

Abstract
Apatinib, a small-molecule inhibitor of VEGFR-2, has attracted much attention due to its encouraging anticancer activity in third-line clinical treatment for many malignancies, including non-small cell lung cancer (NSCLC). Its usage in second-line therapy with chemotherapeutic drugs is still under exploration. In this study we investigated the antitumor effect of apatinib combined with docetaxel against NSCLC and its cellular pharmacokinetic basis. A549 xenograft nude mice were treated with apatinib (100 mg/kg every day for 20 days) combined with docetaxel (8 mg/kg, ip, every four days for 5 times). Apatinib significantly enhanced the antitumor effect of docetaxel and alleviated docetaxel-induced liver damage as well as decreased serum transaminases (ALT and AST). LC-MS/MS analysis revealed that apatinib treatment significantly increased the docetaxel concentration in tumors (up to 1.77 times) without enhancing the docetaxel concentration in the serum, heart, liver, lung and kidney. Furthermore, apatinib decreased docetaxel-induced upregulation of P-glycoprotein in tumors. The effects of apatinib on the uptake, efflux and subcellular distribution of docetaxel were investigated in A549 and A549/DTX (docetaxel-resistant) cells in vitro. A cellular pharmacokinetic study revealed that apatinib significantly increased cellular/subcellular accumulation (especially in the cytosol) and decreased the efflux of docetaxel in A549/DTX cells through P-gp, while apatinib exerted no significant effect on the cellular pharmacokinetics of docetaxel in A549 cells. Consequently, the IC50 value of docetaxel in A549/DTX cells was more significantly decreased by apatinib than that in A549 cells. These results demonstrate that apatinib has potential for application in second-line therapy combined with docetaxel for NSCLC patients, especially for docetaxel-resistant or multidrug-resistant patients.
Keywords: non-small cell lung cancer (NSCLC), docetaxel, apatinib, combined chemotherapy, P-glycoprotein, cellular pharmacokinetics, A549 xenograft nude mice
Introduction
Apatinib, a highly selective small-molecule tyrosine kinase inhibitor of VEGFR21, was approved in China in 2014 as a third-line treatment for patients with advanced gastric cancer. Apatinib can inhibit VEGF-stimulated endothelial cell migration and proliferation and decrease tumor microvascular density. Apatinib is now under study in phase II and III clinical trials for other cancer types, such as non-small cell lung cancer (NSCLC), breast cancer and advanced hepatocellular carcinoma2,3,4. Apatinib has attracted much attention due to its encouraging anticancer activity clinically5,6. However, anti-angiogenic agents have shown modest survival benefits because they exert cytostatic activity rather than cytotoxicity. In recent years, aggressive recurrence and accelerating metastatic tumor growth were observed after the discontinuation of sunitinib in xenograft nude mice or clinical trials7,8. In addition, no prolonged overall survival (OS) was found in some patients because of acquired resistance9,10. Therefore, the combination of two or more anti-tumor agents is a more effective strategy to overcome the acquired resistance and limited efficacy of mono anti-angiogenic therapy11,12.
Lung cancer is the most common primary malignant tumor and leading cause of cancer mortality in the world13. A randomized phase III clinical trial assessed the role of ramucirumab and docetaxel versus docetaxel/placebo in patients with NSCLC who progressed after first-line therapy. This trial demonstrated an OS advantage and progression-free survival (PFS) prolongation with the addition of ramucirumab14. In addition to neutralizing antibodies, the role of anti-angiogenic receptor tyrosine kinase inhibitors (TKIs), including sorafenib15,16, cediranib17, sunitinib18, vandetanib19,20 and nintedanib21,22, in the treatment of NSCLC has been the subject of multiple clinical trials. The results of the LUME - Lung 1 clinical trial showed that PFS was significantly improved with the combination of docetaxel plus nintedanib, indicating that nintedanib in combination with docetaxel was an effective second-line option for patients with advanced NSCLC23. Many clinical trials have confirmed the efficacy of anti-angiogenesis small-molecule compounds in combination with cytotoxic drugs in the treatment of cancer. However, the efficacy and safety of apatinib combined with first- or second-line chemotherapeutic drugs are still under investigation.
Previous studies have shown that apatinib significantly increased the intracellular accumulation of doxorubicin and rhodamine 123 in drug-resistant cells and enhanced the anti-tumor effect of paclitaxel in P-gp resistant KBv200 cancer cell xenografts24. Tong et al also found an enhanced effect of doxorubicin in combination with apatinib in K562/ADR xenograft mice25. The present study focused on the antitumor effects of apatinib combined with docetaxel on NSCLC in vitro and in vivo. The effects of apatinib on the antitumor activity or toxic effect of docetaxel were investigated. Additionally, the material basis of the apatinib and docetaxel combination was explored from the perspective of cellular pharmacokinetics.
Materials and methods
Chemicals and reagents
Apatinib and docetaxel were obtained from Jiangsu Hengrui Medicine Co (Lianyungang, Jiangsu, China). Methanol was HPLC grade and purchased from Merck (Darmstadt, Germany). Purified water was produced using a Milli-Q system purchased from Millipore (Bedford, MA, USA). The test kits for the content of serum transaminases (ALT and AST), alkaline phosphatase (ALP), creatinine (Cr), urea nitrogen (BUN) and phosphocreatine kinase (CK) were purchased from Nanjing Jiancheng Biology Research Institute (Nanjing, Jiangsu, China). Fetal bovine serum and 100 U/mL penicillin and streptomycin were purchased from Invitrogen (Carlsbad, CA, USA). Digoxin, 7-ethyl-10-hydroxycamptothecin (SN-38) and 5(6)-carboxy-2′,7′-dichlorofluorescein diacetate (CDCF) were purchased from Sigma (St Louis, MO, USA). Primary antibodies against P-gp and ATPase Na+/K+ were purchased from Abcam (Cambridge, MA, USA).
Animals and treatments
Male athymic nude mice (BALB/c-nu/nu) aged 4–6 weeks and weighing 18–22 g were purchased from Shanghai Slike Experimental Animals Co (Shanghai, China) and were used for subcutaneous xenograft models under specific pathogen-free conditions. The animals were cared for according to the Animal Facility Guidelines of the China Pharmaceutical University. When the tumor volumes were more than 100 mm3, these A549 xenograft mice were randomized into four groups (n=6 per group): (a) saline (every four days for five times); (b) apatinib (100 mg/kg, every day for 20 days); (c) docetaxel (8 mg/kg, every four days for five times); (d) apatinib (100 mg/kg,) plus docetaxel (8 mg/kg, every four days for five times). Docetaxel and apatinib were administered at intervals and were not administered on the same day in the combined group. Docetaxel was intraperitoneally administered on the first day, and then apatinib was intragastrically administered for three consecutive days. From day 5, apatinib was administered four times (a diagram is shown in Figure 1B). The body weight of the animals as well as the long (a) and short diameters (b) were recorded every day. Tumor volume (TV) was calculated as: a/2×b×b. On the last day (day 21), to avoid transient drug-drug interactions, mice were sacrificed after treatment with docetaxel for 0.5 h without apatinib. Next, plasma and tissues were collected and stored at −80 °C until analysis.
Figure 1.
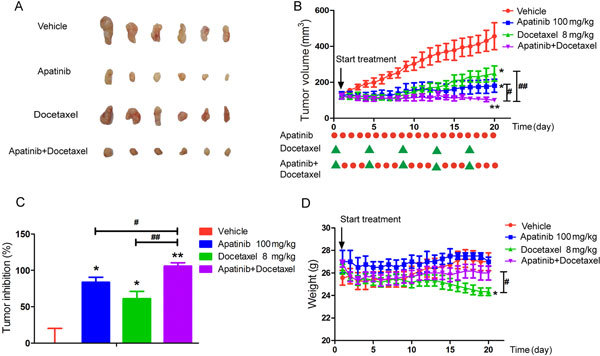
Synergistic antitumor activity of apatinib with docetaxel in A549 xenografts. (A) Tumor size. The picture was taken on day 21 after treatment. (B) Mean tumor volume on different days. The detailed dosing regimen is shown with red dots with green triangles at the bottom. (C) Inhibitory rate of tumor growth. (D) Body weight on different days. The various administrations were as follows: vehicle (vehicle alone); apatinib (100 mg/kg, every day for 20 days); docetaxel (8 mg/kg, every four days for five times); apatinib (100 mg/kg) plus docetaxel (8 mg/kg, every four days for five times). * P<0.05, ** P<0.01 vs vehicle. # P<0.05, ## P<0.01, apatinib+docetaxel vs docetaxel or apatinib.
Hematoxylin and eosin staining and ALT and AST assays
Fresh lung, heart, liver and kidney were fixed in 10% phosphate-buffered formalin immediately and were implanted in paraffin. Next, 5 μm thick slices were cut and stained with hematoxylin and eosin, and 5 μL of serum was used to detect the concentrations of ALT, AST, ALP, Cr, BUN and CK according to the manufacturer's instructions.
Cell culture
Human colon carcinoma Caco-2 cells, human non-small cell lung cancer A549 cells and docetaxel-resistant A549 cells (A549/DTX cells) were purchased from Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China) and were cultured in DMEM and RPMI-1640, respectively, both supplemented with 10% fetal bovine serum and 100 U/mL penicillin and streptomycin at 37 °C with 5% carbon dioxide.
Cell viability assay
A549 and A549/DTX cells (2×105 cells/mL) were cultured in 96-well plates for the 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) colorimetric assay. After 24 h, the control group was incubated with medium containing dimethyl sulfoxide (DMSO). A549 cells were incubated with various concentrations of docetaxel (0.000625, 0.00125, 0.0025, 0.005, 0.01, 0.05, 0.1, 0.5 μmol/L) at 37 °C for 48 h. A549/DTX cells were also treated with various concentrations of docetaxel (0.005, 0.01, 0.05, 0.1, 0.5, 2, 10, 50, 100 μmol/L) at 37 °C for 48 h. A series of concentrations of apatinib (0.125, 0.25, 0. 5, 1, 5, 10, 20, 50 μmol/L) were used to assess the cytotoxicity of A549 and A549/DTX cells. The concentrations of apatinib and docetaxel required to inhibit the growth of A549 and A549/DTX cells by 50% (IC50) were calculated using the SPSS software. In addition, the fold reversal factor of multidrug resistance (MDR) was calculated using the ratio of the IC50 of docetaxel in the absence of apatinib to that in the presence of apatinib or LY335979.
Accumulation of digoxin, SN-38, CDCF and docetaxel in Caco-2 cells
Caco-2 cells were seeded on 24-well cell culture plates evenly. After reaching 80%–90% confluency, the cultured cells were treated with 1 mL of Hanks' solution containing digoxin (a specific substrate of P-gp, 10 μmol/L), digoxin (10 μmol/L) plus apatinib (0.75, 1, 3 μmol/L) or digoxin (10 μmol/L) plus LY335979 (a specific inhibitor of P-gp) (5 μmol/L). The grouping of the accumulations of SN-38 (a specific substrate of BCRP, 10 μmol/L) and CDCF (a specific substrate of BCRP, 10 μmol/L) was the same as that for digoxin but with different positive controls (Ko143 and MK571 are specific inhibitors of BCRP and MRP2). In addition, cultured cells were treated with 1 mL Hanks' solution containing three concentrations of docetaxel (1, 5, 10 μmol/L) with or without specific inhibitors of P-gp, MRP2 and BCRP to investigate whether docetaxel was the substrate of ABC transporters. After incubation for 2 h in a 37 °C incubator, retention was stopped. Cells were lysed in 500 μL of purified water by three freeze-thaw cycles, and 20 μL of the cell suspension was collected for the measurement of protein concentrations using the BCA protein assay kit. Digoxin, SN-38 and docetaxel were determined by LC-MS/MS, and CDCF was detected using a fluorescent microplate reader.
Effect of apatinib on the uptake of docetaxel in A549 and A549/DTX cells
Similar to Caco-2 cells, A549 and A549/DTX cells were treated with 1 mL of Hanks' solution containing docetaxel (10 μmol/L), docetaxel (10 μmol/L) plus apatinib (3 μmol/L) or docetaxel (10 μmol/L) plus LY335979 or Ko143 (5 μmol/L). After incubation for 2 h in a 37 °C incubator, the retention was stopped. We next investigated the uptake progress (0, 0.083, 0.25, 0.5, 1, 2, 4 h) of docetaxel in the absence and presence of apatinib or LY3359799. Cell collection and sample handling were performed as described previously for the drug accumulation study.
Effect of apatinib on the efflux of docetaxel in A549 and A549/DTX cells
Cells were treated as in the docetaxel uptake study and were incubated in a 37 °C incubator for 2 h. Next, each well was washed twice with 1 mL of Hanks' solution to remove impurities from the cell surface, and then, 250 μL of blank serum-free medium was added. After incubation for another 0, 0.25, 0.5, 1, 2, or 4 h, retention was stopped. Cells were lysed in 300 μL of purified water and were treated as described previously for the uptake study.
Effect of apatinib on the subcellular distribution of docetaxel in A549 and A549/DTX cells
A549 and A549/DTX cells were cultured in T-75 culture flasks. After 7 days of culture, the cells were treated with docetaxel (10 μmol/L), docetaxel (10 μmol/L) plus apatinib (3 μmol/L) or docetaxel (10 μmol/L) plus LY335979 (5 μmol/L). After incubation for 0.25, 0.5, 1, or 2 h, the culture flask was washed 3 times with Hanks' solution. Next, the cells were scraped into an Eppendorf tube and centrifuged at 1000 r/min for 5 min. The cells were resuspended in 1 mL of lysis buffer and milled with a glass homogenizer 35–40 times. The cell nucleus, mitochondria and cytoplasm were isolated using the KeyGen Mitochondria/Nuclei Isolation Kit according to the manufacturer's instructions.
Quantitative real-time PCR
Total RNA was extracted from tumor and intestine tissues using the RNAiso Plus reagent according to the manufacturer's protocol. Reverse transcription of 500 ng of total RNA to evaluate MDR1, MRP2 and BCRP in tumor tissue was performed using SuperScript II Reverse Transcriptase (Invitrogen). Quantification of reverse transcription PCR analysis was performed using SYBR green PCR Master Mix and a MyIQ real-time PCR cycler (BioRad, Veenendaal, The Netherlands), and the expression of the targets was normalized to that of β-actin. The primer sequences were as follows: Human MDR1-F: 5′--3′ and 5′--3′; Human MRP2: 5′--3′ and 5′--3′; Human BCRP: 5′--3′ and 5′--3′; Human β-actin: 5′- and -3′; Mouse MDR1: 5′--3′ and 5′--3′; Mouse MRP2: 5′- -3′ and 5′--3′; Mouse BCRP: 5′--3′ and 5′- -3′; Mouse β-actin: 5′- and -3′.
Western blot analysis
Proteins (40 μg) in the membrane extracts were separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to polyvinylidene fluoride membranes (Bio-Rad, Hercules, CA, USA). After blocking with 5% non-fat milk for 1 h, the membrane was incubated with primary antibodies against P-gp (1:1000) and ATPase Na+/K+(1:10000) overnight at 4 °C, followed by incubation with horseradish peroxidase (HRP)-linked secondary antibodies for 2 h at 37 °C. The signals were detected using an enhanced chemiluminescence kit (Thermo Fisher Scientific, MA, USA) and were obtained using the ChemiDoc XRS+ System (Bio-Rad, Hercules, CA, USA). The intensity of the bands was quantified using Image Lab statistical software.
Sample preparation and analysis
Docetaxel was determined by LC-MS/MS. Briefly, 150 μL of methanol containing paclitaxel as an internal standard (200 ng/mL) was added to 50 μL of the sample to precipitate proteins. Tumor, lung, heart, liver and kidney tissues were homogenized in physiological saline (1:10 w/v). After centrifugation at 18 000 r/min for 10 min, 10 μL of the supernatant was injected into the LC-MS/MS system for analysis. The LC-MS/MS system consisted of the Shimadzu HPLC system 20A and AB API4000 quadrupole mass spectrometer with an electrospray ionization (ESI) interface.
Separation of docetaxel was carried out using a Luna C18 column (2.0×150 mm, 5 μm). The column temperature was 40 °C. The mobile phase consisted of solvent A (0.1% formic acid and 2 mg/L sodium acetate) and solvent B (methanol) with the following gradient conditions: the initial proportion was 40% B, followed by a hold for 0.5 min, an increase to 75% B over 0.5 min, an increase to 90% B over 2 min, maintenance for 3 min, a decrease to 40% B over 1 min, and termination at 8.5 min. The flow rate was 0.7 mL/min. The mass spectrometer was used in the positive electrospray ionization (ESI) mode. The MS parameters were as follows: ion spray voltage, 5500 V. Multiple reaction monitoring (MRM) was performed at m/z values of 830.10 →549.14 for docetaxel and 876.5→308.2 for paclitaxel. The collision energies of docetaxel and paclitaxel were 35 eV and 33 eV, respectively. The ion spray voltage was set to 5500 V, and the source temperature was maintained at 550 °C. Detection of digoxin and SN-38 was performed as previously described26,27. Intracellular accumulation of CDCF was detected using the microplate reader Powerwave 200 (Bio-Tek Instruments, VT, USA). The excitation wavelength and emission wavelength were 470 and 529 nm, respectively.
Data analysis
All values are expressed as the mean±SEM. Statistical analysis was analyzed by one-way ANOVA. The difference was considered to be statistically significant if the probability value was less than 0.05 (P<0.05).
Results
Synergistic anti-tumor effect of apatinib in combination with docetaxel in A549 xenograft mice
The efficacy of the combination of docetaxel and apatinib for the treatment of NSCLC was evaluated in nude mice bearing A549 xenografts. The tumor growth curves showed that growth was slower in the treated group than in the vehicle group (Figure 1A and B). On day 20, the average tumor volume (TV) of the vehicle group was 456.42±69.79 mm3, whereas the TVs were 181.00±29.75 mm3 in the apatinib group, 248.91±42.29 mm3 in the docetaxel group, and 100.88±10.10 mm3 in the apatinib+docetaxel group. The tumor inhibitory rate of the combined treatment group was higher than that of the mono treatment group (Figure 1C). However, a significant decrease in the body weight of mice was also observed in the docetaxel and combination groups but not in the apatinib group (Figure 1D), indicating the incidence of toxic side effects of docetaxel to a certain extent.
Protective effect of apatinib on liver damage induced by docetaxel in A549 xenograft mice
Enhancement of the chemotherapeutic effect of docetaxel may increase the risk of side effects. Therefore, we investigate the side effects of docetaxel with or without apatinib on the important organs. As shown in Figure 2A, histopathological abnormality was not evident in the lung, heart, and kidney after treatment with the drugs, indicating both apatinib and docetaxel caused little damage to these tissues. In addition, there was no significant difference in the levels of kidney (Cr and BUN) and cardiac function indicators (CK) in serum, consistent with the histopathological results (Figure 2C and 2D). However, liver injury, such as inflammatory cell infiltration and fiber hyperplasia, occurred after treatment with docetaxel. Moreover, the indicators of liver function (ALT and AST) were increased after administration of docetaxel (Figure 2B), while ALP was not significantly affected. Combined treatment with apatinib significantly alleviated pathological injury and decreased the amount of ALT and AST caused by docetaxel. These results suggest that the combination of apatinib and docetaxel is an effective treatment regimen with reduced side effects.
Figure 2.
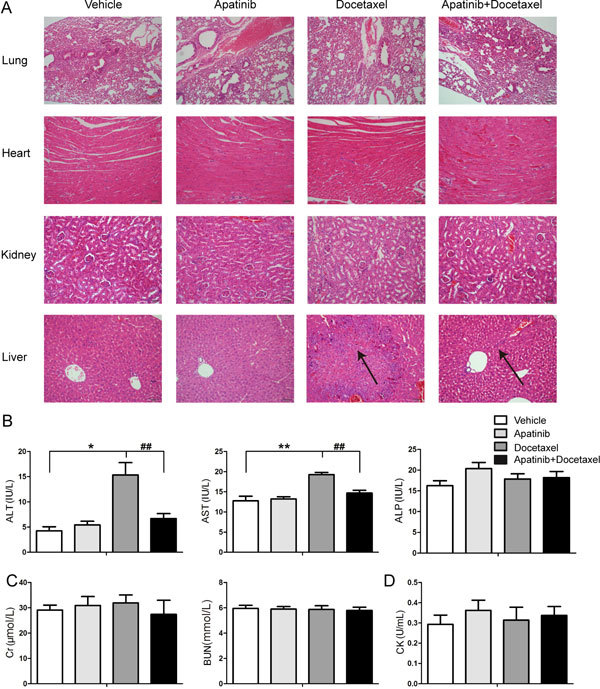
Effect of docetaxel with or without apatinib on the pathology and function of important organs. (A) Representative photographs of H&E staining of tissues. (B) Assays of liver function indicators (ALT, AST and ALP) in serum. (C and D) Assays of kidney function indicators (Cr and BUN) and a cardiac function indicator (CK) in serum. The various administrations were as follows: vehicle (vehicle alone); apatinib (100 mg/kg, every day for 20 days); docetaxel (8 mg/kg, every four days for five times); apatinib (100 mg/kg) plus docetaxel (8 mg/kg, every four days for five times). * P<0.05, ** P<0.01 vs vehicle. ## P<0.01 apatinib+docetaxel vs docetaxel.
Apatinib enhances the tumor concentration of docetaxel without significant effects on the blood, lung, heart and liver distribution in A549 xenograft mice
The distribution of docetaxel in tumor and other main tissues were investigated to clarify whether efficacy enhancement and liver injury alleviation by apatinib were related to the pharmacokinetic changes in docetaxel. Apatinib significantly increased the content of docetaxel in tumors, up to 1.77 times, leading to an improvement in the antitumor efficacy of docetaxel (Figure 3A). However, apatinib did not significantly affect the concentrations of docetaxel in the plasma, liver, kidney, lung and heart (Figure 3B and 3C).
Figure 3.
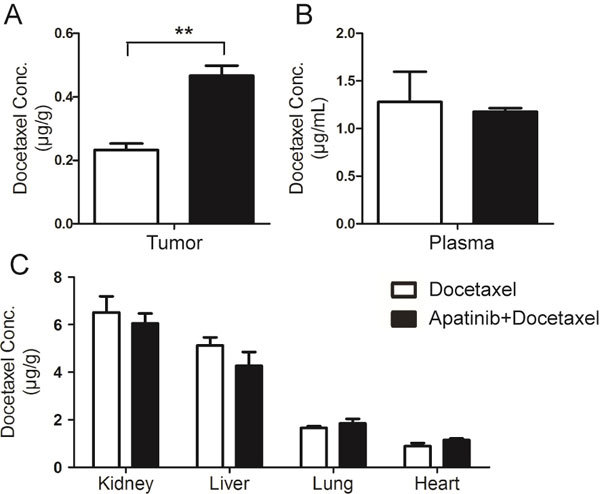
Plasma concentration and tissue distribution of docetaxel in A549 xenograft nude mice with or without apatinib treatment. (A) Concentration of docetaxel in tumors. (B) Concentration of docetaxel in plasma. (C) Tissue distribution of docetaxel in the kidney, liver, lung and heart. ** P<0.01 apatinib+docetaxel vs docetaxel.
Apatinib decreases P-gp expression in tumors induced by docetaxel in A549 xenograft mice
Docetaxel and apatinib are substrates of P-glycoprotein28, and continuous treatment with docetaxel can induce multidrug resistance via P-gp up-regulation. The gene expression levels of the main efflux transporters in tumor and intestinal tissues were determined. Our data showed that gene expression of P-gp in tumor tissue was elevated by 2.13 times compared with that in the vehicle group after administration of docetaxel, a level that was significantly decreased by combined treatment with apatinib (vs the docetaxel group, P<0.01) (Figure 4A). However, the gene expression levels of BCRP and MRP2 were not significantly changed after drug treatments. Moreover, the change in P-gp protein expression in tumors was consistent with that in gene expression (Figure 4B).
Figure 4.
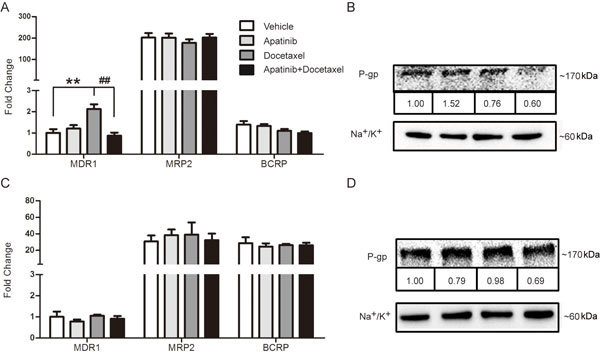
Effects of docetaxel with or without apatinib on efflux transporters. Gene expression levels of efflux transporters in the tumor (A) and intestine (C). Protein levels of P-gp in the tumor (B) and intestine (D). The various administrations were as follows: vehicle (vehicle alone); apatinib (100 mg/kg, every day for 20 days); docetaxel (8 mg/kg, every four days for five times); apatinib (100 mg/kg) plus docetaxel (8 mg/kg, every four days for five times). ** P<0.01 vs vehicle. ## P<0.01, apatinib+docetaxel vs docetaxel.
Because apatinib was orally administered, the gene and protein expression levels of the efflux transporters in intestine tissue were determined. As shown in Figure 4C and 4D, apatinib treatment with or without docetaxel did not affect the expression of P-gp, MRP2 and BCRP.
Increased accumulation of docetaxel induced by apatinib mainly occurs through inhibition of P-gp
Next, we further investigated the effect of apatinib inhibition on the efflux transporters in vitro. Digoxin, SN-38 and CDCF were used as specific substrates for P-gp, BCRP and MRP2, respectively. We found that apatinib could effectively increase the intracellular concentration of digoxin and SN-38, but not that of CDCF, indicating that apatinib could significantly inhibit the function of P-gp and BCRP, but had no effect on MRP2 (Figure 5A). We also studied the effect of specific inhibitors of P-gp, MRP2 and BCRP on docetaxel accumulation in Caco-2 cells. Only LY335979 and Ko143 significantly increased the accumulation of docetaxel (up to 5.2 and 2.3 times), indicating that docetaxel was the main substrate of P-gp and BCRP (Figure 5B). Because the anti-tumor effect was studied in A549 xenograft mice, we used A549 cells (sensitive to docetaxel) and A549/DTX cells (resistant to docetaxel, with highly expressed P-gp) for further cellular pharmacokinetics research. As shown in Figure 5C and 5D, the concentration of docetaxel was approximately three times higher in A549 cells than in A549/DTX cells. Because docetaxel is the substrate of P-gp and BCRP, we investigated the effect of specific inhibitors of P-gp and BCRP on the cellular accumulation of docetaxel in A549 and A549/DTX cells, respectively. The data showed that apatinib significantly increased the content of docetaxel in A549/DTX cells, but not in A549 cells, a finding that was consistent with using LY335979. However, the specific inhibitor of BCRP had no effect on the accumulation of docetaxel in either A549 cells or A549/DTX cells (Figure 5C and 5D). Taken together, these results suggest that P-gp plays an important role in increasing the content of docetaxel by apatinib treatment in A549 xenograft mice.
Figure 5.
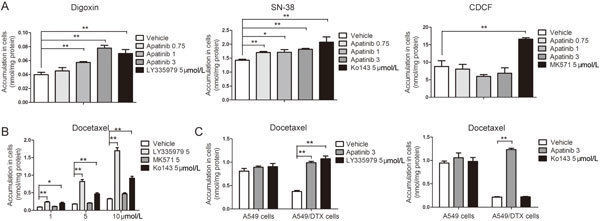
Roles of efflux transporters in the interaction between apatinib and docetaxel. (A) Effect of apatinib on the accumulation of digoxin, SN-38 and CDCF in Caco-2 cells. (B) Effect of LY335979, MK571 and Ko143 on the accumulation of docetaxel in Caco-2 cells. (C) Effect of apatinib and LY335979 on the accumulation of docetaxel in A549/DTX and A549 cells. (D) Effect of apatinib and Ko143 on the accumulation of docetaxel in A549/DTX and A549 cells. LY335979 (5 μmol/L), MK571 (5 μmol/L) and Ko143 (5 μmol/L) are specific inhibitors of P-gp, MRP2 and BCRP, respectively. Digoxin, SN-38 and CDCF are specific substrates of P-gp, BCRP and MRP2, respectively. * P<0.05, ** P<0.01 vs vehicle.
Apatinib increases the cellular accumulation and subcellular distribution of docetaxel in A549/DTX cells but not in A549 cells
Because P-gp is crucial in the increased concentration of docetaxel in the above in vivo and in vitro results, we used LY335979 as a positive control to investigate whether apatinib could affect the cellular pharmacokinetic behavior of docetaxel in A549 and A549/DTX cells. In A549/DTX cells, apatinib and LY335979 both significantly enhanced the accumulation of docetaxel in a time-dependent manner (Figure 6B). The content of docetaxel in A549/DTX cells was rapidly decreased once apatinib or LY335979 was withdrawn and reached a relatively stable level 1 h later, a trend that was still higher in the combined treatment groups than in the docetaxel group (Figure 6D). However, the effect of apatinib or LY335979 on the cellular accumulation of docetaxel was not observed in sensitive A549 cells (Figure 6A and 6C).
Figure 6.
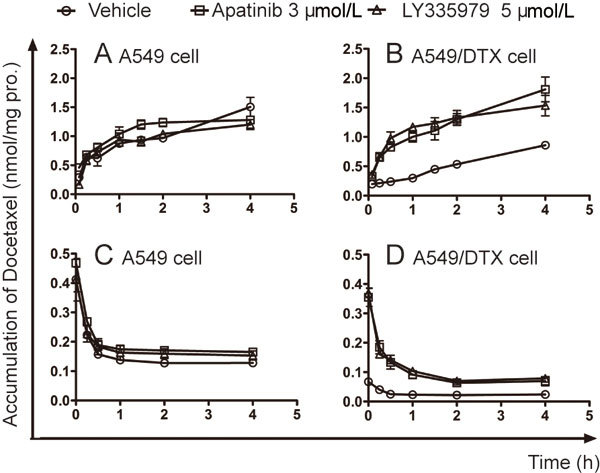
Effects of apatinib on the accumulation of docetaxel in A549 and A549/DTX cells. (A and B) Docetaxel uptake assay in A549 and A549/DTX cells, as indicated by the concentration-time curves of the intracellular accumulation of docetaxel. (C and D) Docetaxel efflux assay in A549 and A549/DTX cells, as indicated by the concentration-time curves of the intracellular retention of docetaxel. LY335979, a specific inhibitor of P-gp, was used as a positive control.
The drug concentration around the target determines its action. Docetaxel, as a microtubule-targeting chemotherapeutic drug, exerts its efficacy in the cytosol. Therefore, the distribution of docetaxel in subcellular organelles (including in the cytosol, nuclei and mitochondria) was detected. The AUC0-2 h of docetaxel in the cytosol was the highest among these subcellular organelles in either A549 or A549/DTX cells (Figure 7G and 7H). Similar to the uptake results, combined treatments with apatinib or LY335979 induced a rapid increase in the subcellular accumulation of docetaxel in the first 0.5 h in A549/DTX cells (Figure 7B, 7D and 7F). Notably, the AUC0-2 h of docetaxel in the cytosol was increased by 6.38 times with apatinib treatment. However, higher accumulation was not observed in the presence of apatinib or LY335979 in sensitive A549 cells (Figure 7A, 7C and 7E).
Figure 7.
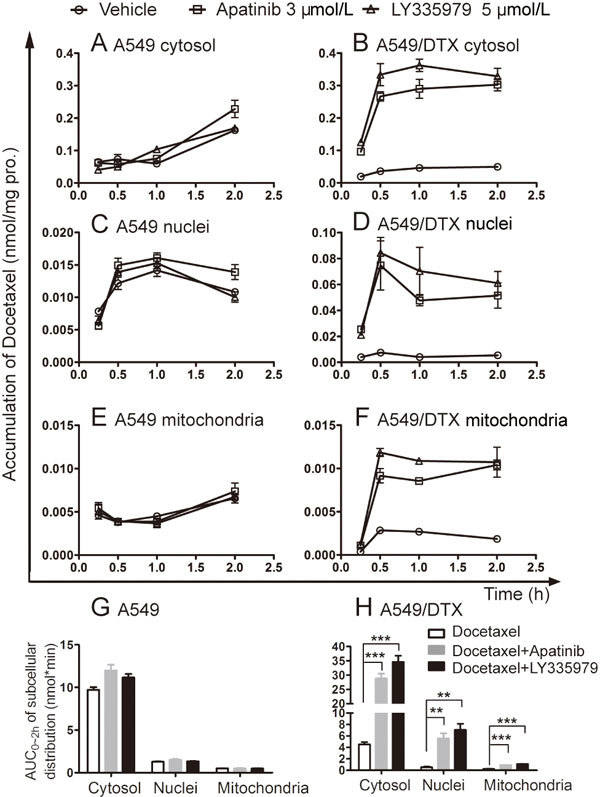
Effects of apatinib on the subcellular distribution of docetaxel in A549 and A549/DTX cells. Time-course analysis of the subcellular accumulation of docetaxel in A549 cells (A, C, E) and A549/DTX cells (B, D, F). The AUC0-2 h of docetaxel in the subcellular organelle (G and H). LY335979, a specific inhibitor of P-gp, was used as a positive control. ** P<0.01 vs vehicle.
Effect of apatinib on the cytotoxicity of docetaxel in A549 and A549/DTX cells
Based on the improved cellular pharmacokinetic behavior of docetaxel, we next studied whether apatinib could enhance the cytotoxicity of docetaxel in A549 and A549/DTX cells. No significant cytotoxicity by apatinib was demonstrated in A549 and A549/DTX cells. The IC50 values of apatinib in A549 and A549/DTX cells were 23.83±1.66 μmol/L and higher than 100 μmol/L, respectively. More than 90% of A549 or A549/DTX cells were viable at 10.0 μmol/L apatinib. Thus, 1.5, 3.0 or 6.0 μmol/L concentrations of apatinib were used in the combined treatment study. The IC50 values of docetaxel in the sensitive and resistant A549 cells and those in combination with different concentrations of apatinib are shown in Table 1. LY335979 was used as the positive control. The IC50 value of docetaxel in A549 cells was 0.049±0.008 μmol/L and that in A549/DTX cells was 40.95±4.51 μmol/L (resistance index=835.7). Apatinib (6 μmol/L) also reversed resistance to docetaxel in A549/DTX cells by 21.00-fold, a trend that was better than the combined treatment with LY335979. Interestingly, we also found that apatinib slightly enhanced the cytotoxicity of docetaxel in A549 cells.
Table 1.
Effect of apatinib (1.5–6 μmol/L) on cytotoxicity of docetaxel in A549 and A549/DTX cells. Data are shown as the mean±SEM (n=5). * P<0.05, ** P<0.01, values obtained in the combined treatment vs the values obtained in docetaxel alone.
| IC50±SEM (μmol/L) (fold-reversal) | ||
|---|---|---|
| A549 | A549/DTX | |
| Apatinib | 23.83±1.66 | >100 |
| Docetaxel | 0.049±0.008 | 40.95±4.51 |
| +1.5 Apatinib | 0.046±0.009 (1.07) | 24.18±3.52* (1.69) |
| +3 Apatinib | 0.021±0.004* (2.33) | 9.28±1.34* (4.41) |
| +6 Apatinib | 0.014±0.007 (3.50) | 1.95±0.41** (21.00) |
| +5 LY335979 | 0.047±0.006 (1.04) | 10.71±3.22* (3.82) |
Cell survival was determined by MTT assays as described in cell viability assay. The fold-reversal of MDR (values in parentheses) was calculated by the ratio of the IC50 for cells treated with docetaxel alone to that obtained in the presence of apatinib or specific inhibitor of P-gp LY335979 5 μmol/L.
Discussion
ATP-binding cassette (ABC) transporters, such as P-gp, MRP and BCRP, are often reported to be highly expressed in cancer, causing active efflux of many chemotherapeutic drugs out of cancer cells and reducing their cytotoxic effect. Additionally, long-term chemotherapy can lead to multi-drug resistance of cytotoxic effects in cancer cells, significantly decreasing the efficacy of cancer chemotherapy. Several anti-angiogenic receptor tyrosine kinase inhibitors (TKIs) were found to interact with major ABC transporters, including P-gp, MRP and BCRP29. TKIs such as gefitinib, sunitinib, and lapatinib could significantly inhibit the function P-gp and BCRP, increasing their intracellular substrate accumulation30,31,32. Apatininb was also found to increase intracellular accumulation of DOX and Rho 123 in MDR cells24. In this study, we investigated whether apatinib could be used in combination with docetaxel in vivo and in vitro, providing more choices for patients with advanced NSCLC.
The present study showed that apatinib enhanced the inhibitory effect of docetaxel on the tumor growth and volume. Pathological data also demonstrated that the lung, heart and kidney were not significantly affected by the drugs in either single or combined treatment. Interestingly, liver damage was alleviated when docetaxel combined with apatinib which was indicated by the histopathologic data and decreased levels of ALT and AST. Clinical studies of liver injury induced by docetaxel have been demonstrated in the treatment of metastatic breast cancer33. Liu et al found that Astragalus polysaccharides could protect against the hepatotoxicity induced by frequently used chemical therapy agents, including docetaxel 34. However, to the best of our knowledge, no other study has reported on the protective effect of combined chemotherapy against docetaxel-induced liver damage. Therefore, alleviation of docetaxel-induced liver injury may be one of the advantages of the combined use of apatinib and docetaxel in clinical NSCLC chemotherapy.
Our pharmacokinetics data showed that apatinib significantly increased the distribution of docetaxel in tumors but exerted no obvious effect on the docetaxel concentrations in the plasma, kidney, lung, heart and liver. This result further confirmed that apatinib enhanced the anti-tumor effect of docetaxel but did not increase docetaxel-induced injury to other organs. P-gp, MRP2 and BCRP are three major ATP-dependent efflux transporters that reduce the intracellular content of many chemotherapeutic agents, possibly resulting in multi-drug resistance35. Our results also showed that multiple administrations of docetaxel induced upregulation of P-gp in tumors, which might be a key mediator of the acquisition of docetaxel resistance36. Zhu et al also found that acquired docetaxel resistance was mediated by the overexpression of P-gp in prostate cancer, while targeting the expression of P-gp could overcome docetaxel resistance by inducing more apoptotic cell death28. The present study found that combined treatment with apatinib down-regulated the expression of P-gp induced by docetaxel, which is in accordance with the increased tumor distribution of docetaxel. Although apatinib was also reported to be a substrate of P-gp, fortunately, continuous oral administration of apatinib in mice did not induce upregulation of P-gp in tumor or intestinal tissue. Several studies have also demonstrated that apatinib could reverse P-gp- and BCRP-mediated multidrug resistance by inhibiting their transport function but did affect their expression25,37. Our in vitro data from Caco-2 cells also showed that docetaxel was the substrate of P-gp and BCRP, the function of which was significantly inhibited by apatinib. However, increased accumulation of docetaxel by apatinib only occurred in A549/DTX cells, which had a similar effect as the P-gp inhibitor LY335979 rather than the BCRP inhibitor Ko143. Taken together, the data indicate that the increased concentration of docetaxel in tumors mainly occurs via the inhibitory effect of apatinib on P-gp.
Cellular pharmacokinetics is an emerging branch of traditional pharmacokinetics and has attracted attention for the evaluation and improvement of anticancer therapy. A previous study showed that 20(S)-ginsenoside Rh2 (20(S)-Rh2) significantly increased the rate and amount of adriamycin entering adriamycin-resistant MCF-7 cellular or subcellular organelles and aggravated adriamycin-induced apoptosis38. Yuan et al also demonstrated the reversal effect of cinobufagin on P-gp-mediated resistance of anticancer drugs regarding cellular pharmacokinetics39. Next, A549/DTX cells were used to study whether apatinib could make a favorable cellular pharmacokinetic change in docetaxel-resistant cells that might subsequently play a role in reversing drug resistance. Obvious enhanced accumulation and decreased efflux of docetaxel were found in the presence of apatinib or LY335979 in A549/DTX cells and subcellular organelles, especially in the target cytosol. In the present study, our data also showed that the apatinib concentration dependently enhanced the cytotoxicity of docetaxel in A549/DTX cells. Interestingly, apatinib also tended to increase the sensitivity of docetaxel in A549 cells. We speculate that the reason for the enhanced efficacy of docetaxel by apatinib may occur via mechanisms other than the increased cellular concentration. Overall, our data provide proof of the effectiveness of the combination of apatinib and docetaxel for treating NSCLC patients, especially patients with acquired docetaxel resistance.
Conclusion
The present study demonstrated that apatinib enhanced the anti-tumor effect of docetaxel and minimized docetaxel-induced side effects in the liver, effects that were closely related to the increased distribution of docetaxel and decreased expression of P-gp in tumors. In vitro, apatinib elevated the accumulation of the cellular/subcellular distribution in docetaxel-resistant A549/DTX cells rather than in A549 cells, and the cytotoxicity of docetaxel was also enhanced by apatinib, especially in A549/DTX cells. Our data are supportive of the clinical combined treatment of apatinib and docetaxel for treating NSCLC, especially for multidrug-resistant patients.
Author contribution
Fang ZHOU, Jiye AA and Guang-ji WANG designed the manuscript. Si-qi FENG, Jing-wei ZHANG and Yuan XIE carried out the experiments. Run-bin SUN, Fei FEI and Jing-qiu HUANG analyzed the data. Fang ZHOU, Si-qi FENG and Ying WANG prepared the manuscript. Fang ZHOU, Guang-ji WANG and Jiye AA critically revised the manuscript. All of the authors have read and approved the final manuscript.
Acknowledgements
The work was supported by the National Natural Science Foundation of China (No 81573496, 81530098, 81573494); Jiangsu Province Nature Science Foundation (BK20160076); China “Creation of New Drugs” Key Technology Projects (2015ZX09501001); the Foundation for Innovative Research Groups of the National Natural Science Foundation of China (No 81421005); the Project Program of State Key Laboratory of Natural Medicines, China Pharmaceutical University (No SKLNMZZCX201608).
Contributor Information
Ji-ye Aa, Email: jiyea@cpu.edu.cn.
Fang Zhou, Email: zf1113@163.com.
References
- 1.Tian S, Quan H, Xie C, Guo H, Lü F, Xu Y, et al. YN968D1 is a novel and selective inhibitor of vascular endothelial growth factor receptor-2 tyrosine kinase with potent activity in vitro and in vivo. Cancer Sci. 2011;102:1374–80. doi: 10.1111/j.1349-7006.2011.01939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang L, Shi M, Huang C, Liu X, Xiong JP, Chen G, et al. A phase II, multicenter, placebo-controlled trial of apatinib in patients with advanced nonsquamous non-small cell lung cancer (NSCLC) after two previous treatment regimens. J Clin Oncol 2012; 30.
- 3.Hu X, Cao J, Hu W, Wu C, Pan Y, Cai L, et al. Multicenter phase II study of apatinib in non-triple-negative metastatic breast cancer. BMC Cancer. 2014;14:820. doi: 10.1186/1471-2407-14-820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin S. Apatinib in Chinese patients with advanced hepatocellular carcinoma: A phase II randomized, open-label trial. ASCO Meeting Abstracts 2014; 32.
- 5.Li CM, Liu ZC, Bao YT, Sun XD, Wang LL. Extraordinary response of metastatic pancreatic cancer to apatinib after failed chemotherapy: A case report and literature review. World J Gastroenterol. 2017;23:7478–88. doi: 10.3748/wjg.v23.i41.7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li F, Liao Z, Zhao J, Zhao G, Li X, Du X, et al. Efficacy and safety of Apatinib in stage IV sarcomas: experience of a major sarcoma center in China. Oncotarget. 2017;8:64471–80. doi: 10.18632/oncotarget.16293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ebos JML, Lee CR, Cruz-Munoz W, Bjarnason GA, Christensen JG, Kerbel RS. Accelerated metastasis after short-term treatment with a potent inhibitor of tumor angiogenesis. Cancer Cell. 2009;15:232–239. doi: 10.1016/j.ccr.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Griffioen AW, Mans LA, De Graaf AMA, Nowak-Sliwinska P, De Hoog CLMM, De Jong TaM, et al. Rapid angiogenesis onset after discontinuation of sunitinib treatment of renal cell carcinoma patients. Clin Cancer Res. 2012;18:3961–71. doi: 10.1158/1078-0432.CCR-12-0002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giuliano S and Pages G. Mechanisms of resistance to anti-angiogenesis therapies. Biochimie. 2013;95:1110–9. doi: 10.1016/j.biochi.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 10.Zhu YJ, Zheng B, Wang HY, and Chen L. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614–22. doi: 10.1038/aps.2017.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miles D, Cameron D, Bondarenko I, Manzyuk L, Alcedo JC, Lopez RI, et al. Bevacizumab plus paclitaxel versus placebo plus paclitaxel as first-line therapy for HER2-negative metastatic breast cancer (MERiDiAN): A double-blind placebo-controlled randomised phase III trial with prospective biomarker evaluation. Eur J Cancer. 2017;70:146–55. doi: 10.1016/j.ejca.2016.09.024. [DOI] [PubMed] [Google Scholar]
- 12.Wong PP, Demircioglu F, Ghazaly E, Alrawashdeh W, Stratford MR, Scudamore CL, et al. Dual-action combination therapy enhances angiogenesis while reducing tumor growth and spread. Cancer Cell. 2015;27:123–37. doi: 10.1016/j.ccell.2014.10.015. [DOI] [PubMed] [Google Scholar]
- 13.Perez-Moreno P, Brambilla E, Thomas R, and Soria JC. Squamous cell carcinoma of the lung: molecular subtypes and therapeutic opportunities. Clin Cancer Res. 2012;18:2443–51. doi: 10.1158/1078-0432.CCR-11-2370. [DOI] [PubMed] [Google Scholar]
- 14.Garon EB, Ciuleanu TE, Arrieta O, Prabhash K, Syrigos KN, Goksel T, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384:665–73. doi: 10.1016/S0140-6736(14)60845-X. [DOI] [PubMed] [Google Scholar]
- 15.Bruix J, Takayama T, Mazzaferro V, Chau GY, Yang JM, Kudo M, et al. Adjuvant sorafenib for hepatocellular carcinoma after resection or ablation (STORM): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2015;16:1344–54. doi: 10.1016/S1470-2045(15)00198-9. [DOI] [PubMed] [Google Scholar]
- 16.Paz-Ares LG, Biesma B, Heigener D, Von Pawel J, Eisen T, Bennouna J, et al. Phase III, randomized, double-blind, placebo-controlled trial of gemcitabine/cisplatin alone or with sorafenib for the first-line treatment of advanced, nonsquamous non-small-cell lung cancer. J Clin Oncol. 2012;30:3084–92. doi: 10.1200/JCO.2011.39.7646. [DOI] [PubMed] [Google Scholar]
- 17.Ruscito I, Gasparri ML, Marchetti C, De Medici C, Bracchi C, Palaia I, et al. Cediranib in ovarian cancer: state of the art and future perspectives. Tumor Biol. 2016;37:2833–9. doi: 10.1007/s13277-015-4781-4. [DOI] [PubMed] [Google Scholar]
- 18.Scagliotti GV, Krzakowski M, Szczesna A, Strausz J, Makhson A, Reck M, et al. Sunitinib plus erlotinib versus placebo plus erlotinib in patients with previously treated advanced non-small-cell lung cancer: a phase III trial. J Clin Oncol. 2012;30:2070–8. doi: 10.1200/JCO.2011.39.2993. [DOI] [PubMed] [Google Scholar]
- 19.Herbst RS, Sun Y, Eberhardt WEE, Germonpre P, Saijo N, Zhou CC, et al. Vandetanib plus docetaxel versus docetaxel as second-line treatment for patients with advanced non-small-cell lung cancer (ZODIAC): a double-blind, randomised, phase 3 trial. Lancet Oncol. 2010;11:619–26. doi: 10.1016/S1470-2045(10)70132-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.De Boer RH, Arrieta O, Yang CH, Gottfried M, Chan V, Raats J, et al. Vandetanib plus pemetrexed for the second-line treatment of advanced non-small-cell lung cancer: a randomized, double-blind phase III trial. J Clin Oncol. 2011;29:1067–74. doi: 10.1200/JCO.2010.29.5717. [DOI] [PubMed] [Google Scholar]
- 21.European Medicines Agency. Vargatef (nintedanib): summary of product characteristics. 23 Mar 2015; Available from: http://www.ema.europa.eu.
- 22.Espinosa Bosch M, Asensi Diez R, Garcia Agudo S, Clopes Estela A. Nintedanib in combination with docetaxel for second-line treatment of advanced non-small-cell lung cancer; GENESIS-SEFH drug evaluation report. Farm Hosp. 2016;40:316–27. doi: 10.7399/fh.2016.40.4.10455. [DOI] [PubMed] [Google Scholar]
- 23.Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M, et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014;15:143–55. doi: 10.1016/S1470-2045(13)70586-2. [DOI] [PubMed] [Google Scholar]
- 24.Mi YJ, Liang YJ, Huang HB, Zhao HY, Wu CP, Wang F, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–91. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tong XZ, Wang F, Liang S, Zhang X, He JH, Chen XG, et al. Apatinib (YN968D1) enhances the efficacy of conventional chemotherapeutical drugs in side population cells and ABCB1-overexpressing leukemia cells. Biochem Pharmacol. 2012;83:586–97. doi: 10.1016/j.bcp.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Zhou F, Niu F, Lu M, Wu X, Sun J, et al. Stereoselective regulations of P-glycoprotein by ginsenoside Rh2 epimers and the potential mechanisms from the view of pharmacokinetics. Plos One. 2012;7:e35768. doi: 10.1371/journal.pone.0035768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan S, Ahmad AW, Wang YF, Abu QA, and Ahmad I. A simple and sensitive LC/MS/MS assay for 7-ethyl-10-hydroxycamptothecin (SN-38) in mouse plasma and tissues: application to pharmacokinetic study of liposome entrapped SN-38 (LE-SN38) J Pharmaceut Biomed. 2005;37:135. doi: 10.1016/j.jpba.2004.09.053. [DOI] [PubMed] [Google Scholar]
- 28.Zhu Y, Liu C, Nadiminty N, Lou W, Tummala R, Evans CP, et al. Inhibition of ABCB1 expression overcomes acquired docetaxel resistance in prostate cancer. Mol Cancer Ther. 2013;12:1829–36. doi: 10.1158/1535-7163.MCT-13-0208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Anreddy N, Gupta P, Kathawala RJ, Patel A, Wurpel JND, and Chen ZS. Tyrosine kinase inhibitors as reversal agents for ABC transporter mediated drug resistance. Molecules. 2014;19:13848–77. doi: 10.3390/molecules190913848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kitazaki T, Oka M, Nakamura Y, Tsurutani J, Doi S, Yasunaga M, et al. Gefitinib, an EGFR tyrosine kinase inhibitor, directly inhibits the function of P-glycoprotein in multidrug resistant cancer cells. Lung Cancer. 2005;49:337–43. doi: 10.1016/j.lungcan.2005.03.035. [DOI] [PubMed] [Google Scholar]
- 31.Shukla S, Robey RW, Bates SE, and Ambudkar SV. Sunitinib (Sutent, SU11248), a small-molecule receptor tyrosine kinase inhibitor, blocks function of the ATP-binding cassette (ABC) transporters P-glycoprotein (ABCB1) and ABCG2. Drug Metab Dispos. 2009;37:359–65. doi: 10.1124/dmd.108.024612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dai CL, Tiwari AK, Wu CP, Su XD, Wang SR, Liu DG, et al. Lapatinib (Tykerb, GW572016) reverses multidrug resistance in cancer cells by inhibiting the activity of ATP-binding cassette subfamily B member 1 and G member 2. Cancer Res. 2008;68:7905–14. doi: 10.1158/0008-5472.CAN-08-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang Z, Liang X, Yu J, Zheng XH, Zhu YL, Yan Y, et al. Non-genetic risk factors and predicting efficacy for docetaxel-drug-induced liver injury among metastatic breast cancer patients. J Gastroen Hepatol. 2012;27:1348–52. doi: 10.1111/j.1440-1746.2012.07131.x. [DOI] [PubMed] [Google Scholar]
- 34.Liu W, Gao FF, Li Q, Lv JW, Wang Y, Hu PC, et al. Protective effect of Astragalus polysaccharides on liver injury induced by several different chemotherapeutics in mice. Asian Pac J Cancer P. 2014;15:10413–20. doi: 10.7314/APJCP.2014.15.23.10413. [DOI] [PubMed] [Google Scholar]
- 35.Dlugosz A, Janecka A. ABC transporters in the development of multidrug resistance in cancer therapy. Curr Pharm Des. 2016;22:4705–16. doi: 10.2174/1381612822666160302103646. [DOI] [PubMed] [Google Scholar]
- 36.Hansen SN, Westergaard D, Thomsen MBH, Vistesen M, Do KN, Fogh L, et al. Acquisition of docetaxel resistance in breast cancer cells reveals upregulation of ABCB1 expression as a key mediator of resistance accompanied by discrete upregulation of other specific genes and pathways. Tumor Biol. 2015;36:4327–38. doi: 10.1007/s13277-015-3072-4. [DOI] [PubMed] [Google Scholar]
- 37.Mi YJ, Liang YJ, Huang HB, Zhao HY, Wu CP, Wang F, et al. Apatinib (YN968D1) reverses multidrug resistance by inhibiting the efflux function of multiple ATP-binding cassette transporters. Cancer Res. 2010;70:7981–91. doi: 10.1158/0008-5472.CAN-10-0111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang JW, Zhou F, Wu XL, Zhang XX, Chen YC, Zha BS, et al. Cellular pharmacokinetic mechanisms of adriamycin resistance and its modulation by 20(S)-ginsenoside Rh2 in MCF-7/Adr cells. Br J Pharmacol. 2012;165:120–34. doi: 10.1111/j.1476-5381.2011.01505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yuan ZT, Shi XJ, Qiu YY, Jia TT, Yuan X, Zou Y, et al. Reversal of P-gp-mediated multidrug resistance in colon cancer by cinobufagin. Oncol Rep. 2017;37:1815–25. doi: 10.3892/or.2017.5410. [DOI] [PubMed] [Google Scholar]