicals and induce antioxidant enzyme upregulation; in neurodegenerative diseases, antioxidant enzymes have the potential for therapeutic use32,33,34. Through its ability to induce the expression of important enzymes involved in defense against ROS, Nrf2 in particular plays a major role in the pathogenesis of various diseases that are known to be accompanied by elevated ROS levels17,18. Most diseases are characterized by extensive inflammation and tissue destruction leading to the formation of ROS, which subsequently causes sustained stress and tissue injury. In AD brains, suppressed levels of Nrf2 have been reported, despite the highly oxidative environment characteristic of the disease; this suppression suggests down-regulation of the Nrf2-ARE pathway35,36,37. APP/PS1 mouse models have been observed to demonstrate reduced expression of Nrf2 and GCLC, which are directly synthesized by the Nrf2-ARE pathway38. Many studies have demonstrated the neuroprotective effects of Nrf2, including its protective effect against Aβ pathology found in AD and increased oxidative stress observed in hippocampal slices from Nrf2 knockout mice39,40.
The PI3K-Akt pathway has been shown to control a variety of cellular processes, including cell survival and proliferation. Activated PI3K catalyzes the phosphorylation of phosphatidylinositol 3,4-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-triphosphate (PIP3)41. Akt is a serine/threonine kinase, necessary for neuronal survival during normal and stress-inducing conditions, especially under conditions involving oxidative stress42,43. Our results showed that pretreatment with cyanidin decreased the expression of p-Akt. Moreover, a specific PI3K inhibitor, LY294002, was found to completely abolish the effect of cyanidin on p-Akt expression. Our results are consistent with those of other studies as regards the mechanism of Aβ induced neurotoxicity, which was used to identify the role of PI3K/Akt in transducing cell death signals after treatment with Aβ25–35. There are several reports demonstrating the pro-inflammatory role of the PI3K/Akt signaling pathway44,45. Activation of the Akt survival signaling pathways leads to phosphorylation of IκBα, and this allows NF-κB to enter the nucleus and regulate NF-κB-dependent gene expression, as well as suppress Nrf2 translocation, resulting in cell oxidative stress43,45,46. We found that cyanidin prevented Aβ25–35-induced NF-κB signaling and enhanced the Nrf2 pathway and that this did not occur through the PI3K/Akt pathway. These results were in contrast with those of other studies in that the mechanism of Aβ involved neurotoxicity only via PI3K/Akt/GSK3 signaling and not through the PI3K/Akt/NF-κB/Nrf2 pathway. Several studies have reported abnormal upregulation of the PI3K/Akt signaling pathway in AD, which could be involved in the development of insulin resistance through feedback mechanisms47. Increased activity of mTOR, a downstream target of PI3K/Akt, also appears to be associated with AD progression48. In this regard, mTOR can upregulate both tau and APP48. Further, mTOR negatively modulates autophagy and clearance of protein aggregates, which may contribute to neurodegeneration. Importantly, Akt can phosphorylate GSK3α/β, strongly inhibiting the activity of this multifaceted enzyme49,50,51. The PI3K/Akt/GSK3 axis is consistently described as being deregulated in AD. GSK3 can regulate the expression of several inflammatory mediators, usually by promoting the expression of pro-inflammatory molecules while decreasing anti-inflammatory factors43,52,53,54. It has been established that this effect depends on the regulatory role of PI3K/Akt/GSK3 in the activation of multiple transcription factors and interaction with signaling pathways, including the NF-κB, STAT3, and JNK pathways55,56. In microglia, GSK3-dependent activation of NF-κB results in increased expression of iNOS and pro-inflammatory cytokines and chemokines, such as TNFα and IL-655,56,57,58,59. It has been demonstrated that GSK3β can directly activate the transcription factors, such as STAT3 and p65 NF-κB, that are involved in the expression of pro-inflammatory genes by phosphorylating those transcription factors55,56,57,58,59. Thus, this finding suggests that cyanidin protects against Aβ-induced neuroinflammation and that it does not involve the PI3K/Akt pathway.
A large number of studies have been performed to explore the role that TLRs play in Aβ recognition; these studies have pointed to the involvement of two specific TLRs: TLR2 and TLR44. Among the cell-surface TLRs present in glia and neurons, several studies in vitro and in vivo have confirmed that TLR4 mediates the induction of neurotoxicity by glia and neurons4,5,10,60,61. The binding of Aβ to these receptors is responsible for the secretion of inflammatory molecules through the activation of signaling cascades such as NF-κB. Furthermore, it has been established through research that TLR4 mediates the binding of Aβ to microglial cells, regulating phagocytosis (in BV-2 microglial cells) and ROS production (in primary murine microglia)3,5,10,60. In primary murine cortical neurons, TLR4 has been demonstrated to mediate Aβ-induced apoptosis via JNK- and caspase-3-dependent mechanisms11,62. Thus, modulation of neuroinflammation via TLRs is a potentially beneficial technique for the treatment of AD. In this study, our results clearly show that Aβ25-35 induced the expression of TLR4, while pretreatment with cyanidin caused the expression of TLR4 to decrease. The present results support the findings of previous studies that TLR4 promotes the production of major inflammatory mediators that are associated with neuronal death. The result showed that cyanidin could prevent Aβ25-35-induced neuroinflammation directly through the TLR4 pathway. Although our results have shown that NF-κB signaling represents one of the downstream pathways of TLR4-induced production of inflammatory factors, there exists the possibility that other pathways such as the MAPK and PI3K/Akt/GSK3 pathways are involved. Moreover, there is evidence of a direct link between Nrf2 and TLR in regulation of inflammatory response and production of ROS. In our study, cyanidin activates the Nrf2 pathway through direct TLR4 pathway activity.
In addition, ROS plays a central role in both the upstream and downstream NF-κB and Nrf2 pathways, which are located at the center of the inflammatory response. It has been shown that NOX4 interacts with TLR4 and induced ROS production63,64. It has also been found that stimulation of TLR4/NOX4 increases the generation of ROS. Therefore, we suggested that Aβ25-35 could stimulate TLR4/NOX4 interaction and then increase the generation of ROS63,64,65,66. Our results show that Aβ25-35 induced neuronal ROS production mediated by TLR4/NOX4 activation. Inhibition of the receptor TLR4 by TAK-242 caused the production of ROS to significantly decrease, whereas inhibition of NOX4 by GKT137831 Aβ25-35 failed to activate ROS production. Our results showed that cyanidin could prevent Aβ25-35-induced oxidation through the TLR/NOX4 signaling pathway. TLR activation has been shown to induce ROS production and NF-κB activation through a direct interaction between TLR and NOX. ROS can activate transcription factors including NF-κB, which, in turn, leads to activation of downstream pathways such as NO production and can suppress the antioxidant pathway via Nrf2, which, in turn, leads to inhibition of downstream targets such as SOD, indicating that the inflammatory response has been activated. Thus, we determined whether the effects of the TLR/NOX4 signaling pathway could involve NO production and SOD activity. We found that both TAK-242 and GKT13783 completely abolished the effects induced by Aβ25-35; our results show that cyanidin protects against Aβ-induced inflammation directly through the TLR4/NOX4 signaling pathway by suppression of NO production and promotion of SOD activity. In the present study, it was found that inhibition of TLR4 signaling led to suppression of NOX4 induction and reduced oxidative stress, resulting in decreased inflammatory response. Taken together, these findings indicate that cyanidin attenuates Aβ-induced inflammation and ROS production mediated by the TLR4/NOX4 signaling pathway.
In summary, our findings demonstrate that cyanidin has a potent neuroprotective effect that slows down inflammatory progression and delays transition from the early stage of AD to the clinical state. Cyanidin is able to attenuate Aβ-induced insults in SK-N-SH cells through the regulation of several mechanisms including 1) suppressing oxidative stress mediated by the TLR4/NOX4 pathway, 2) suppressing NF-κB-mediated TLR4/NOX4 signaling, 3) and activating anti-inflammation by upregulation of the Nrf2 pathway mediated by the TLR4/NOX4 pathway. Thus, TLR4 is a primary receptor for Aβ to trigger neuroinflammation, and our findings suggest that inhibition of TLR4 by cyanidin in SK-N-SH cells could be beneficial in preventing neuronal cell death in the process of Alzheimer's disease. However, our study is focused on cellular, biochemical, and molecular mechanisms in vitro. Therefore, further study should investigate the protective effect of cyanidin in in vivo models and compare them with in vitro models in terms of the molecular mechanisms involved in AD pathogenesis.
Conclusion
The protective effect of cyanidin against Aβ25-35-induced neuroinflammation-mediated TLR4/NOX4 signaling in SK-N-SH cells occurs via inhibition of the NF-κB pathway and activation of the Nrf2 pathway. Furthermore, these findings indicate that inhibition of TLR4/NOX4 signaling is a promising approach to the treatment of AD. Therefore, administration of cyanidin may be a potential pharmacological or functional food therapy against oxidative stress for protection against AD because of the strong antioxidant ability and unique characteristics of cyanidin. It is therefore a priority to investigate further in vivo.
Author contribution
Jiraporn TOCHARUS, Sarinthorn THUMMAYOT, and Apichart SUKSAMRARN conceptualized and designed the study; Sarinthorn THUMMAYOT and Pichaya JUMNONGPRAKHONperformed research; Jiraporn TOCHARUS, Sarinthorn THUMMAYOT, and Chainarong TOCHARUS analyzed data; Jiraporn TOCHARUS, Sarinthorn THUMMAYOT, Chainarong TOCHARUS, and Apichart SUKSAMRARN wrote the manuscript.
Acknowledgements
This study was supported by the Faculty of Medicine, Chiang Mai University; Chiang Mai University; Functional Food Research Center for Well-being, Chiang Mai University and The Thailand Research Fund (DBG5980003). ST acknowledges financial support from the Faculty of Medicine and the Graduate School, Chiang Mai University, Thailand.
References
- 1.Chen JH, Ke KF, Lu JH, Qiu YH, Peng YP. Protection of TGF-beta1 against neuroinflammation and neurodegeneration in Abeta1-42-induced Alzheimer's disease model rats. PLoS One. 2015;10:116549. doi: 10.1371/journal.pone.0116549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kim H, Youn K, Ahn MR, Kim OY, Jeong WS, Ho CT, et al. Neuroprotective effect of loganin against Abeta25-35-induced injury via the NFkappaB- dependent signaling pathway in PC12 cells. Food Funct. 2015;6:1108–16. doi: 10.1039/C5FO00055F. [DOI] [PubMed] [Google Scholar]
- 3.Capiralla H, Vingtdeux V, Zhao H, Sankowski R, Al-Abed Y, Davies P, et al. Resveratrol mitigates lipopolysaccharide- and Abeta mediated microglial inflammation by inhibiting the TLR4/NF-kappa B/STAT signaling cascade. J Neurochem. 2012;120:461–72. doi: 10.1111/j.1471-4159.2011.07594.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding BJ, Ma WW, He LL, Zhou X, Yuan LH, Yu HL, et al. Soybean isoflavone alleviates beta-amyloid 1-42 induced inflammatory response to improve learning and memory ability by down regulation of Toll-like receptor 4 expression and nuclear factor-kB activity in rats. Int J Dev Neurosci. 2011;29:537–42. doi: 10.1016/j.ijdevneu.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 5.Lehnardt S, Massillon L, Follett P, Jensen FE, Ratan R, Rosenberg PA, et al. Activation of innate immunity in the CNS triggers neurodegeneration through a toll-like receptor 4-dependent pathway. Proc Natl Acad Sci U S A. 2003;100:8514–9. doi: 10.1073/pnas.1432609100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the toll-like receptor 4 in neuroinflammation in Alzheimer's disease. Cell Physiol Biochem. 2007;20:947–56. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 7.Kawai T, Akira S. Pathogen recognition with Toll-like receptors. Curr Opin Immunol. 2005;17:338–44. doi: 10.1016/j.coi.2005.02.007. [DOI] [PubMed] [Google Scholar]
- 8.Ojaniemi M, Glumoff V, Harju K, Liljeroos M, Vuori K, Hallman M. Phosphatidylinositol 3-kinase is involved in Toll-like receptor 4-mediated cytokine expression in mouse macrophages. Eur J Immunol. 2003;33:597–605. doi: 10.1002/eji.200323376. [DOI] [PubMed] [Google Scholar]
- 9.Ha T, Hu Y, Liu L, Lu C, McMullen JR, Kelley J, et al. TLR2 ligands induce cardioprotection against ischaemia/reperfusion injury through a PI3K/Akt-dependent mechanism. Cardiovasc Res. 2010;87:694–703. doi: 10.1093/cvr/cvq116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jin JJ, Kim HD, Maxwell JA, Li L, Fukuchi K. Toll-like receptor 4-dependent upregulation of cytokines in a transgenic mouse model of Alzheimer's disease. J Neuroinflammation. 2008;5:23. doi: 10.1186/1742-2094-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tang SC, Lathia JD, Selvaraj PK, Jo DG, Mughal MR, Cheng A, et al. Toll-like receptor-4 mediates neuronal apoptosis induced by amyloid beta-peptide and the membrane lipid peroxidation product 4-hydroxynonenal. Exp Neurol. 2008;213:114–21. doi: 10.1016/j.expneurol.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Calvo-Rodríguez M, 1 de la Fuente C, García-Durillo M, 1 García-Rodríguez C, Villalobos C, Núñez L. Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca2+ responses, and neuron cell death in cultured rat hippocampal neurons. J Neuroinflam. 2017;14:24. doi: 10.1186/s12974-017-0802-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shelat PB, Chalimoniuk M, Wang JH, Strosznajder JB, Lee JC, Sun AY, et al. Amyloid beta peptide and NMDA induce ROS from NADPH oxidase and AA release from cytosolic phospholipase A2 in cortical neurons. J Neurochem. 2008;106:45–55. doi: 10.1111/j.1471-4159.2008.05347.x. [DOI] [PubMed] [Google Scholar]
- 14.Zhao M, Zhou A, Xu L, Zhang X. The role of TLR4-mediated PTEN/PI3K/AKT/NF-κB signaling pathway in neuroinflammation in hippocampal neurons. Neuroscience. 2014;269:93–101. doi: 10.1016/j.neuroscience.2014.03.039. [DOI] [PubMed] [Google Scholar]
- 15.Wu D, Zhang X, Zhao M, Zhou AL. The role of the TLR4/NF-κB signaling pathway in Aβ accumulation in primary hippocampal neurons. Acta Physiol Sin. 2015;67:319–28. [PubMed] [Google Scholar]
- 16.Chen L, Hu L, Zhao J , Hong H , Feng F , Qu W, et al. Chotosan improves Aβ1–42-induced cognitive impairment and neuroinflammatory and apoptotic responses through the inhibition of TLR-4/NF-κB signaling in mice. J Ethnopharmacol. 2016;191:398–407. doi: 10.1016/j.jep.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 17.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37:139–43. doi: 10.5483/bmbrep.2004.37.2.139. [DOI] [PubMed] [Google Scholar]
- 18.Ohtsuji M, Katsuoka F, Kobayashi A, Aburatani H, Hayes JD, Yamamoto M. Nrf1 and Nrf2 play distinct roles in activation of antioxidant response element-dependent genes. J Biol Chem. 2008;283:33554–62. doi: 10.1074/jbc.M804597200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kong JM, Chia LS, Goh NK, Chia TF, Brouillard R. Analysis and biological activities of anthocyanins. Phytochemistry. 2003;64:923–33. doi: 10.1016/S0031-9422(03)00438-2. [DOI] [PubMed] [Google Scholar]
- 20.Harborne JB, Grayer RJ. The anthocyanins. Flavonoids 1994: 1–20.
- 21.Bagchi D, Sen CK, Bagchi M, Atalay M. Anti-angiogenic, antioxidant, and anti-carcinogenic properties of a novel anthocyanin-rich berry extract formula. Biochem. 2004;69:75–80. doi: 10.1023/b:biry.0000016355.19999.93. [DOI] [PubMed] [Google Scholar]
- 22.Thummayot S, Tocharus C, Pinkaew D, Viwatpinyo K, Sringarm K, Tocharus J. Neuroprotective effect of purple rice extract and its constituent against amyloid beta induced neuronal cell death in SK-N-SH cells. Neurotoxicol. 2014;45:149–58. doi: 10.1016/j.neuro.2014.10.010. [DOI] [PubMed] [Google Scholar]
- 23.Thummayot S, Tocharus C, Suksamrarn A, Tocharus J. Neuroprotective effects of cyanidin against Aβ-induced oxidative and ER stress in SK-N-SH cells. Neurochem Int. 2016;101:15–21. doi: 10.1016/j.neuint.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 24.Shahripour RB, Harrigan MR, Alexandrov AV. N-acetylcysteine (NAC) in neurological disorders: mechanisms of action and therapeutic opportunities. Brain Behav. 2014;4:108–22. doi: 10.1002/brb3.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sayed AA. Anti-neuroinflammatory and antioxidant effects of N-acetyl cysteine in long-term consumption of artificial sweetener aspartame in the rat cerebral cortex. J Basic Applied Zool. 2015;72:73–80. doi: 10.1016/j.jobaz.2015.03.002. [DOI] [Google Scholar]
- 26.Wang SW, Wang YJ, Su YJ, Zhou WW, Yang SG, Zhang R, et al. Rutin inhibits β-amyloid aggregation and cytotoxicity, attenuates oxidative stress, and decreases the production of nitric oxide and proinflammatory cytokines. Neurotoxicology. 2012;33:482–90. doi: 10.1016/j.neuro.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 27.Akama KT, Van Eldik LJ. Beta-amyloid stimulation of inducible nitric-oxide synthase in astrocytes is interleukin-1beta- and tumor -necrosis factor-alpha (TNFalpha)-dependent, and involves a TNFalpha receptor-associated factor- and NFkappaB-inducing kinase-dependent signalling mechanism. J Biol Chem. 2000;275:7918–24. doi: 10.1074/jbc.275.11.7918. [DOI] [PubMed] [Google Scholar]
- 28.Pinkaew D, Changtam C, Tocharus C, Govitrapong P, Jumnongprakhon P, Suksamrarn A, et al. Association of neuroprotective effect of di-O-demethylcurcumin on Aβ25–35-induced neurotoxicity with suppression of NF-kB and activation of Nrf2. Neurotox Res. 2016;29:80–91. doi: 10.1007/s12640-015-9558-4. [DOI] [PubMed] [Google Scholar]
- 29.Park SY, Jin ML, Kim YH, Kim Y, Lee SJ. Anti-inflammatory effects of aromatic-turmerone through blocking of NF-κB, JNK, and p38 MAPK signaling pathways in amyloid β-stimulated microglia. Int Immunopharmacol. 2012;14:13–20. doi: 10.1016/j.intimp.2012.06.003. [DOI] [PubMed] [Google Scholar]
- 30.Pergola C, Rossi A, Dugo P, Cuzzocrea S, Sautebin L. Inhibition of nitric oxide biosynthesis by anthocyanin fraction of blackberry extract. Nitric Oxide. 2006;15:30–9. doi: 10.1016/j.niox.2005.10.003. [DOI] [PubMed] [Google Scholar]
- 31.Mittal M, Siddiqui MR, Tran K, Reddy SP, Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxid Redox Signal. 2014;20:1126–67. doi: 10.1089/ars.2012.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih PH, Yeh CT, Yen GC. Anthocyanins induce the activation of phase II enzymes through the antioxidant response element pathway against oxidative stress-induced apoptosis. J Agric Food Chem. 2007;55:9427–35. doi: 10.1021/jf071933i. [DOI] [PubMed] [Google Scholar]
- 33.Cimino F, Speciale A, Anwar S, Canali R, Ricciardi E, Virgili F, et al. Anthocyanins protect human endothelial cells from mild hyperoxia damage through modulation of Nrf2 pathway. Genes Nutr. 2013;8:391–9. doi: 10.1007/s12263-012-0324-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Speciale A, Anwar S, Canali R, Chirafisi J, Saija A, Virgili F, et al. Cyanidin-3-O-glucoside counters the response to TNF-alpha of endothelial cells by activating Nrf2 pathway. Mol Nutr Food Res. 2013;57:1979–87. doi: 10.1002/mnfr.201300102. [DOI] [PubMed] [Google Scholar]
- 35.de Vries HE, Witte M, Hondius D, Rozemuller AJ, Drukarch B, Hoozemans J, et al. Nrf2-induced antioxidant protection: A promising target to counteract ROS-mediated damage in neurodegenerative disease? Free Radic Biol Med. 2008;45:1375–83. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 36.Ramsey CP, Glass CA, Montgomery MB, Lindl KA, Ritson GP, Chia LA, et al. Expression of nrf2 in neurodegenerative disease. J Neuropathol Exp Neurol. 2007;66:75–85. doi: 10.1097/nen.0b013e31802d6da9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hybertson BM, Gao B, Bose SK, McCord JM. Oxidative stress in health and disease: The therapeutic potential of Nrf2 activation. Mol Aspects Med. 2011;32:234–46. doi: 10.1016/j.mam.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 38.Joshi G, Gan KA, Johnson DA, Johnson JA. Increased Alzheimer's disease-like pathology in the APP/PS1ΔE9 mouse model lacking Nrf2 through modulation of autophagy. Neurobiol Aging. 2015;36:664–79. doi: 10.1016/j.neurobiolaging.2014.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rojo AI, Rada P, Egea J, Rosa AO, López MG, Cuadrado A. Functional interference between glycogen synthase kinase-3 beta and the transcription factor Nrf2 in protection against kainate-induced hippocampal cell death. Mol Cell Neurosci. 2008;39:125–32. doi: 10.1016/j.mcn.2008.06.007. [DOI] [PubMed] [Google Scholar]
- 40.Kurzatkowski DM, Trombetta LD. Maneb causes pro-oxidant effects in the hippocampus of Nrf2 knockout mice. Environ Toxicol Pharmacol. 2013;36:427–436. doi: 10.1016/j.etap.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 41.Hemmings BA, Restuccia DF. PI3K-PKB/Akt pathway. Cold Spring Harb Perspect Biol. 2012;4:11189. doi: 10.1101/cshperspect.a011189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang ZZ, Tschopp O, Baudry A, Dümmler B, Hynx D, Hemmings BA. Physiological functions of protein kinase B/Akt. Biochem Soc Trans. 2004;32:350–4. doi: 10.1042/bst0320350. [DOI] [PubMed] [Google Scholar]
- 43.Fu XY, Yang MF, Cao MZ, Li DW, Yang XY, Sun JY. Strategy to suppress oxidative damage-induced neurotoxicity in PC12 cells by curcumin, the role of ROS-mediated DNA damage and MAPKs and AKT pathways. Mol Neurobiol. 2016;53:369–78. doi: 10.1007/s12035-014-9021-1. [DOI] [PubMed] [Google Scholar]
- 44.Bai D, Ueno L, Vogt PK. Akt-mediated regulation of NFkappaB and the essentialness of NFkappaB for the oncogenicity of PI3K and Akt. Int J Cancer. 2009;125:2863–70. doi: 10.1002/ijc.24748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999;9:601–4. doi: 10.1016/S0960-9822(99)80265-6. [DOI] [PubMed] [Google Scholar]
- 46.Romashkova JA, Makarov SS. NF-kappaB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 47.Lee HK, Kumar P, Fu Q, Rosen KM, Querfurth HW. The insulin/Akt signalling pathway is targeted by intracellular beta-amyloid. Mol Biol Cell. 2009;20:1533–44. doi: 10.1091/mbc.e08-07-0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Heras-Sandoval D, Perez-Rojas JM, Hernandez-Damian J, Pedraza-Chaverri J. The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal. 2014;26:2694–701. doi: 10.1016/j.cellsig.2014.08.019. [DOI] [PubMed] [Google Scholar]
- 49.Jimenez S, Torres M, Vizuete M, Sanchez-Varo R, Sanchez-Mejias E, Trujillo-Estrada L, et al. Age-dependent accumulation of soluble amyloid beta (Abeta) oligomers reverses the neuroprotective effect of soluble amyloid precursor protein-alpha (sAPP(alpha)) by modulating phosphatidylinositol 3-kinase (PI3K)/Akt-GSK-3beta pathway in Alzheimer mouse model. J Biol Chem. 2011;286:18414–25. doi: 10.1074/jbc.M110.209718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ryder J, Su Y, Ni B. Akt/GSK3beta serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer's disease mutations. Cell Signal. 2004;16:187–200. doi: 10.1016/j.cellsig.2003.07.004. [DOI] [PubMed] [Google Scholar]
- 51.Kitagishi Y, Nakanishi A, Ogura Y, Matsuda S. Dietary regulation of PI3K/AKT/GSK-3 beta pathway in Alzheimer's disease. Alzheimers Res Ther. 2014;6:35. doi: 10.1186/alzrt265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Beurel E, Jope RS. Glycogen synthase kinase-3 regulates inflammatory tolerance in astrocytes. Neuroscience. 2010;169:1063–70. doi: 10.1016/j.neuroscience.2010.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Koistinaho J, Malm T, Goldsteins G. Glycogen synthase kinase-3beta: a mediator of inflammation in Alzheimer's disease? Int J Alzheimers Dis. 2011;2011:129753. doi: 10.4061/2011/129753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Steinbrecher KA, Wilson W, Cogswell PC, Baldwin AS. Glycogen synthase kinase 3beta functions to specify gene-specific, NF-kappaB-dependent transcription. Mol Cell Biol. 2005;25:8444–55. doi: 10.1128/MCB.25.19.8444-8455.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuskaitis CJ, Jope RS. Glycogen synthase kinase-3 regulates microglial migration, inflammation, and inflammation-induced neurotoxicity. Cell Signal. 2009;21:264–73. doi: 10.1016/j.cellsig.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang MJ, Huang HY, Chen WF, Chang HF, Kuo JS. Glycogen synthase kinase-3beta inactivation inhibits tumor necrosis factor-alpha production in microglia by modulating nuclear factor kappaB and MLK3/JNK signalling cascades. J Neuroinflammation. 2010;7:9. doi: 10.1186/1742-2094-7-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Koh SH, Kim SH, Kwon H, Kim JG, Kim JH, Yang KH, et al. Phospha-tidylinositol-3 kinase/Akt and GSK-3 mediated cytoprotective effect of epigallocatechin gallate on oxidative stress-injured neuronal-differentiated N18D3 cells. Neurotoxicology. 2004;25:793–802. doi: 10.1016/j.neuro.2004.02.001. [DOI] [PubMed] [Google Scholar]
- 58.Samuels Y, Ericson K. Oncogenic PI3K and its role in cancer. Curr Opin Oncol. 2006;18:77–82. doi: 10.1097/01.cco.0000198021.99347.b9. [DOI] [PubMed] [Google Scholar]
- 59.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–5. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 60.Walter S, Letiembre M, Liu Y, Heine H, Penke B, Hao W, et al. Role of the Toll-like receptor 4 in neuroinflammation in alzheimer's disease. Cell Physiol Biochem. 2007;20:947–56. doi: 10.1159/000110455. [DOI] [PubMed] [Google Scholar]
- 61.Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, et al. TLR2 is a primary receptor for Alzheimer's amyloid β peptide to trigger neuroinflammatory activation. J Immunol. 2012;188:1098–107. doi: 10.4049/jimmunol.1101121. [DOI] [PubMed] [Google Scholar]
- 62.Okun E, Griffioen KJ, Lathia JD, Tang SC, Mattson MP, Arumugam TV. Toll-like receptors in neurodegeneration. Brain Res Rev. 2009;59:278–92. doi: 10.1016/j.brainresrev.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mander P, Brown GC. Activation of microglial NADPH oxidase is synergistic with glial iNOS expression in inducing neuronal death: a dual-key mechanism of inflammatory neurodegeneration. J Neuroinflamm. 2005;2:20. doi: 10.1186/1742-2094-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ben Mkaddem S, Pedruzzi E, Werts C, Coant N, Bens M, Cluzeaud F, et al. Heat shock protein gp96 and NAD(P)H oxidase 4 play key roles in Toll-like receptor 4-activated apoptosis during renal ischemia/reperfusion injury. Cell Death Differ. 2010;17:1474–85. doi: 10.1038/cdd.2010.26. [DOI] [PubMed] [Google Scholar]
- 65.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J Immunol. 2004;173:3589–93. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 66.Park HS, Chun JN, Jung HY, Choi C, Bae YS. Role of NADPH oxidase 4 in lipopolysaccharide-induced proinflammatory responses by human aortic endothelial cells. Cardiovasc Res. 2006;72:447–55. doi: 10.1016/j.cardiores.2006.09.012. [DOI] [PubMed] [Google Scholar]