

CAPSULE SUMMARY
Allogeneic HCT practice patterns for PID changed between 1974–2016. Three-year survival improved to >70% for SCID and non-SCID patients after 1999, and further increased to 94% for SCID patients transplanted 2010–2016 following newborn screening diagnosis.
Keywords: Primary Immune Deficiency, Primary Immunodeficiency, Allogeneic Hematopoietic Cell Transplantation, Allogeneic Hematopoietic Stem Cell Transplantation, Bone Marrow Transplant, Severe Combined Immune Deficiency, Chronic Granulomatous Disease, Hemophagocytic Lymphohistiocytosis, Wiskott-Aldrich Syndrome, X-linked Lymphoproliferative Disease
To The Editor,
This year, 2018, marks 50 years since the first allogeneic hematopoietic cell transplantations (HCT) were performed for primary immune deficiencies (PID) in 1968 (severe combined immune deficiency [SCID] and Wiskott-Aldrich Syndrome [WAS]).1, 2 Despite the passage of time and the expansion of allogeneic HCT to many PID diseases, data regarding recent changes in transplant practices and outcomes is lacking. There have been recent sizable studies for specific PIDs such as WAS and SCID.3, 4, 5 Two older European studies described allogeneic HCT for very large groups of SCID and non-SCID patients, but neither included data from the last 10 years.6, 7 In commemoration of this anniversary year, we examined the indications, practice changes, and survival of PID patients treated with allogeneic HCT between 1974–2016 using data reported to the Center for International Blood and Marrow Transplant Research (CIBMTR).
The CIBMTR is a voluntary network of over 500 transplant centers worldwide (https://www.cibmtr.org/About/WhoWeAre/Centers/Pages/index.aspx). All allogeneic transplants in the U.S.A. are now reported to the CIBMTR in fulfillment of the Stem Cell Therapeutic and Research Act of 2005. We categorized transplantations by broad disease category (SCID and non-SCID) based on reported diagnoses and by transplant periods: 1974–1989, 1990–1999, 2000–2009, and 2010–2016. Data was not collected from all centers during all timeframes of this study, therefore comparisons over time periods are limited to data reported to CIBMTR. SCID patients who underwent newborn screening were identified based on their state of residence, date of implementation in their state, and birth date. The Institutional Review Board of the National Marrow Donor Program approved this study. For analysis of overall survival, death from any cause was considered an event and surviving patients were censored at last follow up. The 3-year probabilities of overall survival were calculated using the Kaplan-Meier estimator.8
Nine-hundred and eighty-one SCID patients and 1902 non-SCID patients underwent allogeneic HCT between 1974–2016 (Table 1A–B). The number of transplantations greatly increased over time. There were just over 100 transplants performed for PID from 1974–1989, whereas between 2010–2016, 352 and 816 transplants were performed for SCID and non-SCID patients, respectively. SCID transplants were performed at a median age of 0.5 years. The median age for non-SCID patients was 2 years, and the indication evolved over the decades. Approximately half of transplantations before 1990 were for WAS, whereas the most common recent indications were HLH/XLP and CGD.
Table 1A.
Patient transplant characteristics - SCID.
| Variable | 1974–1989 | 1990–1999 | 2000–2009 | 2010–2016 |
|---|---|---|---|---|
| Number of patients | 93 | 215 | 321 | 352 |
| Age at transplant, years, Median (Inter-quartile range) | 0.53 (0.37–0.78) |
0.55 (0.29–0.89) |
0.55 (0.34–0.94) |
0.38 (0.20–0.99) |
| Donor Type | ||||
| HLA identical sibling | 15 (16) | 46 (21) | 51 (16) | 62 (18) |
| Other relative | 71 (76) | 119 (55) | 95 (30) | 68 (19) |
| Matched other relative | 0 | 0 | 11 | 9 |
| Mismatched other relative | 0 | 0 | 81 | 58 |
| HLA match not reported | 71 | 119 | 3 | 1 |
| Unrelated donor | 7 (8) | 50 (23) | 175 (55) | 222 (63) |
| Graft type | ||||
| Bone marrow | 93 | 196 (91) | 161 (50) | 211 (60) |
| Peripheral blood | 0 | 5 (2) | 50 (16) | 38 (11) |
| Cord blood | 0 | 14 (7) | 110 (34) | 103 (29) |
| Conditioning Intensity | ||||
| No conditioning regimen | 71 (76) | 96 (45) | 61 (19) | 66 (19) |
| Myeloablative | 12 (13) | 81 (38) | 147 (46) | 166 (47) |
| Reduced intensity | 10 (11) | 38 (18) | 113 (35) | 120 (34) |
| In-vivo T-cell depletion | ||||
| No | 83 (89) | 153 (71) | 141 (44) | 124 (35) |
| Yes | 10 (11) | 62 (29) | 180 (56) | 228 (65) |
| GVHD Prophylaxis | ||||
| No GVHD prophylaxis | 17 (18) | 9 (4) | 16 (5) | 13 (4) |
| Ex-vivo T-cell depletion | 63 (68) | 69 (32) | 40 (12) | 44 (13) |
| CD34+ selection | 0 | 0 | 29 (9) | 29 (8) |
| Cyclosporine +/− MMF/MTX | 5 (5) | 108 (50) | 171 (54) | 172 (49) |
| Tacrolimus +/− MMF/MTX | 0 | 0 | 38 (12) | 76 (22) |
| Other | 8 (9) | 29 (13) | 27 (8) | 18 (5) |
HLA, Human Leukocyte Antigen
GVHD, Graft Versus Host Disease
MMF, mycophenolate mofetil
MTX, methotrexate
Table 1B.
Patient transplant characteristics - non-SCID.
| Variable | 1974–1989 | 1990–1999 | 2000–2009 | 2010–2016 |
|---|---|---|---|---|
| Number of patients | 36 | 319 | 731 | 816 |
| Disease sub-types | ||||
| HLH, XLP, CH | 8 (22) | 129 (40) | 381 (52) | 413 (51) |
| Wiskott-Aldrich syndrome | 19 (53) | 120 (38) | 145 (20) | 107 (13) |
| Chronic granulomatous disease | 0 | 11 (3) | 68 (9) | 159 (19) |
| Other Non-SCID PID | 9 (25) | 59 (19) | 137 (19) | 137 (17) |
| Age at transplant, years, Median (Inter-quartile range) | 4 (1–8) | 2 (1–5) | 2 (1–6) | 3 (1–10) |
| Donor Type | ||||
| HLA identical sibling | 16 (44) | 70 (22) | 144 (20) | 142 (17) |
| Other related donor | 10 (28) | 47 (15) | 42 (6) | 60 (7) |
| Matched other relative | 0 | 0 | 12 | 23 |
| Mismatched other relative | 0 | 0 | 30 | 37 |
| HLA-match not reported | 10 | 47 | 0 | 0 |
| Unrelated donor | 10 (28) | 202 (63) | 545 (75) | 614 (75) |
| Graft type | ||||
| Bone marrow | 36 | 256 (80) | 418 (57) | 531 (65) |
| Peripheral blood | 0 | 12 (4) | 68 (9) | 117 (14) |
| Cord blood | 0 | 51 (16) | 245 (34) | 168 (21) |
| Conditioning intensity | ||||
| Myeloablative | 32 (89) | 290 (91) | 579 (79) | 382 (47) |
| Reduced intensity | 4 (11) | 29 (9) | 152 (21) | 434 (53) |
| In-vivo T-cell depletion | ||||
| No | 28 (78) | 142 (45) | 210 (29) | 113 (14) |
| Yes | 8 (22) | 177 (55) | 521 (71) | 703 (86) |
| GVHD Prophylaxis | ||||
| Ex-vivo T-cell depletion | 6 (17) | 35 (11) | 33 (5) | 10 (1) |
| CD34+ selection | 0 | 0 | 15 (2) | 24 (3) |
| Cyclosporine +/− MMF/MTX | 14 (39) | 247 (77) | 533 (73) | 472 (58) |
| Tacrolimus +/− MMF/MTX | 0 | 5 (2) | 104 (14) | 232 (29) |
| Other | 16 (44) | 32 (10) | 46 (6) | 78 (10) |
HLH, Hemophagocytic Lymphohistiocytosis
XLP, X-linked Lymphoproliferative Disease
CH, Chediak-Higashi Syndrome
SCID, Severe Combined Immune Deficiency
PID, Primary Immune Deficiency
HLA, Human Leukocyte Antigen
GVHD, Graft Versus Host Disease
MMF, mycophenolate mofetil
MTX, methotrexate
For SCID patients, non-sibling relatives were the predominant donors prior to 2000, but the number of unrelated donors increased over time to 63% during 20102016. In contrast, HLA-matched siblings were the predominant donor for early non-SCID patients, and unrelated donors were most common after 1990. Bone marrow was the predominant graft for SCID and non-SCID patients. Ex vivo T-cell depletion was common in SCID patients prior to 2000, but thereafter accounted for less than 15% of transplants. CD34+ selected peripheral blood grafts were only used between 2000–2016 and accounted for approximately 10% of SCID transplants. These methods were not commonly used in non-SCID patients.
In regards to conditioning regimen intensity, between 1974–1989, 76% of SCID transplants were performed without conditioning. Myeloablative regimens were common after 1999 and accompanied the increasing use of unrelated donors. By 20102016, over 80% of SCID patients received myeloablative or reduced intensity conditioning (RIC). In contrast, all non-SCID patients received conditioning, though the intensity changed over time. Prior to 2000, RIC accounted for approximately 10% of non-SCID transplantations, but increased to over half of transplantations by 2010–2016. The majority of both SCID and non-SCID patients received in-vivo T-cell depletion in the later time periods, and calcineurin inhibitor-based graft versus host disease prophylaxis was common.
Survival increased over time for all patients. Prior to 2000, 3-year overall survival for SCID was 64% (95% confidence interval [CI] 58–69%), versus 73% from 2000–2009 (CI 68–78%), and 80% from 2010–2016 (CI 76–85%) (p<0.001) (Figure 1A). For patients with non-SCID diseases, survival improved from 62% prior to 2000 (CI 57–67%) to 70% from 2000–2009, but improved to a lesser extent than SCID from 2010–2016, up to 75% (CI 72–79%) (Figure 1B). The introduction of SCID newborn screening likely accounted for the superior increase in SCID survival after 2010, as those diagnosed by newborn screening experienced a 94% 3-year survival probability (CI 89–98%) (Figure 1C). Among non-SCID patients, survival for patients with CGD and WAS appeared to be higher than HLH/XLP and other PIDs (Figure 1D).
Figure 1.
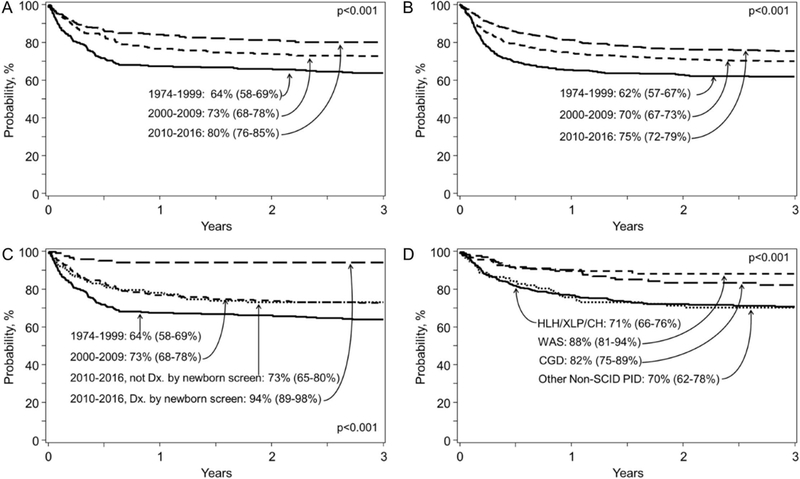
Three-year overall survival for A) SCID patients by time period, B) non-SCID patients by time period, C) SCID patients by time period and by newborn screening diagnosis or not in the time period 2010–2016, and D) non-SCID patients by diagnosis group in the time period 2010–2016.
These CIBMTR data reveal that allogeneic HCT practice patterns have changed considerably over the last 50 years. Unrelated donors are now the most common source of hematopoietic stem cells for all PID patients. An earlier European study reported that 25% of SCID patients and 46% of non-SCID patients received grafts from unrelated donors between 2000–2005,7 but we observed a further increase to 63% of SCID patients and 75% of non-SCID patients transplanted between 2010–2016. We also observed notable changes in conditioning selections over time. For SCID, a previous study reported that approximately half of SCID patients transplanted 20002009 were treated with conditioning,4 while we observed that over 80% of SCID patients transplanted in 2010 and later received conditioning, more in line with a recent report from Heimall et al.9 We also observed a large uptick in the use of RIC in the modern era, which appears to now be used for approximately 1/3 of SCID patients and 1/2 of non-SCID patients.
These data also reveal continued improvements in survival, which is likely multifactorial in nature. Advances in conditioning approaches, HLA typing techniques/experience, donor options, graft manipulation techniques, GVHD prevention, infection prophylaxis/treatment, critical care medicine, and PID diagnostics likely all contribute. Newborn screening for SCID appears to be making a tremendous impact on transplant survival, likely by providing an opportunity for allogeneic HCT before the acquisition of infections. Expected survival following allogeneic HCT in this current era for patients with PID can be estimated based on our data to be at least 70%, and is closer to 80–90% for many patients. It is our hope that ongoing advances in the PID and BMT fields will continue to improve survival outcomes over the next 50 years.
Acknowledgements
The CIBMTR is supported primarily by Public Health Service Grant 5U24-CA076518 from the National Cancer Institute (NCI), the National Heart, Lung and Blood Institute (NHLBI) and the National Institute of Allergy and Infectious Diseases (NIAID); 5U10HL069294 from NHLBI and NCI; a contract HHSH250201200016C with Health Resources and Services Administration (HRSA/DHHS); grants N00014–15-1–0848 and N00014–16-1–2020 from the Office of Naval Research. The views expressed in this article do not reflect the official policy or position of the National Institute of Health, the Department of the Navy, the Department of Defense, Health Resources and Services Administration (HRSA) or any other agency of the U.S. Government.
Footnotes
Disclosures: The authors declare that there are no financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gatti RA, Meuwissen HJ, Allen HD, Hong R, Good RA. Immunological reconstitution of sex-linked lymphopenic immunological deficiency. Lancet 1968; 2:1366–9. [DOI] [PubMed] [Google Scholar]
- 2.Bach FH, Albertini RJ, Joo P, Anderson JL, Bortin MM. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet 1968; 2:1364–6. [DOI] [PubMed] [Google Scholar]
- 3.Moratto D, Giliani S, Bonfim C, Mazzolari E, Fischer A, Ochs HD, et al. Long-term outcome and lineage-specific chimerism in 194 patients with Wiskott-Aldrich syndrome treated by hematopoietic cell transplantation in the period 1980–2009: an international collaborative study. Blood 2011; 118:1675–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pai SY, Logan BR, Griffith LM, Buckley RH, Parrott RE, Dvorak CC, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med 2014; 371:434–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schuetz C, Neven B, Dvorak CC, Leroy S, Ege MJ, Pannicke U, et al. SCID patients with ARTEMIS vs RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood 2014; 123:281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antoine C, Muller S, Cant A, Cavazzana-Calvo M, Veys P, Vossen J, et al. Long-term survival and transplantation of haemopoietic stem cells for immunodeficiencies: report of the European experience 1968–99. Lancet 2003; 361:553–60. [DOI] [PubMed] [Google Scholar]
- 7.Gennery AR, Slatter MA, Grandin L, Taupin P, Cant AJ, Veys P, et al. Transplantation of hematopoietic stem cells and long-term survival for primary immunodeficiencies in Europe: entering a new century, do we do better? J Allergy Clin Immunol 2010; 126:602–10 e1–11. [DOI] [PubMed] [Google Scholar]
- 8.Kaplan EL, Meier P. Nonparametric Estimation from Incomplete Observations. Journal of the American Statistical Association 1958; 53:457–81. [Google Scholar]
- 9.Heimall J, Logan BR, Cowan MJ, Notarangelo LD, Griffith LM, Puck JM, et al. Immune reconstitution and survival of 100 SCID patients post-hematopoietic cell transplant: a PIDTC natural history study. Blood 2017; 130:2718–27. [DOI] [PMC free article] [PubMed] [Google Scholar]