

SUMMARY
It is unknown how the dynamic binding of Transcription Factors (TF) is molecularly linked to chromatin remodeling and transcription. Using SMT, we show that the chromatin remodeler RSC speeds up the search process of the TF Ace1p for its Response Elements (RE) at the CUP1 promoter. We quantified smFISH mRNA data using a gene bursting model, and demonstrated that RSC regulates transcription bursts of CUP1 only by modulating TF occupancy but does not affect initiation and elongation rates. We show by SMT that RSC binds to activated promoters transiently and, based on MNase-seq data, RSC does not affect the nucleosomal occupancy at CUP1. Therefore, transient binding of Ace1p and rapid bursts of transcription at CUP1 may be dependent on short repetitive cycles of nucleosome mobilization. This type of regulation reduces the transcriptional noise and ensures a homogeneous response of the cell population to heavy metal stress.
Keywords: Chromatin remodeling, Transient binding, Transcription factors, RSC complex, CUP1, ACE1, single molecule tracking, smFISH, transcription bursts, Saccharomyces cerevisiae
Graphical Abstract
eTOC Blurb
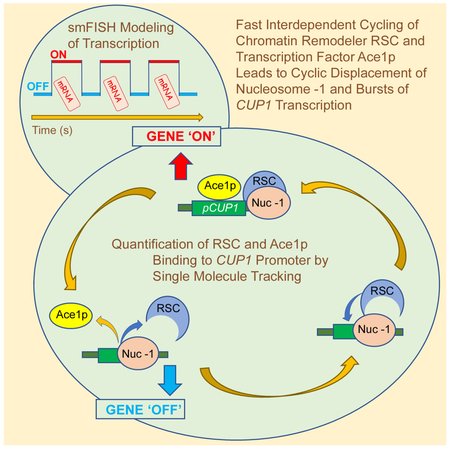
In this study, Mehta et al. show that rapid cycles of the yeast transcription factor Ace1p at CUP1 promoter serve as a pace-maker for the transcription bursts. The chromatin remodeler RSC finetunes this cycle by short repetitive cycles of nucleosome mobilization.
INTRODUCTION
Many transcription factors (TF) bind to specific response elements (RE) with dynamics on the scale of seconds (Voss and Hager, 2014). Biophysical data suggest that fast exchange is a mechanism for optimization of gene activation (Clauss et al., 2017; Lickwar et al., 2012; Stavreva et al., 2004; Van Royen et al., 2014). However, little is known about the regulation of TF transient binding. Chromatin remodelers move and disassemble nucleosomes (Bowman and McKnight, 2017) and were also shown to evict the TF by inducing collisions between a nucleosome and the TF in vitro (Li et al., 2015). Thus, it is plausible that chromatin remodeling may affect TF dynamics. However, nothing is known at the molecular level about the links between in vivo remodeling and TF binding. Single Molecule Tracking (SMT) is a method of choice to quantify in vivo molecular dynamics (Ball et al., 2016; Izeddin et al., 2014; Mazza et al., 2013). However, SMT quantifications are predominantly based on averaging of the data obtained for a population of REs present in the whole nucleus and this may obscure the variability in binding parameters of a TF and its underlying regulatory mechanisms. We characterized by SMT the binding of a TF to a specific promoter. We employed a yeast model consisting of the metallothionein-encoding CUP1 and its copper-dependent transcription activator Ace1p (Furst et al., 1988). Fast cycling of Ace1p at the promoter of CUP1 was previously demonstrated by FRAP and CLK-ChIP (Karpova et al., 2008; Poorey et al., 2013). Activation of CUP1 correlates with a loss of histones (Karpova et al., 2008) and leads to an Ace1p-dependent switch from positioned nucleosomes to a fluid chromatin structure (Shen et al., 2001). By SMT of Ace1p we demonstrated that the chromatin remodeler complex RSC improves the search of Ace1p for its REs at the CUP1 promoter by increasing (a) the fraction of accessible REs, and (b) the molecular ‘on’ rate of Ace1p binding. Contrary to other systems, where transcription is improved by an increase in TF residence time (Loffreda et al., 2017; Morisaki et al., 2014; Paakinaho et al., 2017), for CUP1 improvement in transcription correlates with decrease in Ace1p residence time. To quantify the effect of RSC on transcription we analyzed the smFISH of CUP1 mRNA using a standard model for bursting gene expression. Parameter estimates produced by the model indicate that RSC has no effect on transcription initiation and elongation rates, and changes in transcription induced by RSC may be explained completely by its effect on TF binding. Thus, we have demonstrated how nucleosome displacement by itself can alter transcription dynamics. Consequently, RSC increases the fraction of active cells, reduces the transcriptional noise and ensures a homogeneous response of the cell population to heavy metal stress. As observed by SMT, RSC binds to an activated CUP1 promoter transiently. Based on MNase-seq data, the rapid cyclical activity of RSC does not affect nucleosomal occupancy at CUP1. Our results are summarized in a model proposing that transcription bursts at CUP1 depend on short repetitive TF binding and rapid, repetitive chromatin remodeling.
RESULTS
Assay for specific binding of Ace1p to activated promoters of CUP1
CUP1 encoding yeast metallothionein forms a 10x natural tandem array in yeast Saccharomyces cerevisiae (Zhao et al., 2014). We performed SMT of Ace1p-HaloTag labeled with JF646 (Grimm et al., 2015) at the CUP1 promoter in cells activated with 100 μM Cu2+. Promoters were visualized by Ace1p-3xGFP (Karpova et al., 2008) (Figure 1A, Movie S1). As a control, we also tracked Ace1p over the lacO array, observed by the binding of LacI-GFP, as it has no known binding sites for Ace1p (Figure 1B, Movie S2). We confirmed that all the genetically modified strains for our experiments behave like a wild-type strain in terms of expression levels of ACE1 and CUP1 and their sensitivity to copper (Figure S1, Table S1, S2 and S3). Binding parameters were estimated from survival times of Ace1p (Ball et al., 2016). We plotted a histogram of the times that Ace1p molecules resided over the CUP1 array and the lacO array (Figure 1C). An exponential fit of the distribution of Ace1p survival at CUP1 yielded two binding states with mean residence times of 0.59 ± 0.05 s and 2.96 ± 0.26 s (Figure 1C and 1D). At lacO we observed only a single fraction of Ace1p bound molecules with residence time comparable to that for short-residence Ace1p on CUP1 (0.78 ± 0.05 s). Therefore, the short residence times of Ace1p most likely reflect non-specific binding, while the longer residence times observed only at CUP1 reflect specific binding. The tracking method, and residence time analysis based on the survival distribution is explained in the Method Details (Figure S2).
Fig. 1. Assay for specific binding of Ace1p to activated promoters of CUP1.

Ace1p-3xGFP (A) at the CUP1 array in YTK1498 or GFP-lacI (B) at pes4::LacO array in YTK1528 define the site for tracking of the individual molecules of Ace1p-HaloTag labeled with JF646 at a low density of ≤ 5 particles per nucleus. Molecules of Ace1p-HaloTag (JF646) frequently colocalize with CUP1 array and rarely colocalize with lacO array as demonstrated by single focal plane representative images. Gaussian filter was applied to Ace1p-HaloTag (JF646) channel in Track Record software for better visualization of the particles. Here and in Figure 4B and 4C brightness and contrast were adjusted using linear LUT in Image J and cell contours were drawn manually in Adobe Illustrator. Scale bar = 2 μm. See also Movies S1 and S2. (C) Raw distributions of survival of Ace1p particles are shown with the exponential decay fits. For this Figure and for Figures 2 and 6A, pie charts represent the extracted distributions of diffusing (gray), specific (blue) and non-specific (orange) binding events with their average residence times; the table show the number of molecules / tracks observed for a particular sample. (D) The difference between residence times of Ace1p. ‘***’ - p<0.001 by z-test.
RSC affects transient specific binding of Ace1p at CUP1 promoter
In yeast, specificity of binding of the RSC complex is determined by the mutually exclusive and non-essential subunits Rsc1p and Rsc2p (Cairns et al., 1999). Only Rsc2p, but not Rsc1p affects Ace1p binding (Karpova et al., 2004). Therefore, we tested the effects of Rsc2p on the specific binding of Ace1p to the RE of the CUP1 promoter in conditions of Rsc2p depletion. We took advantage of the Auxin Inducible Degron (AID) system (Morawska and Ulrich, 2013; Nishimura et al., 2009) for the conditional depletion of Rsc2p and characterized this system in our experimental strain (YTK1649) as described in Method Details (Figure S3). The specific-bound fraction of Ace1p is reduced 2.5x times in Rsc2− cells (3.8 ± 0.8%) in comparison with Rsc2+ cells (10.4 ± 2.6%, p<0.001) (Figure 2A). Similarly, the specific-bound fraction of Ace1p is reduced in rsc2Δ (2.7 ± 1.2%) in comparison with RSC2 (14.5 ± 1.8%) (Figure 2B). Wild-type RSC2 introduced into Rsc2− cells compensates for the depletion of the endogenous Rsc2p and restores the specific-bound fraction of Ace1p to 12.1 ± 4.1% (Figure 2C).
Fig. 2. RSC affects transient binding of Ace1p at CUP1 promoter.

SMT of Ace1p at CUP1; charts, graphs and tables as described in the caption to Figure 1. Effect of Rsc2p depletion (A, YTK1649) or rsc2 knock out (B, YTK1526 vs wt YTK1498) on Ace1p binding at CUP1. (C) Compensation for depletion of Rsc2p by Rsc2p expressed from centromeric plasmid in YTK1582. (D) The average residence time of Ace1p at CUP1 from A-C. ‘n.s.’ - not significant 0.09<p<0.94, ‘*’ - p<0.05 by z-test.
RSC associates with active CUP1 locus and modulates its transcription and copper sensitivity of the cells
By ChIP we detected a 60% increase in the accumulation of Rsc2p at activated CUP1 (Figure 3A). By qPCR estimates, depletion of Rsc2p leads to a ~30% reduction in transcription of CUP1 and to higher copper sensitivity (Figures 3B and 3C). Thus, the effect of the RSC complex on Ace1p binding correlates with an improvement in CUP1 transcription and cell survival in the presence of copper. Surprisingly, contrary to other systems, Ace1p residence time increases in Rsc2− cells even though CUP1 transcription is reduced (Figures 2A, 2B and 2D).
Fig. 3. RSC associates with active CUP1 locus and modulates its transcription and copper sensitivity of the cells.

(A) Binding of Rsc2p-AID*−9Myc to promoters of CUP1 upon activation with copper was assayed by ChIP. The effect of Rsc2p depletion on transcription of CUP1 quantified by RT-qPCR (B) and on copper sensitivity of the cells by colony growth assay (C). ‘*’ - p<0.05, ‘**’ - p<0.01, ‘***’ - p<0.001 by t-test.
RSC affects the search of Ace1p for its specific RE
The molecular mechanism of RSC action may be deduced from the SMT estimates of Ace1p binding. The bound fraction of Ace1p (Ceq), defined as the ratio of bound to total molecules, depends on the residence time and on the search time of Ace1p for an RE at CUP1, as described simplistically by (Eq 1):
| (Eq.1) |
The full version of Eq. 1 for the case of the two binding states observed for Ace1p, both short and long, is given in Method Details, Eq. M1. Therefore, an increase in Ceq, observed as a higher occupancy of the RE by TF may result from: (a) decrease in the availability of the TF, Cfree, (b) decrease in the dissociation rate, koff (c) increase in the availability of the RE, Seq, or (d) increase in the association rate, kon. Based on our data, we can eliminate the first two causes. First, there is no difference in total nuclear Ace1p for Rsc2+ and Rsc2− cells (Figure S3D). Second, τres = 1/koff decreases, rather than increases in Rsc2+ cells. Thus, the increase in Ceq observed in Rsc2+ cells must be due to a decrease in the search time. From the estimates for the ratio of Ceq and τres obtained from SMT for the full two-state binding model (Eq. M6), τsearch is ~8 s (Rsc2+) and ~ 25 s (Rsc2−). Therefore, RSC speeds up the search of Ace1p for CUP1 REs, either by an increase in the molecular ‘on’ rate for Ace1p (kon) and/or the concentration of free REs for CUP1 (Seq), as given by:
| (Eq.2) |
RSC affects the “on” rate of Ace1p and the availability of RE at CUP1 promoter
Estimates of the relative impact of kon and Seq may shed light on the molecular mechanism of RSC action. Although both parameters are dependent on the presence of nucleosome-free DNA at the promoter, they depend on slightly different conditions: Seq indicates how many REs are free from nucleosomes, and kon is dependent on the size of the target for the search (Mirny et al., 2009). Predominant regulation of Seq indicates that the area free of nucleosomes may be relatively small, covering only the RE, but predominant regulation of kon indicates that this area should be larger than the RE. However, SMT allows direct estimation of only k*on. Seq may be estimated from the fraction of non-activated CUP1 in the cell population, Fnt. If we assume that binding of Ace1p to any of the four REs in the CUP1 gene occurs equally and independently and results in transcription, then we can relate Fnt to the probability, P, that any one RE is available as described by Method Details Eq M8. From the calculated probability of each RE being available, and providing four RE exist per promoter, we end up with an average number of available RE, and therefore we may calculate Seq based on Avogadro’s number NA and the volume of the yeast nucleus V, as described by Methods Eq M9. From Eq 2 we may then calculate kon.
Estimation of Fnt is complicated by the presence of ten copies of CUP1 in each array, because transcripts observed in individual cells may be produced from any number of active CUP1. To observe transcription of just a single copy of CUP1 per cell by smFISH, we replaced the ORF of a single CUP1 gene within the array by an artificial sequence (CUP1 reporter) (Figures 4A and S4). In the diploid tested, only one of the arrays contained the reporter (Figure 4B). We characterized and validated the use of CUP1 reporter as an adequate test for the expression of the entire CUP1 array (Figure S5). The following conclusions are based on the assumption that all promoters in both arrays and in all the cells perform independently.
Fig. 4. RSC affects the “on” rate of Ace1p and the availability of RE at the CUP1 promoter.

(A) Diagram of the targeted integration of CUP1 reporter into CUP1 array (not to scale). The cassette contains a scrambled sequence derived from mammalian gene (scr. mam) and eight copies of the phage MS2 binding site, that may be transcribed in a single transcript, but cannot be translated. LEU2 is used as a selective marker for integration. Positions of the primers used to amplify the cassette are marked in magenta (T1010) and cyan (T1004). (B) Maximum Intensity projections of z-stack of a diploid cell containing the CUP1 reporter inserted into one of the two CUP1 arrays. TAMRA (red): Reporter mRNA stained by smFISH probes, Ace1p-3xGFP (green): CUP1 locus, DAPI (blue): nucleus. Overlays: CUP1 reporter mRNA colocalizes with only one of the two CUP1 transcription sites (arrowhead). Scale bar = 2 μm. (C) Population of cells with CUP1-reporter: transcribing (yellow circles), i.e. containing > 3 molecules of mature mRNA, and not transcribing (blue circles). Overlays of DAPI (green) and CUP1 reporter mRNA (red). Maximal Intensity Projections of the z-stacks of the images of the cells treated by 100 μM Cu2+ for CUP1 activation and 1 mM auxin for Rsc2 depletion and control cells of YTK1649 are presented. Original 15-bit images were scaled in Metamorph with linear LUT with the same range for brightness and contrast. Scalebar = 2 μm. (D) Counts of mature mRNA in control cells not activated by Cu2+ (upper panel, black bars) are fit to Poisson distribution with mean 0.46. The same Poisson distribution (mean = 0.46) can be used to fit the initial points of the distribution of mature mRNA in cells activated by Cu2+, both Rsc2+ and Rsc2p-depleted, if it is first scaled by a constant factor, α. (E) Histograms of mature mRNA per cell. The population of non-active cells (blue) was obtained by fitting the histogram cut off at 3 mRNA/cell to a scaled Poisson distribution with a fixed mean of 0.46 mRNA/cell estimated from the non-activated control. The scaling parameter in the fit is used to define the fraction of non-active cells, as represented in blue fonts.
We observed that in the control group not treated with Cu2+, cells contain on average very few mature mRNA of the CUP1-reporter (0.46 ± 0.03 (SEM)) per cell, with a maximum of 8 (Figure 4C). Cells that do not contain any mRNA constituted 67.2% of the whole population. The histogram of mature mRNA per cell is fit well to a Poisson distribution, indicating that basal expression of CUP1 is governed by random, uncoordinated initiation, rather than a bursting behavior (Figure 4D, upper panel).
In the cells from the CUP1-reporter sample that were treated with Cu2+, both Rsc2+ and Rsc2p-depleted, the same Poisson distribution (mean = 0.46) can be used to fit the initial points of the distribution of mature mRNA if it is first scaled by a constant factor, a, as described by Method Details Eq M7, indicating that the initial part of the distribution contains cells that are not transcribing above the basal level. Therefore, this fit represents the cells in which the CUP1 reporter is not activated, and the cells treated with Cu2+ contain two distinct sub-populations, one of which is similar to that observed in the population of non-activated cells and can provide an estimate of Fnt. The fittings of this scaled Poisson to the Rsc2+ and Rsc2− mRNA data are shown in the middle and lower panels of Figure 4D, respectively, and the fraction of non-activated cells is highlighted in blue in Figure 4E. Upon depletion of Rsc2p, the fraction of non-active cells, Fnt, in Rsc2− is roughly twice that of Rsc2+, going from 14.4 ± 2.9% to 27.5 ± 11.0%. The calculated probability of an RE being available (P) is 0.38 ± 0.08 for Rsc2+, and 0.28 ± 0.11 for Rsc2− cells (Table 1). Providing four RE exist per CUP1 promoter, we end up with an average number of available RE per promoter, n = 1.54, for Rsc2+, and n = 1.10 for Rsc2− (Table 1). Therefore, Rsc2p stimulates the availability of the RE. From Eq M9, for Rsc2+ Seq = 0.61 ± 0.13 nM, and for Rsc2− Seq = 0.44 ± 0.18 nM. From Seq and k*on (see Eq 2) we can estimate the molecular binding rates as 0.21 ± 0.07 nM−1 s−1 for Rsc2+, and 0.12 ± 0.06 nM−1 s−1 for Rsc2−. Thus, both kon and Seq must change to produce the change in transcription between Rsc2+ and Rsc2p-depleted cell populations. Also, in Monte Carlo simulations of TF binding it is impossible to obtain the differences observed experimentally in Rsc2− and Rsc2+ cells if only one of these parameters is affected (Figure S6 and Method Details). RSC must affect both kon and Seq to speed up the search of Ace1p for CUP1 binding sites.
Table 1.
Comparison of direct measurements and transcription model predictions for the parameters for CUP1 transcription and Ace1p binding
| Parameter Name | Rsc2− | Rsc2+ | p-value |
|---|---|---|---|
| tsearch (s) | 25 ± 3.8 | 7.6 ± 2.0 | <0.001 |
| τres (s) | 5.22 ± 0.76 | 2.18 ± 0.33 | <0.001 |
| RE Available per gene | 1.10 ± 0.44 (0.99 ± 0.06) | 1.54 ± 0.32 (1.49 ± 0.04) | 0.11 (<0.001) |
| koff(s −1) | 0.19 ± 0.03 | 0.46 ± 0.07 | <0.001 |
| k*on (s−1) | 0.05 ± 0.01 (0.05 ± 0.01) | 0.16 ± 0.05 (0.12 ± 0.04) | <0.001 (<0.001) |
| Seq (nM) | 0.44 ± 0.18 (0.39 ± 0.02) | 0.61 ± 0.13 (0.59 ± 0.01) | 0.13 (<0.001) |
| kon (nM−1 s−1) | 0.12 ± 0.06 (0.14 ± 0.01) | 0.21 ± 0.07 (0.21 ± 0.02) | 0.012 (<0.001) |
| PRE | 0.28 ± 0.11 (0.25 ± 0.02) | 0.38 ± 0.08 (0.37 ± 0.01) | 0.15 (<0.001) |
| Ktx (s−1) | 0.24 ± 0.08 (0.20 ± 0.02) | 0.18 ± 0.03 (0.23 ± 0.05) | 0.17 (0.28) |
| tprod(s) | 80.9 ± 43.5 (84.9 ± 30.8) | 70.2 ± 21.0 (76.2 ± 6.2) | 0.68 (0.60) |
| kdeg (s−1) | 2.03 × 10−3 ± 0.38 × 10−3 | 2.03 × 10−3 ± 0.38 × 10−3 | 1 |
| Burst frequency (s −1) | 0.04 ± 0.01 (0.04 ± 0.003) | 0.12 ± 0.03 (0.10 ± 0.02) | <0.001 (<0.001) |
| Burst duration (s) | 5.22 ± 0.76 | 2.18 ± 0.33 | <0.001 |
| Burst amplitude (mRNA/burst) | 1.28 ± 0.47 (1.06 ± 0.19) | 0.39 ± 0.09 (0.52 ± 0.14) | <0.001 (<0.001) |
Parameters measured directly (by SMT and smFISH) and input into the transcription model are in italic. Parameters derived from the directly measured SMT parameters are shown in normal font. Parameters obtained by fitting the smFISH data to the transcription model while varying only ktx and tprod are shown in bold. Finally, parameter values found by fitting the smFISH data to the transcription model by varying Seq, kon, ktx, and tprod are shown in parentheses. Errors represent 95% confidence intervals.
RSC complex modulates CUP1 transcription by affecting Ace1p binding
Potentially, chromatin remodeling induced by RSC may affect any or all stages of transcription, namely pre-initiation, initiation, and elongation (Figure 5A). Our hypothesis is that the binding of Ace1p is necessary and sufficient to switch the gene to the ‘ON’ state. Specifically, we make two assumptions (a) the fraction of Ace1p productively binding to CUP1 promoter is the fraction defined as specifically-bound by SMT; (b) The switch of CUP1 to the ‘ON’ state is triggered by productive binding of at least one Ace1p molecule to the promoter. Therefore, the gene ‘OFF’ rate may be described by the Ace1p residence time, and the gene ‘ON’ rate may be described by kon and Seq of Ace1p. Binding of Ace1p triggers the formation of the Pre-Initiation Complex (PIC). Downstream events such as the initiation, elongation, and degradation are not directly related to Ace1p binding. Thus, in the ‘ON’ state the gene becomes competent for transcription; however, this state does not always lead to the formation of mature mRNA.
Fig. 5. RSC acts at pre-initiation stage.

(A) The production rate, tprod for a single mRNA (pink line) is defined here as the time from initiation “I” through elongation “E” to release “R” of the mature mRNA. Mature mRNA then degrades with an average lifetime of kdeg−1. (B) The gene switches between an ON state and an OFF state (black line). During a burst, transcription is initiated at random (green lines). The rate parameters characterize the frequency of bursts (k*on and koff), duration of a burst (koff), and the amplitude of a burst (ktx and koff). (C) Histogram and bursting gene expression fits (red) of mature mRNA in Rsc2−, and Rsc2+. Fit lines were obtained by simulating the model with the average values of 1000 parameter sets that fit the data well. (D) Schematics of CUP1 burst dynamics in Rsc2+ and Rsc2− cells based on parameters estimated by the gene busting model of transcription.
We applied to smFISH a model for bursting gene expression, in which a gene can switch between an ‘ON’ and an ‘OFF’ state but can only transition to the ‘ON’ state if at least one of its REs are available. The gene may be ‘ON’ (active) even if no transcripts are detected. In the ‘ON’ state transcription may be randomly initiated, and the mRNA elongates. Finally, mature mRNA is allowed to degrade. Therefore, the transcript levels are determined by the number of bursts and burst amplitude (dependent on the amount of time that the gene is ‘ON’, and the probability that the gene is accessible) and the rates of initiation, elongation, and degradation (Figure 5B) (Raj et al., 2006; Senecal et al., 2014; Zenklusen et al., 2008). If RSC acts only through the modulation of TF binding, we expect that changes in the levels of Rsc2p will affect the burst duration, and the burst frequency, but not the transcription initiation rate, ktx, or production time, tprod (Figure 5A and 5B).
We input into the model the parameters for the gene OFF’(koff) and ‘ON’ rate (kon and Seq), and the mRNA degradation rate obtained by measuring a smFISH time-course in fixed cells (Figure S5D). Many sets of the other parameters were then simulated to generate mRNA distributions that were compared to the smFISH distributions. The final parameter values were then found by taking the average of the 1000 parameter sets that generated distributions most similar to the experimental data (Figure 5C). As we always fix the binding koff to the value measured by SMT to constrain the model, it is not possible for us to directly test the assumption that the ‘OFF’ rate is accurately described by koff However, we validated the assumption that the ‘ON’ rate is accurately described by kon by letting all the parameters except koff vary in our fitting procedure for smFISH data. In this case the fitting procedure returns a value of kon that is very similar to the value calculated from SMT data and the fraction of non-active cells estimated from smFISH data (Table 1). If, alternatively, the switch to the ‘ON’ state would be regulated by several factors and the binding of Ace1p alone would be insufficient to switch the gene into the ‘ON’ state, we would expect to see a different value of kon returned, either because the ‘ON’ rate of the gene is not related to the TF binding ‘on’ rate, or because the ‘OFF’ rate is not related to the TF ‘off’ rate, and therefore the ‘ON’ rate needed to change to make the ratio of kon/koff correct. Thus, binding of Ace1p appears to be the rate limiting step for CUP1 to turn both ‘ON’ and ‘OFF’, which is consistent with the assumption that the switch to the ‘ON’ state requires only binding of Ace1p.
We then used the primary parameters from the model to obtain the characteristics of the transcriptional bursts (Table 1 and Figure 5D). Remarkably, the estimates for polymerase initiation (ktx) and elongation (tprod) remain nearly the same for Rsc2+ and Rsc2− cells. The production time is 70.2 ± 21.0 s for Rsc2+ and 80.9 ± 43.5 for Rsc2− (p-value: 0.68; z-test), and the initiation rate is 0.18 ± 0.03 mRNA/s for Rsc2+ and 0.24 ± 0.08 mRNA/s for Rsc2− (p-value: 0.17, z-test). Thus, RSC acts at the pre-initiation step, and does not affect the subsequent stages of transcription. Therefore, we conclude that RSC modulates transcription solely by modulating TF binding. Rsc2p halves the burst duration from 5.22 ± 0.82 s to 2.18 ± 0.33 s and increases the burst frequency by roughly a factor of three, from 0.04 ± 0.01 s−1 to 0.12 ± 0.03 s−1. Due to the constant initiation rate and decreasing burst duration, the amplitude of each burst also decreases from 1.28 ± 0.47 mRNA in Rsc2− to 0.39 ± 0.09 mRNA in Rsc2+ (Table 1). More frequent bursts in Rsc2+ are canceled out by decreased duration and amplitude, and thus the same mean value for mature mRNA per active cell is observed in Rsc2+ and Rsc2− cells.
The role of RSC chromatin remodeler complex in the nucleosomal mobility at CUP1
Interestingly, based on predictions of the random gene bursting model, transcription from an individual CUP1 gene occurs as a batch of very short and fast bursts. This behavior may be explained by cycles of rapid remodeling events that facilitate the cycles of productive TF binding, and thus we decided to quantify the binding of RSC to promoters of CUP1. We compared by SMT Rsc2p-HaloTag-JF646 binding at both CUP1 and lacO (Figure 6A). Consistent with the fact that RSC can bind to chromatin at multiple sites across the genome, we observed a biexponential distribution of residence times for Rsc2p at both CUP1 and lacO. However, a 3x increase in the slow-bound fraction of Rsc2p at the activated CUP1 locus compared to lacO (7.5 ± 3.4% vs 2.5 ± 1.2%; p = 0.007) suggests that more specific binding of Rsc2p occurs at the transcriptionally active CUP1. The residence time of Rsc2p decreases from 3.2 s to 2.1 s. Therefore, as both Ace1p and RSC undergo fast cycling on active CUP1 promoter, our data indicate that preinitiation at CUP1 is dependent on short repetitive remodeling events.
Fig. 6. The role of RSC chromatin complex in nucleosomal mobility at CUP1.

(A) Single molecule tracking of Rsc2p-HaloTag (JF646) at CUP1 (YTK1635) and pes4::lacO (YTK1634). Charts and tables as described in the caption to Figure 1C. (B) Nucleosomal occupancy at CUP1. MNase-seq data for two biological replicate experiments. CUP1-2 - a single copy of CUP1 within the array. UASp,d - proximal and distal Upstream Activating Sequences, each containing two REs for Ace1p. Ovals - approximate positions of nucleosomes before activation. Treatment conditions: Rsc2−, Rsc2+, with or without Cu activation. (C) Proposed sequence of events at CUP1 promoter for an individual molecule of Ace1p: a molecule of RSC recruited to CUP1 mobilizes the −1 nucleosome, promoting binding of Ace1p; next, a bound molecule of Ace1p recruits the preinitiation complex (PIC); next, transcription starts, and PIC is disassembled; next, RSC moves the −1 nucleosome in opposite direction promoting eviction of Ace1p molecule.
Mapping of nucleosomes by MNase-seq (Figure 6B) demonstrates that in cells not treated with Cu2+ Rsc2p makes the chromatin structure of the CUP1 region more regular and makes the distant Upstream Activating Site (UAS) more accessible. Indeed, by ChIP Rsc2p is present at CUP1 promoters even in the absence of activation (Figure 3A). After Cu2+ activation, CUP1 chromatin structure becomes more irregular, which is consistent with heavy transcription, but it is almost unaffected by Rsc2p depletion. This is consistent with RT-qPCR data demonstrating transcription even in the absence of RSC. These data confirm that the remodeling activity of RSC at the activated CUP1 must occur rapidly and cyclically, leading to rapid back and forth movements of the nucleosomes, so that the net effect on nucleosome occupancy in the population (the frequency with which the putative positions are occupied by nucleosomes) cannot be registered in MNase-seq experiments.
DISCUSSION
Binding of TFs to their RE has been quantified in real time by methods of in vivo biophysics. On the other hand, chromatin remodeling of activated promoters has mainly been studied by methods of molecular biology, which do not provide sufficient spatiotemporal resolution. Therefore, it is unknown on what time scale the chromatin remodeling occurs and how it relates to periodic binding of TF. We adopted quantitative single molecule approaches to investigate the molecular links between chromatin remodeling and the fast cycling of TF on activated promoters.
Typically, SMT assays rely on averaging across multiple promoters, which may mask a substantial diversity. We focus on a specific site to reveal promoter-specific mechanisms of regulation. We discovered an alternative mechanism of regulation for Ace1p occupancy by RSC via an increase in molecular binding rate and in the availability of REs. Significantly, improvement of TF search by remodeling is totally sufficient to explain observed effects of the remodeler RSC on the transcription of CUP1, without requiring any modification to other transcriptional parameters such as polymerase initiation or elongation rates.
How can remodelers affect the search and residence time of the TF?
We found that RSC improves the search of Ace1p for specific REs in two ways, first, by increasing the availability of RE from 1.1 to 1.5 sites out of four, and, second, by improving kon. RSC may improve kon by providing larger stretches of nucleosome-free DNA, which may (a) lead to a change in the size of the search target and (b) facilitate search by allowing the non-specifically bound TF to slide along the DNA to its RE (Mirny et al., 2009). In addition, clustering of accessible REs in the same promoter improves the probability that a TF will find at least one. Of course, we cannot exclude the possibility that the improvement in kon is caused by a direct interaction between RSC and Ace1p. However, as the search of Ace1p for an RE depends on the RE availability as much as on kon, RSC must affect the search at least partially or completely by chromatin remodeling. We also observe that RSC reduces the residence time of Ace1p on RE, most likely by remodeling. This idea is supported by published in vitro experiments demonstrating displacement of a TF by a chromatin remodeler (Li et al., 2015; Nagaich et al., 2004).
How does modified binding of TF affect the transcription?
We estimated the transcription parameters in RSC+ and RSC-depleted cells by modeling mRNA output of individual cells measured by smFISH. Our model predicts a batch of ~ 50 frequent short bursts of transcription for CUP1. Note that estimates of the burst amplitude indicate that only one of the three bursts in Rsc2+ cells lead to the production of mature mRNA. Modulation of TF binding effected by RSC leads to changes in burst frequency and duration but doesn’t affect the transcription initiation rate and transcript production rate.
Surprisingly, although RSC depletion increases copper sensitivity (Figure 3C), the net change in transcription is small. Taking into the account the non-activated cells present in the population, the overall output from Rsc2− cells is reduced only by ~ 2%. More efficient production of mature mRNA in Rsc2p-depleted cells, which correlates with longer residence time of Ace1p, partially compensates for the less frequent bursts. In other systems an increase in burst frequency leads to an efficient increase in transcription output (Larson et al., 2013; Senecal et al., 2014). What is the advantage of more frequent bursts, if they do not lead to a significant improvement in the transcription output per cell? RSC makes more cells competent for transcription, and RSC activity reduces the transcriptional noise of the system as can be demonstrated by the reduction in Fano factor from 8.8 in active Rsc2− cells to 6.1 in Rsc2+ cells. Physiologically, this translates into a more uniform response of the population to a heavy metal stress. Notably, data for HIV expression indicate that more frequent bursts may increase the efficiency of the response to an external stimulus, and that the difference in chromatin state may affect the transcription noise (Wong et al., 2018).
Do the rapid and frequent bursts depend on rapid and frequent remodeling?
Interestingly, by MNase-seq RSC has a moderate effect on nucleosomal architecture of CUP1 promoter only in inactive cells. No effect on nucleosomal occupancy and/or changes in the length of nucleosome-depleted regions can be observed in activated cells. However, MNase-seq demonstrates a shift to a fluid chromatin state in activated cells (Figure 6B). Thus, remodeling events initiated by RSC may be short and transient. Indeed, as observed by SMT, RSC is rapidly cycling on CUP1, on the time scale comparable to that of Ace1p (Figure 6A). Repetitive rapid bursts of CUP1 transcription (Figure 5D) point to rapid cycles of chromatin remodeling / TF binding. We propose the following model for the correlation of TF cycling, remodeling, and transcription (Figure 6C). RSC affects Ace1p binding by increasing the rate at which nucleosomes shift from one position to another. Removal of the non-essential Rsc2p subunit of RSC slows down the cycling of Ace1p by decreasing the nucleosomal mobilization rate. The cycle starts when an individual RSC molecule, binding to the Upstream Activation Site (UAS) of CUP1 moves the −1 nucleosome from the partially obstructed RE and facilitates binding of an Ace1p molecule to the UAS. Binding of Ace1p switches the gene to the “ON” state. Ace1p recruits the components of the PIC and facilitates transcription. The next steps of the transcription process are not dependent on Ace1p. RSC removes Ace1p from the UAS by moving the −1 nucleosome into the UAS, and the gene switches to the “OFF” state.
Relevance of the interdependent dynamics of RSC and Ace1p at CUP1 promoter to mammalian genes
The CUP1 promoter is regulated only by two mutually exclusive pathways (heavy metal stress or heat shock response) controlled by either Ace1p or Hsf1p and contains only four REs for Ace1p and two for Hsf1p. Due to the limited number of interacting factors and REs this is an ideal system for modeling dynamics of TFs and chromatin remodelers. Mammalian promoters and enhancers quite often contain multiple REs for multiple TFs, and may be regulated simultaneously by multiple pathways, which makes similar analysis much more complicated. However, we expect that on the molecular level this multi-component regulation may be dissected into simple dynamic interactions analogous to those described in this paper. Specifically, we expect that regulation by search improvement may be important for those mammalian promoters that have to respond quickly and uniformly to environmental stress, and for which it is not critical to quickly turn off the gene by reducing residence time of the TF. Moreover, data of other groups indicate that the same mechanisms may also work for enhancers of multicellular eukaryotes, such as those regulated by the mammalian Glucocorticoid Receptor (GR). Hoffman et al., 2018 demonstrated by ChIP-seq the interdependent facilitation of RE occupancy by GR and the chromatin remodeler BRG1: binding of BRG1 is required to recruit GR, and GR recruits BRG1, which further improves GR binding. We expect the same at CUP1 as we observe that the RSC complex is bound at CUP1 in inactive cells, and after activation, when Ace1p binds, RSC is enriched at CUP1 (Figure 3A). Further support for the interdependent dynamics of TF and chromatin remodelers may be derived from the cycles of remodeling/TF binding/TF release that were observed in vitro for the reconstructed mouse mammary tumor virus (MMTV) enhancer array (Nagaich et al., 2004). In this system SWI/SNF chromatinremodeling complex facilitated not only binding to RE, but also the release of the glucocorticoid receptor (GR) from its RE on the MMTV promoter. Notably, the frequency of the binding/release cycle was dependent on the ratio of GR to SWI/SNF in the reaction mix, thus on the efficiency of remodeling.
In sum, we demonstrate a link between chromatin remodeling and transient binding of transcription factors to specific sites in promoters. Since nucleosome displacement by remodelers is widespread in all eukaryotes, we expect that transcription from many other genes will be similarly regulated by remodeler action.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Karpova Tatiana (karpovat@mail.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains and plasmids
Saccharomyces cerevisiae strains used in this study were derived from the haploids BY4742, BY4741, and diploid BY4743 from the Saccharomyces deletion project, isogenic to S288C (Research Genetics/Invitrogen, Huntsville, AL) (see Table S1 for yeast strains and Table S2 for plasmids). Sequences and maps of plasmids are available upon request.
All strains carrying HaloTag fusions carried pdr5Δ to enhance HaloTag dye retention (Ball et al., 2016). Standard methods were used for yeast transformation (Guldener et al., 1996). For gene deletions and C-terminal tagging of proteins, appropriate PCR fragments (see Table S3 for primers) were introduced into the genome by homologous recombination (Janke et al., 2004; Longtine et al., 1998). All deletions and C-terminal tags were confirmed by either diagnostic PCR or observation of the localization of the appropriate fusions by fluorescent microscopy.
For CUP1 locus visualization we used pTSK405 to amplify the 3xGFP::URA3 cassette for endogenous tagging of ACE1 in (YTK1390). For HaloTag fusions (YTK1391), we used pTSK561 (Ball et al., 2016) to amplify the HaloTag::URA3 cassette. Both fusion proteins were expressed from wild-type ACE1 promoter and thus were not overexpressed. No wild-type Ace1p was present in strains tested. Ace1p-3xGFP fusion and Ace1p-HaloTag fusion bind to CUP1 (Figure S1A). Tagging of Ace1p did not affect the steady state levels of ACE1 mRNA (Figure S1B) and it did not affect either transcription of CUP1 or resistance to copper (Figure S1C, S1D).
In YTK1528, PES4 ORF was replaced with 256 copies of lacO by a two-step PCR based method (Rohner et al., 2008) using pUG73 (Euroscarf, Frankfurt, Germany) and pSR7. To visualize pes4::lacO, GFP-lacI was integrated into LYS2 as a AhdI -BspE1 fragment of pLKL58Y (Tsabar et al., 2016).
For RSC2 deletion, pUG27-borne HIS5 cassette (Euroscarf, Frankfurt, Germany) was amplified using primers T329 and T330 and transformed into YTK1396 and YTK1391.
For auxin-inducible degradation of Rsc2, the endogenous copy of RSC2 was labelled as RSC2-AID*−9MYC or RSC2-AID*−6HA by amplifying the respective cassette from plasmid pHis-AID*−9Myc or pHyg-AID*−6HA (Morawska and Ulrich, 2013) using primers T882, T883. pADH1-AFB2-CYCT1 was expressed constitutively from SwaI-digested pTSK559 integrated into TRP1 locus; pTSK559 was constructed as follows. First, pTSK518 was synthesized by GenScript (Piscataway, NJ, USA). Then LEU2 was amplified from the yeast genome using T894 and T895 and cloned between XbaI-SacI sites of pTSK518 to obtain pTSK554. ThenpADH1-AFB2-CYCT1 was amplified (using primers T896 and T897) from pRS303-pADH1-AFB2 (plasmid #99530, Addgene, Cambridge, MA, USA) and cloned into pTSK554 between SacII-NotI to get pTSK559.
To visualize mRNA from a single copy of CUP1 within the CUP1 array, a CUPl-reporter was synthesized by GeneScript (Piscataway, NJ, USA). It carried a scrambled non-translatable sequence based on the mammalian rat Glucocorticoid Receptor, to which 8 copies of MS2 binding sites were appended (Figure S4). The CUP1-reporter was amplified by PCR from pTSK588 using T1010 and T1004, and the PCR fragment was transformed into YTK1443 replacing the ORF of a single copy of CUP1 within the array. It was expressed from the wild type CUP1 promoter (Figure 4A). We constructed plasmid pTSK588 by cloning the CUP1-reporter into pBSII SK (+) between XhoI-NotI sites and selection marker LEU2 between NotI-SacI sites.
Media and growth conditions
Yeast cells were grown in complete synthetic media (CSM) with respective dropout amino acid mixture (MP Biomedicals, CA, USA). For all microscopy experiments, CSM (+respective dropout amino acid mixture) + 6.5 mg/ml Sodium Citrate was used. To monitor binding of Ace1p-3xGFP to the CUP1 array, cells were treated by 100 μM CuSO4. All experiments were performed at 30 °C at 230 RPM.
For SMT, cultures grown overnight to saturation were diluted to 0.2 OD600 in 3 ml fresh media (in 14 ml polypropylene tubes, Cat no. 352059, Falcon, Maxico). Cells were grown for four hours. Cells were pelleted and resuspended in 1 ml of fresh media with 0.5 nM JF646. Cells were grown further for 30 min and harvested for imaging under the CSM+100 μM CuSO4 containing agarose pads. For auxin-based depletion of Rsc2, Indole-3-acetic acid (IAA, Sigma, Cat no.: I5148) was added to the culture media at 1 mM concentration four hours before harvesting for the complete degradation of the target protein (Nishimura et al., 2009). Transcription of CUP1 was not affected by auxin treatment (Figure S3A). Dynamics of Rsc2p-AID*−9Myc degradation was observed by Western blotting (Figure S3C).
For the Western blots and RT-qPCR, cells were grown exactly like SMT, but JF646 was not added. Copper induction was given in the tubes by adding 100 μM CuSO4 directly to the culture media and shaking the tubes for exactly 10 min.
For smFISH, overnight grown culture was inoculated in 100 ml of fresh media at OD600 0.2 and grown for 240 min in 500 ml flask. Cells were pelleted and resuspended in 35 ml of fresh CSM-LEU media and grown with aeration for 30 min in 500 ml flask. 8 ml samples were harvested as an uninduced condition and fixed with 2 ml of 16% formaldehyde. 100 μM CuSO4 was added to the remaining culture and kept for shaking for exactly 10 min. 8 ml samples were harvested as an induced condition and fixed with 2 ml of 16% formaldehyde. smFISH for the CUP1-reporter mRNA was performed and quantified.
For the quantitative cell viability assay/copper resistance assay (Figure 3C and S1C), cells were grown to O.D.600 0.9. Cell concentration was estimated in a hemocytometer. Cells were diluted to 500/ml−1 in 40 ml of autoclaved distilled water and placed in a sterile Petri dish (D = 90 mm). The cell suspension was plated with a pin replicator onto the plates with solid CSM media (DOBA, MP Biomedicals, CA, USA), containing either auxin (1 mM) and/or CuSO4 (100 μM). Plates were incubated at 30°C for 72 hours and colonies were counted manually. Experiment was repeated thrice, values were normalized to maximum for each replicate and averaged. Error bars indicate standard error of the mean (SEM).
METHOD DETAILS
Chromatin immunoprecipitation (ChIP)
ChIP was performed as in (Mehta et al., 2014) with modifications. Approximately 109 cells were used per sample (four IPs and an Input). Chromatin was cross-linked using 1% formaldehyde for 30 min at 25 °C. Cells were washed twice with ice cold TBS (0.25 M Tris base, 1.37 M NaCl, 0.03 M KCl) and cell lysis was performed in 1.6 ml of lysis buffer (50 mM HEPES-KOH, pH 7.5, 140 mM KCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, freshly added protease inhibitor cocktail from Roche, Germany) and equal volume of 0.5 mm glass beads (Scientific Industries, Bohemia, NY, USA) using MP FastPrep-24 bead beater (MP Biomedicals, USA, 4 cycles of 40 s each with 5 min on watery ice in between). Chromatin was fragmented to an average fragment size of 200–500 base pairs using Diagenode Bioruptor (Diagenode, USA) for 30 cycles of 30 s each at high setting. Sonicated lysates were centrifuged (14000 RμM for 10 min) to remove cell debris. Out of ~1.4 ml of clarified lysate, 100 μl lysate was stored at −20°C as an input (Whole Cell Extract, WCE) and two IPs of 600 μl each were rotated over-night at 4°C with the following beads/antibodies: 20 μl of GFP-TRAP-MA (Chromo Tek, Germany, Cat no. gtma-20) for pulling down Ace1-3xGFP, 5 μg of Anti-Myc tag antibody (abcam, Cat no. ab9132) for pulling down Rsc2-AID*-9Myc, 5 μg of Anti-HaloTag antibody (Promega, G9281) for pulling down Ace1-HaloTag. Following overnight incubation with antibodies / beads, 20 μl of Protein A+G magnetic beads (Millipore-Sigma, Cat no. 16-663) were added to each tube for pulling down Rsc2-AID*−9Myc and Ace1-HaloTag and rotated at 4°C for 2 h. The immunoprecipitated material was washed twice with 1 ml of IP wash buffer I (50 mM HEPESKOH, pH 7.5, 150 mM NaCl, 1 mM EDTA, 0.1% sodium deoxycholate, 1% Triton X-100); once with 1 ml of IP wash buffer II (IP wash buffer I with 500 mM NaCl), once with 1 ml of IP wash buffer III (10 mM Tris pH 8.0, 250 mM LiCl, 0.5% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA) and once with 1 ml of 1 × TE. The beads were resuspended in 100 μl of elution buffer (50 mMTris, pH 8.0, 10 mM EDTA, 1% SDS) and incubated at 65°C for 15 min. The elution step was repeated in 50 μl of elution buffer and both the eluates were mixed to get the total eluate volume of 150 μl per IP and incubated overnight at 65°C for decrosslinking. As input control, 100 μl of WCE (stored at −20°C) was mixed with 50 μl of elution buffer and incubated overnight at 65°C for decrosslinking. DNA from all the samples (IPs and Inputs) were purified using Wizard SV Gel and PCR clean up system (Promega, Cat no. A9282) according to the manufacturer’s instructions and eluted in 50 μl of elution buffer. qPCR was performed in duplicate using a Bio-Rad CFX96 machine in 20 μl SYBR Green reaction mixture (Bio-Rad, USA, Cat no. 170-8882). T509 and T510 primer pair was used to amplifypCUP1 locus (Table S3). Calculations for Enrichment/Input values (%) were made using the following equations: ΔCT = CT(ChIP) –[CT(Input) – logE (Input dilution factor)], where CT is a Cycle Threshold, E is the specific primer efficiency value; Enrichment/Input (%) =E−ΔCT All the ChIP experiments were performed at least twice, values were normalized to maximum for each repeat and averaged. Error bars indicate SEM. YTK1390 strain was used as a No Tag Control.
Quantitative RT-PCR (RT-qPCR)
RNA preparation was performed as described (Ball et al., 2016). cDNA was prepared using the iScript cDNA synthesis kit (BioRad, Inc. Cat no.: 1708891) starting with 1 mg of total RNA. Quantitative real-time PCR (qPCR) was performed as described (Ball et al., 2016). Primers used for the quantification of transcripts were T531, T532 for CUP1, and T1019, T1020 for CUP1 reporter (Table S3). For normalization, the expression of the housekeeping gene TUB1 or ACT1 was quantified using primers T992, T993, or T1055, T1056, respectively (Table S3). To confirm the absence of contaminating genomic DNA in cDNA preparations, reverse transcriptase negative (-RT) samples were used as a control, which produced the Ct value difference of ≥10 cycles between “-RT” and “+RT” samples, indicating negligible amount of genomic DNA contamination in cDNA samples. mRNA extraction, cDNA synthesis and qPCR were repeated at least twice, and qPCR was performed in duplicates for each experiment. Error bars indicate SEM.
Protein concentration by Western blots
Cells of YTK1649 were grown from OD600 0.2 to 0.8 in 3 ml CSM-LEU media. Protein extraction and western blotting were performed by the standard procedure using anti-HA (1:3000 dilution, Roche Mouse Anti-HA (12CA5) Cat no. 11583816001), anti-c-Myc (1:3000 dilution, Roche Mouse Anti-c-Myc (9E10) Cat no. 11667149001) and anti-Tub2 (1:4500 dilution, Rabbit serum) antibodies. Protein extraction was repeated twice, values of band intensities (after background subtraction) were normalized to maximum and averaged. Error bars indicate SEM.
Quantification of Ace1p-GFP
To measure the nuclear brightness of Ace1p-GFP (Figure S3D) or the brightness of the individual arrays (Figure S2E), cells were imaged with the 100× 1.4 NA oil immersion objective using the FITC filter set (Ex 475/28; Em 525/48 Chroma Technology Corp, Bellows Falls, VT) on a DeltaVision Elite imaging system (GE Healthcare, IL, USA) based on an Olympus IX71 inverted microscope (Olympus America, Inc. Melville, NY) equipped with LED illumination and a sCMOS pco.edge camera (PCO AG, Kelheim, Germany), and controlled by SoftWoRX software. Z-stacks of 9-11 focal planes were acquired with a Z step size of 350 - 400 nm, and XY pixel size of 65 nm (bit depth: 15-bit). Exposure was set to 150-300 ms. For the nuclear measurements different fields of cells were collected. For the array measurements, movies with a 30 s time lapse were acquired. Measurements of the nuclear brightness or of the array brightness were performed in > 150 cells or 38 arrays, correspondingly. Fluorescence intensity was measured from the projection images using Metamorph (Molecular Devices, CA, USA). Average image background intensity was subtracted from the average intensity. In time lapse movies brightness of the arrays was divided by the brightness of Ace1p in the nucleoplasm to correct for the photobleaching due to imaging. The brightness curves were normalized to the first time point, where no arrays were yet observed.
Custom-built Microscope for SMT
For SMT, we used a custom wide-field microscope capable of simultaneous imaging in three channels that is based on designs previously described (English and Singer, 2015; Morisaki et al., 2016). Briefly, three solid-state excitation lasers at wavelengths 488, 561, and 647 nm (OBIS, Coherent Inc., CA, USA) are combined, expanded to provide more even illumination at the sample, and focused at the back focal plane of a 100x, 1.49 N.A., oil immersion objective (Olympus Scientific Solutions, MA, USA). To reduce background, HILO illumination (Tokunaga et al., 2008) is achieved by moving the radial position of the beam in the objective back aperture with a movable mirror, and the thickness of the excitation is adjusted by use of a manual diaphragm. Fluorescence emission is separated from scattered laser light by use of a quad-band dichroic (ZT405/488/561/647, Chroma Technology Corp., VT, USA), and the emission bands are separated by two long-pass filters (T588lpxr, T660lpxr), and emission filters (525/50, 609/58 and 736/128, Semrock, Inc., NY, USA) before being focused on separate EMCCD cameras (iXon Ultra 888, Andor Technology Ltd., UK), with 200 mm tube lenses. The combination of objective lens and tube lens results in a total magnification of 111x, corresponding to a XY pixel size of 117 nm. The sample is held on a motorized XY translation stage with piezo Z (PZ-2000 XYZ, Applied Scientific Instrumentation, OR, USA), which is mounted on a Rapid Automated Modular Microscope System (RAMM, Applied Scientific Instrumentation). All lasers and cameras are synchronized using a microcontroller board (Arduino UNO), and images are collected using the open source microscope control software, Micro-Manager (Edelstein et al. 2014).
SMT: staining, imaging and quantification
The protein of interest (Ace1p-HaloTag or Rsc2p-HaloTag) was labelled with JF646 at 0.5 nM concentration as described in (Ball et al., 2016). Live cells were imaged under CSM+100 μM CuSO4 agarose pads using LabTek II chambers. We simultaneously imaged GFP and JF646 channels on the custom-built imaging system at room temperature. Time-lapse movies were acquired as 16-bit single focal plane images with 30 ms laser irradiation (Ex: 100 μW 488 nm; Ex: 1 mW 647nm) and 200 ms time interval for a total of 2 min.
In our experiments we were interested in assaying the TF binding to a specific promoter. Tracking of Ace1p in the whole nucleus may measure binding not only to CUP1 but also to the other four promoters that are regulated by Ace1p (Gross et al., 2000). To ensure that we are measuring the TF binding either at CUP1 promoter, or at the region where there are reliably no known REs for Ace1p, Ace1p-HaloTag-JF646 or Rsc2p-HaloTag-JF646 were tracked only at the ROI defined by the green spot of the CUP1 or lacO array. We compared binding over the CUP1 array to binding over lacO (Figure 1) to offset any unforeseen effects of measuring the region smaller than the whole nucleus. We also tracked Ace1p in the whole nucleus and compared it with Ace1p at the CUP1 array from the same cells (Figure S2B). We found that the parameters for non-specific binding remain the same, hence tracking volume doesn’t affect non-specific binding. SMT for Figure 1, 2 and 6 were performed with ROIs manually drawn around individual arrays based on the GFP images. The sizes of the ROIs were similar for Rsc2p+ and Rsc2p−, with mean areas of 0.93 ± 0.03 (S.E.M.) μM2, and 1.16 ± 0.05 μM2, respectively (p-value = 0.009, by Kolmogorov-Smirnov test), while the lacO arrays were slightly smaller with a mean area of 0.5 ± 0.2 μM2. This variability may be due to the different level of decompactization of the DNA. Heavily transcribed arrays of loci may become bigger due to looping out of the transcribed DNA. In addition, it is known that in RSC2-depleted cells the sister chromatids are prematurely separated, which may contribute to the larger perceived size of the array.
Tracking was performed automatically with the MATLAB-based (The Mathworks, Natick, MA). Track Record software (freely available at https://sourceforge.net/projects/single-molecule-tracking). Track Record links molecule positions in two consecutive planes using a nearest-neighbor approach (Mazza et al., 2012). Molecules are considered bound if they move less than rmax = 220 nm from one frame to the next (Ball, et al 2016) for at least Nmin = 4 frames (Figure S2A). Assuming a moderate diffusion coefficient, Df, of 0.5 μM2/s, with our frame interval, Δt, of 200 ms, these parameters reduce the probability that a freely diffusing molecule will be classified as bound to P = 1.7×10−4 based on the equation (Mazza et al., 2012):
The total bound fraction, Ceq, is found by summing up the number of time-points in all bound track segments and dividing by the total number of particles found. The fraction of diffusing molecules is therefore given by (1- Ceq).
The survival distribution (1 – CDF) is generated from the bound track segments. Photobleaching is measured within each experiment by counting the number of particles found in each frame, and fitting to a bi-exponential decay by least-squares. The survival distribution is then divided by the best-fit photobleaching curve to correct for photobleaching and normalized to the total bound fraction Ceq. This procedure was validated in Monte Carlo simulations (Ball et al., 2016).
Mean residence times and the fraction of total molecules within each population were estimated by fitting by least squares the photobleaching-corrected survival plots to mono- and biexponential functions. Typically, the plots are fit best with bi-exponential decay. The fastest (short-residence) component represents nonspecific binding to DNA, i.e. sampling the DNA for binding sites. See (Ball et al., 2016) for further discussion of nonspecific binding of Ace1p. The second (longer-residence) component represents binding to specific RE (specific binding). The third sub-population shown in Figures 1 and 2 arises from those track segments that were considered unbound based on the criteria described above, and particles that were not tracked and represent diffusing molecules. Diffusion coefficients for all the fractions for Ace1p and Rsc2p are presented in Figure S2C. Due to the long exposure time relative to molecular diffusion, we likely underestimate the number of freely diffusing TFs. However, the focus of this study is on the bound molecules. Comparisons between the various strains remain valid, as all were imaged under the same conditions.
The track analysis method that we adopted (fitting of the Cumulative Distribution Function, or CDF) is based on survival rates (Morisaki et al, 2014; Loffereda et al, 2017; Presman et al., 2017). Other methods available are Probability Density Function (PDF) fitting (Das et al 2009; Liao et al 2015) or variational Bayes Single-particle tracking (vbSPT, Persson et al., 2013). As evaluated in detail in Presman et al., 2017, CDF fitting provides better estimates for the residence times of bound molecules than PDF fitting and thus it is a better match for our scientific focus on bound, rather than diffusing TF. In this paper we further evaluated comparative applicability of CDF fitting and vbSPT specifically to our data range. We generated artificial tracks based on parameters extracted from SMT of Ace1p in RSC+ cells activated by Cu2+, i.e. based on data of Figure 1C and1D. Then we applied CDF fitting and vbSPT analysis to the simulated tracks (Figure S2D). Ideally, a good analysis method would return the parameters close to those used to simulate the tracks. The vbSPT analysis, as opposed to CDF fitting, returned dramatically incorrect residence times for both short-residence and long-residence molecules. Interestingly, correct estimates for the fraction of diffusing molecules are returned by vbSPT only for the homogeneous population (lacO) but this fraction is grossly underestimated if a second, specifically-bound fraction is present (CUP1 array). Thus, we adopted CDF fitting for our data.
Quantification by survival rates adopted in this paper assumes that the Ace1p binding is in steady state. We ensured that we perform SMT at the Ace1p binding-unbinding equilibrium. Ace1p-GFP binding measured by fluorescent intensity of the array indicates that for about ~ 5 min it remains relatively constant, i.e. the system is in steady state. This plateau in fluorescent intensity is preceded by a rapid rise in fluorescence after the addition of copper and is also followed by a rapid decline in fluorescence (Figure S2E). These phases of fluorescence increase or decrease were avoided in all of our SMT measurements, as we started measurements after 5 min of Cu activation and performed them on bright arrays for 2 min total time.
smFISH: staining, imaging and quantification
By smFISH one can observe mature transcripts and identify CUP1 transcription sites (TS) containing nascent mRNA. Due to the multiple copies of CUP1 in the genome, smFISH of native CUP1 does not allow for the monitoring of the transcriptional output of an individual gene in the array. We therefore replaced one ORF within the array with a synthetic CUP1-reporter. This reporter was derived from the scrambled sequence of the rat Glucocorticoid Receptor (GR), to which 8 MS2-binding sites were appended (Figure 4A, and S4). As the sequence of this reporter is unique, its transcripts may be observed separately from the transcripts from the remaining copies of CUP1. The sequence contains multiple stop codons and cannot be translated, preventing any toxicity from the expressed protein.
Cells were stained with TAMRA-labeled custom-made Stellaris smFISH probes (synthesized by LGC Biosearch Technologies, Petaluma, CA, USA) specific for the transcripts of CUP1-reporter (see Figure S4 for the position of the probes). Briefly, we fixed the cells with 3.7% formaldehyde after 10 min of Cu induction (or control cells with or without Cu/Auxin induction). Fixed cells were stained with smFISH probes as described in (Raj and Tyagi, 2010). After staining, cells were resuspended in 30 μl of Antifade buffer with glucose oxidase and catalase. 5 μl cell suspension was layered on a poly-L-lysine coated cover glass. A drop of Vectashield mounting medium (+DAPI, Vector Laboratories, UK) was placed in the center of a glass slide and this slide was placed upside down on top of the coverglass and pressed gently to mix the cells in suspension with Vectashield. Cells were imaged with the 100× 1.4 NA oil immersion objective on a DeltaVision Elite imaging system. Z-stacks of 50 focal planes were acquired with a step size of 200 nm, and XY pixel size of 65 nm (bit depth: 15-bit). Images of ~500 cells were captured using filter sets for DAPI (Ex 390/18; Em 435/48) and TRITC (Ex 542/27; Em 597/45, Chroma Technology Corp, Bellows Falls, VT) and deconvolved. Exposure was set to 300 ms for TRITC channel and 10 ms for DAPI channel.
Images were analyzed using FISH-Quant software (Mueller et al., 2013). Mature RNAs for measurements were identified in deconvolved images as 3D gaussian spots with intensity higher than an arbitrary threshold, which was held constant for all acquisitions belonging to the same experiment. The number of mRNA per cell, number of nascent transcripts per transcription site and the brightness of the individual mRNA spot were measured from raw images. The spots that we measured were indeed single molecules of mRNA, because of the narrow distribution of the brightness of the individual mRNA spots (Figure S5A). Quantification of the transcription of the reporter by smFISH and by RT-qPCR demonstrate that both methods applied to the same reporter register the same proportion in transcription reduction in absence of RSC. Thus, smFISH is an adequate method for quantification of the expression of the CUP1 reporter (Figure S5B). Transcriptional output of the CUP1 reporter inserted under a natural CUP1 promoter is representative of the other CUP1 genes of the same array because quantification of CUP1 reporter transcription provides results similar to those obtained for the whole array of the endogenous CUP1 in the same culture (Figure S5C). Experiment was repeated twice and error bars indicate SEM (Figure S5B, S5C).
mRNA degradation rate
The degradation rate of the CUP1 reporter mRNA was measured in YTK1649 by smFISH as above. 200 cells were measured every 5 minutes after washing out the Cu2+ from the growth medium (Figure S5D, upper panel). The degradation rate of the endogenous CUP1 was measured by RT-qPCR, as transcription of ten copies of CUP1 results in very large amounts of mature mRNA in the cytoplasm uncountable by smFISH due to overlapping spots. Cells for RT-qPCR were harvested every 5 minutes after washing out Cu2+. In both cases, the degradation rate was calculated by fitting by least squares the mean mRNA values of the time-course to a monoexponential decay (middle and lower panel). We can measure the activity of individual promoters only by reporter transcription, therefore we have to input the degradation rate of the reporter into our model. The endogenous CUP1 mRNA is more stable than mRNA of the reporter (12.1 ± 3.4 min vs 8.2 ± 1.6 min). However, this difference does not affect our conclusions. If the degradation rate changes, then all the parameters would be scaled proportionally, but this would not affect the ratio between those.
CUP1 nucleosome occupancy by MNase-seq
Nucleosome occupancy was determined for the following four conditions: (1) Rsc2+ (No Auxin), no Cu activation, (2) Rsc2+ (No Auxin), with Cu activation, (3) Rsc2− (With Auxin), no Cu activation, (4) Rsc2− (With Auxin), with Cu activation. Cells of haploid YTK1460 were grown overnight in 10 ml CSM-URA and diluted to OD600 0.2 in 400 ml of the fresh CSM-URA. Cells were grown for 270 min to OD600 0.8 at 30°C and 230 RPM. For auxin-induced Rsc2p depletion IAA was added to the culture media 240 min before Cu treatment to a final concentration of 1 mM. For copper activation, cells were treated for 10 min with 100 and 4 μM CuSO4. Cells were harvested by filtering the culture through 0.45 μm Nalgene Rapid Flow Nylon Filter unit (Thermo Fisher Scientific, Cat no. 151-4045) and washing the cells with ice-cold autoclaved distilled water into 50 ml Falcon tubes. Pellets were frozen at −80°C.
Nuclei were prepared and digested by MNase as described (Cole et al., 2012). DNA was purified from digests composed predominantly of mono-nucleosomes. Nucleosomal DNA (0.5 - 1 μg) was repaired using the PreCR repair kit (NEB M0309). Paired-end libraries were prepared from the repaired DNA using NEB Illumina kits E7370 and E7335, with 7 PCR cycles, according to the manufacturer’s instructions. The samples were multiplexed and sequenced by an Illumina HiSeq2500 (2x 50 nt reads). Nucleosome occupancy plots were constructed as described (Chereji et al., 2017). The sequencing data from this study, including biological replicates, have been submitted to the NCBI Gene expression Omnibus under accession number GSE112685 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?token =mdcragqebhopnob&acc=GSE112685).
The yeast genome sequence used for alignment includes two identical copies of the CUP1 locus, named CUP1-1 and CUP1-2. Our data show that the coverage of CUP1 locus is about 5X of the flanking sequences, indicating that there are about ten CUP1 repeats in this strain. When cells are depleted of Rsc2p, the Transcription Start Site (TSS), initiation element and TATA box show decreased nucleosome occupancy, whereas the UAS elements show a small increase in nucleosome occupancy. The latter effect is primarily due to an upstream shift of ~40 bp in the position of a particular +1 nucleosome such that it now covers the distal UAS as well as the proximal UAS. The net effect of Rsc2 is to increase the accessibility of the distal UAS.
QUANTIFICATION AND STATISTICAL ANALYSIS
Relationship between SMT parameters
In the main text, we use a simplified system (Eq. 1) to illustrate the interdependence of off-rate (koff), pseudo ‘on’-rate (k*on, and bound fraction (Ceq). When a TF transiently binds to random DNA sequences and more strongly to its target sequence, Eq. 1 becomes (Loffreda et al., 2017)
| (Eq M1) |
where Fs is the fraction of bound molecules that are associated to their target sequence, k*onS, is the pseudo-‘on’ rate for the specific binding, k*onN is the pseudo-‘on’ rate for non-specific binding, and koffS and koffN are the ‘off’ rates for the specific and non-specific bound molecules, respectively.
The fraction of bound molecules that are at the target sequence can also be expressed solely in terms of the pseudo ‘on’-rates
| (Eq M2) |
Solving Eq M1 and M2 for k*onS and noting that the average lifetime of all bound molecules, the pseudo ‘on’-rate for the specifically bound population can be calculated based on the values obtained from fitting the SMT data for total bound fraction, Ceq, slow-bound fraction, Fs, and the lifetimes of the two states, τS and τN
| (Eq M3) |
In addition, if we note that defines the average time between two binding events, Eq. Ml becomes
| (Eq M4) |
Rearranging Eq. M4 yields
| (Eq M5) |
On average, it will take 1/FS trials for the TF to locate its target sequence, with each trial requiring a time equal to τ3D + τN, with the exception of the final trial, which requires only τ3D. The total time required to locate the target sequence is therefore
| (Eq M6) |
Note that if there is only specific binding, FS = 1, and therefore , which is a rearranged version of Eq. 1 from the main text.
Estimating fraction of non-active cells
The diploid YTK1649 cells were treated the same as described above for smFISH without the addition of Cu2+ to obtain the basal transcription output of CUP1. In this control group, cells contain on average very few mature mRNA of CUP1-reporter (0.46 ± 0.03 (SEM)) per cell, with a maximum of 8. Cells that do not contain any mRNA constitute 67.2% of the whole population (Fig. 4D and4E). The histogram of mature mRNA is fit well to a Poisson distribution with a mean of 0.46, indicating that basal expression of CUP1 is governed by random, uncoordinated initiation, rather than a bursting behavior.
To estimate the fraction of non-active cells, Fnt, in the Cu2+-treated populations of Rsc2+ and Rsc2−, the initial part of each distribution (cells containing three or fewer transcripts) was fit to a scaled Poisson with the mean, γ, fixed at the mean of the control cells (0.46 transcripts/cell), and varying only the scaling parameter α:
| (Eq M7) |
The scaled Poisson with a fixed mean was used instead of directly fitting to a separate Poisson distribution to avoid overestimation of Fnt due to active cells with low mRNA levels.
Estimation of Seq and kon
If we assume that binding of Ace1p to any of the four binding sites in the CUP1 gene occurs equally and independently and results in transcription, then we can relate Fnt to the probability, P, that any one RE is available:
| (Eq M8) |
By knowing how many RE are available for each gene and assuming a nuclear radius of 1 μM, corresponding to a volume of V = 4.19 × 10−15 1, we may calculate Seq
| (Eq M9) |
where NA is Avogadro’s constant.
Monte Carlo Simulations of TF binding
To examine the relative impact of changes to the RE availability or ‘on’ rate on transcriptional output, we performed Monte Carlo simulations of a random telegraph model in MATLAB (The Mathworks, Natick, MA) using Gillespie’s SSA algorithm (Gillespie, 1977). Promoters of CUP1 contain four REs for Ace1p (Shen et al., 2001). Thus, we assigned four REs to simulated CUP1 promoters, and we assumed that binding of Ace1p to any of the four REs resulted in equally efficient transcription. The simulation of each cell was initialized with a fixed fraction of available binding sites, by selecting a binomially distributed random number with the number of trials equal to the total binding sites, and the probability equal to the specified fraction of available sites. Once initialized, the availability of each site remained fixed throughout the time-course. Each available RE was either unbound or bound by a TF and switched to these states with exponentially distributed waiting times with means 1/koff and 1/k*on, respectively. When any of the RE was in the ‘ON’ state, transcription was initiated with an exponentially distributed waiting time with mean, 1/ktx. At all time-points of the simulation, each transcript was classified as either nascent if it had existed for less than the production time (tprod, or mature if it had existed for longer. Mature transcripts could degrade with an exponentially distributed lifetime with mean of 1/kdeg.
When assessing the role of the RE availability, probabilities between 1-99% were set in the model, while keeping all other parameters fixed. Similarly, to determine the impact of the ‘on’ rate, values between 1.35 χ108 and 5.4 χ 108 M−1 s−1 were used with other parameters held constant. For clarity, we plot the mean number of transcripts in active cells vs. the fraction of non-active cells of these scans (Figure S6A). Neither of the curves fits the experimental data. Although the experimental data (black dots) are close to the simulated plot for the variability of RE (green curve), the difference between the mean mRNA value for Rsc2+ and the value on the green line for the same fraction of non-active cells is statistically significant based on a Kolmogorov-Smirnov test (p-value <0.001). We also varied the ‘on’ rate over a greater range (from 104 – 1012 M−1s−1) to determine if a much faster (or slower) ‘on’ rate could recapitulate the experimentally obtained mean mRNA in active cells (Figure S6B, black line), and the fraction of non-active cells (Figure S6C, black line). In this case, all REs were made available by setting the probability to 1. The result of this parameter scan showed that there exist 3 regimes: (1) with an ‘on’ rate below ~107 M−1s−1 only the fraction of non-active cells changed, while the mean number of mRNAs in active cells remained fixed at a very low abundance (< 2 mRNAs/cell); (2) between ~107-~1010 M−1s−1, only the mean number of mRNA in active cells changes; (3) above 1010 M−1s−1, both the fraction of non-active cells, and the mean number of mRNA in active cells remain constant. Increasing the number of bound REs required to start transcription to two (red curves in Figure S6B and C) or three (blue curves in Figure S6B and C) only changes the value of kon required to obtain the observed mean mRNA in active cells (~20 per cell), but still does not provide a regime where only the fraction of inactive cells changes. Therefore, it is not possible to simultaneously obtain different fractions of active cells and the same (high) mean number of mRNA by changing only the ‘on’ rate.
To further test that, we looked at the impact of changing the probability of RE availability (P) alone, or of all three parameters: P, kon, and koff. Experimentally, for CUP1 we observe that in Rsc2+ the fraction of the cells with non-activated CUP1 decreases, and the trancriptional output of activated CUP1 stays the same: 19 ± 0.5 (S.E.M) for Rsc2− and 18 ± 0.4 for Rsc2+.
Simulations show that maintaining the same level of output requires changes not only in P but also in kon, and koff. Specifically, if a gene contains four REs (such as CUP1), then increasing only P from 0.28 to 0.38 resulted not only in a reduction in the fraction of the cells with non-activated CUP1 similar to that observed in experimental data, but also increased the mean simulated transcriptional output of active cells, which contradicts the experimental data (Figure S6D). Thus, we cannot reproduce this result if only the availability of RE changes. Only by increasing P, kon, and koff were we able to see something similar to our experiments (Figure S6E). Namely, the fraction of cells with non-activated CUP1 decreases the same as in Figure S6D, but the mean mRNA in cells with active CUP1 is relatively unchanged. In contrast, when a gene contains only a single RE, the results of changing only P (figure S6F), or changing P, kon, and koff (Figure S6G) are indistinguishable.
Our model also indicates that all four REs for CUP1 actively participate in binding. Assuming the gene contains four RE, simulations of the distribution of mRNA/cell are similar to the experimental (compare Figure S6D and E with Figure 5C). In contrast, if the gene is assumed to contain only one RE, the distribution of mRNA/cell is much narrower than experimental distributions (compare Figure S6F and G with Figure 5C), which provides further evidence that CUP1 gene contains multiple (most likely four) RE.
Finally, we tested the assumptions of our model. We assume that all four binding sites for Ace1p in the CUP1 promoter are equal, and productive binding of at least one Ace1p converts the gene to ‘ON’ state. Our assumption that Ace1p does not bind cooperatively is based on the following: (a) a single binding site is enough to activate other genes of the copper regulon by Ace1p (Pena et al., 1998); (b) Ace1p does not dimerize prior to binding (Buchman et al., 1990). We decided to test the outcome of fitting of experimental data based on a requirement for transcription of multiple bound Ace1p molecules. Assuming a requirement for multiple binding events dramatically affected predictions for the ‘on’ rate. In the case where there is no cooperativity and only a single RE binding is sufficient, the predicted ‘on’ rate is 2.7×108 M−1s-1. If transcription of CUP1 requires two REs to be occupied by TFs the returned ‘on’ rate will be ~3× faster (8×108 M−1s−1), and ~100× faster (3×1010 M− s−1) if CUP1 requires three REs to be occupied by TFs to begin the transcription. The high ‘on’ rate found assuming multiple occupancy is in agreement with measurements of the lac repressor in bacteria cells (Riggs, et al 1970). However, most studies of DNA-binding proteins have found rates that are on the order of 108 M− s−1 (Kim, et al 1987, Kleinschmidt, et al, 1988, Hoopes, et al 1992). We therefore chose the model with a requirement of a single RE-TF complex. Importantly, regardless of the required number of RE-TF complexes our conclusion remains unchanged: observed behavior, namely, that the number of active cells increases with Rsc2p, but the mean number of mRNA in active cells stays the same, may be explained only by simultaneous changes both in ‘on’ rate and RE availability (Figure S6B). Thus, simulations showed that our data can be fit well without the addition of cooperative binding. However, we do not exclude cooperative binding completely. We just argue that cooperative binding is not necessary to explain our results.
Our model takes into account the summary result of the extrinsic and intrinsic noise, although it is not possible for us to completely separate these sources of noise as we only observe the output of one gene (CUP1 reporter) per cell. Assuming four REs, we model a system that displays a combination of extrinsic and intrinsic noise. The cell to cell variability in the model with four REs arises because in every cell, each of the four REs are either open or closed and remain in that state throughout the simulation. In a real cell this would lead to intrinsic variation between several copies of CUP1, which we cannot observe. However, in the model it results in different simulated cells having a different number of available REs and thus in extrinsic variation. A cell with more available REs will have a higher probability of binding a TF, and therefore produce more mRNA. In contrast, when only a single RE is present, each cell either has one available RE or none. Therefore, all active cells behave more similarly, and this situation models a system with only intrinsic noise. Based on comparison of the distributions in Figure S6E and G, the CUP1 system displays both intrinsic and extrinsic noise. When the model includes 4 REs, we accurately reproduce the variability in the experimental data as characterized by the Fano factor (ratio of variance to mean). For Rsc2+, the Fano factor of active cells is 6.1, while for Rsc2− it is 8.8. If the model only uses a single RE, the Fano factor is reduced to 1.8, which is close to Poisson-like noise (Fano factor = 1), and therefore indicative of purely intrinsic noise. Therefore, as four REs allow us to fit the data much better than one RE, it indicates that in experimental data we observe a mix of extrinsic and intrinsic noise, although the relative impact of intrinsic and extrinsic noise cannot be derived from the existing experimental data for the single reporter.
Curve-fitting of mature transcript distributions
We fit the mature mRNA distributions of the CUP1 -reporter obtained by smFISH for both Rsc2+ and Rsc2− using the above random telegraph model. During the fitting, kof was held constant at the appropriate value: 0.19 for Rsc2−; 0.46 for Rsc2+, while Seq and kon were kept at the values described above, and the mRNA degradation rate was a constant obtained by measuring a smFISH time-course on fixed cells (Figure S3C). We then extracted ktx and tprod following the fitting strategy of (Senecal et al., 2014). Briefly, we used a custom random sampling method to explore parameter space, in which new possible parameters were selected to be close to linear combinations of previously found well-fitting parameters. Experimental mRNA distributions were compared to the model using the sum of the squared residuals. This process was repeated until 1000 parameter sets were obtained, and we report the mean and 95% confidence intervals of the best-fitting parameters. Table 1 shows the values of all parameters used to obtain the fits for the data shown in Figure 5C.
We then used the primary parameters from the model to obtain the characteristics of the transcriptional bursts. The bursting dynamics of transcription are typically characterized by the burst duration, , which is the average time the gene is ON, the frequency, , which is how often the gene turns on, and the amplitude, , which is the average number of transcripts produced in a single burst. A schematic representation of the model parameters and burst characteristics is presented in Figure 5B, 5D.
DATA AND SOFTWARE AVAILABILITY
The sequencing data for MNase-seq experiments, including biological replicates, have been submitted to the NCBI Gene expression Omnibus under accession number GSE112685 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi7token =mdcragqebhopnob&acc=GSE112685). Original images of SMT, smFISH and western blot are uploaded to Mendeley data (for SMT and smFISH, only two images of each experimental condition were uploaded due to database size limitations) (http://dx.doi.org/10.17632/hp9bwtnbgc.1). TrackRecord (MatlabTrack_v5.4.4) can be downloaded from https://sourceforge.net/projects/single-molecule-tracking.
Supplementary Material
Ace1-HaloTag-JF646 over the CUP1 array, related to figures 1A, 1C, 1D, 2 Time lapse images with 200 ms interval and 600 frames. Playback speed: 30 fps (6x faster than real time), Red channel: Ace1-HaloTag-JF646, green channel: Ace1-3xGFP, Scale bar: 2 μm. Left: Raw data, right: filtered data.
Ace1-HaloTag-JF646 over the CUP1 array, related to figures 1A, 1C, 1D, 2 Time lapse images with 200 ms interval and 600 frames. Playback speed: 30 fps (6x faster than real time), Red channel: Ace1-HaloTag-JF646, green channel: Ace1-3xGFP, Scale bar: 2 μm. Left: Raw data, right: filtered data.
Highlights (within 85 characters limit, including spaces).
SMT and smFISH reveal the link between chromatin remodeling and TF binding.
Cycles of TF Ace1p binding at the promoter lead to transcription bursts of CUP1.
CUP1 transcription bursts depend on short cycles of nucleosome mobilization by RSC.
RSC ensures a homogeneous response of the cell population to heavy metal stress.
ACKNOWLEDGMENTS
This work was supported by Intramural Research Program of National Institutes of Health (NCI, NICHD, CCR). Funding for the open access charge: Intramural Research Program of NIH. We thank Brian English and Tatsuya Morisaki for their advice in the design of the custom microscope, NHLBI Core Facility for paired-end sequencing, Florian Mueller for the advice on using FISH-quant, Brad Cairns and Helle Ulrich for the plasmids, Luke Lavis for the JF dyes, David Auble, Yuri Chernoff and Matthew Ferguson for the critical reading of the manuscript. This study utilized the high performance computational capabilities of the Biowulf Linux cluster at the National Institutes of Health.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES:
- Ball DA, Mehta GD, Salomon-Kent R, Mazza D, Morisaki T, Mueller F, McNally JG, and Karpova TS (2016). Single molecule tracking of Ace1p in Saccharomyces cerevisiae defines a characteristic residence time for non-specific interactions of transcription factors with chromatin. Nucleic acids research 44, e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman GD, and McKnight JN (2017). Sequence-specific targeting of chromatin remodelers organizes precisely positioned nucleosomes throughout the genome. Bioessays 39, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchman C, Skroch P, Dixon W, Tullius TD, and Karin M (1990). A single amino acid change in CUP2 alters its mode of DNA binding. Molecular and cellular biology 10, 4778–4787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns BR, Schlichter A, Erdjument-Bromage H, Tempst P, Kornberg RD, and Winston F (1999). Two functionally distinct forms of the RSC nucleosome-remodeling complex, containing essential AT hook, BAH, and bromodomains. Mol Cell 4, 715–723. [DOI] [PubMed] [Google Scholar]
- Chereji RV, Ocampo J, and Clark DJ (2017). MNase-Sensitive Complexes in Yeast: Nucleosomes and Non-histone Barriers. Mol Cell 65, 565–577.e563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clauss K, Popp AP, Schulze L, Hettich J, Reisser M, Escoter Torres L, Uhlenhaut NH, and Gebhardt JCM (2017). DNA residence time is a regulatory factor of transcription repression. Nucleic acids research 45, 11121–11130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole HA, Howard BH, and Clark DJ (2012). Genome-wide mapping of nucleosomes in yeast using paired-end sequencing. Methods in enzymology 513, 145–168. [DOI] [PubMed] [Google Scholar]
- Das R, Cairo CW, and Coombs D (2009). A Hidden Markov Model for Single Particle Tracks Quantifies Dynamic Interactions between LFA-1 and the Actin Cytoskeleton. PLOS Computational Biology 5, e1000556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelstein AD, Tsuchida MA, Amodaj N, Pinkard H, Vale RD, and Stuurman N (2014). Advanced methods of microscope control using muManager software. Journal of biological methods 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- English BP, and Singer RH (2015). A three-camera imaging microscope for high-speed single-molecule tracking and super-resolution imaging in living cells. Proc SPIE Int Soc Opt Eng 9550, 955008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furst P, Hu S, Hackett R, and Hamer D (1988). Copper activates metallothionein gene transcription by altering the conformation of a specific DNA binding protein. Cell 55, 705–717. [DOI] [PubMed] [Google Scholar]
- Gillespie DT (1977). Exact stochastic simulation of coupled chemical reactions. The Journal of Physical Chemistry 81, 2340–2361. [Google Scholar]
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, et al. (2015). A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature methods 12, 244–250, 243 p following 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Kelleher M, Iyer VR, Brown PO, and Winge DR (2000). Identification of the copper regulon in Saccharomyces cerevisiae by DNA microarrays. The Journal of biological chemistry 275, 32310–32316. [DOI] [PubMed] [Google Scholar]
- Güldener U, Heck S, Fielder T, Beinhauer J, and Hegemann JH (1996). A new efficient gene disruption cassette for repeated use in budding yeast. Nucleic acids research 24, 2519–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman JA, Trotter KW, Ward JM, and Archer TK (2018). BRG1 governs glucocorticoid receptor interactions with chromatin and pioneer factors across the genome. eLife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopes BC, LeBlanc JF, and Hawley DK (1992). Kinetic analysis of yeast TFIID-TATA box complex formation suggests a multi-step pathway. The Journal of biological chemistry 267, 11539–11547. [PubMed] [Google Scholar]
- Izeddin I, Recamier V, Bosanac L, Cisse II, Boudarene L, Dugast-Darzacq C, Proux F, Benichou O, Voituriez R, Bensaude O, et al. (2014). Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. eLife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janke C, Magiera MM, Rathfelder N, Taxis C, Reber S, Maekawa H, Moreno-Borchart A, Doenges G, Schwob E, Schiebel E, et al. (2004). A versatile toolbox for PCR-based tagging of yeast genes: new fluorescent proteins, more markers and promoter substitution cassettes. Yeast 21, 947–962. [DOI] [PubMed] [Google Scholar]
- Karpova TS, Chen TY, Sprague BL, and McNally JG (2004). Dynamic interactions of a transcription factor with DNA are accelerated by a chromatin remodeller. EMBO reports 5, 1064–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova TS, Kim MJ, Spriet C, Nalley K, Stasevich TJ, Kherrouche Z, Heliot L, and McNally JG (2008). Concurrent fast and slow cycling of a transcriptional activator at an endogenous promoter. Science (New York, N.Y.) 319, 466–469. [DOI] [PubMed] [Google Scholar]
- Kim JG, Takeda Y, Matthews BW, and Anderson WF (1987). Kinetic studies of Cro repressor-operator DNA interaction. J Mol Biol 196, 149–158. [DOI] [PubMed] [Google Scholar]
- Kleinschmidt C, Tovar K, Hillen W, Porschke D (1988). Dynamics of repressor-operator recognition: the Tn10-encoded tetracycline resistance control. Biochemistry 27, 1094–1104. [DOI] [PubMed] [Google Scholar]
- Larson DR, Fritzsch C, Sun L, Meng X, Lawrence DS, and Singer RH (2013). Direct observation of frequency modulated transcription in single cells using light activation. eLife 2, e00750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Hada A, Sen P, Olufemi L, Hall MA, Smith BY, Forth S, McKnight JN, Patel A, Bowman GD, et al. (2015). Dynamic regulation of transcription factors by nucleosome remodeling. eLife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Schroeder JW, Gao B, Simmons LA, and Biteen JS (2015). Single-molecule motions and interactions in live cells reveal target search dynamics in mismatch repair. Proceedings of the National Academy of Sciences 112, E6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lickwar CR, Mueller F, Hanlon SE, McNally JG, and Lieb JD (2012). Genome-wide protein-DNA binding dynamics suggest a molecular clutch for transcription factor function. Nature 484, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffreda A, Jacchetti E, Antunes S, Rainone P, Daniele T, Morisaki T, Bianchi ME, Tacchetti C, and Mazza D (2017). Live-cell p53 single-molecule binding is modulated by C- terminal acetylation and correlates with transcriptional activity. Nature communications 8, 313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR- based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Mazza D, Abernathy A, Golob N, Morisaki T, and McNally JG (2012). A benchmark for chromatin binding measurements in live cells. Nucleic acids research 40, e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazza D, Ganguly S, and McNally JG (2013). Monitoring dynamic binding of chromatin proteins in vivo by single-molecule tracking. Methods in molecular biology (Clifton, N.J.) 1042, 117–137. [DOI] [PubMed] [Google Scholar]
- Mehta GD, Agarwal M, and Ghosh SK (2014). Functional characterization of kinetochore protein, Ctf19 in meiosis I: an implication of differential impact of Ctf19 on the assembly of mitotic and meiotic kinetochores in Saccharomyces cerevisiae. Molecular microbiology 91, 1179–1199. [DOI] [PubMed] [Google Scholar]
- Mirny Leonid SM, Wunderlich Zeba, Tafvizi Anahita, Leith Jason, Kosmrlj Andrej (2009). How a protein searches for its site on DNA: the mechanism of facilitated diffusion. Journal of Physical A: Mathematical and Theoretical 42, 1–23. [Google Scholar]
- Morawska M, and Ulrich HD (2013). An expanded tool kit for the auxin-inducible degron system in budding yeast. Yeast 30, 341–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morisaki T, Lyon K, DeLuca KF, DeLuca JG, English BP, Zhang Z, Lavis LD, Grimm JB, Viswanathan S, Looger LL, et al. (2016). Real-time quantification of single RNA translation dynamics in living cells. Science (New York, N.Y.) 352, 1425–1429. [DOI] [PubMed] [Google Scholar]
- Morisaki T, Muller WG, Golob N, Mazza D, and McNally JG (2014). Single-molecule analysis of transcription factor binding at transcription sites in live cells. Nature communications 5, 4456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller F, Senecal A, Tantale K, Marie-Nelly H, Ly N, Collin O, Basyuk E, Bertrand E, Darzacq X, and Zimmer C (2013a). FISH-quant: automatic counting of transcripts in 3D FISH images. Nat Meth 10, 277–278. [DOI] [PubMed] [Google Scholar]
- Mueller F, Stasevich TJ, Mazza D, and McNally JG (2013b). Quantifying transcription factor kinetics: at work or at play? Critical reviews in biochemistry and molecular biology 48, 492–514. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Walker DA, Wolford R, and Hager GL (2004). Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell 14, 163–174. [DOI] [PubMed] [Google Scholar]
- Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, and Kanemaki M (2009). An auxinbased degron system for the rapid depletion of proteins in nonplant cells. Nature methods 6, 917–922. [DOI] [PubMed] [Google Scholar]
- Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, Mazza D, Morisaki T, Karpova TS, and Hager GL (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nature communications 8, 15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pena MM, Koch KA, and Thiele DJ (1998). Dynamic regulation of copper uptake and detoxification genes in Saccharomyces cerevisiae. Mol Cell Biol 18, 2514–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson F, Linden M, Unoson C, and Elf J (2013). Extracting intracellular diffusive states and transition rates from single-molecule tracking data. Nature methods 10, 265. [DOI] [PubMed] [Google Scholar]
- Poorey K, Viswanathan R, Carver MN, Karpova TS, Cirimotich SM, McNally JG, Bekiranov S, and Auble DT (2013). Measuring chromatin interaction dynamics on the second time scale at single-copy genes. Science (New York, N.Y.) 342, 369–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Presman DM, Ball DA, Paakinaho V, Grimm JB, Lavis LD, Karpova TS, and Hager GL (2017). Quantifying transcription factor binding dynamics at the single-molecule level in live cells. Methods (San Diego, Calif.) 123, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, Peskin CS, Tranchina D, Vargas DY, and Tyagi S (2006). Stochastic mRNA synthesis in mammalian cells. PLoS biology 4, e309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raj A, and Tyagi S (2010). Detection of individual endogenous RNA transcripts in situ using multiple singly labeled probes. Methods in enzymology 472, 365–386. [DOI] [PubMed] [Google Scholar]
- Riggs AD, Bourgeois S, and Cohn M (1970). The lac repressor-operator interaction. 3. Kinetic studies. J Mol Biol 53, 401–417. [DOI] [PubMed] [Google Scholar]
- Rohner S, Gasser SM, and Meister P (2008). Modules for cloning-free chromatin tagging in Saccharomyces cerevisae. Yeast 25, 235–239. [DOI] [PubMed] [Google Scholar]
- Senecal A, Munsky B, Proux F, Ly N, Braye FE, Zimmer C, Mueller F, and Darzacq X (2014). Transcription factors modulate c-Fos transcriptional bursts. Cell reports 8, 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, and Clark DJ (2001). DNA sequence plays a major role in determining nucleosome positions in yeast CUP1 chromatin. The Journal of biological chemistry 276, 35209–35216. [DOI] [PubMed] [Google Scholar]
- Shen CH, Leblanc BP, Alfieri JA, and Clark DJ (2001). Remodeling of yeast CUP1 chromatin involves activator-dependent repositioning of nucleosomes over the entire gene and flanking sequences. Molecular and cellular biology 21, 534–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen CH, Leblanc BP, Neal C, Akhavan R, and Clark DJ (2002). Targeted histone acetylation at the yeast CUP1 promoter requires the transcriptional activator, the TATA boxes, and the putative histone acetylase encoded by SPT10. Molecular and cellular biology 22, 6406–6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavreva DA, Muller WG, Hager GL, Smith CL, and McNally JG (2004). Rapid glucocorticoid receptor exchange at a promoter is coupled to transcription and regulated by chaperones and proteasomes. Molecular and cellular biology 24, 2682–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga M, Imamoto N, and Sakata-Sogawa K (2008). Highly inclined thin illumination enables clear single-molecule imaging in cells. Nature methods 5, 159–161. [DOI] [PubMed] [Google Scholar]
- Tsabar M, Haase J, Harrison B, Snider CE, Eldridge B, Kaminsky L, Hine RM, Haber JE, and Bloom K (2016). A Cohesin-Based Partitioning Mechanism Revealed upon Transcriptional Inactivation of Centromere. PLoS genetics 12, e1006021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Royen ME, van Cappellen WA, Geverts B, Schmidt T, Houtsmuller AB, and Schaaf MJ (2014). Androgen receptor complexes probe DNA for recognition sequences by short random interactions. Journal of cell science 127, 1406–1416. [DOI] [PubMed] [Google Scholar]
- Voss TC, and Hager GL (2014). Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat Rev Genet 15, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VC, Bass VL, Bullock ME, Chavali AK, Lee REC, Mothes W, Gaudet S, and Miller-Jensen K (2018). NF-kappaB-Chromatin Interactions Drive Diverse Phenotypes by Modulating Transcriptional Noise. Cell reports 22, 585–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zenklusen D, Larson DR, and Singer RH (2008). Single-RNA counting reveals alternative modes of gene expression in yeast. Nature structural & molecular biology 15, 1263–1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Strope PK, Kozmin SG, McCusker JH, Dietrich FS, Kokoska RJ, and Petes TD (2014). Structures of naturally evolved CUP1 tandem arrays in yeast indicate that these arrays are generated by unequal nonhomologous recombination. G3 (Bethesda, Md.) 4, 2259–2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Ace1-HaloTag-JF646 over the CUP1 array, related to figures 1A, 1C, 1D, 2 Time lapse images with 200 ms interval and 600 frames. Playback speed: 30 fps (6x faster than real time), Red channel: Ace1-HaloTag-JF646, green channel: Ace1-3xGFP, Scale bar: 2 μm. Left: Raw data, right: filtered data.
Ace1-HaloTag-JF646 over the CUP1 array, related to figures 1A, 1C, 1D, 2 Time lapse images with 200 ms interval and 600 frames. Playback speed: 30 fps (6x faster than real time), Red channel: Ace1-HaloTag-JF646, green channel: Ace1-3xGFP, Scale bar: 2 μm. Left: Raw data, right: filtered data.