

SUMMARY
Alternative polyadenylation generates numerous 3’ mRNA isoforms that can vary in biological properties such as stability and localization. We developed methods to obtain transcriptome-scale structural information and protein binding on individual 3’ mRNA isoforms in vivo. Strikingly, near-identical mRNA isoforms can possess dramatically different structures throughout the 3’UTR. Analyses of identical mRNAs in different species or refolded in vitro indicate that structural differences in vivo are often due to trans-acting factors. The level of Pab1 binding to poly(A) containing isoforms is surprisingly variable, and differences in Pab1 binding correlate with the extent of structural variation for closely-spaced isoforms. A pattern encompassing singlestrandedness near the 3’ terminus, double-strandedness of the poly(A) tail, and low Pab1 binding is associated with mRNA stability. Thus, individual 3’ mRNA isoforms can be remarkably different physical entities in vivo. Sequences responsible for isoform-specific structures, differential Pab1 binding, and mRNA stability are evolutionarily conserved, indicating biological function.
Keywords: RNA structure, mRNA half-life, mRNA, DMS, Pab1, yeast, alternative polyadenylation, RNA folding, DREADS, CLIP-READS
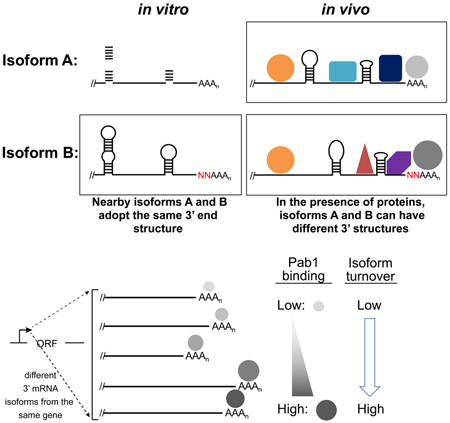
eTOC
Alternative polyadenylation generates numerous 3’ mRNA isoforms. Moqtaderi et al. demonstrate that near-identical 3’ mRNA isoforms can have dramatically different structures in vivo, largely due to differential mRNA-protein interactions. Sequences responsible for isoform-specific structures, differential association with poly(A)-binding protein (Pab1), and mRNA stability are evolutionarily conserved, indicative of biological function.
INTRODUCTION
Alternative polyadenylation generates multiple 3’ mRNA isoforms per eukaryotic gene (Ozsolak et al., 2010; Wu et al., 2011; Moqtaderi et al., 2013). Regulation of this process is critical in a number of biological processes, including development (Weill et al., 2012; Elkon et al., 2013; Tian and Manley, 2013) and oncogenesis (Mayr and Bartel, 2009; Li and Lu, 2013; Masamha et al., 2014). Depending on the organism, individual 3’ mRNA isoforms from the same gene may differ by many kilobases or by as little as one nucleotide (Ozsolak et al., 2010; Sherstnev et al., 2012; Moqtaderi et al., 2013; Pelechano et al., 2013). Same-gene isoforms can vary widely in biological properties such as stability, subcellular localization, and translational efficiency (Geisberg et al., 2014; Berkovits and Mayr, 2015; Floor and Doudna, 2016). Differences with respect to these important biological properties raise the possibility that multiple mRNA isoforms may exist to play distinct roles under specific cellular conditions.
In the yeast S. cerevisiae, mRNA isoforms arising from the same gene—even ones that differ in length by a single nucleotide—can exhibit marked differences in stability (Geisberg et al., 2014; Gupta et al., 2014). Computational modeling suggests that variation in transcript turnover may be influenced by structural differences close to 3’ termini (Geisberg et al., 2014). Isoform-specific structure has also been hypothesized to be important for translational efficiency (Floor and Doudna, 2016) and is implicated in 3’ end processing and stability of mammalian transcripts (Wu and Bartel, 2017). Mechanistically, long intervening sequences between isoform endpoints could affect folding of the longer isoform via the formation of secondary structures with upstream sequences. Longer isoforms could also contain binding sites for mRNA-binding proteins (and in mammalian cells, miRNAs) that are absent in shorter transcripts. By either mechanism, the extra sequences in longer isoforms could alter their folding and/or functional properties.
Several general approaches have been used either singly or in combination to analyze RNA structure in vivo. Computational modeling based on the primary sequence of the RNA strives to predict which intramolecular base-pairing interactions will yield a thermodynamically optimal secondary structure. Secondary and some tertiary structure can be mapped experimentally by chemical cleavage or by enzymatic probing with specific ribonucleases such as RNase T1 and V1 (Ziehler and Engelke, 2001). In such methods, solvent-exposed surfaces unobstructed by secondary or tertiary structure are typically identifiable by their availability as substrates. More recently, deep sequencing methods combined with the use of suitable RNA-modifying chemicals have allowed genome-wide probing of RNA structure in vivo (Ding et al., 2014; Rouskin et al., 2014; Kubota et al., 2015; Bevilacqua et al., 2016). In methods such as DMS-Seq, SHAPE-Seq, and Structure-Seq, the general strategy is to treat with a chemical that targets particular structural features of the RNA (e.g. solvent-exposed surfaces), followed by library construction in a manner that preserves information about modified RNA positions.
A major drawback of these methods is that they do not provide any 3’ isoform specificity, but rather yield an ensemble structure of all transcripts from a given gene (Rouskin et al., 2014). To address this issue, we developed DREADS, a method based on DMS modification in vivo, to probe the structures of individual 3’ mRNA isoforms over the entire yeast transcriptome. We also developed a related method, SHREADS (SHAPE-READS), that is based on the NAI reagent (Spitale et al., 2013), as well as a transcriptomescale technique (CLIP-READS) to measure protein binding to individual 3’ mRNA isoforms.
We demonstrate extensive structural diversity across same-gene 3’ mRNA isoforms, including many that differ by only a few nucleotides, in multiple yeast species. Individual 3’ mRNA isoforms display distinct structures in vivo; remarkably, these structural differences are not restricted to the 3’ termini but often occur throughout the entire 3’UTR. We show that much of this structural variation is due to RNA-binding proteins that differentially associate with the 3’UTRs of same-gene isoforms in vivo. We demonstrate that the poly(A)-binding protein (Pab1) exhibits unexpected variability in binding to poly(A)-containing isoforms. Thus, individual 3’ mRNA isoforms and their associated proteins can be remarkably different physical entities in vivo. Importantly, sequences responsible for isoform-specific structures, differential Pab1 binding, and mRNA stability are mechanistically linked and evolutionarily conserved, indicating biological function.
RESULTS
A DMS-based method for probing the structures of individual 3’ mRNA isoforms on a transcriptome scale
We developed DREADS (DMS Region Extraction and Deep Sequencing), an adaptation of DMS-seq, Structure-Seq and 3’ READS (Hoque et al., 2013; Ding et al., 2014; Rouskin et al., 2014), to perform structural analysis of 3’ mRNA isoforms on a transcriptome scale (Figure 1A). DMS modifies single-stranded RNA at A and C residues, thus providing a marker for unstructured regions in vivo (Wells et al., 2000; Liebeg and Waldsich, 2009). During sequencing library construction, DMS-modified positions block the progress of reverse transcriptase. When the library is subjected to paired-end sequencing, the upstream fragment endpoint reveals the site of the DMS modification, while the downstream endpoint links the modification to a specific 3’ isoform (Figure 1B). We performed DREADS on duplicate cultures of DMS-treated and untreated S. cerevisiae cells. The relative frequencies of mRNA 3’ isoforms exhibit good correlation with those obtained with previous techniques, both at the global level and at individual loci (R = 0.42 - 0.71; Figure S1 A-C).
Figure 1. DREADS schematic.

(A) Exponentially-growing yeast cells are treated with DMS, which methylates exposed A and C residues in cellular RNAs. During library construction, reverse transcriptase (RT) stalls at DMS-modified positions (blue asterisks) as well as at naturally occurring stall sites on mRNA molecules. Each resulting library fragment contains information on both the specific poly(A) isoform (downstream end) and the endpoint of the RT reaction (upstream end). Paired-end deep sequencing thus makes it possible to link multiple RT stops with individual 3’ isoforms. Subtraction of the naturally occurring (non-DMS) RT stops in the untreated control allows for precise mapping of DMS-reactive, accessible A and C residues in each 3’ isoform.
(B) Example of a library fragment subjected to paired-end sequencing. Gray segments at the fragment ends represent adapter sequences added during library construction. The R1 sequence read encompasses the junction between the poly(A) tail remnant and the last nt of genomically-encoded 3’UTR sequence; this allows mapping of the specific 3’ isoform. The R2 sequence read supplies information about where the reverse transcription reaction stopped (naturally or at potential DMS-modified site) on the same mRNA molecule during library construction.
For every 3’ isoform, the DMS reactivity profile is defined by the “net” reads terminating at every A and C position upstream of the poly(A) site. Net reads for a given A or C residue are obtained by subtracting the number of read counts in the non-DMS treated control from the number of read counts in the DMS-treated sample (Ding et al., 2014). This approach corrects for natural termination preferences of the reverse transcriptase, and it is not confounded by any potential variation in enzyme performance at a particular locus. Hence, the DMS reactivity profile of an individual isoform represents a structural blueprint that is independent of its expression level. Two independent replicates of S. cerevisiae for both DMS-treated cells and untreated cells are highly correlated with each other (R = 0.93; Figure S2 A-D) and moderately correlated with a previously published dataset (R = 0.3-0.4; Figure S2E) obtained without isoform specificity and using completely different methodology (Rouskin et al., 2014). We compared DMS reactivity profiles of distinct isoforms from the same 3’ UTR by considering only nucleotides common to both isoforms (i.e., the comparison window ends at the 3’ terminus of the shorter isoform).
Extensive structural diversity among isoforms of the same gene
While reactivity profiles for individual 3’ isoforms across two biological replicates are highly correlated (> 50% of isoforms have a Pearson correlation greater than 0.9), different isoforms arising from the same gene often possess strikingly different reactivity profiles (Figure 2A and B; the two different isoforms are shown with their identical sequences aligned). Remarkably, structural differences occur over an extended region upstream of the poly(A) sites (Figure 2A). Only 30% of same-gene isoforms compared pairwise exhibit a correlation coefficient over 0.9, and 20% correlate very poorly (R < 0.3; Figure 2B). Unexpectedly, mRNA isoforms can possess remarkably different DMS reactivity profiles even if their 3’ ends are closely spaced, and this dissimilarity increases as a function of interisoform distance (Figures 2C, S2F). These different DMS reactivity profiles cannot be attributed to different transcriptional start sites for the isoforms, because 5’ end selection varies independently of poly(A) site selection (Pelechano et al., 2013). Same-gene isoforms (limited here to isoforms terminating ≤ 100 nt from one another) are far more likely than biological replicates of the same isoform (P < 10−100) to exhibit extensive differences in their DMS reactivity profiles, providing direct evidence that related (and in some cases even adjacent) isoforms can have dramatically different structures.
Figure 2. Closely-related mRNA isoforms can possess radically different DMS reactivity profiles.

(A)Examples of DMS reactivity profiles (limited to 200nt upstream of the poly(A) tail for display clarity) of biological replicates and closely related (2 nt apart) neighboring isoforms. Reactivities are scaled relative to the isoform’s highest observed reactivity value (100). Top: A and C reactivities for two biological replicates of the RPS31 mRNA “+48” isoform arising from polyadenylation 48 nt downstream of the ORF. Bottom: A and C reactivities (average of two replicates) for the same +48 isoform compared to those of the isoform ending 2 nt away, at 50 nt downstream of the ORF. The indicated coordinates for both isoforms are defined with respect to the same +48 position and hence represent the identical nucleotides in the two isoforms.
(B)Genome-wide percentile distribution of Pearson correlation coefficients for DREADS isoform reactivity profiles. Gray bars: correlations for the same isoform’s reactivity profiles in two biological replicates. Black bars: correlations of every isoform’s reactivity profile with every other same-gene isoform’s reactivity profile.
(C)Percentage of poorly correlating DREADS isoform reactivity profiles increases as a function of inter-isoform spacing.
(D)Genome-wide percentile distribution of Pearson correlation coefficients for isoform reactivity profiles (SHREADS). Gray bars: correlations for the same isoform’s reactivity profiles in two biological replicates. Black bars: correlations of every isoform’s reactivity profile with every other same-gene isoform’s reactivity profile
(E) DREADS and SHREADS identify a highly overlapping subset of structurally distinct isoform pairs. Same gene isoform pairs exhibiting substantial structural differences (ΔR ≥ 0.3) are far more likely to be identified by both methods than by chance alone (P = 10−51).
Isoform structural diversity confirmed by in vivo SHAPE
For independent confirmation of our results, we developed SHREADS (SHAPE-READS), a procedure that utilizes a cell-permeable alkylating reagent (NAI) that reacts with the solvent-exposed (i.e. single-stranded) sugar backbone of all four ribonucleotides (Spitale et al., 2013). As in DREADS, alkylated RNA residues block the progress of reverse transcriptase, and are identified by and linked to individual 3’ isoforms via pair-end sequencing. SHREADS isoform expression levels and reactivity profiles of biological replicates are highly correlated (Figure 2D; Figure S2 G and H). The correlation between the isoform reactivity profiles obtained from SHREADS and DREADS analyses is highly significant (P = 8.5 × 10−31Figure S2I). Although this correlation appears quantitatively modest (R = 0.35), it is in broad agreement with previously observed differences between SHAPE and DMS profiles on multiple structures (Fang et al., 2015; Somarowthu et al., 2015; Smola et al., 2016).
As observed with DREADS, SHREADS reactivity profiles for biological replicates of individual isoforms are highly correlated (43% of isoforms possess R > 0.9; Figure 2D), but a significant proportion of neighboring isoforms exhibit very poor correlation (R < 0.3; P < 10−100). Importantly, DREADS and SHREADS have remarkably similar distributions of reactivity profile correlations for same gene isoform pairs (compare Figures 2B and2D), and isoform pairs that are structurally dissimilar (R < 0.3) under one assay are far more likely to be found structurally dissimilar in the other (P = 1.1 × 10−51; Figure 2E). Thus, dramatic structural diversity among same gene isoforms is readily observed using two different chemical probes.
Nucleotide substitutions can yield dramatic changes in DMS reactivity
Extensive differences in reactivity profiles can also be observed among alleles of the same 3’ isoform whose sequences differ by 1-4 base substitutions near the poly(A) tail (Figure 3). We generated such allelic variants by randomizing short stretches of 3’UTR sequences just upstream of highly-expressed isoforms of selected genes. Collections of strains harboring plasmids from these libraries were treated (or mock-treated) with DMS, and specific loci were then enriched by a modified hybridization capture protocol using locked nucleic acid probes complementary to ~20 nt regions immediately upstream of the randomized sequences. Comprehensive analysis of these full libraries will be published elsewhere, but representative alleles of one such locus, SOD1, are shown in Figure 3. While a single mutation (A66U) near the cleavage/polyadenylation site has only a modest effect on the reactivity profile (Figure 3, top), a triply substituted allele (A66U, U67G, U70G) near the poly(A) site of SOD1 +71 shows extensive reactivity profile changes (Figure 3, bottom).
Figure 3. Single point mutations can alter an isoform’s structural profile.

DMS reactivity profile comparison of the wild-type SOD1 +71 (SOD +71wt) isoform to two mutants possessing an identical cleavage/polyadenylation site (SOD1 +71A66U, SOD1 +71UGG). Reactivities for each isoform are scaled relative to that isoform’s highest observed reactivity value (100). Top: A comparison of A and C reactivities for SOD +71wt and the single substitution mutant SOD1 +71A66U reveal modest differences in reactivity profiles. Bottom: Extensive differences in reactivity profiles between SOD +71wt and the triple substitution mutant SOD1 +71UGG.
Structural differences in similar isoforms can occur far from the poly(A) site
Differential DMS reactivity profiles, and hence structures, of any two same-gene neighboring 3’ isoforms are ultimately due to sequences adjacent to the poly(A) tail that are present in one isoform and lacking in the other. Remarkably, there are numerous examples in which closely related 3’ mRNA isoforms show major differences in DMS reactivity that occur >200 nt upstream of the poly(A) tail. Similar effects are observed in the mutated versions of otherwise wild-type isoforms. Importantly, these long-range structural effects are identified exclusively by differential DMS reactivity of identical residues across isoforms and are completely independent of any structural modeling.
Mechanistically, structural differences between same-gene isoforms might be due to cis features (e.g. secondary/tertiary RNA structure or covalent ribonucleotide modifications) and/or trans factors. For simplicity, we will consider trans factors to be RNA-binding proteins; trans-acting RNAs are also possible candidates, although these are very rare in yeast (Aw et al., 2016). As described below, we assessed the contributions of cis and trans factors to isoform structure.
Isoform structure differences are much more pronounced in vivo than in vitro
Nucleotides with poor DMS reactivity might not necessarily be base-paired, but could instead be rendered inaccessible by bound proteins (or other factors). We performed several lines of investigation to examine the possible role of RNA-binding proteins in mediating structural differences of same-gene isoforms. First, to assess the extent to which RNA-binding proteins influence DMS reactivity, we compared the structural relationships of isoform pairs in vivo and in vitro. Specifically, we compared the reactivity correlations of neighboring isoform pairs in vivo with the reactivity correlations for the identical isoform pairs in vitro after mRNA isolation, denaturation, and re-folding (Figure 4A). A meaningful proportion (2%–9%) of neighboring isoforms ≤ 6 nt apart show major structural differences in vivo. By contrast, only a tiny percentage (0% to < 0.5%) of the corresponding isoform pairs from the in vitro data set differ structurally, arguing that nearly all of the observed DMS reactivity differences among these closely-related isoforms are mediated by RNA-binding proteins. As expected, the percentage of poorly correlated isoforms increases with greater inter-isoform spacing, underscoring the important role that sequences immediately adjacent to cleavage/poly(A) sites play in the overall folding of mRNA 3’ ends. However, the proportion of poorly correlating isoforms is substantially greater in the in vivo samples irrespective of inter-isoform spacing, indicating that RNA-binding proteins are still likely to play an important role in shaping overall reactivity profiles in same-gene isoform pairs with longer (≥ 9 nt) median spacing.
Figure 4. DMS reactivity profiles are influenced by cellular factors.

(A) Percentage of poorly correlating mRNA isoform reactivity profiles (R < 0.3) in in vivo (from Figure 2C, black bars) and in vitro (gray bars) refolded samples as a function of the distance between isoform endpoints.
(B) Left panel: DMS reactivity profile comparison of identical mRNA isoforms from native D. hansenii and from a D. hansenii chromosome segment in an S. cerevisiae host strain. ΔR is the difference by which an isoform’s reactivity profiles correlate in biological replicates compared to the correlation of that isoform’s reactivity profiles in the native species (D. hansenii or K. Lactis) versus when transplanted into S. cerevisiae (Rreplicates – Rcross-species). Blue bars represent isoforms for which the interspecies Pearson correlation is worse (three different ΔR thresholds shown) than the same-species biological replicate correlation. Red bars represent isoforms for which the interspecies correlation is better than the biological replicate; these bars represent the experimental error. Right panel: the same analysis with identical K. lactis isoforms from native and S. cerevisiae-YAC strains.
Identical mRNA sequences often exhibit different structures in different yeast species
To directly assess the influence of factors other than the sequence itself on mRNA structure in vivo, we obtained DREADS data for native D. hansenii and K. lactis strains, as well as for S. cerevisiae strains each harboring a large genomic region from either D. hansenii or K. lactis (Hughes et al., 2012; Moqtaderi et al., 2013; Jin et al., 2017) (Figures 4B, S3A and B). Transplanting large chromosomal regions from another yeast species on a yeast artificial chromosome (YAC) into an S. cerevisiae host allows direct in vivo structural comparison of identical mRNA isoforms in two different cellular contexts. DREADS analysis of native D. hansenii and K. lactis mRNAs yields similar results to those seen in S. cerevisiae (Figure S3C). Strikingly, reactivity profiles of identical D. hansenii isoforms assayed in the native species and the YAC-bearing S. cerevisiae strain are four times more likely to differ than those of same-species biological replicates (Figure 4B, left panel; P = 2.2 × 10−5 to 1.2 × 10−3, depending on the threshold for similarity). In contrast, the number of K. lactis identical isoforms that exhibit differences in reactivity profiles between native and YAC contexts is essentially indistinguishable from the experimental error (Figure 4B, right panel).
These observations indicate that species-specific trans-acting factors contribute to in vivo structural differences. The host-specific differences observed for identical D. hansenii isoforms, but not for K. lactis isoforms, are consistent with the much greater evolutionary distance between D. hansenii and S. cerevisiae as compared to K. lactis and S. cerevisiae (Dujon et al., 2004). This observation also suggests that RNA binding trans-factors are largely conserved between S. cerevisiae and K. lactis.
Isoform protein-binding differences track with structural differences
As an independent way to address whether differential protein binding to pairs of same-gene isoforms could explain some of the structural variation at their 3’ ends, we overlaid genome-scale 3’UTR protein-binding data (Freeberg et al., 2013) onto our isoform structure data. We paid particular attention to protein binding within the non-overlapping region unique to the longer of two isoforms in each pair. Most isoform pairs for which the observed protein binding occurred within sequences common to both isoforms are structurally similar (ΔR < 0.1 for the isoform pair vs same-isoform replicates; see methods; Figure 5A). By contrast, > 70% of all isoform pairs with protein binding to the sequence unique to the longer isoform exhibit at least some structural variation (ΔR > 0.1; P = 3.6 × 10−8). Highly different structures (ΔR > 0.3) are twice as frequent among isoform pairs with protein binding in the non-overlapping region as they are among pairs with protein binding to upstream common sites (Figure 5A). These results provide further evidence for an important role for proteins in governing isoform-specific mRNA 3’UTR structure at sequences proximal to binding sites.
Figure 5. Relationship of isoform-specific protein binding and structure.

(A) Structural similarity of same-gene isoform pairs (compared to structural similarity of same-isoform biological replicates) when protein binding sites are present in common sequences (gray bars) or in the extra sequence unique to the longer of the two isoforms (black bars). ΔR is a measure of the difference in reactivity profiles of two same-gene isoforms compared to the biological replicates of those isoforms (see methods); small ΔR values (≤ 0.1) indicate that the two same-gene isoforms are structurally similar, while larger ΔR values (> 0.3) are indicative of widespread structural differences.
(B) The sequence unique to the longer of two 3’ isoforms is more likely to be evolutionarily conserved when the isoforms are structurally different. For pairs of isoforms with similar folding (left, ΔR < 0.1), the extra sequence unique to the longer isoform is more frequently non-conserved (median PhastCons score in the sequence element < 0.33, black bar) than conserved (median PhastCons score in element > 0.67, gray bar). For structurally dissimilar isoform pairs (right, ΔR > 0.3), this sequence is more frequently conserved than non-conserved.
(C) CLIP-READS schematic. Exponentially growing cells are irradiated with UV light (254 nm) to crosslink RNA-protein complexes, after which the protein of interest is immunoprecipitated from the extract. Immunopurified protein:mRNA complexes are treated with proteinase K to digest bound proteins and mRNAs are captured on oligo(dT) beads. This mRNA population is subjected to isoform-specific deep sequencing by the 3’ READS method (Jin et al., 2015), which allows identification and quantification of the individual mRNA 3’ isoforms bound to the protein of interest (in this example, Pab1) in vivo. An input sample consisting of a portion of the same extract is also subjected to deep sequencing in parallel to the CLIP-READS sample. Isoform frequencies obtained for the CLIP-READS sample are then divided by frequencies obtained in the input sample to arrive at isoform-specific protein binding levels.
(D) Isoform-specific Pab1 binding exhibits considerable variation. Expression-corrected relative Pab1 binding levels for isoforms in two representative genes, with the value of 1 representing the median expression-corrected Pab1 binding for the entire dataset of > 25,000 isoforms.
(E) Correlation of differential Pab1 binding with extensive structural differences in same-gene isoforms ≤ 20 nt apart.
Sequences between isoform 3’ endpoints are more frequently conserved for structurally distinct isoform pairs
Biologically important protein binding sites are likely to be maintained by selective pressure. Drawing upon the published database of nucleotide conservation for every yeast genomic position (Siepel et al., 2005), we examined pairs of 3’ isoforms for the degree to which the non-overlapping sequence between the isoform endpoints is conserved across evolution in yeast. For pairs of structurally similar (ΔR < 0.1) same-gene isoforms, non-conserved nucleotides predominate in the non-overlapping sequence unique to the longer isoform (Figure 5B, left side; Figure S4A). Conversely, for isoform pairs with different structures (ΔR > 0.3), this non-overlapping sequence is more frequently conserved across evolution, suggesting that maintenance of the structural differences between same-gene isoforms is biologically important (Figure 5B, right side; Figure S4A).
A transcriptome-wide, isoform-specific method to assay protein binding reveals variations in Pab1 association that correlate with structural differences
Because the poly(A)-binding protein (Pab1) binds poly(A) tails and potentially other A-rich sequences found near 3’ ends, we developed the CLIP-READS technique to measure isoform-specific binding of Pab1 in vivo (Figure 5C). In this method, yeast cells are UV irradiated to generate covalent adducts between proteins and mRNAs, epitope-tagged Pab1-RNA complexes are immunoprecipitated from cellular extracts, and isoform-specific libraries are generated via 3’ READS (Jin et al., 2015). Three biological replicates exhibited excellent reproducibility (R = 0.84 to 0.96; Figure S4 B and C) and a good correlation (R = 0.55) to a non-isoform-specific Pab1 dataset (Tuck and Tollervey, 2013), despite considerable differences in methodologies, strain genotypes, and growth conditions (Figure S4D).
Expression-corrected, isoform-specific Pab1 binding can vary several-fold in same-gene isoforms (Figure 5D), and ranges over nearly two orders of magnitude across the transcriptome (Figure S4E). We explored the relationship between relative Pab1 binding and structural differences within pairs of same-gene isoforms (Figure 5E). Same-gene isoform pairs ≤ 20 nt apart that exhibit ≥ 2-fold differential Pab1 binding are much more likely to exhibit substantial differences in DMS reactivity profiles, and this disparity in Pab1 binding is directly correlated with the extent of the structural differences between these isoforms (R = 0.45; P = 9.4 × 10−5; Figure 5E). No such correlation is seen for same-gene isoforms spaced > 20 nt apart. Thus, differential binding of Pab1 to same-gene isoforms is likely to contribute significantly to structural differences among isoform pairs that are ≤ 20 nt apart, presumably because sequences immediately adjacent to the poly(A) tail affect Pab1 binding.
A structural motif associated with transcript stabilization and its biological relevance
We previously showed that same-gene mRNA 3’ isoforms can have different half-lives and provided suggestive, but not direct, evidence that this was due to structural differences (Geisberg et al., 2014). By combining DMS reactivity and half-life measurements (Geisberg et al., 2014) of individual isoforms, we now identify a structural motif in which two distinct short regions at the 3’ terminus of the mRNA are significantly associated with transcript stabilization. Predicted single-strandedness between 24 and 18 nt upstream of the poly(A) addition site and double-strandedness between the 8th and 17th positions in the poly(A) tail are correlated both individually and jointly with increased transcript stability (P = 6.3 × 10−5 for poly(A) positions +8 to +16 and 4.1 × 10−4 for positions −24 to −18) (Figures 6A, S5A). The median half-life is < 23 minutes for isoforms with 0 to 3 positions matching over both regions of the structural motif, but 32 minutes for isoforms with 8 or more matching positions (P = 4.3 × 10−9 ; Figure 6A). Pab1 binding is also inversely correlated with isoform half-life (Figure S5B), and the combination of low Pab1 binding with the presence of this structural motif is even more predictive of isoform stability (Figure S5C). The important roles of both the structural motif and reduced Pab1 binding do not exclude potential contributions to mRNA stability from RNA-binding proteins, RNA modifications, and interactions with other nucleic acids (Aw et al., 2016; Lu et al., 2016).
Figure 6. Relationship of isoform properties with stability.

(A) A two-part structural motif consisting of a predicted unstructured region (−24 to −18) and a double-stranded region in the poly(A) tail (+8 to +16) correlates with increased isoform stability.
(B) Relationship between half-life, evolutionary conservation and predicted structural character in sequence windows near the poly(A) site. +1 is the first A of the poly(A) tail; −1 is the last nt before the poly(A) tail. Blue bars denote the subset of isoforms with unstructured sequences (≥ 6 predicted unstructured residues) in the indicated sequence window, while red bars denote isoforms in which this sequence is structured (≥ 6 predicted structured residues).
To provide independent evidence that this structural motif is functionally relevant, we examined the evolutionary conservation of 7-nt windows upstream of the poly(A) tail with respect to whether residues within these windows are structured (non-reactive to DMS) or unstructured (DMS reactive). Interestingly, the −24 to −18 region is unique in that it is more conserved when it is structured than when it is unstructured (Figure 6B, left). Lack of structure in the −24 to −18 region is linked to increased mRNA stability (Figure 6B, right). Conversely, structure in this 7-nt stretch is associated with greater evolutionary conservation and is linked with increased mRNA turnover. As structured (non-reactive) residues often reflect association with RNA-binding proteins, we suggest that there is evolutionary pressure for RNA-binding proteins to interact with this region and facilitate mRNA decay.
DISCUSSION
DREADS, SHREADS, and CLIP-READS, transcriptome-scale methods for determining structure and protein association of 3’ mRNA isoforms
The DREADS and SHREADS techniques provide transcriptome-scale analysis of 3’ mRNA structure at the level of individual isoforms. In contrast, previous genome-scale methods for RNA structure analysis obtain data at the level of genes, without regard for differences among individual isoforms. Because a DMS- or NAI-modified base blocks the progress of reverse transcriptase, the sequence from a modified mRNA molecule reflects a single modified nucleotide that is closest to the poly(A) tail. As the DMS or NAI concentration is kept low and reaction times short, the reactivity profile of a given isoform represents an average of all molecules of that isoform. An alternate method with the potential to study isoform-specific structure uses the TGIRT reverse transcriptase that can read through DMS modifications and hence can permit the structural analysis of single mRNA molecules (Wu and Bartel, 2017; Zubradt et al., 2017). However, as TGIRT-mediated read-through of DMS modifications is very inefficient, this enzyme is best suited for analyzing the structure of short regions of RNA in close proximity to the 3’ end. In contrast, DREADS AND SHREADS provide structural information at least 300 nt from the poly(A) tail.
CLIP-READS is a method for measuring protein binding to individual 3’ mRNA isoforms on a transcriptome scale. In contrast, conventional methods for measuring mRNA-protein interactions rely on fragmentation of RNA and do not provide 3’ isoform specificity. In addition, CLIP-READS does not require the use of ribonucleotide analogs such as 4-thiouridine, which can alter mRNA structure. CLIP-READS measures the relative protein binding level for each 3’ isoform, but it does not identify the actual binding site of the associated protein. In yeast, ≥1,000 proteins (~20% of all protein-coding genes) have been identified in systematic screens for mRNA-binding proteins (Mitchell et al., 2013; Beckmann et al., 2015). For the vast majority of these polypeptides, binding specificities and biological functions in the context of mRNA binding are unknown. CLIP-READS, in conjunction with conventional methods to determine binding sites of these proteins, should allow one to draw more precise structure-function parallels and to gain insights into isoform-specific functions.
Closely related 3’ mRNA isoforms exhibit structural differences over an extended region
DREADS analysis reveals a remarkable diversity of DMS reactivity profiles for 3’ mRNA isoforms arising from the same gene. Strikingly, such diversity of mRNA structures occurs for many very closely spaced isoforms, including some differing by a single nucleotide. Likewise, nucleotide substitutions in the region between closely-related isoforms can also cause extensive isoform-specific structural changes. In both cases, this wide range of structures is demonstrated directly from the DMS data itself, without dependence on any particular computational algorithm for structure prediction. Furthermore, these structural differences often extend hundreds of nucleotides upstream of the poly(A) site. This remarkable structural diversity among isoforms is confirmed by SHREADS analysis, which involves an orthogonal structural probe.
Our demonstration that very subtle changes at 3’ termini of mRNAs can exert profound effects on structure at a considerable distance is conceptually novel and very unexpected. In vivo, subtle changes within an RNA can alter the local structure, inhibit protein binding to a target site, or possibly cause unfolding of a stable tertiary structure. The larger-scale structural differences we observe among closely-related isoforms are distinct from local effects, nor can they be due to global unfolding of the 3’UTR, because many residues throughout the 3’UTRs of these isoforms are protected from DMS. Furthermore, the experiments here are performed in vivo and hence are strongly influenced by the entire constellation of RNA-binding proteins in the cell (see below), unlike experiments carried out in vitro. Finally, it is important to note that the individual 3’ mRNA isoforms are not mutated versions of a wild-type entity, but rather true physiological entities that contribute to biological phenotypes in living cells
Differential protein binding is a major factor underlying isoform-specific structural differences
In principle, A and C residues in RNA are protected from DMS modification either because they exist in base-paired form and/or they are bound by trans-acting factors. Such trans-acting factors are almost certainly RNA-binding proteins, although it is formally possible that separate RNA molecules, yet-to-be identified ligands, or other small molecules may be involved. However, trans-acting RNAs rare in yeast (Aw et al., 2016) and hence are unlikely to play a significant role in isoform structural diversity.
Several lines of evidence indicate that differential protein binding is a major factor for isoform-specificity of DMS-reactivity profiles. First, purified mRNA isoforms differing by one or a few nucleotides rarely show meaningful differences in DMS profiles in vitro, whereas the same isoforms are much more likely to show large differences in vivo. Thus, intrinsic RNA folding per se cannot account for the structural diversity of closely related isoforms. Second, identical mRNA isoforms exhibit different DMS profiles when transplanted into another yeast species, demonstrating the role of host-specific factors. Third, previously documented protein binding (Freeberg et al., 2013) in regions between isoform pairs is strongly associated with structural differences between the corresponding isoforms. Fourth and most directly, Pab1 displays striking isoform-specific binding, and differences in Pab1 binding levels are strongly correlated with differences in isoform structure.
As closely-spaced isoforms can exhibit very different reactivity profiles over hundreds of nucleotides, protein-binding sites located well upstream of poly(A) sites -- and hence common to both isoforms -- can be structurally distinct. These structural differences in shared sequences might reflect binding of different proteins (see below). Many RNA-binding proteins, including Roquin, STAU1, SLBP, and ZNF326, do not simply bind to an optimal sequence, but rather require a precise three-dimensional configuration of select mRNA sequences for efficient binding (Tan et al., 2013; Schlundt et al., 2014; Sugimoto et al., 2015; Dominguez et al., 2018). Thus, long-range structural differences between same-gene isoforms may reflect differences associated proteins and/or in tertiary structure. By either of these mechanisms, individual 3’ mRNA isoforms can be remarkably different structural entities in vivo.
Potential mechanisms for high structural diversity of 3’ mRNA isoforms
In this paper, we use the word “structure” to refer to the pattern of DMS accessibility, which is determined both by intrinsic RNA folding and by the influence of proteins. Intrinsic RNA folding and protein binding are highly intertwined molecular events that mutually influence each other. The secondary and tertiary structure of an RNA plays a key role in protein binding, and conversely, protein binding affects RNA structure. Thus, the DMS reactivity profile represents the combined effects and mutual influences of protein binding and RNA folding that occur throughout the region of investigation.
Ultimately, structural differences between two isoforms must be due to nucleotides adjacent to the poly(A) tails that are unique to the longer isoform and absent in the shorter one (Figure 7). These structural differences could be due to the extra sequences per se or to the particular nucleotides forming the junction with the poly(A) tail. It is also possible that isoform-specific differences in poly(A) tail length could affect isoform structures. In vitro, differences of only a few nucleotides generally have little effect on intrinsic RNA folding, implying that pronounced structural differences in vivo between closely spaced isoforms stem from differential protein interactions.
Figure 7. Potential models for differential structures of 3’ mRNA isoforms in vivo.

The model depicts a subset of all same-gene isoforms that exhibit extensive structural differences in vivo; for simplicity, many common structural parts are not shown. Identically positioned residues with varying DMS reactivities are circled and numbered. For each panel, a representation of the DMS reactivity pattern across the five numbered positions is depicted in the upper right-hand corner, with DMS-reactive positions in green and unreactive positions in black. Left panel: In vitro, extra sequences (red N’s) or mutations (blue asterisks) near the poly(A) site have no effect on RNA folding or isoform reactivity profiles. In an environment devoid of RNA-binding proteins, the three isoforms possess identical structures. Right panel: In vivo, the presence of mutations or additional sequences near poly(A) sites results in differential association of Pab1 and/or other RNA-binding proteins that alter RNA-RNA interactions within the mRNA that in turn can influence protein binding, leading to a cascade of differential structural and binding effects over an extensive region upstream of the poly(A) tail. For isoforms that are not closely spaced, the cascade of structural effects may also stem from intrinsic differences in RNA-RNA interactions.
A few differing nucleotides between isoforms are unlikely to directly affect protein binding throughout the 3’ UTR, but they can influence the efficiency of protein interaction with the 3’ end. For example, Pab1 binds the poly(A) tail, but the residues immediately upstream, whether by their sequence per se or by influence on local structure (e.g. hybridization of U-rich sequences to the poly(A) tail and adjacent region), can affect the efficiency of Pab1 binding (Figure 7). More generally, binding of any protein near the 3’ end might require, or be inhibited by, the extra sequences in the longer isoform. Alternatively, the extra sequence could interact with an upstream protein binding site common to both isoforms, thereby resulting in differential protein binding.
Figure 7 illustrates ideas for how differential protein binding at the 3’ end can bring about much larger-scale structural changes. In principle, Pab1 binding differences influenced by the particular nucleotides adjacent to the poly(A) tail can alter RNA-RNA interactions within the mRNA. This in turn will influence protein binding (via protein-RNA and/or protein-protein interactions), leading to a cascade of differential structural and binding effects much further from the poly(A) tail where Pab1 actually binds. In general, any RNA-binding protein whose interaction is affected by the region between two isoforms could cause a domino effect on RNA structure and protein binding, resulting in isoform-specific differences in structure over an extended region of the RNA.
Functional importance and evolutionary implications of isoform structural diversity
3’ mRNA isoforms of the same gene can be functionally different, and there is growing recognition of the importance of 3’ UTR elements and structures in mRNA stability (Geisberg et al., 2014; Gupta et al., 2014), regulation of translation (Floor and Doudna, 2016), mRNA processing, cellular localization, and macromolecular assembly (Berkovits and Mayr, 2015; Mitra et al., 2015; Taliaferro et al., 2016; Wu and Bartel, 2017). Likewise, structural elements highly conserved in other RNA species (e.g. lncRNA, snoRNA, etc.) also play critical roles in biological processes ranging from transcriptional regulation to cell signaling and oncogenesis (Brown et al., 2014; Li et al., 2016).
The remarkable structural diversity observed here among 3’ isoforms, including some differing by only one or a few nucleotides, is likely to be biologically important. In particular, sequences between the 3’ endpoints of structurally different isoforms display greater evolutionary conservation than comparable sequences of structurally similar isoforms. In addition, the −24 to −18 region of the motif linked to mRNA half-lives is more conserved when it is structured than when it is unstructured. Structure in this region is correlated with mRNA instability, suggestive of evolutionary selection of interactions with RNA-binding proteins that facilitate mRNA decay. This is consistent with the role of 3’ UTR mRNA-binding proteins as negative regulators of transcript stability (Schoenberg and Maquat, 2012). Structural differences among closely-related isoforms are linked to differences in isoform stability, but it is unclear whether the function of these evolutionarily conserved residues is influencing mRNA stability per se and/or some other process.
In accord with the remarkable structural diversity of native 3’ isoforms that differ by only one or a few nucleotides, individual or closely clustered mutations near the poly(A) site can dramatically affect isoform structure over an extended region of the mRNA. Our in vitro experiments indicate that the intrinsic RNA structures of closely related isoforms are generally indistinguishable, strongly suggesting that subtle mutational changes would also have minimal effects on intrinsic RNA structure. Thus, the large-scale changes in DMS reactivity profiles that arise from subtle mutations are very likely due to multiple changes in protein binding and RNA structure. We suggest that some mutations that arise during evolution can cause major structural differences (intrinsic or due to protein binding) in the 3’ UTRs, with possible functional consequences contributing to biological phenotypes.
STAR METHODS
METHODS
Cell culture for DREADS and RNA isolation
Two independent 50 ml cultures of yeast strain JGY2000 (Moqtaderi et al., 2013) were grown at 30°C in YPD medium to OD600 = 0.5. Duplicate 50 ml cultures of strains NCYC 2572 (D. hansenii) and CliB 209 (K. lactis)(Hughes et al., 2012) were grown in foreign yeast medium (1% peptone, 1.5% yeast extract, 2% glucose, 1x SC mix [100mg/l each of arginine, aspartic acid, glutamic acid, isoleucine, lysine, methionine, phenylalanine; 800mg/l serine; 400mg/l threonine; 20mg/l tyrosine; 300mg/l valine], 100mg/l histidine, 100mg/l leucine, 50mg/l tryptophan, 25mg/l adenine and 25mg/l uracil) to OD600 = 0.5. Strains JYAC2 and JYAC7, harboring artificial chromosomes derived from K. lactis and D. hansenii, respectively (Hughes et al., 2012), were grown in duplicate in foreign yeast medium at 30°C without tryptophan and uracil to OD600 = 0.5.
For each of these cultures, cells were harvested and mixed with 4 × 106 of S. pombe cells as a spike-in control. Cells were then processed and treated with DMS as described below.
DMS treatment
15 ml of harvested culture were treated with 500μl DMS for 2 minutes at room temperature as described (Rouskin et al., 2014). At this DMS concentration, we observed no meaningful bias in reactivity as a function of distance from the poly(A) site. DMS was quenched with 30 ml stop solution (30% beta-mercaptoethanol, 25% isoamyl alcohol)(Rouskin et al., 2014) and cells were collected by centrifugation as above. Quenching was repeated once, and cells were washed with 1 ml of water prior to freezing at −80°C.
NAI treatment
25 ml cells were resuspended in a total volume of 380μl of pre-warmed YPD. Cells were incubated with either 20μl of 2M NAI (in DMSO; +NAI sample) or with 20ml DMSO (−NAI control) at 30°C for ~ 3 minutes, centrifuged and washed twice with water prior to freezing at −80°C.
RNA isolation
20 ml of the harvested culture were centrifuged at 3,000 rpm (1,800 × g) in a tabletop centrifuge for 2 minutes at 4°C. The collected cells were washed with distilled water, recentrifuged and frozen at −80°C. Total RNA was isolated from the frozen cell pellets by the hot acid phenol method (Collart and Oliviero, 1993). RNA obtained was further purified using Qiagen RNeasy columns with DNAse I treatment according to the manufacturer’s instructions.
DREADS/SHREADS library construction
25 μg mRNA was purified from total RNA samples with oligo dT(25) magnetic beads (NEB), and was then bound to beads pre-coated with a U45T5 biotinylated mRNA capture oligonucleotide (Jin et al., 2015). Immobilized mRNA tails were trimmed with RNase H (NEB) to generate shortened poly(A) sequences averaging ~5 A’s as described (Jin et al., 2015). After ligation of 5’ pre-adenylated adapter A to the trimmed mRNA 3’ ends, mRNAs were reverse transcribed with Superscript III (Life Technologies) using primer B (Jin et al., 2015). cDNAs were ligated with T4 DNA Ligase (NEB) to splint adapter C, and the single-stranded DNA products (110 – 500 nt) were purified on 6% polyacrylamide-urea gels. DNA fragments were amplified by 18 cycles of PCR with barcoded oligonucleotides as described (Jin et al., 2015). Amplified libraries were purified with SizeSelector-I beads (Aline Biosciences) as described (Jin et al., 2015). Purified barcoded libraries were then examined and quantified via Bioanalyzer (Agilent Technologies), pooled, and further gel purified by 8% PAGE.
Hybridization capture of CHS7 and SOD1 RNAs
We constructed plasmid libraries that contained the CHS7 and SOD1 genomic loci (from 500 nt upstream of the ATG to 400 nt downstream of the stop codon) randomized in the 3’ UTRs at positions +78 to +81 (CDS1) and +66 to +70 (SOD1) relative to the stop codon (full details will be described elsewhere). Plasmid libraries were separately transformed into S. cerevisiae JGY2000, and the transformants were grown selectively in casamino acid medium lacking uracil, diluted into 50 ml of YPD and grown at 30°C to OD600 = 0.5 as described above. DMS treatment, RNA isolation and DREADS were performed as described. 500 ng of pooled barcoded DREADS libraries were blocked with 5 μg Cot-1 DNA (Invitrogen) and 1 μl each of p5 and p7 custom blocker small RNA (IDT). Hybridization of locked nucleic acid probes specific to CHS7 and SOD1 (Exiqon) was carried out using the xGen hybridization and wash kit (IDT) according to the manufacturer’s instructions, except that the hybridization and was carried out at 55°C for 18 hours. Subsequent capture, washes and purification were performed exactly as described in the kit’s user manual, with the exception of all the steps that require incubation at 65°C were done at 55°C. Captured fragments were eluted from the beads in 50 μl EB for 5 minutes 75°C and amplified by 15 cycles of PCR with Illumina p5 and p7 primers. Libraries were purified by 8% PAGE and subjected to deep sequencing.
In vitro DMS modification of refolded RNA
25 μg of RNA (QIAGEN purified and treated with DNase I) was resuspended in 100 μl of H2O, denatured for 2 minutes at 95°C and snap-cooled on ice. After the addition of 20 μl of 6X renaturation buffer (60 mM Tris pH 8.0, 900 mM KCl, 36 mM MgCl2), the RNA was allowed to re-fold for 30 minutes at 30°C. 4 μl of DMS were then added, and the samples were incubated for 2 minutes at 30°C. DMS was quenched by the addition of 360 μl of 2-mercaptoethanol and samples were immediately placed on ice. The RNA was precipitated by the addition of 120 μl of 3M sodium acetate, 650 μl of isopropanol, and 2 μl of GlycoBlue. Samples were collected by centrifugation at 20,000 × g for 30 minutes at 4°C, washed with 70% ethanol and air-dried. Pellets were resuspended in 100 μl of H2O and frozen at −80°C. Non-DMS-treated samples were processed in the exact same way, except that 4 ml of DMS was replaced by an equal volume of 1 × renaturation buffer.
CLIP-READS
CLIP-READS was performed in triplicate, with two biological replicates (IP A and IP B) of Pab1 tagged with three copies of the myc epitope and one replicate of an isogenic strain containing Pab1 tagged with three copies of the V5 epitope (IP C)(Moqtaderi and Struhl, 2008). Cultures were grown to OD600 = 0.5 in YPD. Cells were resuspended in 40 ml of fresh pre-warmed YPD, transferred to 15-cm petri dishes, and subjected to 1.2 J/cm2 UV irradiation at 254 nm in a Stratalinker (Agilent). Cells were collected by centrifugation, and extracts were immunoprecipitated with either anti-myc (9E10; SantaCruz Biotechnology) or anti-V5 (Abd Serotec) antibodies for 4 hours at 4°C. Antibody:RNA:protein complexes were collected by incubation with magnetic protein G-Dynabeads (Life Technologies) and were extensively washed with FA low detergent buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.2% Triton X-100, 0.02% sodium deoxycholate, 0.02% sodium dodecyl sulphate) prior to elution. Protein:RNA complexes were treated with 400 mg proteinase K, and RNA was purified by phenol-chloroform extraction and ethanol precipitation. 3’ ends of immunoprecipitated mRNA were quantified by 3’ READS as described (Jin et al., 2015). Separate input samples were prepared by directly subjecting a portion (2%) of the extracts of all three samples to 3’ READS deep sequencing as described (Jin et al., 2015).
Sequencing
Except for NCYC 2572, CliB 209, JYAC2 and JYAC7 DREADS libraries (see below), all sequencing was performed on an Illumina HiSeq 2500 instrument. S. cerevisiae (JGY2000) DREADS multiplexed libraries were pair-end sequenced twice (once in a single lane of a rapid flow cell and once across an entire 2-lane rapid flow cell) and combined. Multiplexed, in vitro re-folded S. cerevisiae (JGY2000) DREADS libraries were pair-end sequenced on an individual 2-lane rapid flow cell. CLIP-READS samples and inputs were single-end sequenced in a single lane on a rapid flow cell. NCYC 2572, CliB 209, JYAC2 and JYAC7 libraries were pair-end sequenced on individual NextSeq High 2 × 75 bp full flow cells on an Illumina NextSeq 500 platform.
Analysis of DREADS/SHREADS sequence data
De-multiplexed individual libraries were analyzed as follows: Before mapping, for the R1 end of the paired sequence reads, the four random nucleotides corresponding to those added during library construction were removed from the beginning of the sequence. (Due to ligation bias, we observed strong preferences in the distribution of the 4-mers ligated to the enzymatically shortened poly(A) sequence at R1 ends; these distributions were essentially identical in the no-DMS and DMS-treated samples). Any initial T residues at the 5’ end of the sequence read (corresponding to the poly(A) tail and any genomically encoded A residues at the 3’ end of the RNA) were first counted and then removed. An integer corresponding to the number of initial T nucleotides was appended to both the R1 and R2 read identifiers for future reference. Reads lacking initial T nucleotides, reads with ambiguous bases, and reads with fewer than 9 nt remaining after removal of Ts were excluded from further analysis. For all others, the first 17 nt were used for mapping, to maximize unique mapping while minimizing overlap with the primer on the far side of short sequences. For the R2 member of the read pair, the sequences corresponding to the four possible 4-mers were removed, and the sequence from the second nt to the 18th were retained for mapping. In our experience, the first nt, representing the final nt added by reverse transcriptase during library construction, is frequently a mismatch in DMS-treated samples; we therefore used the following nt as the starting point for mapping and compensated for this shift after mapping. Mapping of the remaining paired sequence reads to a mixed reference genome consisting of the S. cerevisiae and S. pombe genomes was performed using bowtie (Langmead et al., 2009), accepting unique genomic matches only and allowing no more than one mismatched nt. Next, uniquely mapped pairs were screened as follows to remove any that were not demonstrably from poly(A) addition sites: we discarded any R1 read that did not have more initial Ts than the number of genomically encoded As at the corresponding position. The 5’ (R2) boundary for each mapped pair was adjusted backwards two nt: one to compensate for our having started the mapping from the second nt, and another to identify the potentially DMS-modified position. The total number of completely processed reads was scaled by a small multiplier so as to give equal numbers of reads in DMS and non-DMS samples. This scaling factor was not large; for example, for the two replicates of the main data sets, the initial un-scaled poly(A)-RNA-derived read counts for the DMS sample and the untreated control differed by less than 10%.
For every gene, we thus identified all 3’ RNA endpoints in the sequenced library, and for each of these 3’ endpoints, we tabulated all corresponding paired 5’ fragment ends representing the endpoints of the reverse transcription reactions. Frequencies of these associated 5’ endpoints for untreated and DMS-treated cells were compared to identify positions with DMS-induced modifications capable of inhibiting the progress of reverse transcriptase.
For D. hansenii native and JYAC7 (containing the D. hansenii YAC) strains, mapping was performed using bowtie to a combined reference file consisting of the D. hansenii Deha2, S. cerevisiae SacCer3, and S. pombe EF2 genomes. For K. lactis native and JYAC2 (containing the K. lactis YAC) strains, mapping was performed using bowtie to a combined reference file consisting of the K. lactis Klla0, S. cerevisiae SacCer3, and S. pombe EF2 genomes. To eliminate any ambiguity arising from possible cross-species-matching of homologous genes in the YAC strains, only positions mapping uniquely over the entire combined reference genome were considered. All subsequent data preparation and normalization steps were performed as described above.
Computation of net reads, isoform reactivity profiles, and Winsorized reactivity values
For every 3’ isoform, we tabulated the number of RT stops at every A and C position in a 300nt window upstream of the cleavage/polyadenylation site. For every A or C position in this 300nt window, the net read count was obtained by subtracting the count in the control sample from that of the corresponding position in the DMS sample. Negative values were assigned a value of 0.
The reactivity profile of an individual isoform consists of the net read counts at all A and C positions in a 300nt window upstream of the poly(A) tail. For subsequent analyses, we considered only those isoforms with a minimum read count, typically 500 reads per replicate. Read counts at each position were normalized to the residue with the highest read count, which was defined as 100%. For every isoform the net DMS signal of the highest peak is highly related to the expression level (total sequence reads for that isoform); with respect to the median ratio, >75% of the isoforms give ratios ±40%, and >95% of the isoforms within a factor of 2. Pearson correlation coefficients between any two isoform reactivity profiles were computed by correlating position-specific net reads for each isoform over the common window that the two isoforms share.
To convert net reads into Winsorized reactivity values for use in structure prediction, the average of the net reads values of the top 15% of positions was assigned a value of 1, and the net reads at all other A or C positions in the profile were linearly scaled to this top value (Hastings et al., 1947).
Computation of NAI net reads profiles
For the SHREADS experiment, net NAI reads were determined by background subtraction, as above for the DMS samples, except that all four positions (A,C,G, and U) were considered instead of only A and C.
Calculation of Pearson correlations and ΔR values for isoform reactivity profiles
For structural comparison of same-gene isoform pairs, we treated the two biological replicates separately. We computed Pearson correlation coefficients for reactivity profiles of biological replicates of each is (RIsoform 1 rep. A vs Isoform 1 rep. B, RIsoform 2 rep. A vs Isoform 2 rep. B) as well as the correlation coefficients for same-gene isoform pair reactivity profiles (RIsoform 1 rep. A vs Isoform 2 rep. B , RIsoform 1 rep. B vs Isoform 2 rep.A). Distributions of these correlation coefficients are plotted in Figure 2B, 2D, S2, and S3; a subset of these values is also used in Figures 2B and4A.
In Figures 2E, 5A and 5B, ΔR represents the extent to which reactivity profiles of two same-gene isoforms differ from each other and is equal to the average correlation of biological replicate reactivity profiles minus the average correlation of same-gene isoform pair reactivity profiles (ΔR = Rreplicates - Rpaired isoforms). ΔR values approach zero for same-gene isoforms whose reactivity profiles are similar, while a large ΔR indicates that the reactivity profiles of the isoform pair exhibit extensive differences.
For Figure 4B, ΔR represents the difference between the correlation of a given isoform’s reactivity profiles in biological replicates and the same isoform’s correlation of reactivity profiles between native species and when expressed in S. cerevisiae (ΔR = Rreplicates – Rcross-species). ΔR values are small for isoforms whose reactivity profiles are similar when transplanted, while large ΔR’s indicate that the reactivity profiles of the isoform exhibit extensive structural differences in the two different hosts.
Comparison to the gPAR-CLIP dataset of (Freeberg et al., 2013)
Protein binding sites were mapped onto isoform pairs with inter-isoform spacing of ≤ 30 nt by requiring that (1) the 3’ ends of the binding sites exactly match or be downstream of the cleavage/poly(A) site of the longer isoform, (2) the 5’ ends be at or upstream of the cleavage/poly(A) site of the shorter isoform. Significance of the differences in the two distributions was assessed using the Mann-Whitney U test.
Structure prediction
Folding of RNAs was performed with RNAStructure (Reuter and Mathews, 2010). Unless otherwise stated, all RNAs were assumed to contain a 30 nt poly(A) tail (Subtelny et al., 2014) and were folded at 30°C with a maximum base pairing window distance of 200 nucleotides and incorporating DMS reactivity values (see above).
Evolutionary conservation analysis
Individual nucleotide PhastCons scores (Siepel et al., 2005) converted to SacCer3 were looked up and assigned for every nucleotide contained within the sequence window of interest. For the analysis in Figure 5B, a conservation score was computed for each region by taking the median of all PhastCons scores within that window. In Figure S4, all nucleotide PhastCons scores are treated as individual data points and are displayed for all closely spaced (≤ 20 nt) isoform pairs. Individual and median PhastCons scores were classified as conserved (score ≥ 0.67), non-conserved (score ≤ 0.33), or neither (scores > 0.33 and < 0.67).
Functional evolutionary (YAC) analysis
Due to the limited number of isoforms contained in YAC sequences, we reduced our minimum total net reads requirement for each isoform to 25. As many of these isoforms are rather poorly expressed, the data are inherently quite noisy and individual reactivity profiles are of little value for structure prediction. However, increasing the sample size at the expense of data quality allows us to draw statistically meaningful conclusions that are otherwise impossible to draw with only a handful of highly expressed isoforms.
For every isoform with 25 or more reads (both in the native species and when expressed on a YAC), we computed the average correlation of biological replicate reactivity profiles (Rreplicates = average correlation of same-species biological replicates A vs B). We also computed the average cross-species correlation of the same isoform (Rcross-species) by averaging the correlation of reactivity profiles of native species replicate A vs S. cerevisiae replicate B and native species replicate B vs S. cerevisiae replicate A. We then calculated ΔR by subtracting Rcross-species from Rreplicates (ΔR = Rreplicates - Rcross-species). ΔR represents a measure by which isoform reactivity profiles differ when expressed in native species (D. hansenii or K. lactis) as compared to when transplanted into S. cerevisiae. For isoforms whose reactivity profiles are very similar in both native species and S. cerevisiae, ΔR approaches zero. Conversely, isoforms whose reactivity profiles differ greatly when expressed in native species as compared to S. cerevisiae will exhibit a large ΔR. Isoforms bearing negative ΔR values represent experimental error. The extent to which isoforms with positive ΔR values outnumber those with negative ΔR’s determines the significance, which is calculated by Fisher’s Exact test.
CLIP-READS data analysis
Single-end sequence reads were prepared for mapping by removal of initial Ts as described for above for DREADS. Reads were mapped to a combined reference genome consisting of S. cerevisiae SacCer3 and S. pombe EF2, allowing 1 mismatch and considering only uniquely mapped reads. Mapped reads not representing bona fide polyadenylated transcripts were discarded as described above. Read counts were tabulated for every 3’ isoform in all CLIP-READS and input samples.
Isoforms with ≥ 10 reads in each of the three CLIP-READS replicates and each of the three corresponding input samples were selected for further analysis. Individual isoform read counts in CLIP-READS replicates were normalized relative to true poly(A) read totals. Similarly, read counts for the same isoforms in input samples were normalized relative to true poly(A) read totals. Averaged CLIP-READS read values for each isoform were then divided by the corresponding averaged input read values in order to obtain expression-corrected relative Pab1 binding.
Motif scoring
For each isoform, we obtained three different scores: one score for the sub-region −24 to −18 upstream of the cleavage/polyadenylation site, another for the sequence from +8 to +16 of the poly(A) tail, and the third for the overall motif. For the −24 to −18 sub-region, the isoform’s score is the sum of all predicted single-stranded nts within this stretch (Figure S5A, top). For the +8 to +16 section of the poly(A) tail, the isoform’s score is the sum of all predicted double-stranded nts between +8 and +16 (Figure S5A, bottom). The overall motif (Figure 6F) is the sum of these two sub-scores.
Because Pab1 binding is inversely correlated with transcript stability (Figure S5B), we assigned every isoform a score from 0 to 16, reflecting the relative absence of Pab1 (16 being the lowest Pab1 binding, 0 being the strongest). For each isoform, this Pab1 binding value was added to the overall structural motif score to arrive at a combined Pab1 binding/structural motif score (Figure S5C).
Oligonucleotides
Adapter A: 5′-/5rApp/NNNNGATCGTCGGACTGTAGAACTCTGA
AC/3ddC/-3′
Primer B: 5′ -GTTCAGAGTTCTACAGTCCGACGATC-3′
Splint Adapter: This is a double-stranded adapter where Z is the double-stranded equivalent of N.
5’ – ZZZZTGGAATTCTCGGGTGCCAAGG – 3’
3’ – NNNNNNZZZZACCTTAAGAGCCCACGGTTCC – 5’
Custom p7 blocker small RNA: 5’-
CAAGCAGAAGACGGCATACGAGATXXXXXXGTGACTGGAGTTCCTTGGCACCCGAGAATTCCA-3’
Custom p5 blocker small RNA: 5’-
AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACGAT
C/3CSpC3/-3’
CHS7 locked nucleic acid capture oligo: 5’-
/5Biosg/GT+AATAA+GCA+GG+ACTAT+GAC-3’
SOD1 locked nucleic acid capture oligo: 5’-/5Biosg/AGTAAGCGCAT+CAATT+CAT+TC-3’
Strains
JGY2000, JYAC2, JYAC7, NCYC 2572 and CliB 209 were previously described (Hughes et al., 2012; Geisberg et al., 2014). ZMY506 (Pab1 bearing three copies of the myc epitope at the C-terminus) and ZM516 (Pab1 bearing three copies of the V5 epitope at the C-terminus) were constructed by the PCR tagging and loopout methods (Moqtaderi and Struhl, 2008). Tagged PAB1 loci were verified by sequencing.
JGY2000: MATa RPO21-FRB-Kan fpr1::KanMX RPL13A-FKBP12-HIS his3Δ1 met15Δ0 (Geisberg et al., 2014)
ZMY506: MATa RPO21-FRB-Kan fpr1::KanMX RPL13A-FKBP12-HIS his3Δ1 met15Δ0 PAB1-3myc
ZMY516: MATa RPO21-FRB-Kan fpr1::KanMX RPL13A-FKBP12-HIS his3Δ1 met15Δ0 PAB1-3V5
JYAC2: MATa ura3-52 trp1-289 lys2-1 ade2-1 can1-100 his5 ρ+ ψ+ +143kb YAC (K. lactis chromosome C 339793-482935)
JYAC7: MATa ura3-52 trp1-289 lys2-1 ade2-1 can1-100 his5 ρ+ ψ+ +216kb YAC (D. hansenii chromosome D 1148162-1364529).
Data and software availability
Data sets have been deposited with Gene Expression Omnibus (GEO) under record GSE95788.
Supplementary Material
Highlights.
Methods to determine structures of and protein binding to 3’ mRNA isoforms Closely related mRNA isoforms can differ dramatically in structure and Pab1 binding A structural pattern near the 3’ terminus is associated with mRNA isoform stability Isoform-specific structures, Pab1 binding, and mRNA stability are conserved
ACKNOWLEDGEMENTS
We thank Yi Jin for helpful discussion. This work was supported by a grant to K.S. from the National Institutes of Health (GM30186).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information includes 5 figures and can be found with this article online at https://doi.org/10.1016/j.molcel.2018.08.044.
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aw JG, Shen Y, Wilm A, Sun M, Lim XN, Boon KL, Tapsin S, Chan YS, Tan CP, Sim AY, et al. (2016). In vivo mapping of eukaryotic RNA interactomes reveals principles of higher-order organization and regulation. Mol. Cell 62, 603–617. [DOI] [PubMed] [Google Scholar]
- Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume AM, Schwarzl T, Curk T, Foehr S, Huber W, et al. (2015). The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun. 6, 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkovits BD, and Mayr C (2015). Alternative 3' UTRs act as scaffolds to regulate membrane protein localization. Nature 522, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua PC, Ritchey LE, Su Z, and Assmann SM (2016). Genome-wide analysis of RNA secondary structure. Annu. Rev. Genet. 50, 235–266. [DOI] [PubMed] [Google Scholar]
- Brown JA, Bulkley D, Wang J, Valenstein ML, Yario TA, Steitz TA, and Steitz JA (2014). Structural insights into the stabilization of MALAT1 noncoding RNA by a bipartite triple helix. Nat. Struct. Mol. Biol. 21, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collart MA, and Oliviero S (1993). Preparation of yeast RNA. Curr. Protoc. Mol. Biol. 23, 13 12 11–15. [DOI] [PubMed] [Google Scholar]
- Ding Y, Tang Y, Kwok CK, Zhang Y, Bevilacqua PC, and Assmann SM (2014). In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 505, 696–700. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. (2018). Sequence, Structure, and Context Preferences of Human RNA Binding Proteins. Mol. Cell 70, 854–867 e859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujon B, Sherman D, Fischer G, Durrens P, Casaregola S, Lafontaine I, De Montigny J, Marck C, Neuveglise C, Talla E, et al. (2004). Genome evolution in yeasts. Nature 430, 35–44. [DOI] [PubMed] [Google Scholar]
- Elkon R, Ugalde AP, and Agami R (2013). Alternative cleavage and polyadenylation: extent, regulation and function. Nat. Rev. 14, 496–506. [DOI] [PubMed] [Google Scholar]
- Fang R, Moss WN, Rutenberg-Schoenberg M, and Simon MD (2015). Probing Xist RNA structure in cells using targeted Structure-Seq. PLoS Genet. 11, e1005668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floor SN, and Doudna JA (2016). Tunable protein synthesis by transcript isoforms in human cells. Elife 5, e10921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeberg MA, Han T, Moresco JJ, Kong A, Yang YC, Lu ZJ, Yates JR, and Kim JK (2013). Pervasive and dynamic protein binding sites of the mRNA transcriptome in Saccharomyces cerevisiae. Genome Biol. 14, R13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geisberg JV, Moqtaderi Z, Fan X, Ozsolak F, and Struhl K (2014). Global analysis of mRNA isoform half-lives reveals stabilizing and destabilizing elements in yeast. Cell 156, 812–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta I, Clauder-Munster S, Klaus B, Jarvelin AI, Aiyar RS, Benes V, Wilkening S, Huber W, Pelechano V, and Steinmetz LM (2014). Alternative polyadenylation diversifies post-transcriptional regulation by selective RNA-protein interactions. Mol. Syst. Biol. 10, 719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings C, Mosteller F, Tukey JW, and Winsor CP (1947). Low moments for small samples: a comparative study of order statistics. Ann. Math. Stat. 18, 413–426. [Google Scholar]
- Hoque M, Ji Z, Zheng D, Luo W, Li W, You B, Park JY, Yehia G, and Tian B (2013). Analysis of alternative cleavage and polyadenylation by 3' region extraction and deep sequencing. Nat. Methods 10, 133–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes A, Jin Y, Rando OJ, and Struhl K (2012). A functional evolutionary approach to identify determinants of nucleosome positioning: A unifying model for establishing the genome-wide pattern. Mol. Cell 48, 5–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Geisberg JV, Moqtaderi Z, Ji Z, Hoque M, Tian B, and Struhl K (2015). Mapping 3' mRNA isoforms on a genomic scale. Curr. Protoc. Mol. Biol. 110, 4.23.21–24.23.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Eser U, Struhl K, and Churchman LS (2017). The ground state and evolution of promoter regions directionality. Cell 170, 889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota M, Tran C, and Spitale RC (2015). Progress and challenges for chemical probing of RNA structure inside living cells. Nat. Chem. Biol. 11, 933–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, and Lu X (2013). The emerging roles of 3' untranslated regions in cancer. Cancer Lett. 337, 22–25. [DOI] [PubMed] [Google Scholar]
- Li R, Zhu H, and Luo Y (2016). Understanding the functions of long non-coding RNAs through their higher-order structures. Int. J. Mol. Sci. 17, E685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liebeg A, and Waldsich C (2009). Probing RNA structure within living cells. Methods Enzymol. 468, 219–238. [DOI] [PubMed] [Google Scholar]
- Lu Z, Zhang QC, Lee B, Flynn RA, Smith MA, Robinson JT, Davidovich C, Gooding AR, Goodrich KJ, Mattick JS, et al. (2016). RNA duplex map in living cells reveals higher-order transcriptome structure. Cell 165, 1267–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, and Wagner EJ (2014). CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature 510, 412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr C, and Bartel DP (2009). Widespread shortening of 3'UTRs by alternative cleavage and polyadenylation activates oncogenes in cancer cells. Cell 138, 673–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell SF, Jain S, She M, and Parker R (2013). Global analysis of yeast mRNPs. Nat. Struct. Mol. Biol. 20, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitra M, Johnson EL, and Coller HA (2015). Alternative polyadenylation can regulate post-translational membrane localization. Trends Cell. Mol. Biol. 10, 37–47. [PMC free article] [PubMed] [Google Scholar]
- Moqtaderi Z, and Struhl K (2008). Expanding the repertoire of plasmids for PCR-mediated epitope tagging in yeast. Yeast 25, 287–292. [DOI] [PubMed] [Google Scholar]
- Moqtaderi Z, Geisberg JV, Jin Y, Fan X, and Struhl K (2013). Species-specific factors mediate extensive heterogeneity of mRNA 3' ends in yeasts. Proc. Natl. Acad. Sci. U.S.A. 110, 11073–11078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F, Kapranov P, Foissac S, Kim SW, Fishilevich E, Monaghan AP, John B, and Milos PM (2010). Comprehensive polyadenylation site maps in yeast and human reveal pervasive alternative polyadenylation. Cell 143, 1018–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, and Steinmetz LM (2013). Extensive transcriptional heterogeneity revealed by isoform profiling. Nature 497, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter JS, and Mathews DH (2010). RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouskin S, Zubradt M, Washietl S, Kellis M, and Weissman JS (2014). Genome-wide probing of RNA structure reveals active unfolding of mRNA structures in vivo. Nature 505, 701–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlundt A, Heinz GA, Janowski R, Geerlof A, Stehle R, Heissmeyer V, Niessing D, and Sattler M (2014). Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat. Struct. Mol. Biol. 21, 671–678. [DOI] [PubMed] [Google Scholar]
- Schoenberg DR, and Maquat LE (2012). Regulation of cytoplasmic mRNA decay. Nat. Rev. 13, 246–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherstnev A, Duc C, Cole C, Zacharaki V, Hornyik C, Ozsolak F, Milos PM, Barton GJ, and Simpson GG (2012). Direct sequencing of Arabidopsis thaliana RNA reveals patterns of cleavage and polyadenylation. Nat. Struct. Mol. Biol. 19, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hiller LW, Richards S, et al. (2005). Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15, 1034–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smola MJ, Christy TW, Inoue K, Nicholson CO, Friedersdorf M, Keene JD, Lee DM, Calabrese JM, and Weeks KM (2016). SHAPE reveals transcript-wide interactions, complex structural domains, and protein interactions across the Xist lncRNA in living cells. Proc. Natl. Acad. Sci. U.S.A. 113, 10322–10327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somarowthu S, Legiewicz M, Chillon I, Marcia M, Liu F, and Pyle AM (2015). HOTAIR forms an intricate and modular secondary structure. Mol. Cell 58, 353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spitale RC, Crisalli P, Flynn RA, Torre EA, Kool ET, and Chang HY (2013). RNA SHAPE analysis in living cells. Nat. Chem. Biol. 9, 18–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subtelny AO, Eichhorn SW, Chen GR, Sive H, and Bartel DP (2014). Poly(A)-tail profiling reveals an embryonic switch in translational control. Nature 508, 66–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto Y, Vigilante A, Darbo E, Zirra A, Militti C, D'Ambrogio A, Luscombe NM, and Ule J (2015). hiCLIP reveals the in vivo atlas of mRNA secondary structures recognized by Staufen 1. Nature 519, 491–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro JM, Vidaki M, Oliveira R, Olson S, Zhan L, Saxena T, Wang ET, Graveley BR, Gertler FB, Swanson MS, et al. (2016). Distal Alternative Last Exons Localize mRNAs to Neural Projections. Mol. Cell 61, 821–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan D, Marzluff WF, Dominski Z, and Tong L (2013). Structure of histone mRNA stem-loop, human stem-loop binding protein, and 3'hExo ternary complex. Science 339, 318–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian B, and Manley JL (2013). Alternative cleavage and polyadenylation: the long and short of it. Trends iBiochem.Sci. 38, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tuck AC, and Tollervey D (2013). A transcriptome-wide atlas of RNP composition reveals diverse classes of mRNAs and lncRNAs. Cell 154, 996–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weill L, Belloc E, Bava FA, and Mendez R (2012). Translational control by changes in poly(A) tail length: recycling mRNAs. Nat. Struct. Mol. Biol. 19, 577–585. [DOI] [PubMed] [Google Scholar]
- Wells SE, Hughes JM, Igel AH, and Ares M Jr. (2000). Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 318, 479–493. [DOI] [PubMed] [Google Scholar]
- Wilkening S, Pelechano V, Jarvelin AI, Tekkedil MM, Anders S, Benes V, and Steinmetz LM (2013). An efficient method for genome-wide polyadenylation site mapping and RNA quantification. Nucl. Acids Res. 41, e65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Liu M, Downie B, Liang C, Ji G, Li QQ, and Hunt AG (2011). Genomewide landscape of polyadenylation in Arabidopsis provides evidence for extensive alternative polyadenylation. Proc. Natl. Acad. Sci. U.S.A 108, 12533–12538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, and Bartel DP (2017). Widespread influence of 3’-end structures on mammalian mRNA processing and stability. Cell 169, 905–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziehler WA, and Engelke DR (2001). Probing RNA structure with chemical reagents and enzymes. Curr. Protoc. Nucleic Acid Chem Chapter 6, Unit 6 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zubradt M, Gupta P, Persad S, Lambowitz AM, Weissman JS, and Rouskin S (2017). DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 14, 75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.