

Abstract
Neuronal nicotinic acetylcholine receptors containing α6 subunits (α6*-nAChRs) show highly restricted distribution in midbrain neurons associated with pleasure, reward, and mood control, suggesting an important impact of α6*-nAChRs in modulating mesolimbic functions. However, the function and pharmacology of α6*-nAChRs remain poorly understood because of the lack of selective agonists for α6*-nAChRs and the challenging heterologous expression of functional α6*-nAChRs in mammalian cell lines. In particular, the α6 subunit is commonly co-expressed with α4*-nAChRs in the midbrain, which masks α6*-nAChR (without α4) function and pharmacology. In this study, we systematically profiled the pharmacology and function of α6*-nAChRs and compared these properties with those of α4β2 nAChRs expressed in the same cell line. Heterologously expressed human α6/α3 chimeric subunits (α6 N-terminal domain joined with α3 trans-membrane domains and intracellular loops) with β2 and β3 subunits in the human SH-EP1 cell line (α6*-nAChRs) were used. Patch-clamp whole-cell recordings were performed to measure these receptor-mediated currents. Functionally, the heterologously expressed α6*-nAChRs exhibited excellent function and showed distinct nicotine-induced current responses, such as kinetics, inward rectification and recovery from desensitization, compared with α4β2-nAChRs. Pharmacologically, α6*-nAChR was highly sensitive to the α6 subunit-selective antagonist α-conotoxin MII but had lower sensitivity to mecamylamine and dihydro-β-erythroidine. Nicotine and acetylcholine were found to be full agonists for α6*-nAChRs, whereas epibatidine and cytisine were determined to be partial agonists. Heterologously expressed α6*-nAChRs exhibited pharmacology and function distinct from those of α4β2-nAChRs, suggesting that α6*-nAChRs may mediate different cholinergic signals. Our α6*-nAChR expression system can be used as an excellent cell model for future investigations of α6*-nAChR function and pharmacology.
Keywords: nicotinic acetylcholine receptor, nicotine, acetylcholine, SH-EP1 cells, patch-clamp
Introduction
Nicotinic acetylcholine receptors (nAChRs) in mammals exist as a diverse family of molecules composed of different combinations of subunits derived from at least sixteen genes1,2,3. Nicotinic receptors containing α6 subunits are not widely expressed in the brain, but they are prevalent in midbrain dopaminergic (DA) regions associated with pleasure, reward, and mood control4,5. This suggests that α6*-nAChRs play critical roles in nicotine dependence and in modulating mood and emotion attributed to nicotine exposure6. The functional and pharmacological properties of α6*-nAChRs are largely unknown due to the lack of selective α6*-nAChR agonists. Furthermore, α6 subunits are typically co-expressed with other α nAChR subunits (eg, α4) to form natural α4α6*-nAChRs, which could mask the properties of α6 subunits alone. Because the heterologous expression of functional α6 nAChRs has been difficult to achieve, α6-nAChRs were initially given “orphan subunit” status. Lindstrom's laboratory first reported heterologous expression in oocytes of a functional α6*-nAChR with poor and variable function7. Kuryatov et al observed functional expression of human α6 plus β4 plus β3 subunits in an oocyte system; however, the functional expression of that system was poor for other combinations and was unremarkable for responses to nicotine when compared with those for ACh8. An efficient heterologous expression of the α6 subunit in any combination involving β2 subunits (resulting in functional ion channels) has proven extremely difficult. Grinevich et al succeeded in establishing a cell line stably transfected with α6, α5, β3 and β4 subunits that yielded radioligand-binding nAChRs with such properties as α-conotoxin MII sensitivity (as expected for α6*-nAChRs) and identified candidate ligands selective for the expressed receptor9. However, chimeric subunits where the N-terminal (ligand-binding) domain of α6 is fused to the transmembrane domain of α3 or α4 (α6/α3 or α6/α4) were found to produce functional receptors when co-expressing β2 or β4 subunits8. The β3 subunit appears to play a significant part in stabilizing/promoting correctly assembled α6-nAChR complexes in mammalian cells and impacting expression in Xenopus oocytes10,11. For other heteromeric nAChRs, encouraging results for heterologous receptor expression levels have been achieved: by introducing a single point mutation in the 9'-position of the pore-lining transmembrane region of the β3 subunit (V9'S), a conserved hydrophobic valine residue in the second transmembrane domain of the subunit was changed to a hydrophilic serine12,13. This evidence supports the idea that a chimeric construct comprising α6/α3, β2 and β3V9'S may produce functional receptors which could be considered surrogates of natural α6β2β3 nAChRs. The development of a high-throughput Ca2+ imaging (FLIPRTM) assay based on a HEK293 cell line stably expressing such an α6/α3β2β3V9'S construct was reported, and a number of well-known, as well as novel, nAChR agonists were evaluated in this assay14. Studies by Dash and co-workers have identified subunit interactions and α6 subunit modifications that promote functional expression in oocytes of different α6*-nAChR subtypes10,15,16. Using electrophysiological approaches, the native and functional α6*-nAChRs in midbrain dopamine (DA) neurons have been reported17,18,19,20,21,22. However, these α6*-nAChRs are often assembled with α4 subunit18,19.
We have established functional α6*-nAChRs by co-transfecting a α6/α3 chimera and β2 and β3 subunits into the human SH-EP1 cell line. This transfected α6*-nAChR exhibits excellent function (Figure 1) and has been used for drug screening due to its ability to avoid enhanced agonist potency and efficacy effects associated with a 9'S mutant subunit23. However, detailed functions and pharmacology of this α6*-nAChR have not been described. Considering that the native α6 subunit is commonly co-assembled with α4*-nAChRs in the rodent midbrain, the aim of this study was to systematically evaluate the function and pharmacology of the α6*-nAChR and compare these with those of α4β2-nAChRs transfected in the same human SH-EP1 cell line using patch-clamp whole-cell recording techniques. Our results demonstrated that the heterologously expressed α6/α3 chimera (α6 N-terminal extracellular domain plus α3 trans-membrane domains and intracellular loops) with β2 and β3 subunits formed functional α6*-nAChRs that exhibited distinct current kinetics, desensitization, and recovery from desensitization when compared with α4β2-nAChR. These results suggested that this α6*-nAChR can serve as an excellent cell model to investigate α6*-nAChR function and pharmacology and can be used to screen for new compounds that modulate α6*-nAChRs.
Figure 1.
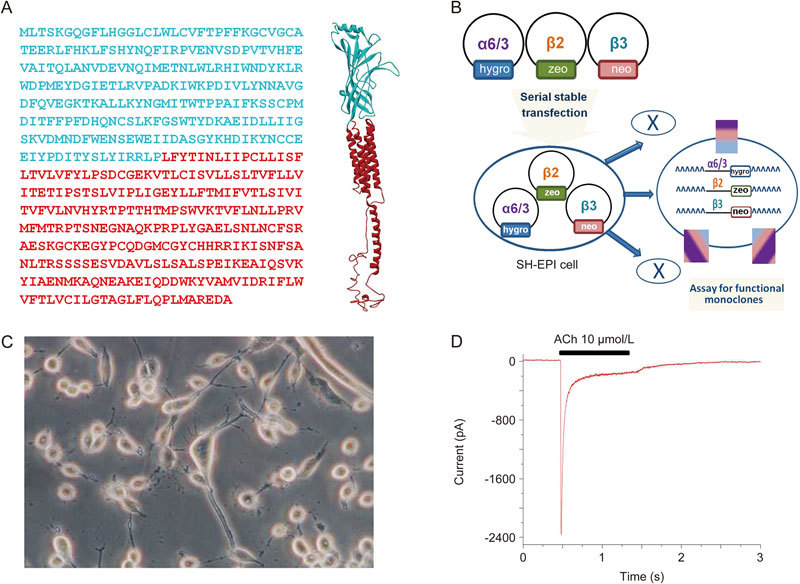
Heterologous expression of α6α3β2β3-nAChR in human SH-EP1 cells. (A) α6/α3 chimera: Left, the chimera sequence with the ligand-binding N-terminal domain of the human α6, colored cyan, and C-terminal domain of the human α3, colored red. Right, the homology model of the α6/α3 chimera without the signal peptide with the colors matching the colors in the sequence. (B) A cartoon figure shows the process of the stably transfected α63β2β3 cell line. (C) A phase-contrast picture of SH-EP1-α6*-nAChR cells before patch-clamp recording. (D) A typical trace shows an ACh-induced inward whole-cell current in SH-EP1 cell transfected with this chimeric α6*-nAChR.
Materials and methods
Expression of human neuronal α6/α3β2β3-nAChR in human SH-EP1 cells
Construction of the cell line expressing α6/α3β2β3-nAChR was first described by Breining et al 23. α6/α3 denotes a chimeric subunit composed of the extracellular, ligand-binding domain of the human α6 subunit fused to the first transmembrane domain, following the sequence of the human α3 nAChR subunit (Figure 1A). This approach reproducibly increased expression compared with that seen for native α6 subunits while retaining α6-like pharmacology8. Consensus-sequence β2 and β3 human nAChR subunit clones were also used. To summarize the salient details, wild-type SHEP1 cells were transfected with nAChR subunit clones using a cationic polymer (Qiagen, Valencia, CA, USA). nAChR subunit genes optimized for mammalian expression were synthesized by GeneArt, Inc (Thermo Fisher Scientific, Inc, Waltham, MA, USA) and were delivered in pcDNA3.1 expression vectors (Invitrogen, Thermo Fisher Scientific, Inc, Waltham, MA, USA. pcDNA3.1zeo for the α6/α3 subunit, pcDNA3.1hygro for the β2 subunit, and pcDNA3.1neo for the β3 subunit). Triple transfectants expressing α6/α3, β2, and β3 subunits (Figure 1B) were selected for using zeocin (0.25 mg/mL, Invitrogen), hygromycin B (0.4 mg/mL, 0.13 mg/mL biologically active hygromycin, Calbiochem, EMD Millipore, Billerica, MA, USA), and G418 sulfate at a final concentration of 0.6 mg/mL (AG Scientific, San Diego, CA, USA). The polyclonal pool of survivors from this selection round was then used to pick monoclones. Clones were screened for radioligand binding using 3H]epibatidine binding and for function using 86Rb+ efflux assays24,25. A clone exhibiting consistently high functional nAChR expression was selected. These cells were maintained in Dulbecco's modified Eagle's medium (Invitrogen) with 10% horse serum (Invitrogen), 5% fetal bovine serum (HyClone, GE Healthcare Life Sciences, Pittsburg, PA, USA), 1 mmol/L sodium pyruvate, and 4 mmol/L L-glutamine supplemented with 0.25 mg/mL zeocin, 0.13 mg/mL hygromycin B, and G418 at 0.6 mg/mL, ensuring positive selection of transfectants. Low-passage-number (1−40 from our frozen stocks) cultures were used to ensure stable phenotype expression. Cells were typically passaged once weekly by splitting just-confluent cultures 1/20−1/40 to maintain cells in proliferative growth phase.
Patch-clamp recordings
Conventional patch-clamp whole-cell current recordings coupled with a computer-controlled fast-drug application and removal system were implemented in this study as previously described26,27,28. Briefly, cells plated on 35-mm culture dishes were placed on the stage of an inverted microscope (Olympus IX7, Lake Success, NY, USA) and continuously superfused with standard external solution (2 mL/min). Glass microelectrodes (1.5 mm×100 mm, Narishige, East Meadow, NY, USA) were made in two steps using a vertical electrode puller (P83, Narishige, East Meadow, NY, USA). Electrodes with a resistance of 3–5 MΩ were used to form tight seals (>1 GΩ) on the cell surface until suction was applied to break the membrane and convert to conventional whole-cell recording. Thereafter, the recorded cell was lifted and voltage-clamped at a holding potential (VH) of −40 mV (unless specifically mentioned), and ionic currents in response to the application of nicotinic ligands were measured (Axopatch 200B amplifier, Axon Instruments, Foster City, CA, USA). The whole-cell access resistance was less than 20 MΩ before series resistance compensation and monitored throughout the experiment. If access resistance varied by more than 20%, the data were discarded. Pipette and whole-cell current capacitances were minimized, and series resistance was routinely compensated to 80%. Typically, the current output was filtered at 2 kHz, displayed and digitized at 10 kHz on-line (Digidata 1440 series A/D board, Axon Instruments, Foster City, CA, USA), and streamed to disk. Data acquisition and analyses of whole-cell currents were done using Clampex v10.2 (Axon Instruments, Foster City, CA, USA), and results were plotted using Origin 8.0 (Microcal, North Hampton, MA, USA) or Prism 5.0 (GraphPad Software, Inc, San Diego, CA, USA). Concentration-response curves were fitted to the Hill equation. Acute nAChR desensitization was analyzed for decay time constant (τ), peak current (Ip), and steady-state current (Is) using fits to a single exponential function: I = [(Ip - Is) e-t/τ] + Is, or to its double-exponential variant as appropriate, using data from 90% to 10% of the period between the peak amplitude of the inward current and the termination of the typical 1-s period of agonist exposure. Replicate determinations of τ (a measure of the rate of acute desensitization) and peak current amplitude are presented as the mean±standard error of the mean (SEM) and were analyzed for significance using Student's t-test (paired or independent). All experiments were performed at room temperature (22±1 ° C).
Solutions and drug application
The standard external solution contained 120 mmol/L NaCl, 3 mmol/L KCl, 2 mmol/L MgCl2, 2 mmol/L CaCl2, 25 mmol/L D-glucose, and 10 mmol/L HEPES at pH 7.4 with Tris-base. In some experiments using ACh as an agonist, 1 μmol/L atropine sulfate was added to the standard solution to exclude any possible influences of muscarinic receptors. The pipette solutions used for conventional whole-cell recordings were (in mmol/L): Tris phosphate dibasic 110, Tris base 28, EGTA 11, MgCl2 2, CaCl2 0.1, Na-ATP 4, pH 7.3. This “Tris+” electrode solution was used by Huguenard & Prince29 to prevent nAChR receptor functional run-down28. To study the current (I) and voltage (V) relationship of nAChR-mediated whole-cell currents, the internal pipette solution was a high K+ solution containing (mmol/L) 140 KCl, 4 MgSO4, 0.1 EGTA, 4 Mg-ATP and 10 HEPES adjusted to pH 7.2 with Tris-base.
To initiate whole-cell current responses under constant perfusion in the recording chamber, nicotinic drugs were rapidly delivered to the recorded cell using a computer-controlled 'U-tube' application system. The applied drug completely surrounded the recorded cell within 20 ms. The intervals between drug applications (1 min) were specifically adjusted to ensure the stability of nAChR responsiveness (absence of functional rundown). The selections of pipette solutions used in most of the studies described here were made with the same objective. The drugs used in the present study were (-) nicotine, ACh, epibatidine, cytisine, lobeline, dihydro-β-erythroidine (DHβE), and mecamylamine (MEC) (Sigma Chemical Co, St Louis, MO, USA). α-Conotoxin MII was synthesized as previously described30.
Homology modeling
A homology model of the α6/α3 chimera built using the sequence in Figure 1A without signal peptide was submitted to the I-TASSER server (http://zhanglab.ccmb.med.umich.edu/I-TASSER/). One of the five top-scored models was used for presentation. The 3D structural presentation of the α6/α3 chimera was made using Discovery Studio Visualizer 4.0 (Accelrys, San Diego, CA, USA).
Data analysis and statistics
Data are reported as the mean±SEM with numbers shown in parentheses (n). Probability levels P<0.05 were considered significant. Significant differences were determined using the two-tailed Student's t-test or one-way ANOVA as appropriate with Origin 8.0 (Microcal Software, Inc, Northampton, MA, USA) or GraphPad Prism 5.0 (GraphPad Software, Inc, La Jolla, CA, USA).
Results
Nicotine concentration-response relationship of α6*-nAChRs
Initial experiments were designed to compare the affinity of α6*- or α4β2-nAChRs for nicotine. To obtain the concentration-response relationship of nicotine-induced current, α6*-nAChR (Figure 2A)- or α4β2-nAChR (Figure 2B)-mediated whole-cell currents induced by different concentrations of nicotine were recorded using Tris+ electrodes at a holding potential (VH) of −40 mV (the resting membrane potential of SH-EP1 cell is close to -40 mV). Normalized (to 100 μmol/L nicotine as indicated with an asterisk; Figure 2C) peak whole-cell current responses to nicotine, when plotted as a function of nicotine concentrations, were sigmoidal and were fit by a single-site logistic equation model. Fits to the nicotine data yielded EC50 values and Hill coefficients of 1.34±0.02 μmol/L and 0.7 for α6*-nAChR (n=12) and 3.3±0.6 μmol/L and 0.8 for α4β2-nAChR (n=10). Unpaired t-test analyses revealed that the difference between these two subtype receptor EC50 values was highly significant (P<0.01). These results demonstrated a higher affinity for nicotine acting on α6*-nAChRs when compared with α4β2 nAChRs. Therefore, in the following experiments, the EC50 concentration of nicotine used was 1 μmol/L for α6*-nAChRs and 3 μmol/L for α4β2-nAChR.
Figure 2.
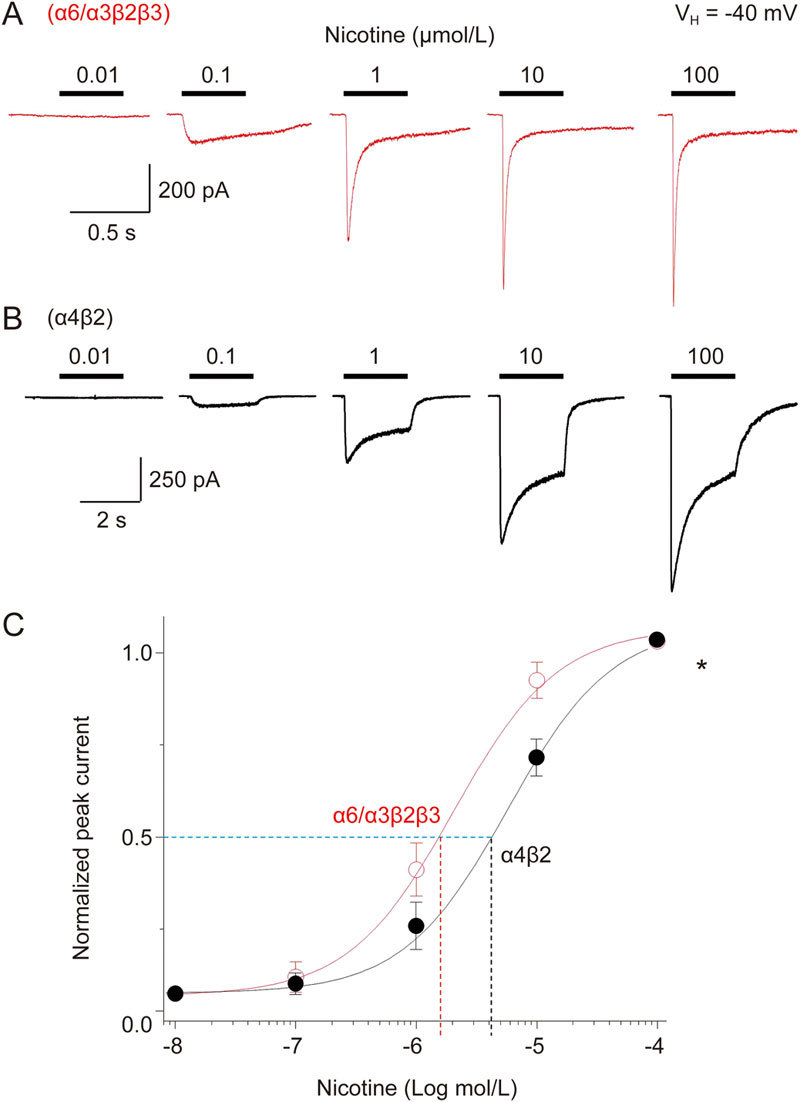
Nicotine concentration-response relationships for α6*-nAChR-mediated whole-cell current in SH-EP1 cells. Five typical whole-cell current traces elicited in response to nicotine exposure (0.01–100 μmol/L) are shown for cells expressing either α6*-nAChR (A) or α4β2-nAChR (B). (C) Nicotine concentration-response curves plotted for peak currents normalized to those evoked in response to 100 μmol/L nicotine show differences at α6*-nAChRs and α4β2-nAChRs in agonist potency. Each symbol represents the average from 10–12 cells, and vertical bars represent standard errors.
Nicotinic agonist properties of α6*-nAChRs
To profile the efficacy and potency of α6*-nAChR agonists, including NIC, ACh, epibatidine (EPBD), cytosine and lobeline (Figure 3A), we constructed agonist concentration-response relationship curves and normalized them to the maximal response to nicotine (at 100 μmol/L; Figure 3A). These profiles indicated ACh, similar to NIC, was a full agonist for α6*-nAChRs, whereas EPBD (∼25%) and cytisine (∼20%) were partial agonists (Figure 3B). The lobeline-induced current was nearly undetectable with α6*-nAChRs even at high concentrations (Supplementary Figure S1). When responses to individual agonists were normalized to the maximum effect at a given α6*-nAChR (Figure 3C), it became evident that the EC50 and Hill coefficient values of ACh, epibatidine, and cytisine were 1.36±0.15 μmol/L and 1.1 (n=11), 10.7±5.7 nmol/L and 0.72 (n=6), and 89.1±33.8 nmol/L and 0.84 (n=6), respectively. These results indicated that for transfected α6*-nAChRs, nicotine and ACh were full agonists, whereas epibatidine and cytisine were partial agonists with high receptor binding affinity.
Figure 3.
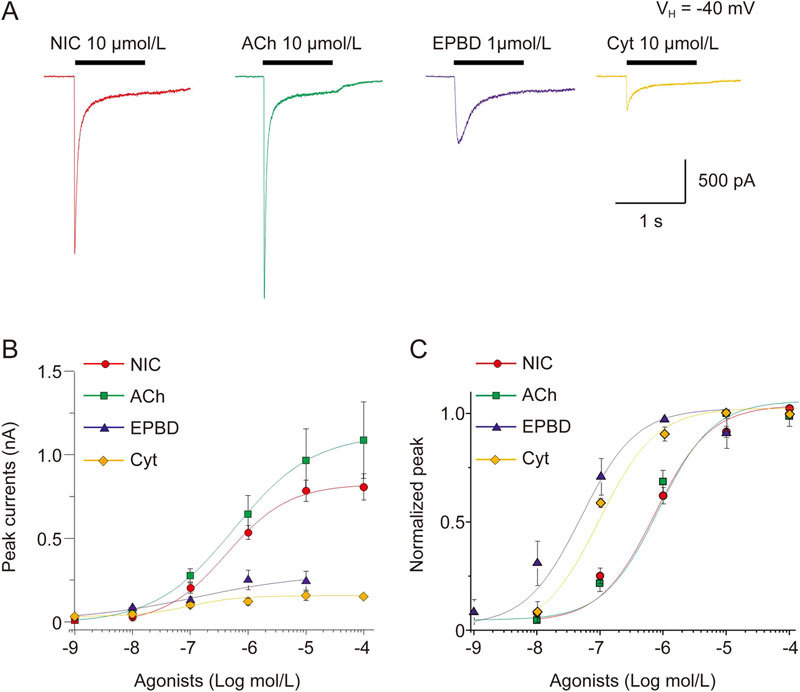
Concentration-response curves for NIC, ACh, epibatidine (EPBD) and cytisine acting at α6*-nAChR. (A) Representative traces showing α6*-nAChR-mediated whole-cell currents elicited by select nAChR agonists nicotine (NIC), ACh, EPBD, and cytisine. (B) Peak current amplitudes of α6*-nAChR evoked by NIC, ACh, EPBD and cytisine are plotted to the real current peak current amplitudes, and show the different efficacy of these nAChR agonists tested. (C) α6*-nAChR-mediated whole-cell currents responses evoked by those agonists were normalized to their maximal response. Each symbol represents the average from 6-8 cells tested, and vertical bars represent standard errors.
Nicotinic antagonist properties of α6*-nAChRs
A main pharmacological feature of α6*-nAChRs is their sensitivity to snail toxins, such as α-Ctx MII. Thus, we examined the effects of α-Ctx MII on 1 μmol/L nicotine-induced current. As shown in Figure 4A, a 5-min pretreatment with 100 nmol/L α-Ctx MII nearly abolished nicotine-induced currents. After a 5-min washout of α-Ctx MII, the nicotine-induced current was partially recovered. When plotted as a function of α-Ctx MII concentrations, the curve showed a typical sigmoidal shape and was fit well with a single-site model of the logistic equation for nicotine-induced currents (Figure 4B). Fits to the data yielded IC50 values and Hill coefficients of 10.3±1.2 nmol/L and 0.9 for nicotine-induced current plus α-Ctx MII (Figure 4B). These results demonstrated that the heterologously expressed α6*-nAChRs were highly sensitive to the selective α6*-nAChR blocker α-Ctx MII. To further evaluate the antagonist profile of α6*-nAChRs, we examined the inhibitory effects of two other nAChR antagonists, dihydro-β-erythroidine (DhβE, Figure 4B) and mecamylamine (MEC, Figure 4C) on α6*-nAChR-mediated current (Figure 4C). We found that the IC50 values and Hill coefficients of DHβE and MEC to 1 μmol/L nicotine-induced currents were -5.9±0.05 mol/L and 0.6±0.06 (n=6–11) and -4.9±0.09 mol/L and 0.7±0.09 (n=6-22), respectively. Figure 4D summarizes the concentration-inhibition curves of α-Ctx MII, DHβE and MEC. Therefore, transfected α6*-nAChRs in SH-EP1 cells were highly sensitive to α-Ctx MII but relatively insensitive to DHβE and MEC, suggesting the chimeric receptor exhibited typical α6*-nAChRs antagonist properties.
Figure 4.
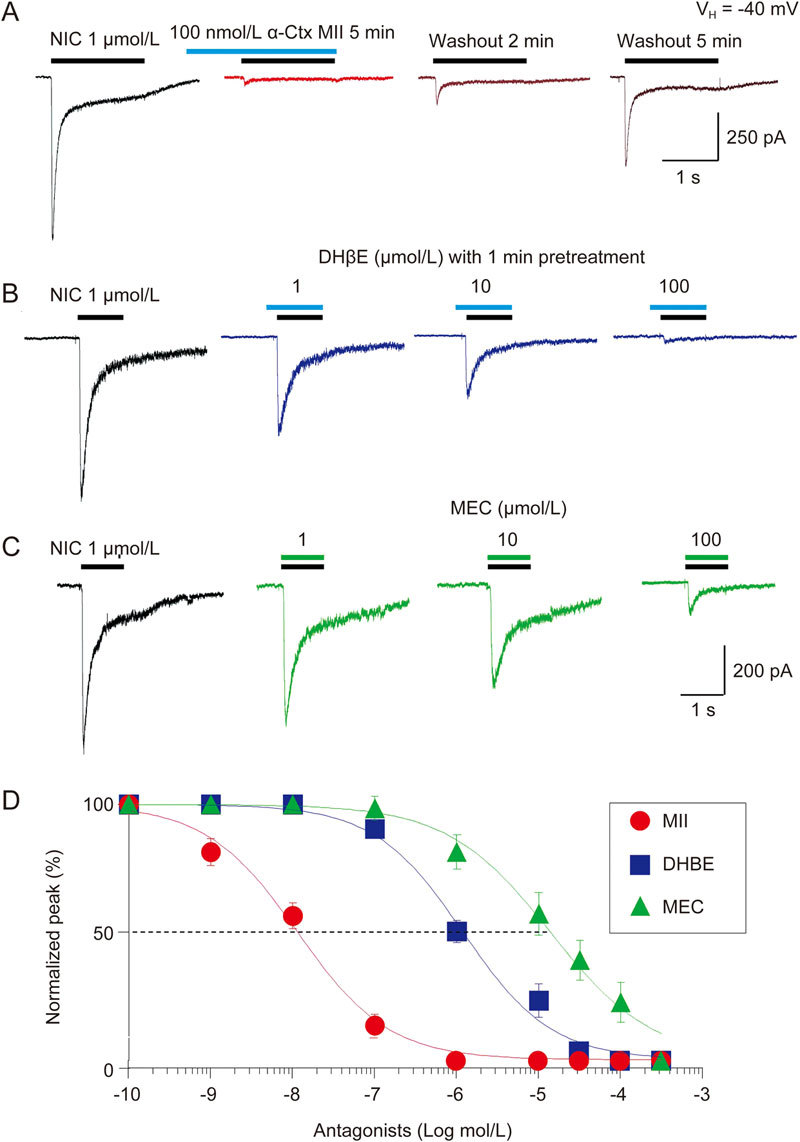
Effects of α6*-nAChR antagonists. Representative typical traces showing antagonism of α6*-nAChR-mediated currents (induced by 1 μmol/L NIC) by the α-Ctx MII (100 nmol/L with 5-min pretreatment (A), the DHβE (0.1, 1, 10 μmol/L with 1-min pretreatment (B), and the MEC (1, 30, 300 μmol/L without pretreatment (C). After washout for 5 min, the inhibition by α-Ctx MII can be partially recovered (A). (D) three nAChR antagonists, α-Ctx MII, DHβE and MEC are superimposed and are plotted as concentration-response curves. Each symbol represents the average from 6–8 cells, and vertical bars represent standard errors.
Kinetics of α6*- nAChR-mediated whole-cell current
Comparisons of whole-cell current kinetics between α6*-nAChR (1 μmol/L nicotine, close to the EC50 concentration) and α4β2-nAChR (3 μmol/L nicotine, close to the EC50 concentration) expressed in SH-EP1 cells revealed that the rate of current decay of α6*-nAChRs induced by 1 μmol/L nicotine was faster, as represented by a significantly smaller decay time constant compared with that of the 3 μmol/L nicotine-induced whole-cell current in α4β2-nAChR (Figure 5A and 5B, right panel). Based on previous reports, α6*-nAChR-mediated whole-cell currents exhibited two components of decay constant, ie, fast and slow components21. We examined whether our α6*-nAChR-mediated currents exhibited these two decay components. In the 22 cells tested, 1 μmol/L nicotine induced inward whole-cell currents showed the fast and slow decay constants (τ values were measured using Clampfit, a double standard exponential fitting). The fast and slow τ values were 149.3±9.2 ms and 1621.6±240.6 ms, respectively. Another difference was the whole-cell current rise time. The α6*-nAChR-mediated current (induced by 1 μmol/L nicotine) had a shorter duration than the α4β2-nAChR-mediated current (induced by 3 μmol/L nicotine). The whole-cell current amplitudes of α6*-nAChR and α4β2-nAChR were not different (Figure 5B, left panel). These results indicated the more rapid acute desensitization of α6*-nAChR compared with α4β2-nAChR.
Figure 5.
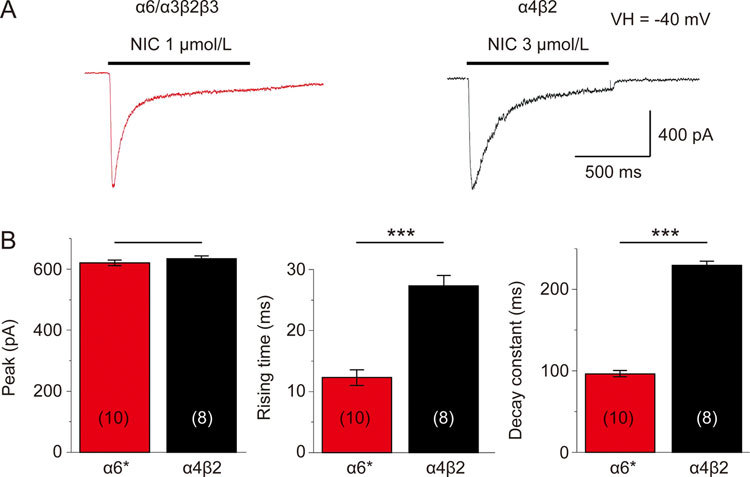
Kinetics of α6*-nAChR-mediated whole-cell current in human SH-EP1 cells. EC50 (1 μmol/L for α6*-nAChR, (A), or 3 μmol/L for α4β2-nAChR, (B) concentrations of nicotine were applied to induce whole-cell inward currents in transfected SH-EP1 cells stably expressing either the human α6*-nAChR (A) or the human α4β2-nAChR (B). (C) Bar graphs summarizing results of replicate studies (10 cells for α6*-nAChR and 8 cells for α4β2-nAChR) illustrating differences between the α6*-nAChR (red columns) and the α4β2-nAChR (black columns) nAChR responses in the rising times of whole-cell currents (B, middle panel) and whole-cell current decay constants (B, right panel). There was no significant difference in peak nicotine current amplitudes, but there was a significant difference in the rise time (B, left panel). Data were represented as the means, and vertical bars represent SEMs. Asterisks *** represent significance level P<0.001 between α6*- and α4β2-nAChRs.
Recovery from desensitization of α6*-nAChR-mediated whole-cell current
To determine the kinetics of α6*-nAChR recovery from acute desensitization, we applied 1 μmol/L nicotine for 5 s followed by 1 s nicotine exposures at intervals of 5, 10, 20 and 40 s for α6*-nAChR (Figure 6A) or application of 3 μmol/L nicotine for 5 s followed by 1 s nicotine exposure at the intervals of 5, 10, 20 and 40 s for α4β2-nAChR (Figure 6B). When fit with a single exponential function, the results showed a time constant (τ) of 22.6 ms for α6*-nAChR-mediated current recovery from desensitization, whereas the τ value for recovery from desensitization in heterologously expressed α4β2-nAChR was 7.3 ms (n=30, and α6*-nAChR vs α4β2-nAChR P<0.01, Figure 6C). These results suggested that α6*-nAChR readily desensitized and required longer intervals to recover when compared with α4β2-nAChR.
Figure 6.
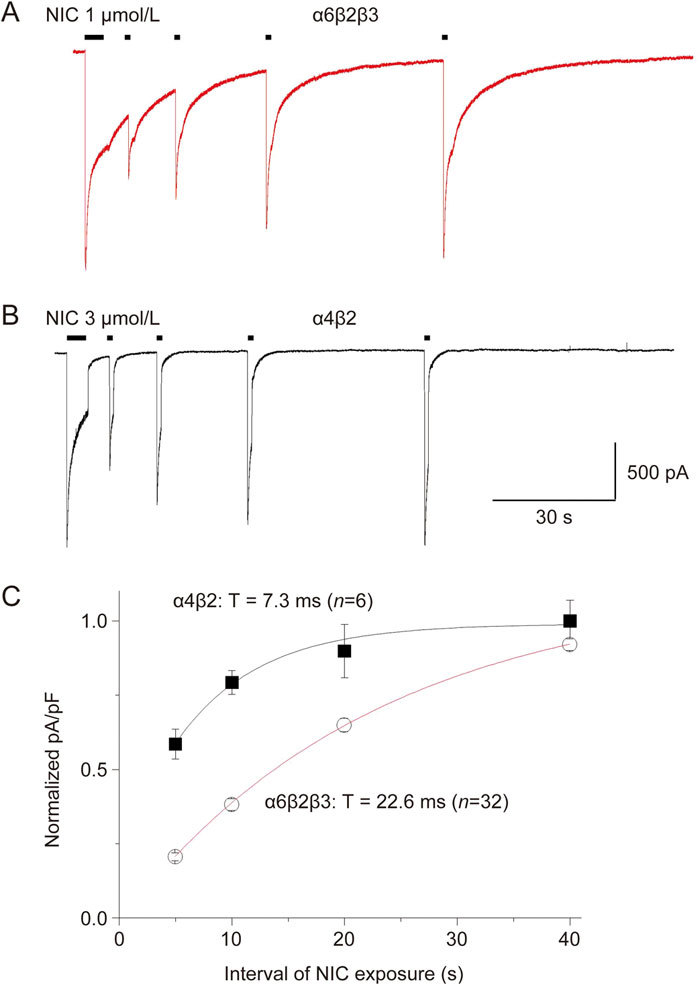
Recovery from desensitization of α6*-nAChR-mediated whole-cell current in SH-EP1 cells. Time course for recovery from desensitization of α6*-nAChRs after a 5-s desensitizing pulse. Typical whole-cell current responses are illustrated for SH-EP1- α6*-nAChR cells subjected to paired 5 s desensitizing pulses of 1 μmol/L NIC followed by 1 s test pulses of the same agonist and dose after an inter-pulse interval of 5, 10, 20 and 40 s (A) or 3 μmol/L NIC using the same protocol for α4β2-nAChR (B). Recordings were obtained at a holding potential of -40 mV. Averaged current net charge (pA/pF) for responses to test pulses normalized to the amplitude of the desensitizing pulse response (ordinate) are plotted for NIC responses to α6*-nAChR (C, red curve) or α4β2-nAChR (C, black curve) as a function of inter-pulse interval (sec; abscissa). Data were fit to mono-exponential equations yielding values for extent of rate constants for recovery indicated in Figure 6C.
Current-voltage (I-V) relationships for α6*-nAChRs
Whole-cell current traces recorded using a K+ electrode solution (see methods) and obtained with a 100 μmol/L nicotine-induced activation of α6*-nAChRs after step changes (-80 to +60 mV) in holding potential (VH) showed an inward rectification at positive VH (Figure 7A). α4β2-nAChR-mediated current (100 μmol/L nicotine) using the same experimental protocol showed a stronger whole-cell current inward rectification (Figure 7B). Figure 7C summarizes the pooled data of steady-state current-voltage (I-V) relationship curves obtained from α6*-nAChRs (red)- and α4β2 (black)-nAChR-mediated currents held at different holding potentials. One-way ANOVA showed high significance between α6*- and α4β2-nAChR-mediated I-V curves (F1,14=197.8, P<0.001). Newman-Keuls comparisons between α6*- and α4β2-nAChR-mediated currents at each holding potential showed significant differences at VH values of -20 mV (P<0.05), 0 mV (P<0.05) and +60 mV (P<0.001). These results indicated that nicotine-induced α6*-nAChR-mediated currents exhibited weaker inward rectification than α4β2-nAChR-mediated currents.
Figure 7.
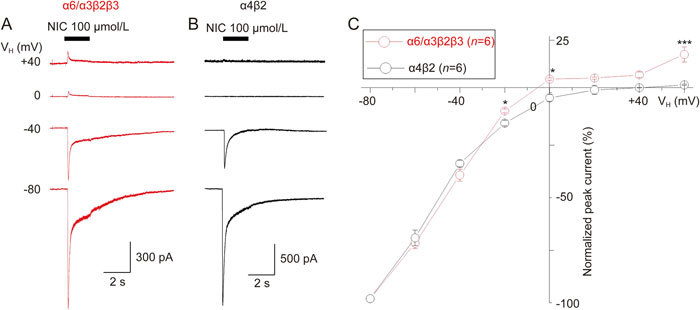
Current-voltage (I-V) relationship curve for α6*-nAChR-mediated whole-cell current in SH-EP1 cells. (A) Typical traces of current-voltage (I-V) relationship using K+ electrodes at a different (from -80 to +60 mV) holding potentials (VH), 100 μmol/L NIC was repetitively applied for 2 s at 2-min intervals to SH-EP1 cells expressing α6*-nAChR (A). (B) Under the same experimental protocol, 100 μmol/L NIC was repetitively applied for 5 s at 2-min intervals to SH-EP1 cells expressing α4β2-nAChR (B). Peak nicotine currents from whole-cell current traces were normalized to the peak current amplitude at the VH of -80 mV (indicated as #) and plotted against the peak currents at different holding potentials. Each symbol represents the average from 6 cells tested, and vertical bars represent standard errors. Asterisks * and *** represent significance levels of P<0.05 and P<0.001, respectively, for α6*-nAChR (red symbols) vs α4β2-nAChR (black symbols).
Discussion
The present study systematically evaluated and compared the pharmacology and physiology of heterologously expressed α6*- and α4β2-nAChRs in a human SH-EP1 cell line. Pharmacologically, α6*-nAChRs demonstrated approximately 3-fold higher affinity for nicotine than α4β2-nAChRs. Nicotine and ACh are full agonists, whereas EPBD and cytisine are partial agonists for α6*-nAChRs. α6*-nAChRs were highly sensitive to the selective antagonist, α-Ctx MII but less sensitive to DHβE and MEC. Functionally, these stably expressed α6*-nAChRs exhibited nicotine-induced inward whole-cell currents with more rapid kinetics of current decay and slower recovery from receptor desensitization compared with α4β2-nAChRs expressed in the same cell line. The α6*-nAChR current-voltage (I-V) curve showed slightly lower inward rectification when compared with that of α4β2-nAChR. Collectively, these results suggested that stably transfected α6*-nAChR in SH-EP1 cell line was an ideal cell model for evaluating α6*-nAChR function and for developing new compounds for pharmacological manipulations of α6*-nAChRs. Additionally, α6*-nAChRs exhibited some functional and pharmacological properties different from those of α4β2-nAChRs, suggesting that this type of nAChR may play a special role in cholinergic modulations in midbrain dopamine-associated signaling, behavior and diseases.
α6*-nAChRs are highly expressed in midbrain DA neurons, but their functions remain poorly understood31. Thus far, there have been three experimental approaches for evaluating α6*-nAChR function: gene manipulation (ie, the transgenic knockout of the α6 subunit or the transgenic expression of highly sensitive α6*-nAChR); the heterologous expression of α6*-nAChRs in recombinant expression systems, which has been successfully demonstrated in Xenopus oocytes; and the use of α6*-nAChR selective antagonists α-conotoxin (α-Ctx) MII, MIIH9L15A or PIA. Ideally, the heterologous expression of α6*-nAChRs is a good model that allows the direct measurements of α6*-nAChR-mediated currents. Lindstrom's laboratory first reported the heterologous expression of nAChR containing α6 subunits in oocytes7. Studies from Clementi's and Gotti's laboratories reported interesting properties of naturally expressed α6*-nAChRs in chick retina, suggesting a variety of subunit combinations for α6*-nAChRs; these subtypes exhibited high affinity agonist binding and interactions with α-Ctx MII antagonist. These data suggested that there are functional α6-containing nAChRs in vivo 32. In general, wild-type (WT) nAChR α6 expressed with β subunits (eg, β2, β3, and β4) in oocytes does not express high levels of functional receptors. To improve α6*-nAChR function, the beta subunit (eg, β3V9'S) was incorporated. The combination of the α6 subunit with β4 and β3V9'S subunits produced approximately 10-fold enhanced nicotine efficacy10,33. α6*-nAChR function was also increased by using an α6 subunit chimera with the α3 subunit; the α6 subunit N-terminal ligand-binding domain was combined with the α3 transmembrane domain plus β2β3V9'S, resulting in enhanced α6*-nAChR function in oocytes23 in transiently expressed HEK 293 cells34 and tsA201 cells35.
In the present study, we applied the same approach to form chimeric, stably expressed human α6*-nAChRs in human SH-EP1 cells without the V9'S mutation in the β3 subunit. Our approach avoided enhanced agonist potency and efficacy effects associated with the 9'S mutant subunit23. Patch-clamp recording demonstrated robust sensitivity to nicotinic agonists. Whole-cell currents showed faster decay constant kinetics, more profound desensitization and slower recovery from receptor desensitization compared with those of α4β2 nAChRs expressed in the same SH-EP1 cell line. These functional properties were consistent with previous reports using patch-clamp recordings from similar chimeric combinations of α6/α3β2β3V9'S nAChRs expressed in HEK 293 cells34. These data suggested that our stably transfected α6*-nAChR was a good model for studying α6*-nAChR pharmacology and function.
Of the five nAChR agonists tested, (-) nicotine, ACh, EPBD, and cytisine induced inward currents mediated by human α6*-nAChRs. Lobeline induced only a small current even at high concentrations. The EC50 for a nicotine-induced current of ∼1 μmol/L was approximately 3-fold higher than that (∼3 μmol/L) for human α4β2-nAChR-mediated whole-cell current heterologously expressed in the same SH-EP1 cell line (Figure 2C). Compared with heterologously expressed α6/α3β2β3V9'S-nAChRs in HEK-293 cells (EC50=0.14 μmol/L)34, our α6*-nAChR cells exhibited approximately 10-fold lower affinity21, suggesting that the β3 subunit mutation (β3V9'S) increased agonist affinity when co-expressed with α6/α3 and β2 subunits. Therefore, compared with α6/α3β2β3V9'S, our transfected α6/α3β2β3-nAChR may be closer to the native α6*-nAChR. Nicotine and ACh displayed comparable peak current efficacies (at maximum agonist dose) in human α6*-nAChRs expressed in SH-EP1 cells, whereas the EPBD- and cytisine-induced maximum current amplitude was approximately 20% of the ACh maximum response, suggesting that EPBD and cytisine were partial agonists for α6*-nAChR. Interestingly, the α4β2-nAChR partial agonist lobeline failed to induce a detectable current response in the α6*-nAChRs. This new finding will allow us to distinguish α6*-nAChRs from α4β2-nAChR based on agonist property. Collectively, these results indicated that heterologously expressed human α6*-nAChRs in SH-EP1 cells exhibited some agonist properties that differed from those of α6/α3β2β3V9'S-nAChRs heterologously expressed in HEK 293 cells34.
Previous studies have shown that natural α6*-nAChRs exhibit high affinities for α-Ctx MII or PIA31, which are major characteristics of functional α6*-nAChRs. Our studies show, at least for human α6*-nAChRs heterologously expressed in SH-EP1 cells, that the highest functional inactivation potencies for α-Ctx MII required pre-exposure of receptors to the antagonists prior to agonist challenge. The pretreatment time of α-Ctx MII was approximately 5 min for 1 μmol/L nicotine-induced currents. From the concentration-inhibition curve, the IC50 value of α-Ctx MI was approximately 10 nmol/L, which was lower than that of α6/α3β2β3V9'S-nAChRs in HEK-293 cells (IC50=44 nmol/L)34. α-Ctx MII was recognized to have slow on-rate kinetics for α6*-nAChR. In our experiments, we applied a 5-min α-Ctx MII pretreatment. An efficient inhibition under our experimental conditions was observed. However, we remain unsure whether a 5-min pretreatment was of sufficient duration to reach α-Ctx MII equilibrium.
In the present study, α6*-nAChR-mediated nicotine responses were also inhibited by MEC (IC50 ∼35 μmol/L), but the affinity was 3500-fold lower than that of α-Ctx MII. The relatively selective α4β2-nAChR antagonist DHβE showed an IC50 value of 0.18 μmol/L, which had 18-fold lower affinity than α-Ctx MII. Collectively, these antagonist properties suggested that while the α6*-nAChR-mediated nicotine response can be blocked by different nAChR antagonists, α-Ctx MII showed the highest affinity.
Although the α6-containing subtype of nAChRs is highly expressed in midbrain dopamine neurons, its detailed function in the modulation of the mesolimbic DA system activity and roles in diseases remain largely unknown. In this study, we heterologously expressed human α6/α3β2β3-nAChRs into human epithelial SH-EP1 cells and obtained a transfected α6*-nAChR with robust function. By using this α6*-nAChR cell model, we provided a detailed description of α6*-nAChR function and pharmacology and compared these to α4β2-nAChR expressed in the same SH-EP1 cell line. These functional and pharmacological properties of α6*-nAChR suggested different roles played in native cholinergic modulations. For example, due to a higher agonist affinity, α6*-nAChR may mediate modulation effects at nicotinic concentrations low enough to not activate α4β2-nAChR. The kinetics of rapid receptor desensitization and slow recovery implied that brain nicotine levels produced by cigarette smoking may readily desensitize α6*-nAChRs. Considering that native α6*-nAChRs in the nucleus accumbens play a critical role in the modulation of DA release, the high affinity of α6*-nAChRs to nicotine was consistent with an important role of this type of nAChR in nicotine reward and dependence. Collectively, our data suggested that the artificial α6*-nAChRs may serve as an excellent cell model to investigate α6*-nAChR function and pharmacology and may enable the development of new compounds for α6*-nAChRs. However, we realize that a limitation of this study was that this transfected α6*-nAChR did not contain a full-length α6 subunit. Instead, we used a α6 subunit N-terminal ligand-binding domain with an α3 subunit transmembrane domain chimera co-transfected with β2 and β3 subunits. To date, the use of full-length α6 co-transfected with β subunits in mammalian systems has not yielded functional α6*-nAChRs. However, functional α6*-nAChRs have been produced in a Xenopus oocyte expression system, which contained co-transfections of the full-length α6 subunit with one mutation in the transmembrane domain and with the second internal loop of the α3 subunit plus β2 and β3 subunits36. Therefore, whether our described function and pharmacology of α6*-nAChR can behave like natural α6*-nAChRs in animal or even human brain remains unclear. Further research is necessary to heterologously express more physiological (full-length) α6*-nAChRs in mammalian cells.
Electronic supplementary material
Supplementary Figures
Acknowledgements
Work toward this project was supported by NIH R01 DA035958, NIH R21 DA026627, NIH R01 GM103801, P01 GM48677, R01 DA042749, Barrow Neuroscience Foundation, Philips Morris External Research Grant, and Special Innovation Project of Education Department of Guangdong Province. Production of the α6/3β2β3-nAChR cell line was sponsored by Targacept.
Electronic supplementary material
Supplementary Information is available at website of Acta Pharmacologica Sinica.10.1038/aps.2017.209
References
- 1.Jensen AA, Frolund B, Liljefors T, Krogsgaard-Larsen P. Neuronal nicotinic acetylcholine receptors: structural revelations, target identifications, and therapeutic inspirations. J Med Chem. 2005;48:4705–45. doi: 10.1021/jm040219e. [DOI] [PubMed] [Google Scholar]
- 2.Lukas RJ, Changeux JP, Le Novere N, Albuquerque EX, Balfour DJ, Berg DK, et al. International Union of Pharmacology. XX. Current status of the nomenclature for nicotinic acetylcholine receptors and their subunits. Pharmacol Rev. 1999;51:397–401. [PubMed] [Google Scholar]
- 3.Wu J, Lukas RJ. Naturally-expressed nicotinic acetylcholine receptor subtypes. Biochem Pharmacol. 2011;82:800–7. doi: 10.1016/j.bcp.2011.07.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Azam L, Winzer-Serhan UH, Chen Y, Leslie FM. Expression of neuronal nicotinic acetylcholine receptor subunit mRNAs within midbrain dopamine neurons. J Comp Neurol. 2002;444:260–74. doi: 10.1002/cne.10138. [DOI] [PubMed] [Google Scholar]
- 5.Klink R, de Kerchove d'Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21:1452–63. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shytle RD, Silver AA, Wilkinson BJ, Sanberg PR. A pilot controlled trial of transdermal nicotine in the treatment of attention deficit hyperactivity disorder. World J Biol Psychiatry. 2002;3:150–5. doi: 10.3109/15622970209150616. [DOI] [PubMed] [Google Scholar]
- 7.Gerzanich V, Kuryatov A, Anand R, Lindstrom J. “Orphan” alpha6 nicotinic AChR subunit can form a functional heteromeric acetylcholine receptor. Mol Pharmacol. 1997;51:320–7. doi: 10.1124/mol.51.2.320. [DOI] [PubMed] [Google Scholar]
- 8.Kuryatov A, Olale F, Cooper J, Choi C, Lindstrom J. Human alpha6 AChR subtypes: subunit composition, assembly, and pharmacological responses. Neuropharmacology. 2000;39:2570–90. doi: 10.1016/S0028-3908(00)00144-1. [DOI] [PubMed] [Google Scholar]
- 9.Grinevich VP, Letchworth SR, Lindenberger KA, Menager J, Mary V, Sadieva KA, et al. Heterologous expression of human {alpha}6{beta}4{beta}3{alpha}5 nicotinic acetylcholine receptors: binding properties consistent with their natural expression require quaternary subunit assembly including the {alpha}5 subunit. J Pharmacol Exp Ther. 2005;312:619–26. doi: 10.1124/jpet.104.075069. [DOI] [PubMed] [Google Scholar]
- 10.Dash B, Bhakta M, Chang Y, Lukas RJ. Modulation of recombinant, alpha2*, alpha3* or alpha4*-nicotinic acetylcholine receptor (nAChR) function by nAChR beta3 subunits. J Neurochem. 2012;121:349–61. doi: 10.1111/j.1471-4159.2012.07685.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tumkosit P, Kuryatov A, Luo J, Lindstrom J. Beta3 subunits promote expression and nicotine-induced up-regulation of human nicotinic alpha6* nicotinic acetylcholine receptors expressed in transfected cell lines. Mol Pharmacol. 2006;70:1358–68. doi: 10.1124/mol.106.027326. [DOI] [PubMed] [Google Scholar]
- 12.Boorman JP, Groot-Kormelink PJ, Sivilotti LG. Stoichiometry of human recombinant neuronal nicotinic receptors containing the b3 subunit expressed in Xenopus oocytes. J Physiol. 2000;529:565–77. doi: 10.1111/j.1469-7793.2000.00565.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Broadbent S, Groot-Kormelink PJ, Krashia PA, Harkness PC, Millar NS, Beato M, et al. Incorporation of the beta3 subunit has a dominant-negative effect on the function of recombinant central-type neuronal nicotinic receptors. Mol Pharmacol. 2006;70:1350–7. doi: 10.1124/mol.106.026682. [DOI] [PubMed] [Google Scholar]
- 14.Capelli AM, Castelletti L, Chen YH, Van der Keyl H, Pucci L, Oliosi B, et al. Stable expression and functional characterization of a human nicotinic acetylcholine receptor with alpha6beta2 properties: discovery of selective antagonists. Br J Pharmacol. 2011;163:313–29. doi: 10.1111/j.1476-5381.2011.01213.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dash B, Chang Y, Lukas RJ. Reporter mutation studies show that nicotinic acetylcholine receptor (nAChR) alpha5 Subunits and/or variants modulate function of alpha6*-nAChR. J Biol Chem. 2011;286:37905–18. doi: 10.1074/jbc.M111.264044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dash B, Bhakta M, Chang Y, Lukas RJ. Identification of N-terminal extracellular domain determinants in nicotinic acetylcholine receptor (nAChR) alpha6 subunits that influence effects of wild-type or mutant beta3 subunits on function of alpha6beta2*- or alpha6beta4*-nAChR. J Biol Chem. 2011;286:37976–89. doi: 10.1074/jbc.M111.263673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Drenan RM, Grady SR, Whiteaker P, McClure-Begley T, McKinney S, Miwa JM, et al. In vivo activation of midbrain dopamine neurons via sensitized, high-affinity alpha 6 nicotinic acetylcholine receptors. Neuron. 2008;60:123–36. doi: 10.1016/j.neuron.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engle SE, Shih PY, McIntosh JM, Drenan RM. alpha4alpha6beta2* nicotinic acetylcholine receptor activation on ventral tegmental area dopamine neurons is sufficient to stimulate a depolarizing conductance and enhance surface AMPA receptor function. Mol Pharmacol. 2013;84:393–406. doi: 10.1124/mol.113.087346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berry JN, Engle SE, McIntosh JM, Drenan RM. alpha6-Containing nicotinic acetylcholine receptors in midbrain dopamine neurons are poised to govern dopamine-mediated behaviors and synaptic plasticity. Neuroscience. 2015;304:161–75. doi: 10.1016/j.neuroscience.2015.07.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Henderson BJ, Wall TR, Henley BM, Kim CH, Nichols WA, Moaddel R, et al. Menthol alone upregulates midbrain nAChRs, alters nAChR subtype stoichiometry, alters dopamine neuron firing frequency, and prevents nicotine reward. J Neurosci. 2016;36:2957–74. doi: 10.1523/JNEUROSCI.4194-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drenan RM, Grady SR, Steele AD, McKinney S, Patzlaff NE, McIntosh JM, et al. Cholinergic modulation of locomotion and striatal dopamine release is mediated by alpha6alpha4* nicotinic acetylcholine receptors. J Neurosci. 2010;30:9877–89. doi: 10.1523/JNEUROSCI.2056-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang K, Buhlman L, Khan GM, Nichols RA, Jin G, McIntosh JM, et al. Functional nicotinic acetylcholine receptors containing alpha6 subunits are on GABAergic neuronal boutons adherent to ventral tegmental area dopamine neurons. J Neurosci. 2011;31:2537–48. doi: 10.1523/JNEUROSCI.3003-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Breining SR, Melvin M, Bhatti BS, Byrd GD, Kiser MN, Hepler CD, et al. Structure-activity studies of 7-heteroaryl-3-azabicyclo[3.3.1]non-6-enes: a novel class of highly potent nicotinic receptor ligands. J Med Chem. 2012;55:9929–45. doi: 10.1021/jm3011299. [DOI] [PubMed] [Google Scholar]
- 24.Eaton JB, Peng JH, Schroeder KM, George AA, Fryer JD, Krishnan C, et al. Characterization of human alpha 4 beta 2-nicotinic acetylcholine receptors stably and heterologously expressed in native nicotinic receptor-null SH-EP1 human epithelial cells. Mol Pharmacol. 2003;64:1283–94. doi: 10.1124/mol.64.6.1283. [DOI] [PubMed] [Google Scholar]
- 25.Gentry CL, Lukas RJ. Local anesthetics noncompetitively inhibit function of four distinct nicotinic acetylcholine receptor subtypes. J Pharmacol Exp Ther. 2001;299:1038–48. [PubMed] [Google Scholar]
- 26.Wu J, Liu Q, Yu K, Hu J, Kuo YP, Segerberg M, et al. Roles of nicotinic acetylcholine receptor beta subunits in function of human alpha4-containing nicotinic receptors. J Physiol. 2006;576:103–18. doi: 10.1113/jphysiol.2006.114645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu J, Kuo YP, George AA, Xu L, Hu J, Lukas RJ. beta-Amyloid directly inhibits human alpha4beta2-nicotinic acetylcholine receptors heterologously expressed in human SH-EP1 cells. J Biol Chem. 2004;279:37842–51. doi: 10.1074/jbc.M400335200. [DOI] [PubMed] [Google Scholar]
- 28.Zhao L, Kuo YP, George AA, Peng JH, Purandare MS, Schroeder KM, et al. Functional properties of homomeric, human alpha 7-nicotinic acetylcholine receptors heterologously expressed in the SH-EP1 human epithelial cell line. J Pharmacol Exp Ther. 2003;305:1132–41. doi: 10.1124/jpet.103.048777. [DOI] [PubMed] [Google Scholar]
- 29.Huguenard JR, Prince DA. A novel T-type current underlies prolonged Ca2+-dependent burst firing in GABAergic neurons of rat thalamic reticular nucleus. J Neurosci. 1992;12:3804–17. doi: 10.1523/JNEUROSCI.12-10-03804.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McIntosh JM, Azam L, Staheli S, Dowell C, Lindstrom JM, Kuryatov A, et al. Analogs of alpha-conotoxin MII are selective for alpha6-containing nicotinic acetylcholine receptors. Mol Pharmacol. 2004;65:944–52. doi: 10.1124/mol.65.4.944. [DOI] [PubMed] [Google Scholar]
- 31.Yang KC, Jin GZ, Wu J. Mysterious alpha6-containing nAChRs: function, pharmacology, and pathophysiology. Acta Pharmacol Sin. 2009;30:740–51. doi: 10.1038/aps.2009.63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vailati S, Hanke W, Bejan A, Barabino B, Longhi R, Balestra B, et al. Functional alpha6-containing nicotinic receptors are present in chick retina. Mol Pharmacol. 1999;56:11–9. doi: 10.1124/mol.56.1.11. [DOI] [PubMed] [Google Scholar]
- 33.Dash B, Li MD, Lukas RJ. Roles for N-terminal extracellular domains of nicotinic acetylcholine receptor (nAChR) beta3 subunits in enhanced functional expression of mouse alpha6beta2beta3- and alpha6beta4beta3-nAChRs. J Biol Chem. 2014;289:28338–51. doi: 10.1074/jbc.M114.566018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rasmussen AH, Strobaek D, Dyhring T, Jensen ML, Peters D, Grunnet M, et al. Biophysical and pharmacological characterization of alpha6-containing nicotinic acetylcholine receptors expressed in HEK293 cells. Brain Res. 2014;1542:1–11. doi: 10.1016/j.brainres.2013.10.024. [DOI] [PubMed] [Google Scholar]
- 35.Jensen AB, Hoestgaard-Jensen K, Jensen AA. Pharmacological characterisation of alpha6beta4 nicotinic acetylcholine receptors assembled from three chimeric alpha6/alpha3 subunits in tsA201 cells. Eur J Pharmacol. 2014;740:703–13. doi: 10.1016/j.ejphar.2014.06.005. [DOI] [PubMed] [Google Scholar]
- 36.Ley CK, Kuryatov A, Wang J, Lindstrom JM. Efficient expression of functional (alpha6beta2)2beta3 AChRs in Xenopus oocytes from free subunits using slightly modified alpha6 subunits. PLoS One. 2014;9:e103244. doi: 10.1371/journal.pone.0103244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures