

Abstract
Background:
Transcript levels for cytokines and the viral restriction factor interferon-induced transmembrane protein (IFITM) are markedly higher in the prefrontal cortex in schizophrenia. These gene products are regulated by the nuclear factor-κB (NF-κB) transcriptional complex. NF-κB activity, which requires the formation of NF-κB family member heterodimers, is regulated by activation receptors, kinases, and inhibitors. Whether any of these factors are altered in schizophrenia is not known. It is also unclear whether NF-κB-related disturbances reflect ongoing cortical immune activation or a long-lasting response to a prenatal immune-related insult.
Methods:
Transcript levels for NF-κB pathway markers were assessed using quantitative PCR in the prefrontal cortex from 1) 62 matched pairs of schizophrenia and unaffected comparison subjects, 2) antipsychotic-exposed monkeys, and 3) adult mice exposed prenatally to maternal immune activation or in adulthood to the immune stimulant poly(I:C).
Results:
In schizophrenia subjects, but not antipsychotic-exposed monkeys, we found higher mRNA levels for 1) most NF-κB family members, 2) all NF-κB activation receptors, 3) several kinases, and 4) one inhibitor (IκBα) whose transcript level is itself regulated by NF-κB activity. A similar pattern of elevated NF-κB-related mRNA levels was seen in adult mice that received daily poly(I:C) injections, but not in those subjected to maternal immune activation in utero.
Conclusions:
Higher NF-κB activity, evidenced by elevated transcript levels for NF-κB family members, activation receptors, and kinases, may contribute to increased markers of cortical immune activation in schizophrenia.
Keywords: prefrontal cortex, cytokine, NF-κB, inflammation, maternal immune activation, microglia, postmortem
Introduction
Molecular evidence of immune activation has been consistently reported in the prefrontal cortex (PFC) in schizophrenia, particularly at the transcriptome level (1, 2). For example, RNA-Seq, microarray, quantitative PCR and in situ hybridization studies have repeatedly found elevated transcript levels for immune markers, such as interferon-induced transmembrane protein (IFITM), in schizophrenia (3–8). Furthermore, several-fold elevations in mRNA levels for cytokines, such as interleukin-6 (IL-6), IL-8, IL-1β, and interferon-β, have also been consistently found in the PFC of large cohorts of schizophrenia subjects (9–11). Interestingly, the nuclear factor-κB (NF-κB) complex provides direct transcriptional control over the expression of the IFITMs and cytokines (3, 4, 12) that are over-expressed in schizophrenia (9–11). In the PFC of schizophrenia subjects, elevated mRNA levels for two NF-κB family members, NF-κB1 and NF-κB2, and lower mRNA levels for Schnurri-2, an NF-κB site-binding protein that inhibits NF-κB function (12), have also been reported (11). These findings suggest that higher NF-κB transcriptional activity may play an important role in elevating IFITM and cytokine mRNA levels in the PFC in schizophrenia.
Understanding the nature of elevated NF-κB activity, and the subsequent effects on the expression of immune markers, in schizophrenia requires investigating the factors that regulate NF-κB transcriptional activity. For example, because NF-κB1 and NF-κB2 lack transactivation domains, transcriptional activity requires the formation of heterodimers with other NF-κB family members that also contain Rel homology domains, such as RelA, RelB, and cRel (4). Furthermore, NF-κB transcriptional activity is initiated and regulated through canonical and non-canonical pathways that involve a diverse collection of activation receptors, several kinases that remove inhibition of and/or enhance NF-κB activity, and inhibitors that block NF-κB binding to DNA (Figure 1). Consequently, we tested the hypothesis that signaling pathway components that enhance, or reflect higher, NF-κB transcriptional activity are elevated at the transcript level in the PFC in schizophrenia.
Figure 1. Canonical and non-canonical NF-κB activation pathways and their alterations in schizophrenia.
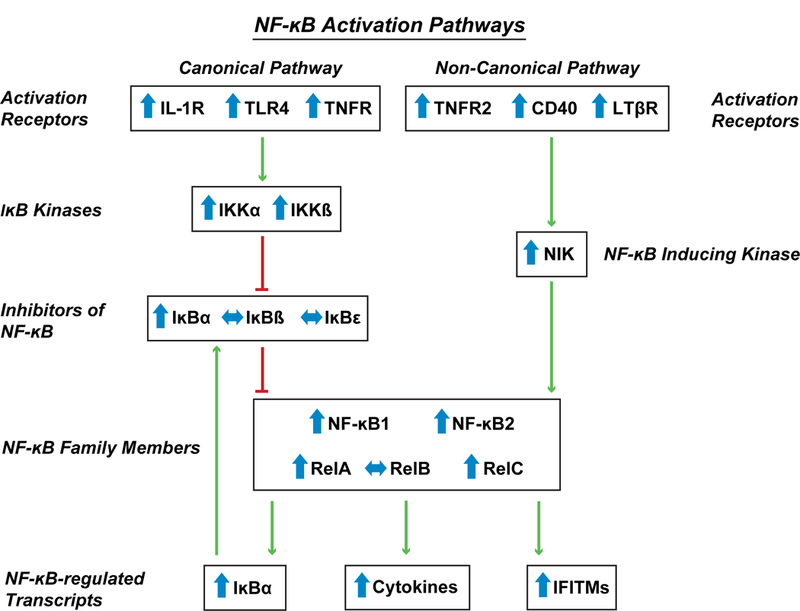
The NF-κB transcriptional complex is composed of heterodimers between NF-κB family members, including NF-κB1, NF-κB2, and other members of the NF-κB family that contain Rel homology domains including RelA, RelB, and cRel. In the canonical pathway, activation of receptors (IL-1R, TLR4, TNFR) increases activity of IκB kinases (IKKα, IKKβ) which in turn phosphorylate, and lead to the degradation of, inhibitors of NF-κB (IκBα, IκBβ, IκBϵ) which reduces the “brakes” on NF-κB transcriptional activity. In the non-canonical pathway, receptor activation increases the activity of NF-κB-inducing kinase (NIK) which then leads to activation of NF-κB2. Green arrows indicate increased and red lines indicate decreased activity or function. Transcript levels in schizophrenia subjects relative to unaffected comparison subjects are indicated by the direction of the blue arrows.
Furthermore, maternal immune activation has been identified as a risk factor for schizophrenia (13) and can lead to long-lasting transcriptome changes (14). However, evidence from murine models suggests that the pattern of elevated immune-related transcriptome changes in schizophrenia is not attributable to prenatal exposure to maternal immune activation, but rather is consistent with an active, though not yet characterized, process that is present throughout adulthood (11). Therefore, we also used murine models to test the hypothesis that immune stimulation occurring in adulthood, but not prenatally, leads to a pattern of elevated cortical NF-κB transcriptional activity similar to that seen in schizophrenia.
Methods and Materials
Human subjects
Brain specimens were obtained during routine autopsies conducted at the Allegheny County Medical Examiner’s Office after consent was obtained from next-of-kin. All subjects died out-of-hospital with minimal to no agonal state events. An independent committee of experienced research clinicians made consensus DSMIV (15) diagnoses, or confirmed the absence of a psychiatric diagnosis, for each subject using structured interviews with family members and review of medical records (16). To reduce biological variance between groups, subjects with schizophrenia (n=39) or schizoaffective disorder (n=23) were matched individually to one unaffected comparison subject for sex and as closely as possible for age (Supplemental Table S1). To control for experimental variance, samples from paired subjects were processed together. The mean age, postmortem interval, RNA integrity number (RIN), and freezer storage time did not differ between subject groups (t(122) ≤0.45, p ≥0.65) (Table 1). Mean (± standard deviation) brain pH, an indicator of agonal state, was statistically different between the schizophrenia (6.6 ± 0.3) and comparison groups (6.7 ± 0.2; t(122) =2.6, p=0.01), but the difference was small and of uncertain significance. All procedures were approved by the University of Pittsburgh’s Committee for the Oversight of Research and Clinical Training Involving the Dead and Institutional Review Board for Biomedical Research.
Table 1.
Summary of demographic and postmortem characteristics of human subjects
| Parameter | Unaffected Comparison | Schizophrenia |
|---|---|---|
| N | 62 | 62 |
| Sex | 47M / 15F | 47M / 15F |
| Race | 52W / 10B | 46W / 16B |
| Age (years) | 48.7 ± 13.8 | 47.7 ± 12.7 |
| Postmortem Interval (hours) | 18.8 ± 5.5 | 19.2 ± 8.5 |
| Freezer Storage Time (months) | 145.4 ± 56.4 | 141.7 ± 60.9 |
| Brain pH | 6.7 ± 0.2 | 6.6 ± 0.3 |
| RNA Integrity Number | 8.2 ± 0.6 | 8.1 ± 0.6 |
| Medications At Time of Death* | ||
| Antipsychotic | - | 54 |
| Antidepressant | - | 27 |
| Benzodiazepine/Anticonvulsant | - | 24 |
| NSAID | 13 | 16 |
| Substance Use Disorder History* | ||
| Lifetime | - | 40 |
| Current at Time of Death | - | 25 |
For age, postmortem interval, freezer storage time, brain pH, and RNA integrity number, values are group means ± standard deviation. For brain pH, t(122) =2.6, p=0.01. For all others, t(122) ≤0.45, p ≥0.65.
For medications at time of death and substance use disorder history, number of subjects in each applicable category are provided.
NSAID: non-steroidal anti-inflammatory drug.
Quantitative PCR
Quantitative PCR (qPCR) was performed with RNA from PFC area 9 gray matter homogenates using the comparative cycle threshold (CT) method with Power SYBR Green dye and the ViiA-7 Real-Time PCR System (Applied Biosystems) as previously described (17) (Supplemental Methods). Three reference genes (beta actin, cyclophilin A, and GAPDH) which we previously reported to be stably expressed in the present cohort of schizophrenia and comparison subjects (18), were used to normalize target mRNA levels. The difference in CT (dCT) for each target transcript was calculated by subtracting the geometric mean CT for the three reference genes from the CT of the target transcript (mean of four replicate measures). Because dCT represents the log2-transformed expression ratio of each target transcript to the reference genes, the relative level of the target transcript for each subject is reported as 2-dCT (19, 20).
Antipsychotic-exposed monkeys
Young adult male monkeys (Macaca fascicularis) received oral doses of haloperidol, olanzapine or placebo (n=6 monkeys/group) twice daily for 17–27 months, as previously described (21). RNA was isolated from PFC area 9, and qPCR was conducted for the same three reference genes and NF-κB-related markers included in the human study (Supplemental Table S2) with all monkeys from a triad processed together on the same plate.
Poly(I:C)-exposed mice
As previously described (11) (Supplemental Methods), timed pregnant C57BL/6J mice (n=12/condition) received intraperitoneal injections of poly(I:C) 20 mg/kg or an equivalent volume of normal saline daily for three days at mid- (E11–13) or late- (E15–17) gestation (Supplemental Table S3) (22–24) in parallel with non-pregnant adult female mice (n=8). The non-pregnant mice were euthanized 3 hours after the last injection (random estrous cycle). RNA was isolated from frontal cortex homogenates from one 8-week-old male and/or female offspring per poly(I:C)-injected mother (n=7–8/sex/condition; Supplemental Table S3) and from the non-pregnant adult mice that received daily injections. QPCR assessment of NF-κB-related markers (Supplemental Table S2) was performed as described for the human studies with the following exception: primer efficiency testing found that transcript levels for TLR4, TNFR2, CD40, and IκBϵ in mouse frontal cortex were insufficient for qPCR analysis.
All animal studies followed the NIH Guide for the Care and Use of Laboratory Animals and were approved by the University of Pittsburgh’s Institutional Animal Care and Use Committee.
Statistical analysis
The ANCOVA model reported for the schizophrenia study includes mRNA level as the dependent variable, diagnostic group as the main effect, and sex, age, postmortem interval, brain pH, RIN, and freezer storage time as covariates. Because each schizophrenia subject was individually matched to a comparison subject to account for the parallel processing of tissue samples from a pair and to balance diagnostic groups for sex and age, a second ANCOVA model with subject pair as a blocking factor and including postmortem interval, brain pH, RIN, and storage time was also used. Because both ANCOVA models produced similar results, we only report the first model. Because 15 different NF-κB-related mRNAs were quantified in the human study, the Bonferroni correction for multiple comparisons was employed to establish the statistical significance level at α=.0033 (i.e. .05/15). Subsequent analyses of differences in mRNA levels between subgroups of schizophrenia subjects were then conducted using the first ANCOVA model, as described in the Results section (25). Spearman’s rank correlation coefficients (rs) were calculated to assess the relationships between quantified mRNA levels. For the antipsychotic-exposed monkey study, an ANCOVA model with mRNA level as the dependent variable, treatment group as the main effect, and triad as a blocking factor was employed. For the mouse studies, measures of mRNA levels were compared between groups using t tests with α=0.05.
Results
NF-κB-related markers in the PFC in schizophrenia
NF-κB transcriptional activity requires the formation of heterodimers between NF-κB1 and NF-κB2 and other NF-κB family members, such as RelA, RelB, and cRel (Figure 1) (4). Therefore, we determined whether mRNA levels for RelA, RelB, and cRel were elevated in the PFC of the same schizophrenia subjects in whom we had previously reported higher NF-κB1 and NF-κB2 mRNA levels using the same qPCR methodology (11). Schizophrenia subjects had higher mean mRNA levels for RelA (+27%; F(1,116)=33.1, p<.0001; Figure 2) and cRel (+19%; F(1,116)=27.3, p<.0001), but not RelB (+3%; F(1,116)=0.03, p=.87), relative to unaffected comparison subjects. RelA and cRel mRNA levels were higher in 81% and 74%, respectively, of schizophrenia subjects relative to their matched comparison subject. Furthermore, mRNA levels for RelA and cRel were positively correlated with mRNA levels for NF-κB1 (rs=.60 and rs=.68, respectively, p<.0001) and NF-κB2 (rs=.74 and rs=.61, respectively, p<.0001) across all subjects as well as in either schizophrenia (all rs≥.75, all p<.0001) or comparison subjects alone (all rs≥.48, all p<.0001; Supplemental Figure S1). These associations are consistent with a higher rate of formation of NF-κB heterodimers.
Figure 2. Transcript levels of NF-κB signaling pathway markers in the PFC in schizophrenia.
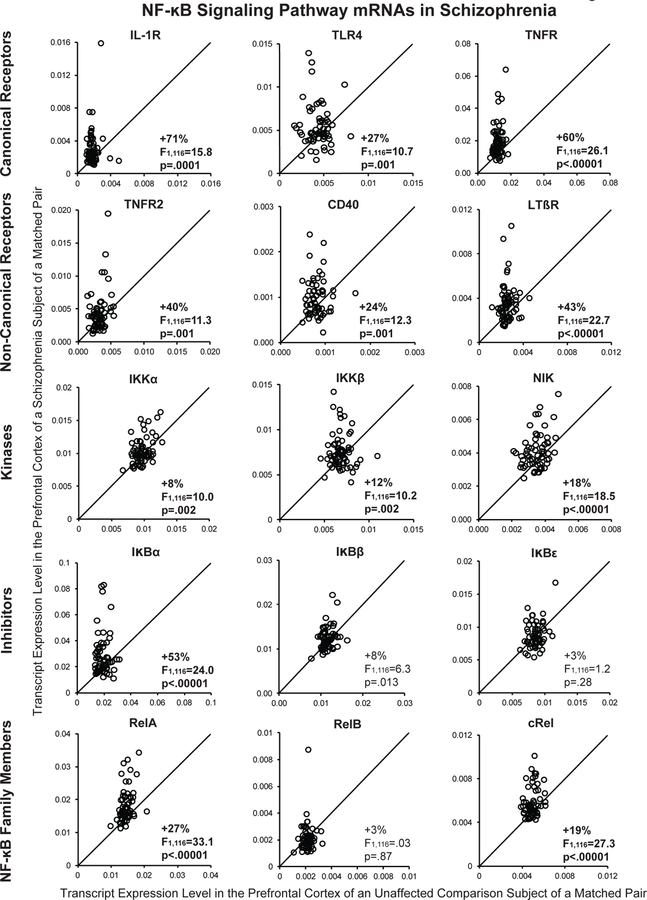
Transcript levels for each schizophrenia subject relative to the matched unaffected comparison subject are indicated by open circles. Data points to the left of the unity line indicate higher mRNA levels in the schizophrenia subject relative to the unaffected comparison subject. Percent difference in diagnostic group means and primary statistical analysis results are provided for each transcript. Mean (± standard deviation) transcript levels for schizophrenia and unaffected comparison subjects (respectively) were: IL-1R 0.0031 ± 0.0022 and 0.0018 ± 0.0006; TLR4 0.0055 ± 0.0024 and 0.004 3 ± 0.0012; TNFR 0.0194 ±0.0104 and 0.0121 ± 0.0022; TNFR2 0.0045 ± 0.0030 and 0.0032 ± 0.0009; CD40 0.0010 ± 0.0004 and 0.0008 ± 0.0002; LTβR 0.0036 ± 0.0017 and 0.0025 ± 0.0006; IKK α 0.0104 ± 0.0020 and 0097 ± 0.0012; IKKβ 0.0076 ± 0.0020 and 0.0068 ± 0.0010; NIK 0.0041 ± 0.0010 and 0.0035 ± 0.0006; IκBα 0.0292 ± 0.0160 and 0.0191 ± 0.0042; I κBβ 0.0124 ± 0.0025 and 0.0115 ± 0.0014; IκBϵ 0.0088 ± 0.0020 and 0.0086 ±0.0010; RelA 0.0182 ± 0.0052 and 0.0144 ± 0.0017; RelB 0.0022 ± 0.0010 and 0.0021 ± 0.0004; and cRel 0.0059 ± 0.0013 and 0.0049 ± 0.0005.
We previously reported positive correlations between NF-κB1 and NF-κB1 and several immune-related mRNAs that are regulated by NF-κB transcriptional activity, including IL-6, IFITM1, and IFITM2/3 (11). In the present study, we also found positive correlations between mRNA levels for RelA and IL-6, IFITM1, and IFITM2/3 across all subjects (all rs≥.47, all p<.0001) and in schizophrenia subjects alone (all rs≥.59, all p<.0001); however, only the correlation between RelA and IFITM1 was statistically significant in comparison subjects alone (rs=.35, p=.006). Similarly, we found positive correlations between mRNA levels for cRel and IL-6, IFITM1, and IFITM2/3 across all subjects (all rs≥.44, all p<.0001) and in schizophrenia subjects alone (all rs≥.45, all p≤.0003); however, only the correlation between cRel and IFITM2/3 was statistically significant in comparison subjects alone (rs=.25, p=.046). These positive correlations are at least consistent with the idea that higher levels of NF-κB heterodimers may lead to higher transcription rates of NF-κB-regulated immune-related mRNAs in schizophrenia.
NF-κB transcriptional activity can be initiated through a canonical activation pathway involving interleukin-1 receptor (IL-1R), Toll-like receptors (e.g., TLR4), and tumor necrosis factor receptor (TNFR) (4) (Figure 1). In schizophrenia, mean mRNA levels were higher for IL-1R (+71%; F(1,116)=15.8, p=.0001), TLR4 (+27%; F(1,116)=10.7, p=.001), and TNFR (+60%; F(1,116)=26.1, p<.00001) in the PFC relative to comparison subjects (Figure 2). When subject pairs with a potential outlier (i.e., mRNA level more than three standard deviations from the group mean) were excluded from the analysis (11), the differences in mRNA levels in the schizophrenia subjects for IL-1R (F(1,114)=25.6, p<.0001; N=1 outlier), TLR4 (F(1,112)=8.2, p=.005; N=2 outliers), and TNFR (F(1,114)=31.8, p<.0001; N=1 outlier) remained statistically significant. In addition, mRNA levels for these receptors were higher in 81% (IL-1R), 65% (TLR4), and 82% (TNFR) of schizophrenia subjects relative to their matched comparison subject. Elevated levels of these receptors suggest that NF-κB transcriptional activity is increased through the canonical activation pathway in schizophrenia.
Activation of receptors in the canonical NF-κB signaling pathway leads to activation of the IκB kinase complex (e.g., IKKα, IKKβ) which in turn phosphorylates inhibitors of NF-κB (e.g., IκBα, IκBβ, IκBϵ), leading to their proteosomal degradation (Figure 1). Because IκBs are normally bound to NF-κB dimers and render the NF-κB complex transcriptionally inactive, this degradation of IκBs results in greater levels of NF-κB transcriptional activity (4). In schizophrenia, transcript levels were slightly higher for the IκB kinases IKKα (+8%; F(1,116)=10.0, p=.002) and IKKβ (+12%; F(1,116)=10.2, p=.002) relative to comparison subjects (Figure 2). Transcript levels were also higher for the NF-κB inhibitor IκBα (+53%; F(1,116)=24.0, p<.00001), but not for IκBϵ (+3%; F(1,116)=1.2, p=.28) or IκBβ (+8%; F(1,116)=6.3, p=.013) which did not reach statistical significance using the Bonferroni correction (α=.0033) in schizophrenia subjects. Furthermore, IKKα, IKKβ, and IκBα mRNA levels were higher in the schizophrenia subject in 55%, 63%, and 74%, of the subject pairs, respectively. Because transcript levels for IκBα are themselves regulated by NF-κB transcriptional activity as part of a negative feedback loop (26), higher IκBα mRNA levels are consistent with higher NF-κB activity. Furthermore, higher levels of IKKs could lead to increased phosphorylation and subsequent degradation of IκBα protein. Taken together, elevated transcript levels for multiple NF-κB family members, activation receptors, and kinases may contribute to the marked elevations in levels of transcripts that are regulated by NF-κB activity, including cytokine, IFITM, and IκBα mRNAs, in schizophrenia (5–7, 9–11).
NF-κB transcriptional activity can also be initiated through a non-canonical activation pathway involving other TNF receptor superfamily members (e.g., TNFR2, CD40, lymphotoxin beta receptor [LTβR]) which results in activation of NF-κB-inducing kinase (NIK) which leads to higher NF-κB activity (Figure 1) (4). Mean mRNA levels were higher for TNFR2 (+40%; F(1,116)=11.3, p=.001), CD40 (+24%; F(1,116)=12.7, p=.001), LTβR (+43%; F(1,116)=22.7, p<.00001), and NIK (+18%; F(1,116)=18.5, p<.0001) in schizophrenia subjects (Figure 2). When subject pairs with a potential outlier were excluded, differences in mRNA levels in schizophrenia for TNFR2 (F(1,114)=12.4, p=.001; N=1 outlier), CD40 (F(1,114)=11.5, p=.001; N=1 pair removed), LTβR (F(1,112)=27.6, p<.0001; N=2 outliers), and NIK (F(1,114)=19.1, p<.0001; N=1 outlier) remained statistically significant. In addition, mRNA levels were higher in the schizophrenia subject in 65% (TNFR2), 61% (CD40), 69% (LTβR), and 71% (NIK) of subject pairs. These findings suggest that NF-κB transcriptional activity is also increased through the non-canonical activation pathway in schizophrenia.
Transcript levels of NF-κB-related markers did not differ in schizophrenia subjects as a function of antipsychotics (typical only, atypical only, both, or none: all F(3,51) ≤1.3, p≥0.29; Table 1, Supplemental Table S1), antidepressants (all F(1,54)≤2.3, p≥0.13), benzodiazepines and/or valproate (all F(1,54)≤1.8, p≥0.18 except for NIK: F(1,54)=6.7, p=0.01), NSAIDs (all F(1,54)≤1.7, p≥0.20); or smoking (all F(1,46)≤1.1, p≥0.16) at time of death. NF-κB-related markers also did not differ between schizophrenia subjects with or without a history of an immune/inflammation-related illness (25), either chronic or as a cause of death (i.e., type I diabetes [subject 1712], psoriasis [1088], alopecia [1211], peritonitis [781, 1455], myocarditis [933], pneumonia [904, 1296, 1734], anaphylaxis [1706] (all F(1,54)≤3.3, p≥0.07) or as a function of living in a dependent setting such as a group home at time of death (F(1,54)≤2.9, p≥0.10;Supplemental Table S1) (25). While levels of NF-κB-related mRNAs did not differ in schizophrenia subjects as a function of a lifetime diagnosis of a substance use disorder (all F(1,54)≤1.5, p≥0.22; Table 1), transcript levels for IκBα (+24%; F(1,54)=4.5, p=.038), TNFR (+27%; F(1,54)=5.2, p=.026), IL-1R (+62%; F(1,54)=9.4, p=.003), and LTβR (+25%; F(1,55)=4.3, p=.043), but not for the 11 other NF-κB-related markers (all F(1,54)≤3.7, p≥.06), were elevated in schizophrenia subjects with a substance use disorder that was current at time of death relative to other schizophrenia subjects. However, schizophrenia subjects without a substance use disorder current at time of death still had elevated transcript levels for IκBα (+40%; F(1,66)=9.6, p=.003), TNFR (+46%; F(1,66)=14.8, p=.0003), IL-1R (+37%; F(1,66)=9.7, p=.003), and LTβR (+32%; F(1,66)=10.5, p=.002) relative to their comparison subjects.
NF-κB-related markers in antipsychotic-exposed monkeys
In the PFC, none of the transcripts for NF-κB family members, receptors in the canonical or non-canonical activation pathways, kinases, or inhibitor of NF-κB that were elevated in schizophrenia differed between monkeys chronically exposed to olanzapine, haloperidol, or placebo (all F(2,10)≤1.9, p≥.20; Figure 3).
Figure 3. Transcript levels of NF-κB signaling pathway markers in the PFC of antipsychotic-exposed monkeys.
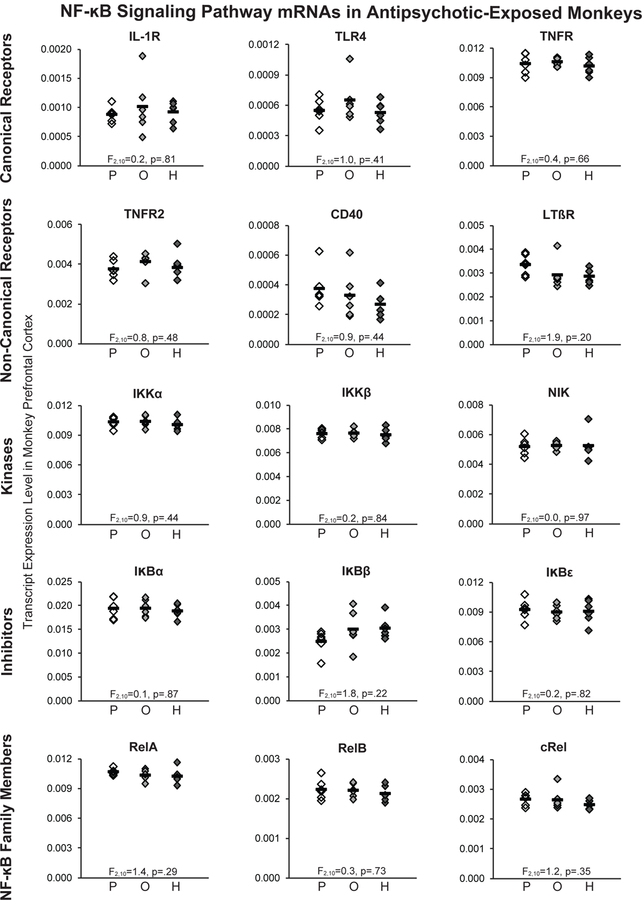
QPCR analysis revealed no statistically significant differences in transcript levels for any NF-κB family members, receptors in the canonical and non-canonical NF-κB activation pathways, kinases, or inhibitors in PFC area 9 of monkeys chronically exposed to either olanzapine or haloperidol compared to placebo. Mean values are shown as horizontal black bars.
Effects of maternal and adult immune activation on NF-κB-related marker expression in mouse frontal cortex
We next determined whether maternal immune activation, a risk factor for schizophrenia (13), could lead to a pattern of changes in NF-κB signaling pathway mRNAs similar to that seen in schizophrenia. However, no differences in NF-κB-related mRNA levels were found in the frontal cortex of male and/or female adult offspring of pregnant mice exposed to poly(I:C) for 3 days at middle (E11–13; all t26≤|0.9|, p≥0.39) or late (E15–17; all t30≤|0.9|, p≥0.39) gestation relative to the adult offspring of normal saline-exposed pregnant mice (Figure 4; Supplemental Table S4). In contrast, adult non-pregnant female mice that were exposed to poly(I:C) daily for 3 days and euthanized three hours after the final injection had a pattern of alterations in NF-κB-related mRNA levels that was similar, though not identical, to that seen in schizophrenia. Higher transcript levels for IL-1R (+88%; t14=5.9, p=.0004), TNFR (+40%; t14=5.5, p=.00008), IκBα (+89%; t14=4.0, p=.001), and RelA (+18%; t14=4.8, p=.0003) were found in the frontal cortex of adult mice exposed to poly(I:C) relative to adult mice exposed to normal saline (Figure 4). Interestingly, higher mRNA levels for RelB (+29%; t14=2.6, p=.02), but not cRel (+6%; t14=1.0, p=.36), were found in the frontal cortex of mice with adult poly(I:C) exposure, whereas in schizophrenia, mRNA levels for cRel, but not RelB, were elevated in the PFC (Figure 2). However, frontal cortex mRNA levels for LTβR, IKKα, IKKβ, NIK, and IκBβ were not statistically significantly different between adult mice exposed to poly(I:C) or normal saline (all t14≤|1.8|, p≥0.09). To determine if residual blood cells in brain parenchyma may have contributed to these findings, an additional group of adult non-pregnant female mice was exposed to poly(I:C) daily for three days then underwent transcardial perfusion with normal saline three hours after the last injection (Supplemental Figure S2). The pattern of altered NF-κB-related mRNA levels seen in the saline-perfused mice with daily poly(I:C) exposure (Supplemental Figure S2) was highly similar to that seen in mice that were not saline-perfused. Taken together, these findings suggest that subacute exposure to poly(I:C) only partially recapitulates NF-κB signaling pathway alterations in schizophrenia.
Figure 4. Transcript levels for NF-κB signaling pathway markers in the frontal cortex of adult mice exposed either prenatally or in adulthood to immune stimulation.
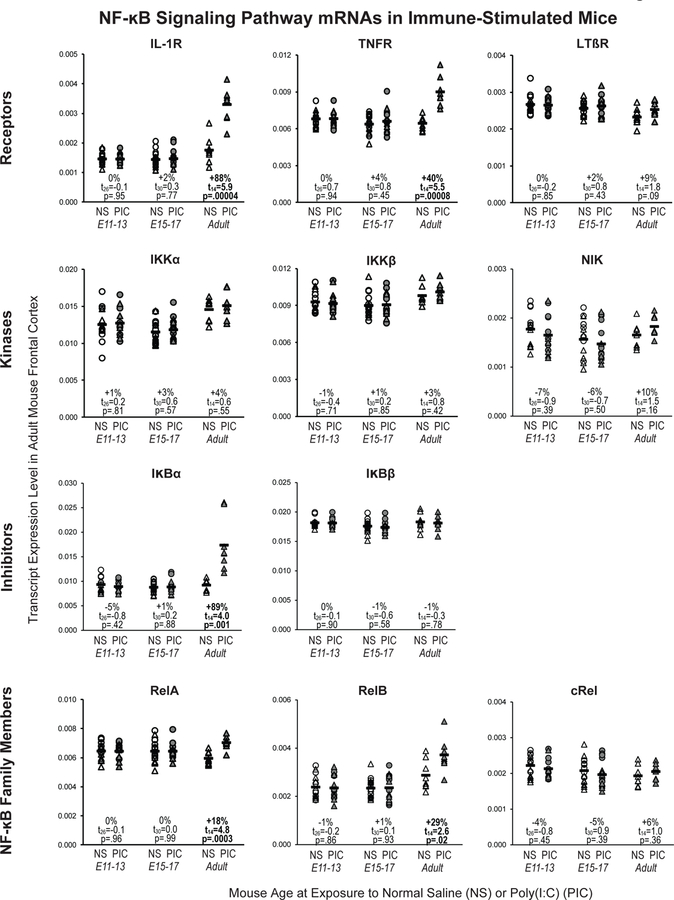
Transcript levels for NF-κB family members, activation receptors, kinases, and inhibitors were quantified in the frontal cortex of 1) adult offspring (male: circle; female: triangle) of pregnant mice exposed to either normal saline (NS) or poly(I:C) (PIC) daily for three days of gestation (E11–13: n=7 males and 7 females per condition; E15–17: n=8 males and 8 females per condition), and 2) adult female mice exposed to NS or PIC (n=8 per condition) daily for three days. Black bars indicate mean mRNA levels for each condition.
Discussion
Here, we investigated NF-κB activation pathways in the PFC in schizophrenia by quantifying transcript levels of NF-κB family members, receptors, kinases, and inhibitors in the canonical and non-canonical NF-κB activation pathways (Figure 1). First, we found higher mRNA levels for the NF-κB family members, RelA and cRel, in schizophrenia subjects. Transcript levels for these NF-κB family members were strongly positively correlated with previously reported transcript levels for NF-κB1 and NF-κB2 (11), indicating that the same schizophrenia subjects have the highest levels of these NF-κB family members across studies. These findings are at least consistent with the presence of greater levels of functional NF-κB heterodimers in the disorder. Second, we found markedly higher mRNA levels for multiple receptors that initiate the canonical and non-canonical NF-κB activation pathways. Third, transcript levels for kinases (e.g., IKKα, IKKβ, and NIK) that are activated by these receptors and remove inhibition of and/or enhance NF-κB activity were also elevated in schizophrenia. Fourth, one inhibitor (i.e. IκBα) that blocks the binding of the NF-κB transcriptional complex to DNA was also elevated in schizophrenia. However, transcript levels for IκBα are themselves regulated by NF-κB transcriptional activity (26), and consequently, higher IκBα mRNA levels are also an indication of higher NF-κB activity. Higher levels of IKKs could also lead to increased phosphorylation and subsequent degradation of IκBα protein. Fifth, we also found that NF-κB-related markers were higher in the majority (55–82%) of schizophrenia subjects relative to their matched comparison subjects. Sixth, transcript levels for multiple NF-κB family members were positively correlated with levels of transcripts that are regulated by NF-κB activity, including cytokines and IFITMs, in schizophrenia. Seventh, although the potential contribution of antipsychotic medication effects cannot be entirely ruled out, no statistically significant effects of antipsychotic medications were found on any NF-κB-related mRNA levels in human or monkey subjects. Finally, exposure to immune stimulation in adult mice resulted in higher frontal cortical mRNA levels for NF-κB-related mRNAs that were similar, though not identical, to that seen in schizophrenia. Although additional studies are needed to determine the extent to which these changes in transcript levels are associated with changes in protein levels, these findings together suggest that NF-κB activity is greater in the PFC in schizophrenia and may contribute to marked elevations in levels of immune-related transcripts that are regulated by NF-κB activity in the disorder (5–7, 9–11).
We also mined unpublished RNA-Seq data from the CommonMind Consortium that was obtained from the prefrontal cortex of many of the same schizophrenia and comparison subject pairs (n=57 pairs) and found results that were highly similar to the present study (27). For example, levels of eight transcripts (IL-1R, TNFR, TNFR2, LTβR, IKKβ, NIK, RelA, and cRel) that were found to be higher in schizophrenia subjects in the present study were also similarly higher in the same schizophrenia subjects using unpublished CommonMind Consortium RNA-Seq data at a false discovery rate of 0.05. Levels of two transcripts (IκBβ and IκB ϵ) were found to be unchanged in schizophrenia subjects in both the present study and by RNA-Seq data, and levels of two transcripts (CD40, RelB) were considered too low for analysis in the RNA-Seq data. Another recent RNA-Seq study in a separate large cohort of schizophrenia subjects also found significantly altered expression of multiple immune-related genes in the prefrontal cortex, although NF-κB-related mRNAs were not among the transcripts identified to have differential expression in schizophrenia using this approach (8).
Chronic exposure to drugs of abuse has been reported to increase NF-κB activity (28–31). We found that NF-κB-related mRNA levels did not differ in schizophrenia subjects as a function of a lifetime diagnosis of a substance use disorder. However, transcript levels for several receptors that initiate NF-κB activation pathways (i.e. IL-1R, TNFR, and LTβR) and for IκBα were even higher in the PFC of schizophrenia subjects with a diagnosis of a substance use disorder that was current at time of death relative to other schizophrenia subjects. Importantly, several lines of evidence indicate that higher levels of NF-κB activity in the PFC reflects the disease process of schizophrenia. First, exclusion of schizophrenia subjects with a substance use disorder current at time of death still revealed elevated transcript levels for these receptors and IκBα in schizophrenia subjects relative to comparison subjects. Second, levels of most NF-κB-related mRNAs did not differ in schizophrenia subjects as a function of a substance use disorder current at time of death. Nonetheless, these findings indicate that substance abuse, to a certain extent, may further exacerbate elevated levels of NF-κB activity in the illness, and reinforce the importance of investigating potential effects of drug abuse on future studies of NF-κB-related markers in psychiatric illness. It remains possible that individuals with schizophrenia had a greater accumulative lifetime exposure to immune and inflammation-related illnesses relative to comparison subjects because individuals with schizophrenia are more likely to experience homelessness, live in group homes, and spend more time in hospitals. However, the cohort of schizophrenia subjects in the present study comes from a community-based population. Furthermore, we found that transcript levels of NF-κB-related markers did not differ in schizophrenia subjects as a function of the presence or absence of an immune or inflammation-related illness at death or living independently versus in a dependent setting such as a group home.
Maternal immune activation has been reported to be a risk factor for schizophrenia in offspring (13) and can induce epigenetic modifications that can affect gene expression postnatally (14). However, we found that several days of maternal immune activation during middle or late gestation in mice did not lead to higher levels of NF-κB-related mRNA levels in the frontal cortex of young adult offspring. Consistent with this finding, we previously reported that maternal immune activation also did not affect levels of NF-κB-regulated immune-related mRNAs, such as IL-6 and IFITMs (11). Taken together, these findings suggest that maternal immune activation may not be sufficient to cause long-lasting changes in NF-κB-related mRNAs in the PFC in schizophrenia.
An important limitation of the present study is that qPCR in gray matter homogenates does not permit identification of the specific cell types that express higher levels of NF-κB-related mRNAs in the PFC in schizophrenia. However, recent cell-type specific RNA-Seq studies in human brain found that several activation receptors (i.e., TNFR2, CD40), kinases (i.e., NIK), and NF-κB family members (i.e., cRel, NF-κB1) are selectively and highly expressed in microglia, but not in astrocytes, neurons, or oligodendrocytes, while the remaining NF-κB-related markers are most commonly found to be heavily enriched in both microglia and astrocytes, but not in neurons or oligodendrocytes (32, 33). It is also possible that residual white blood cell contamination may partly contribute to our findings, and unfortunately, peripheral serum samples are not available for any of the human subjects. However, this explanation seems unlikely as even with the use of multiple different primer sets, we could not quantify well-known white blood cell-specific markers (i.e. CD3 for T lymphocytes; CD19 for B lymphocytes; CD66b for granulocytes) in our gray matter homogenates. Furthermore, a highly similar pattern of elevated NF-κB-related mRNA levels was seen in adult mice with daily poly(I:C) exposure regardless of whether the mice underwent transcardial saline perfusion or not. Taken together these lines of evidence suggest that microglia and/or astrocytes are the most likely source of elevated NF-κB activity in the PFC in schizophrenia. Future studies are needed to determine the effects of elevated NF-κB transcriptional activity on cortical circuitry dysfunction in schizophrenia.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (MH100066 to Dr. Volk; MH043784 and MH084053 to Dr. Lewis) and by funds from the VISN 4 Mental Illness Research, Education, and Clinical Center (MIRECC, Director: D. Oslin; Associate Director: G. Haas), VA Pittsburgh Healthcare System. The contents of this article do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Financial Disclosures
David A. Lewis currently receives investigator-initiated research support from Pfizer. In 2015–2017, he served as a consultant in the areas of target identification and validation and new compound development to Sunovion and Merck. All other authors report no biomedical financial interests or potential conflicts of interest.
References
- 1.Horvath S, Mirnics K (2014): Immune system disturbances in schizophrenia. Biol Psychiatry 75:316–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Volk DW (2016): Role of microglia disturbances and immune-related marker abnormalities in cortical circuitry dysfunction in schizophrenia. Neurobiol Dis 99:58–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tornatore L, Thotakura AK, Bennett J, Moretti M, Franzoso G (2012): The nuclear factor kappa B signaling pathway: integrating metabolism with inflammation. Trends in cell biology 22:557–566. [DOI] [PubMed] [Google Scholar]
- 4.Hoesel B, Schmid JA (2013): The complexity of NF-kappaB signaling in inflammation and cancer. Molecular cancer 12:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arion D, Unger T, Lewis DA, Levitt P, Mirnics K (2007): Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry 62:711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saetre P, Emilsson L, Axelsson E, Kreuger J, Lindholm E, Jazin E (2007): Inflammation-related genes up-regulated in schizophrenia brains. BMC Psychiatry 7:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siegel BI, Sengupta EJ, Edelson JR, Lewis DA, Volk DW (2014): Elevated viral restriction factor levels in cortical blood vessels in schizophrenia. Biol Psychiatry 76:160–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Birnbaum R, Jaffe AE, Chen Q, Shin JH, BrainSeq C, Kleinman JE, et al. (2018): Investigating the neuroimmunogenic architecture of schizophrenia. Mol Psychiatry 23:1251–1260. [DOI] [PubMed] [Google Scholar]
- 9.Fillman SG, Cloonan N, Catts VS, Miller LC, Wong J, McCrossin T, et al. (2013): Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol Psychiatry 18:206–214. [DOI] [PubMed] [Google Scholar]
- 10.Fillman SG, Sinclair D, Fung SJ, Webster MJ, Shannon Weickert C (2014): Markers of inflammation and stress distinguish subsets of individuals with schizophrenia and bipolar disorder. Translational psychiatry 4:e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Volk DW, Chitrapu A, Edelson JR, Roman KM, Moroco AE, Lewis DA (2015): Molecular mechanisms and timing of cortical immune activation in schizophrenia. Am J Psychiatry 172:1112–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takao K, Kobayashi K, Hagihara H, Ohira K, Shoji H, Hattori S, et al. (2013): Deficiency of schnurri-2, an MHC enhancer binding protein, induces mild chronic inflammation in the brain and confers molecular, neuronal, and behavioral phenotypes related to schizophrenia. Neuropsychopharmacology 38:1409–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown AS, Derkits EJ (2010): Prenatal infection and schizophrenia: A review of epidemiologic and translational studies. Am J Psychiatry 167:261–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang B, Jia H, Kast RJ, Thomas EA (2013): Epigenetic changes at gene promoters in response to immune activation in utero. Brain, behavior, and immunity 30:168–175. [DOI] [PubMed] [Google Scholar]
- 15.American Psychiatric A (1994): DSM-IV. Diagnostic and Statistical Manual of Mental Disorders. Fourth Edition. Washington, D.C.: American Psychiatric Association. [Google Scholar]
- 16.Volk DW, Eggan SM, Lewis DA (2010): Alterations in metabotropic glutamate receptor 1alpha and regulator of G protein signaling 4 in the prefrontal cortex in schizophrenia. Am J Psychiatry 167:1489–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Volk DW, Edelson JR, Lewis DA (2014): Cortical inhibitory neuron disturbances in schizophrenia: role of the ontogenetic transcription factor Lhx6. Schizophr Bull 40:1053–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Volk DW, Chitrapu A, Edelson JR, Lewis DA (2015): Chemokine receptors and cortical interneuron dysfunction in schizophrenia. Schizophr Res 167:12–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, et al. (2002): Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol 3:3431–34.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Volk DW, Radchenkova PV, Walker EM, Sengupta EJ, Lewis DA (2012): Cortical opioid markers in schizophrenia and across postnatal development. Cereb Cortex 22:1215–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dorph-Petersen KA, Pierri JN, Perel JM, Sun ZX, Sampson AR, Lewis DA (2005): The influence of chronic exposure to antipsychotic medications on brain size before and after tissue fixation: A comparison of haloperidol and olanzapine in macaque monkeys. Neuropsychopharmacology 30:1649–1661. [DOI] [PubMed] [Google Scholar]
- 22.Garay PA, Hsiao EY, Patterson PH, McAllister AK (2013): Maternal immune activation causes age- and region-specific changes in brain cytokines in offspring throughout development. Brain Behav Immun 31:54–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith SE, Li J, Garbett K, Mirnics K, Patterson PH (2007): Maternal immune activation alters fetal brain development through interleukin-6. J Neurosci 27:10695–10702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao EY, Patterson PH (2011): Activation of the maternal immune system induces endocrine changes in the placenta via IL-6. Brain Behav Immun 25:604–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benros ME, Pedersen MG, Rasmussen H, Eaton WW, Nordentoft M, Mortensen PB (2014): A nationwide study on the risk of autoimmune diseases in individuals with a personal or a family history of schizophrenia and related psychosis. Am J Psychiatry 171:218–226. [DOI] [PubMed] [Google Scholar]
- 26.Pahl HL (1999): Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene 18:6853–6866. [DOI] [PubMed] [Google Scholar]
- 27.Fromer M, Roussos P, Sieberts SK, Johnson JS, Kavanagh DH, Perumal TM, et al. (2016): Gene expression elucidates functional impact of polygenic risk for schizophrenia. Nat Neurosci 19:1442–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crews FT, Sarkar DK, Qin L, Zou J, Boyadjieva N, Vetreno RP (2015): Neuroimmune Function and the Consequences of Alcohol Exposure. Alcohol research : current reviews 37:331–341, 344–351. [PMC free article] [PubMed] [Google Scholar]
- 29.Nennig SE, Schank JR (2017): The Role of NFkB in Drug Addiction: Beyond Inflammation. Alcohol and alcoholism (Oxford, Oxfordshire) [DOI] [PMC free article] [PubMed]
- 30.Wang Z, Wu W, Tang M, Zhou Y, Wang L, Xu W, et al. (2013): NF-kappaB pathway mediates vascular smooth muscle response to nicotine. The international journal of biochemistry & cell biology 45:375–383. [DOI] [PubMed] [Google Scholar]
- 31.Wongtrakool C, Grooms K, Bijli KM, Crothers K, Fitzpatrick AM, Hart CM (2014): Nicotine stimulates nerve growth factor in lung fibroblasts through an NFkappaB-dependent mechanism. PLoS One 9:e109602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. (2016): New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113:E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y, Sloan SA, Clarke LE, Caneda C, Plaza CA, Blumenthal PD, et al. (2016): Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 89:37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.