

Abstract
Oysters are keystone species in estuarine ecosystems and are of substantial economic value to fisheries and aquaculture worldwide. Contending with disease and environmental stress are considerable challenges to oyster culture. Here we report a draft genome of the Sydney Rock Oyster, Saccostrea glomerata, an iconic and commercially important species of edible oyster in Australia known for its enhanced resilience to harsh environmental conditions. This is the second reference genome to be reported from the family Ostreidae enabling a genus-level study of lophotrochozoan genome evolution. Our analysis of the 784-megabase S. glomerata genome shows extensive expansions of gene families associated with immunological non-self-recognition. Transcriptomic analysis revealed highly tissue-specific patterns of expression among these genes, suggesting a complex assortment of immune receptors provide this oyster with a unique capacity to recognize invading microbes. Several gene families involved in stress response are notably expanded in Saccostrea compared with other oysters, and likely key to this species’ adaptations for improved survival higher in the intertidal zone. The Sydney Rock Oyster genome provides a valuable resource for future research in molluscan biology, evolution and environmental resilience. Its close relatedness to Crassostrea will further comparative studies, advancing the means for improved oyster agriculture and conservation.
Keywords: Oyster, genome, bivalve adaptation, gene family evolution, resilience
1. Introduction
Oysters of the family Ostreidae are a group of bivalve molluscs that include over 70 extant members considered to be keystone species widely distributed in estuarine ecosystems, performing important roles in mitigating turbidity and improving water quality.1,2 Edible oysters have established commercial significance in fisheries and aquaculture industries being among the most highly produced mollusc species in the world.3 Having evolved an extraordinary resilience to the harsh conditions of intertidal marine environments, oysters are capable of tolerating wild fluctuations in temperature and salinity, extended emersion and the persistent exposure to microbes encountered by filter-feeding.4 A rich and diverse set of immune and stress response genes in the oyster genome are thought to be pivotal to the remarkably effective host defence system that enables these animals to thrive in estuaries and coastal oceans worldwide.4,5 Despite these adaptations, oyster populations both wild and captive are threatened with mass mortalities caused by epizootic infections6 and by factors associated with environmental change.1 Understanding and ameliorating susceptibility to these threats is essential for the establishment of secure mariculture and effective conservation.
The Sydney Rock Oyster (Saccostrea glomerata) is an economically important species of edible oyster in Australia, naturally populating the shorelines of its eastern coast and extending across the Tasman Sea to the northern regions of New Zealand. Its cultivation contributes substantially to an aquaculture industry in Australia dating back to the 19th century, supported by selective breeding programmes that have been operating for over 25 years.1,7 The Sydney Rock Oyster, in contrast to the more widely distributed and invasive Pacific oyster (Crassostrea gigas), grows ∼60% slower under favourable conditions, yet has a higher tolerance to abiotic stress, surviving up to three times longer out of water.8 It also appears to be resistant to the devastating viral disease Pacific Oyster Mortality Syndrome caused by OsHV-1.9 These characteristics provoke inquiry into the nature of the higher resilience observed in S. glomerata and offer a unique opportunity for comparative studies given the recent availability of the Pacific oyster genome. A lack of sequenced genomes from closely related species of lophotrochozoa have limited the extent of comparative studies within this highly diverse superphylum of species. Decoding a complete Saccostrea genome enables a deeper understanding of the biology and evolution of the Ostreidae and will serve as a valuable resource for genetic improvement within the oyster farming industry. Here, we present an annotated draft genome for S. glomerata and explore comparisons between genes relevant to resilience among a close relative and other more evolutionarily distant molluscs.
2. Materials and methods
2.1. Generation of sequence data
Mantle and gill tissues were dissected from a single female oyster for high molecular weight DNA extraction and library preparation. PCR-free short-insert libraries of 210 and 450 bp along with mate-pair libraries of 3, 6 and 9 kb were sequenced on the illumina HiSeq 2500 (Supplementary Table S1). A Chicago library was produced from an additional oyster and sequenced on the illumina platform.
2.2. Sequence assembly
Raw reads were quality filtered and trimmed using either Trimmomatic10 or Skewer11 to produce over 200 Gb of clean data (Supplementary Table S2). Clean reads were assembled and initially scaffolded de novo using Meraculous2.12 For detailed information see Supplementary data. Construction of a primary haploid assembly was performed using HaploMerger213 and further scaffolding performed by Dovetail Genomics (Santa Cruz, CA, USA).14 Read mapping of genomic reads was performed using Bowtie2.15 Rate of heterozygosity was estimated using the SAMtools/BCFtools16 (mpileup) pipeline to call SNPs and short InDels. Estimates of completeness were undertaken using both the CEGMA17 pipeline and BUSCO18 searches of the OrthoDB metazoan library.
2.3. Genome annotation
Automated gene annotation was performed using MAKER219 (see Supplementary data). Repetitive sequences were soft-masked with RepeatMasker (http://www.repeatmasker.org/RMDownload.html (13 September 2018, date last accessed)) using the RepBase20 library and a custom repeat library generated with RepeatModeler (http://www.repeatmasker.org/RepeatModeler/ (13 September 2018, date last accessed)). Protein-coding sequences from the genomes of C. gigas, Pinctada fucata, Lottia gigantea, Octopus bimaculoides, Drosophila melanogaster and Homo sapiens were used for homology-based gene prediction. Stranded RNA-Seq data were aligned to the genome for use with the BRAKER121 pipeline for training an AUGUSTUS22 model that was included with a stranded transcriptome assembly produced by Ertl et al.,23 CEGMA17 derived proteins and the features of the RNA-Seq alignment in the MAKER2 pipeline. Predictions were filtered from the final gene set if they displayed no alignment to either the protein-coding genes from other species using BLAST (E-value <10−10) or to the Pfam24 database.
2.4. Phylogenetic analysis
Amino acid sequences of 1,205 genes were concatenated to create a super gene (see Supplementary data). The concatenated sequences from 13 species were aligned using MUSCLE25 by selecting default settings. After removing alignment gaps from all sequences 247,779 amino acids were available for further analysis. This multiple sequence alignment was used to infer the phylogenetic relationship between the species and the maximum likelihood based RAxML26 was used for this purpose. A γ distribution was used to model the rate variation among sites and four rate categories were chosen. To model substitutions between amino acids we opted the LG (Le and Gascuel) substitution matrix27 and used the empirical amino acid frequencies. The species Nematostella vectensis was set as the outgroup. A bootstrap resampling procedure with 100 pseudo-replicates was used to obtain statistical confidence for each bifurcation (node) of the phylogenetic tree. The software FigTree (http://tree.bio.ed.ac.uk/software/figtree/ (13 September 2018, date last accessed)) was used to view and print the tree generated by RAxML.
2.5. Divergence time estimation
In order to estimate the divergence times between molluscan species a Bayesian statistics based MCMCtree28 method was employed. The amino acid sequence alignment was used for this analysis and the maximum likelihood tree obtained from the RAxML program was used as the guide tree. The following fossil ages were used to calibrate the tree: 306–581 million years (MY) for the spilt between Capitella teleta and Helobdella robusta29 470–532 MY for Aplysia californica–L. gigantea divergence,29 532–549 MY for the first appearance of the molluscs30 and 550–636 MY for the first appearance of the Lophotrochozoa and Eumetazoa.30 We also fixed a maximum age of 650 MY for the root of the tree (root age). To obtain the Hessian matrix for the protein data, the codeml program of the software PAML31 was used. Using the WAG+Gamma32 model of amino acid substitution matrix and the four calibration times listed above the divergence times were estimated. The results of MCMCtree were checked for convergence using the program Tracer (http://tree.bio.ed.ac.uk/software/tracer/ (13 September 2018, date last accessed)) and the time-tree generated by this program was viewed using FigTree. The divergence times for each bifurcating node are given in Fig. 1a.
Figure 1.

Divergence time and rate of non-synonymous substitutions between bivalves. (a) A time tree based on protein sequences from 16 metazoan genomes. Divergence times were estimated using a Bayesian MCMC method. A maximum likelihood based phylogenetic tree was calibrated based on seven well-defined fossils (see Methods). (b) Genetic divergence measured by the non-synonymous substitution rate (dN) between bivalves and the outgroup Lottia gigantea using 3,269 orthologous genes. The relative divergences with respect to the outgroup reveal the differences in the rate of protein evolution among bivalves.
2.6. Rate of protein evolution and gene expression
To examine the correlation between gene expression and rate of protein evolution we used 11,388 orthologous genes for the Sydney Rock-Pacific Oyster comparison. Using the codeml module of software program PAML31 we obtained the likelihood based pairwise non-synonymous divergence for each gene. We then obtained the expression levels of each gene in five tissues (gill, mantle, muscle, haemolymph and digestive system). Our analysis revealed a highly significant negative correlation (P < 0.0001) between expression level and rate of protein evolution (Supplementary Fig. S7a). We also sorted protein-coding genes based on their expression levels and grouped them into 12 categories containing equal number (949) of genes. The average estimates of gene expression levels and rate of protein evolution were computed for genes belonging to each category (Supplementary Fig. S7b). The relationship based on the mean estimates was also highly significant (P < 0.0001). To examine the rate of protein evolution across bivalves we used 3,269 orthologous genes from the 6 bivalves and L. gigantea, which was used as the outgroup. Using the codeml module of software program PAML31 we obtained the likelihood based pairwise non-synonymous divergence between L. gigantea and bivalve genomes. Since the time of divergence between L. gigantea and each bivalve is expected to be the same, any difference in the pairwise divergence suggest the variation in the rate of evolution between different bivalves (Fig. 1b).
2.7. Gene family analysis
Protein sequences from S. glomerata, C. gigas, P. fucata and Patinopecten yessoensis were compared for orthology using all-against-all BLASTP alignment (E-value of 10−5) and clustered using OrthoMCL33 (inflation value of 1.5). Protein family domain analysis was performed with Hidden Markov Model (HMM) searches of the Pfam24 database (see Supplementary data). Phylogenetic trees were generated using full length protein sequences aligned with MUSCLE25 and constructed with FastTree34 using the Jones–Taylor–Thornton model then visualized with FigTree v1.4.3 and MEGA 7.35 Functional annotation of the protein-coding genes from the S. glomerata genome was undertaken using BLASTp 2.5.0+ (E-value 10−5) to search for homologs with a local copy of the NCBI non-redundant database (nr). Protein-coding sequences were aligned against the NCBI KOG database (version 28 March 2017) using RPSBLAST v2.2.15 performed via the WebMGA36 server and for analysis of biological pathways present within the S. glomerata genome, KEGG orthologous gene information was obtained using the KEGG Automatic Annotation Server (KAAS)37 using a bi-directional best hit approach with the eukaryote representative gene set as reference.
2.8. Quantifying gene expression
Paired-end transcriptome libraries from gill, mantle, male gonad, female gonad, muscle, haemolymph and digestive system used in this study were generated previously.7,23 Expression levels were measured by aligning quality processed RNA-Seq reads to the genome assembly using HiSat.38 Mapped reads were sorted with SAMtools39 and counts reported as transcripts per one million mapped reads (TPM) using StringTie.40 TPM values visualized in heatmaps were transformed to log2 (TPM + 1) and normalized across tissues using the scale function in R.
3. Results
3.1. Genome sequencing and de novo assembly
The genome of S. glomerata was estimated to be 784 Mb in size based on an analysis of k-mer frequency distribution. It was sequenced to over 300-fold coverage based on this estimate by a whole-genome shotgun approach using the illumina HiSeq platform (Supplementary Table S1). Over 200 Gb of quality filtered short-insert and mate-pair read data (Supplementary Table S2) was initially assembled and scaffolded in accordance with the strategy outlined in Supplementary Fig. S1. Further contiguity improvements were made by Chicago library sequencing, HiRise scaffolding and gap filling (Supplementary data). The final S. glomerata draft assembly included 788 Mb in 10,107 scaffolds with a scaffold N50 of 804.2 kb and a contig N50 of 39.8 kb (Supplementary Tables S3 and S4).
This genome is ∼44% larger than that estimated for the closely related Pacific oyster C. gigas (545 Mb)5 yet smaller than the pearl oyster P. fucata (1.14 Gb).41 Many invertebrate genomes, including that of oysters, exhibit high levels of heterozygosity and repetitiveness which can complicate the assembly process. The repeat content of the draft genome was estimated to be 45.03% (Supplementary Table S5). The S. glomerata genome exhibits a relatively high level of heterozygosity based on the observation of 3.2 million SNPs and 354,373 short insertion/deletions (indels) in 703,199,470 eligible positions. This resulted in a polymorphism rate of 0.51%, comparable with the inbred sequenced C. gigas (0.73%)5 though, in contrast with the octopus, O. bimaculoides (0.08%).42 Almost 82% complete and 96% partial matches to the 248 core eukaryotic gene set could be identified in the draft assembly using the CEGMA pipeline (Supplementary Table S6), which is comparable with other invertebrate genome assemblies reported previously.43 Using the BUSCO tool we could detect a total of 787 (93.3%) of the 843 genes in the metazoan library with 672 (79%) of these being complete matches. Over 91% of the clean short-insert read data could be aligned to the draft assembly. Half the assembly was contained in the longest 241 scaffolds ranging from 0.8 to 7.1 Mb. Over 90% of the assembled bases were covered by the longest 1,321 (13%) scaffolds. The assembly metrics of the S. glomerata genome are comparable to those of other published mollusc genomes5,42,44,45 and together with completeness measures, indicate the production of a comprehensive draft.
3.2. Genome annotation and comparative analysis
A total of 29,738 protein-coding genes from the S. glomerata genome were annotated using homology-based and ab initio predictions (Supplementary data). Almost 88% of these were supported by RNA-Seq evidence derived from six different tissues. The gene content spanned one-third of the genome with a mean gene length of 8,737 bp averaging 8 exons per transcript (Supplementary Table S7). The number of gene predictions presented here is similar to that reported for the two published oysters C. gigas (28,027) and P. fucata (29,353).5,44 Comparing BUSCO search results with the gene models from C. gigas and P. fucata show that the predictions from this study have the greatest number of complete (87.2%) and least number if missing genes (5.6%) of the three, indicating this study has produced the most complete gene model set for an oyster species reported to date (Supplementary Table S8).
Phylogenetic analysis using 1,205 conserved orthologues with 247,779 amino acid positions from 16 metazoan species show that Saccostrea diverged from Crassostrea ∼77 million years ago (Ma) (Fig. 1a and Supplementary Fig. S6). The time estimates obtained for the other nodes of the tree was comparable to those of other previous studies.5,46,47Saccostrea glomerata has the slowest non-synonymous substitution rate in protein sequences of the six bivalve assemblies reported to date, suggestive of a slowly evolving genome, a rate slightly lower than the scallop (Fig. 1b). The rate of protein evolution has a particularly strong negative correlation with the level of gene expression indicating strong selection pressures in genes expressed highly across the genome (Supplementary Fig. S7).
Comparison of orthologous gene groups shared among bivalves S. glomerata, C. gigas, P. fucata and the scallop P. yessoensis show a core set of 8,838 gene groups and unique set of 1,111 that are Saccostrea-specific. Saccostrea glomerata shares 13,106 gene groups with the most closely related C. gigas yet has a larger number of unique genes (Fig. 2a and b). Gene ontology (GO) enrichment analysis of the gene groups unique to Saccostrea reveal an overrepresentation of GO terms associated largely with binding and metabolism (Supplementary Table S9).
Figure 2.

Gene family representation analysis. (a) Numbers of shared and unique gene groups in four species of molluscs. Gene groups were constructed by clustering of orthologous groups using OrthoMCL software. (b) Genome-wide orthology based on OrthoMCL gene clustering among nine species of molluscs. (c) A selection of expanded Pfam domains in S. glomerata. Associated gene families were considered expanded with a corrected P value of <0.01. Multiple domains in a given gene model were counted only once. Sgl, S. glomerata; Cgi, C. gigas: Pfu, P. fucata; Pye, P. yessoensis; Mph, Modiolus philippinarum; Bpl, Bathymodiolus platifrons; Lgi, Lottia gigantea; Aca, Aplysia californica; Obi, O. bimaculoides; Lan, Lingula anatina; Cte, Capitella teleta; Dme, Drosophila melanogaster; Spu, Strongylocentrotus purpuratus; Cel, Caenorhabditis elegans; Dre, Danio rerio; Xtr, Xenopus tropicalis; Hsa, Homo sapiens; Nve, Nematostella vectensis. *Pfam domain enriched in each species of bivalves; #Pfam domains not found expanded in Crassostrea.
3.3. Gene family expansion
To better understand the genome-wide similarities among molluscs, we examined the distribution of 8,629 Pfam domains across a diverse set of 26 metazoan genomes identifying a number of significant gene family expansions in Saccostrea. These include toll-like receptors (TLRs); PF01582, immunoglobulin domain-containing genes; PF00047, thrombospondins; PF00090, complement (C1q) subunits; PF00386, G-protein-coupled receptors (GPCRs); PF00002 and heat-shock proteins; PF00012, many of which are not found expanded in Crassostrea (Fig. 2c). A group of 42 gene families were determined to be significantly expanded in the S. glomerata genome and not so in any of the other molluscs examined. Almost a third (13) of these appear uniquely in Saccostrea, that is to say, not considered expanded in any of the other 25 species included in this analysis (Supplementary Table S10). Of these gene family expansions, some of the most notable have occurred in gene families associated with non-self-recognition and other components of the immune response. Expansions of some immune-related gene families has been described previously in studies of Crassostrea and other bivalves, and has been attributed to a pathogen-rich and dynamic intertidal habitat.5,48 However, several protein families in Saccostrea greatly exceed the domain counts found in the other oysters. Saccostrea glomerata also retain a considerable inventory of some of the important and well-studied immune-related gene families compared with other lophotrochozoans (Table 1), even though these families do not appear expanded when compared with the metazoan genomes used in this study.
Table 1.
Distribution of protein families associated with immune response
| Pfam | Domain | Sgl | Cgi | Pfu | Pye | Mph | Bpl | Lgi | Aca | Obi | Lan | Cte |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PF00335 | Tetraspanin | 63 | 54 | 53 | 52 | 4 | 42 | 39 | 49 | 31 | 43 | 50 |
| PF00061 | Lipocalin | 12 | 13 | 14 | 11 | 1 | 11 | 14 | 17 | 0 | 17 | 2 |
| PF02798 | GST_N | 33 | 25 | 37 | 29 | 2 | 23 | 24 | 56 | 22 | 44 | 22 |
| PF00255 | GSHPx | 6 | 8 | 9 | 6 | 1 | 7 | 4 | 3 | 4 | 10 | 3 |
| PF08210 | APOBEC_N | 7 | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 6 | 0 |
| PF00080 | Sod_Cu | 8 | 10 | 8 | 8 | 2 | 6 | 9 | 9 | 4 | 6 | 6 |
| PF01823 | MACPF | 20 | 13 | 9 | 5 | 10 | 13 | 4 | 3 | 0 | 8 | 1 |
| PF11648 | RIG-I_C-RD | 9 | 8 | 6 | 5 | 2 | 12 | 3 | 5 | 1 | 6 | 3 |
| PF02898 | NO_synthase | 1 | 1 | 7 | 1 | 1 | 2 | 1 | 3 | 1 | 3 | 2 |
| PF16673 | TRAF_BIRC3_bd | 2 | 2 | 2 | 1 | 1 | 1 | 0 | 0 | 0 | 0 | 1 |
| Total | 161 | 136 | 147 | 119 | 25 | 118 | 98 | 145 | 63 | 143 | 90 |
Number of proteins containing the specific Pfam domain for each family. GST_N, glutathione S-transferase; GSHPx, glutathione peroxidase; APOBEC_N, apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like N-terminal domain, N-terminal domain; Sod_Cu, Copper/zinc superoxide dismutase (SODC); MACPF, membrane attack complex/perforin (MACPF) domain; RIG-I_C-RD, C-terminal domain of RIG-I; NO_synthase, nitric oxide synthase, oxygenase domain; TRAF_BIRC3_bd, TNF receptor-associated factor BIRC3 binding domain.
TLRs are characterized by an intracellular toll/interleukin-1 receptor (TIR) domain and have a well described association with innate immunity in animals and plants.49 The S. glomerata genome contains 182 TIR domain-containing genes that are markedly expanded beyond the relatively broad repertoire of 61 and 91 found in C. gigas and P. fucata, respectively (Fig. 3a). This level of expansion has extended to other families of non-self-recognition proteins, such as C-type lectins and fibrinogen-related proteins (FREPs). The diversification of TLRs is thought to be important for the recognition of Pathogen Associated Molecular Patterns (PAMPs) and the subsequent activation of an immunological response via MYD88 signalling.50,51 The expansions of TLRs indicate a highly specialized recognition and signalling system in Saccostrea. The abundant expression in the male gonad implies a number of these genes may have a role in reproduction (Fig. 3c). TLRs have various functions in addition to immunity in other invertebrates52 and mammalian studies have suggested a role for TLRs in spermatogenesis53 and the protection of spermatozoa.54 The clear tissue specificity of groups of TIR domains in S. glomerata may reflect distinctions or possible interactions between reproductive and immune processes.
Figure 3.

Expansion of Toll/interleukin-1 receptor domain containing genes in S. glomerata. (a) Phylogenetic tree of TIR domain-containing genes among five molluscs, S. glomerata (blue), C. gigas (red), P. fucata (purple), P. yessoensis (green), L. gigantea (gold). I indicates an expansion that includes the largest cluster of 13 genes located on a single scaffold (Scaffold SL_29), II & III indicate the major Saccostrea expansions. (b) Scaffold containing the largest cluster of TIR domain-containing genes. The 13 blue regions indicate TIR genes with 3 non-TIR genes interspersed in gold. (c) Expression profile of 154 TIR genes in 7 different tissues. Cell colours indicate the number of standard deviations from the mean expression level.
The fibrinogen_C gene family is the C-terminal globular domain of FREPs that are known to exhibit extremely high levels of sequence variability55 and can function as a molecular recognition unit in immunological defence.56 There are a total of 576 genes in the S. glomerata genome containing at least one fibrinogen_C domain, more than in any of the 25 genomes included in the gene family analysis. This number is triple that of C. gigas (192) and P. fucata (163) and over 100 more than B. floridae which has the highest compliment of FREPs (395) of any other species included in the comparison (Fig. 4a). The S. glomerata genes containing fibrinogen_C domains vary considerably in their length, composition and domain arrangement and the majority appear to be indiscriminately distributed across the genome apart from a few main clusters, the largest being a set of 17 genes arranged in the same orientation ranging over 478 kb on a single scaffold (Fig. 4b). The majority of FREPs were not found to be expressed among the 7 tissue types used in this study; however, the expression profile of the 262 FREPs that were detected suggests these genes are distinctly tissue-specific, occurring to a lesser extent within the haemolymph (Fig. 4c). The broadest expression of FREPs can be seen in gill tissue, likely due to the increased exposure of this organ to exogenous microbes. Also observed is an expansion of C-type lectin domain-containing genes that are similarly important for non-self-recognition and display comparable tissue-specific patterns of expression (Supplementary Fig. S9).
Figure 4.

Expansion and expression of S. glomerata fibrinogen_C domain-containing genes. (a) Distribution of fibrinogen_C domain-containing gene models detected using hmmsearch among 22 selected metazoan genomes. (b) Scaffold SGL_242 and Scaffold SGL_221 contain the 2 largest clusters of fibrinogen_C domain-containing genes. The largest cluster is on Scaffold SGL_242 and contains 17 genes that vary in genomic span and are each orientated in the anti-sense direction. Scaffold SGL_221 contains 15 genes and is orientated with the majority in a set in the sense direction flanked by 5 genes in the anti-sense direction. (c) Expression profiles of 262 fibrinogen_C domain containing genes in 7 S. glomerata tissues. Expression of the remaining 314 fibrinogen_C domain containing genes was not detected in any of the 7 tissues sequenced.
Phenoloxidases are copper containing proteins including tyrosinase and laccase57 that have important roles in the innate immune mechanisms in invertebrates.58 Laccases contain multicopper oxidase domains (PF00394) and along with tyrosinase domains (PF00264) are both expanded in S. glomerata. Phenoloxidase has been shown to be important in disease resistance in S. glomerata59 and laccases and have been shown to have antibacterial activity in C. gigas.60 Phylogenetic analysis of genes in S. glomerata that contain at least three multicopper oxidase domains form two clusters and are expressed most highly in the digestive system and mantle and with a pattern suggestive of tissue-specific functionality (Supplementary Fig. S10).
In addition to the expansion of immune-related gene families, oysters have stress response adaptations that enable them to persist in challenging environments. Comparisons of anti-apoptosis and stress-related genes reveal a broadening of these gene families in S. glomerata compared with C. gigas (Fig. 5). A similar pattern is shared between the mussel B. platifrons which endures the stress associated with deep sea vents and the shallow-water mussel M. philippinarum.46 Expansion of these gene families appears to be a critical response for building resilience in these more stress-adapted species.
Figure 5.
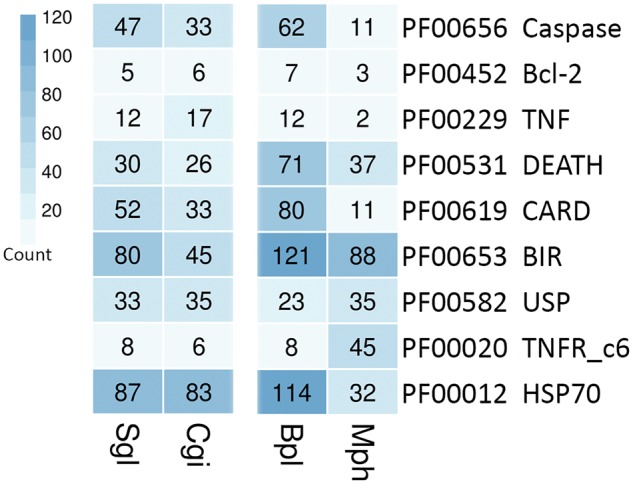
Comparison of domains associated with stress between oysters and mussels. Number of proteins containing the specific Pfam domain for each family. Bcl-2, apoptosis regulator proteins Bcl-2 family; TNF, Tumour necrosis factor family; CARD, Caspase recruitment domain; BIR, inhibitor of apoptosis domain; USP, universal stress protein family; TNFR_c6, tumour necrosis factor receptor cysteine-rich region.
Precise regulation of apoptosis is essential for an organism’s ability to adapt to changing environments.61 Inhibitor of apoptosis proteins (IAPs) are characterized by the presence of at least one copy of the baculovirus IAP repeat (BIR) domain which facilitate binding between IAPs and caspases, controlling apoptotic signalling.62 Oysters are thought to maintain a powerful anti-apoptosis system given the repertoire of 48 IAPs annotated from the genome of Crassostrea,5 and further supported by the 61 found in Pinctada in this study. The S. glomerata genome encodes 80 IAP genes, almost half of which form two clusters, based on phylogenetic analysis, suggesting a recent expansion (Supplementary Fig. S8a). The expression of these 80 IAPs in 7 oyster tissues appears tissue-specific, suggesting the possibility of functional specializations (Supplementary Fig. S8b). Although, the phylogenetic relationships of these IAPs do not appear to accord with tissue-specific patterns of expression. Expansions of genes containing the IAP repeat were found only within the 6 bivalves included in this study. The enrichment of IAP genes unique to the bivalve lineage is likely a fundamental component of their extraordinary survivability.
3.4. Analysis of molluscan genes associated with reproduction
Reproductive activities of all eumetazoans are controlled by the neuroendocrine system.63 In vertebrates, this includes a hypothalamic–pituitary–gonadal (HPG) axis. Several homologs of the HPG axis, such as gonadotropin-releasing hormone (GnRH) and glycoprotein hormones,64 have been identified among invertebrates. However, the molecular components of reproductive regulation in invertebrates are still largely unresolved and can be variable across phyla.65 We found 40 genes in the S. glomerata genome that have previous been linked to invertebrate reproductive processes. A similar distribution was observed for these genes among six other molluscan genomes (Fig. 6). The majority of non-neuropeptide genes associated with reproduction were highly expressed in the gonads, both testis and ovary. However, abundant expression of some of these genes within non-reproductive tissues (haemolymph, gill, mantle, muscle and digestive gland) indicates they may fulfil additional roles. For example, the high abundance of NPY and GPR54-2 within the digestive gland suggests a functional association of these two proteins in the physiological regulation of digestive system and feeding. Putative receptors for GnRH/corazonin, tachykinin and NPY, and a homologue of vertebrate GPR54, which is a receptor for a vertebrate reproductive neuropeptide ‘kisspeptin’,66 were abundant in the gonad tissues. This provides strong evidence for a role of these receptors, with their cognate ligands, in Saccostrea reproduction.
Figure 6.

Genes associated with reproduction and their distribution in 6 molluscs and their expression in S. glomerata. Genes that were more abundant in the testis compared with ovary includes the putative receptor genes (5HTR1, TACRs, GnRHR/CrzR, and NPYR) and testis-specific protein genes (TSSKs). Conversely, expression of genes encoding reproductive signalling factors, such as CDA-A, β-catenin, Phb2, nanos, FoxL2 and vitellogenin, were higher in ovary tissue. Neuropeptides, including CCAP, buccalin, APGWamide and ELH, were found at relative low abundance in the gonads, but higher in the gill and mantle tissues. In the muscle and digestive tissues, there was overall low expression of all reproductive-related genes, except for NPY and GPR54-2, which were highly expressed and potentially specific to the digestive gland.
4. Discussion
This study presents the sequenced and assembled genome of the Sydney Rock Oyster, S. glomerata, an important species for Australian aquaculture and a focus of conservation due to the severe decline in shellfish reef habitats. The S. glomerata genome is the second reference genome to be reported from the family Ostreidae (edible oysters), enabling for the first time whole-genome comparative studies between ostreid species. This resource can improve our understanding of the mechanisms these organisms have evolved for survival in the highly stressful intertidal environment, facilitating selective breeding and the enhancement of oyster cultivation practices. The draft assembly achieved comparably high levels of completeness and contiguity with respect to other published mollusc genomes. The statistics for scaffold and contig N50 improved upon the two other oyster genomes previously reported due largely to technological advances in sequencing. The use of 250 bp read lengths and ‘Chicago’ libraries contributed heavily to the production of a highly contiguous assembly, offering a more affordable alternative to BAC or fosmid sequencing.
The larger genome size of S. glomerata did not appear to contain a proportionately higher number of protein-coding genes, nor larger average exon or intron sizes. However, 354 Mb of repetitive sequences (45% of the genome) were identified compared with only 202 Mb (36% of the genome) found in C. gigas.5 An expansion of nuclease and transposase domains such as PF01498 (15 out of 89 total genes across 23 species) in S. glomerata, suggests that this may be the result of increased transposable element activity, adding further support to a role for these mobile sequences in shaping genome variation.5
The types of gene families found expanded in the S. glomerata genome are not dissimilar to those in the other bivalve genomes. In fact, there is a noteworthy collection of genes associated with stress and immune defence in the genome of C. gigas that are thought to have expanded in response to the environmental variability and the exposures of filter-feeding confronted by the oyster.44 It appears that these features of the oyster genome are not unique to C. gigas, despite its invasive capacity, and are rather a characteristic of oysters more generally and their adaptation to a sessile existence in harsh environments. However, some of the immune-associated gene families are present up to 3-fold higher in Saccostrea than in the other published molluscs and are more abundant than in any of the other 26 species investigated here, suggesting an evolutionary history of extensive selective pressure from invading microbes. Given the relative short divergence time from Crassostrea and a slower rate of protein evolution yet a distinctly larger, more repetitive genome, it may be that the mechanisms for gene duplications are intrinsically more active in Saccostrea and are, at least in part, contributing to a phenotype of higher environmental resilience.
Genes associated with molecular pattern recognition are collectively the most significantly expanded in the S. glomerata genome. Large expansions of FREPs have been extensively studied in the snail Biomphalaria and are speculated to be due to lineage-specific selective pressure from trematode parasites like Schistosoma mansoni.48,67 Protozoan parasites, among other microbes, are major threats for a variety of bivalve species.68 Commercial production of S. glomerata has been severely impacted by mortalities arising from infection with the protozoan pathogens, such as Marteilia sydneyi causing QX disease, which infect S. glomerata exclusively.69,70 The substantial expansion of FREPs in Saccostrea may be due to increased selective pressure from persistent challenge by disease-causing microbes and a need for broad recognition capacity. Clustering of these genes within the genome offers some evidence of local tandem duplications and the clear pattern of tissue-specific expression indicates these genes may have evolved to perform specialized functions. Interestingly, the expression of FREPs appears somewhat concentrated in the gill tissue that coincides with the site of infection for M. sydneyi and other important pathogens.59,71
In Australia, wild populations of S. glomerata have been under pressure from the introduction of the faster growing C. gigas, which can rapidly overgrow and displace the native oyster at the low to mid-intertidal zones.72 At higher intertidal areas, however, S. glomerata is able to survive due to its greater tolerance to thermal stress and emersion.8 A highly developed anti-apoptosis system is thought to be important for the endurance of oysters to abiotic stress ascribed to the expansion of IAP genes and the propensity of air exposure to induce the dramatic upregulation of IAPs in C. gigas.5,62 The expansion of anti-apoptosis and stress response genes in the S. glomerata genome appears essential to the resilience observed in this species.
The draft S. glomerata genome presented here provides a valuable resource for further studies in molluscan biology and would accelerate genetic enhancement programs for commercially produced oyster species.
Supplementary Material
Acknowledgements
We gratefully acknowledge the University of the Sunshine Coast for a Post-Graduate Research Scholarship to D.P. and Genecology, the Centre for Genetics, Ecology and Physiology for financial support.
Accession numbers
The Saccostrea genome project has been deposited at the NCBI under the BioProject number PRJNA414259 including the clean reads from whole genome sequencing data deposited in the Sequence Read Archive (SRA) database along with the genome assembly. The predicted gene nucleotide and protein annotations are available from the web database http://soft.bioinfo-minzhao.org/srog/# (13 September 2018, date last accessed).
Conflict of interest
None declared.
References
- 1. Thompson J., Stow A., Raftos D.. 2017, Lack of genetic introgression between wild and selectively bred Sydney rock oysters Saccostrea glomerata, Mar. Ecol. Prog. Ser., 570, 127–39. [Google Scholar]
- 2. Salvi D., Macali A., Mariottini P.. 2014, Molecular phylogenetics and systematics of the bivalve family Ostreidae based on rRNA sequence-structure models and multilocus species tree, PLoS One, 9, 19–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Saavedra C., Bachère E.. 2006, Bivalve genomics, Aquaculture, 256, 1–14. [Google Scholar]
- 4. Guo X., He Y., Zhang L., Lelong C., Jouaux A.. 2015, Immune and stress responses in oysters with insights on adaptation, Fish Shellfish Immunol., 46, 107–19. [DOI] [PubMed] [Google Scholar]
- 5. Zhang G., Fang X., Guo X.. 2012, The oyster genome reveals stress adaptation and complexity of shell formation, Nature, 490, 49–54. [DOI] [PubMed] [Google Scholar]
- 6. Dégremont L., Garcia C., Allen S.K.. 2015, Genetic improvement for disease resistance in oysters: a review, J. Invertebr. Pathol., 131, 226–41. [DOI] [PubMed] [Google Scholar]
- 7.In, Van V., Ntalamagka N., O’Connor W.. 2016, Reproductive neuropeptides that stimulate spawning in the Sydney Rock Oyster (Saccostrea glomerata), Peptides, 82, 109–19. [DOI] [PubMed] [Google Scholar]
- 8. Krassoi F.R., Brown K.R., Bishop M.J., Kelaher B.P., Summerhayes S.. 2008, Condition-specific competition allows coexistence of competitively superior exotic oysters with native oysters, J. Anim. Ecol., 77, 5–15. [DOI] [PubMed] [Google Scholar]
- 9. Jenkins C., Hick P., Gabor M., et al. 2013, Identification and characterisation of an ostreid herpesvirus-1 microvariant (OsHV-1 µ-var) in Crassostrea gigas (Pacific oysters) in Australia, Dis. Aquat. Org., 105, 109–26. [DOI] [PubMed] [Google Scholar]
- 10. Bolger A.M., Lohse M., Usadel B.. 2014, Trimmomatic: a flexible trimmer for Illumina sequence data, Bioinformatics, 30, 2114–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jiang H., Lei R., Ding S.-W., Zhu S.. 2014, Skewer: a fast and accurate adapter trimmer for next-generation sequencing paired-end reads, BMC Bioinformatics, 15, 182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chapman J.A., Ho I., Sunkara S., Luo S., Schroth G.P., Rokhsar D.S.. 2011, Meraculous: de novo genome assembly with short paired-end reads, PLoS One, 6, e23501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Huang S., Kang M., Xu A.. 2017, HaploMerger2: rebuilding both haploid sub-assemblies from high-heterozygosity diploid genome assembly, Bioinformatics, 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Putnam N.H., O’Connell B., Stites J.C., et al. 2015, Chromosome-scale shotgun assembly using an in vitro method for long-range linkage, arXiv, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Langmead B., Salzberg S.L.. 2012, Fast gapped-read alignment with Bowtie 2, Nat. Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Li H. 2011, A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data, Bioinformatics, 27, 2987–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parra G., Bradnam K., Korf I.. 2007, CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes, Bioinformatics, 23, 1061–7. [DOI] [PubMed] [Google Scholar]
- 18. Simão F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M.. 2015, BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs, Bioinformatics, 31, 3210–2. [DOI] [PubMed] [Google Scholar]
- 19. Holt C., Yandell M.. 2011, MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects, BMC Bioinformatics, 12, 491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bao W., Kojima K.K., Kohany O.. 2015, Repbase update, a database of repetitive elements in eukaryotic genomes, Mob. DNA, 6, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hoff K.J., Lange S., Lomsadze A., Borodovsky M., Stanke M.. 2016, BRAKER1: unsupervised RNA-Seq-based genome annotation with GeneMark-ET and AUGUSTUS, Bioinformatics, 32, 767–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Stanke M., Schöffmann O., Morgenstern B., Waack S.. 2006, Gene prediction in eukaryotes with a generalized hidden Markov model that uses hints from external sources, BMC Bioinformatics, 7, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ertl N.G., O’Connor W.A., Papanicolaou A., Wiegand A.N., Elizur A.. 2016, Transcriptome analysis of the Sydney Rock Oyster, Saccostrea glomerata: insights into molluscan immunity, PLoS One, 11, e0156649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Finn R.D., Bateman A., Clements J., et al. 2014, Pfam: the protein families database, Nucl. Acids Res., 42, D222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Edgar R.C. 2004, MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res., 32, 1792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stamatakis A. 2014, RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies, Bioinformatics, 30, 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Le S.Q., Gascuel O.. 2008, An improved general amino acid replacement matrix, Mol. Biol. Evol., 25, 1307–20. [DOI] [PubMed] [Google Scholar]
- 28. Reis M. d., Yang Z.. 2011, Approximate likelihood calculation on a phylogeny for Bayesian estimation of divergence times, Mol. Biol. Evol., 28, 2161–72. [DOI] [PubMed] [Google Scholar]
- 29. Benton M., Donoghue P.C.J., Asher R.J.. 2009, Calibrating and constraining molecular clocks, The Timetree of Life, p 35–86. [Google Scholar]
- 30. Benton M.J., Donoghue P.C.J., Asher R.J., Friedman M., Near T.J., Vinther J.. 2015, Constraints on the timescale of animal evolutionary history, Palaeontol. Electron., 1–107. [Google Scholar]
- 31. Yang Z. 2007, PAML 4: phylogenetic analysis by maximum likelihood, Mol. Biol. Evol., 24, 1586–91. [DOI] [PubMed] [Google Scholar]
- 32. Whelan S., Goldman N.. 2001, A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach, Mol. Biol. Evol., 18, 691–9. [DOI] [PubMed] [Google Scholar]
- 33. Li L., Stoeckert C.J., Roos D.S.. 2003, OrthoMCL: identification of ortholog groups for eukaryotic genomes, Genome Res., 13, 2178–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Price M.N., Dehal P.S., Arkin A.P.. 2010, FastTree 2—approximately maximum-likelihood trees for large alignments, PLoS One, 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kumar S., Stecher G., Tamura K.. 2016, MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets, Mol. Biol. Evol., 33, 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wu S., Zhu Z., Fu L., Niu B., Li W.. 2011, WebMGA: a customizable web server for fast metagenomic sequence analysis, BMC Genomics, 12, 444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Moriya Y., Itoh M., Okuda S., Yoshizawa A.C., Kanehisa M.. 2007, KAAS: an automatic genome annotation and pathway reconstruction server, Nucleic Acids Res., 35, W182–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim D., Langmead B., Salzberg S.L.. 2015, HISAT: a fast spliced aligner with low memory requirements, Nat. Methods, 12, 357–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li H., Handsaker B., Wysoker A., et al. 2009, The sequence alignment/map format and SAMtools, Bioinformatics, 25, 2078–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pertea M., Pertea G.M., Antonescu C.M., Chang T.-C., Mendell J.T., Salzberg S.L.. 2015, StringTie enables improved reconstruction of a transcriptome from RNA-seq reads, Nat. Biotechnol., 33, 290–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Takeuchi T., Kawashima T., Koyanagi R., et al. 2012, Draft genome of the pearl oyster Pinctada fucata: a platform for understanding bivalve biology, DNA Res., 19, 117–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Albertin C.B., Simakov O., Mitros T., et al. 2015, The octopus genome and the evolution of cephalopod neural and morphological novelties, Nature, 524, 220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Luo Y.-J., Takeuchi T., Koyanagi R., et al. 2015, The Lingula genome provides insights into brachiopod evolution and the origin of phosphate biomineralization, Nat. Commun., 6, 8301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Takeuchi T., Koyanagi R., Gyoja F., et al. 2016, Bivalve-specific gene expansion in the pearl oyster genome: implications of adaptation to a sessile lifestyle, Zoological Lett., 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Simakov O., Marletaz F., Cho S.-J., et al. 2013, Insights into Bilaterian evolution from three spiralian genomes, Nature, 493, 526–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sun J., Zhang Y., Xu T., et al. 2017, Adaptation to deep-sea chemosynthetic environments as revealed by mussel genomes, Nat. Ecol. Evol., 1, 121. [DOI] [PubMed] [Google Scholar]
- 47. Luo Y.-J., Kanda M., Koyanagi R., et al. 2018, Nemertean and phoronid genomes reveal lophotrochozoan evolution and the origin of bilaterian heads, Nat. Ecol. Evol., 2, 141–51. [DOI] [PubMed] [Google Scholar]
- 48. Zhang L., Li L., Guo X., Litman G.W., Dishaw L.J., Zhang G.. 2015, Massive expansion and functional divergence of innate immune genes in a protostome, Sci. Rep., 5, 8693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Fornarino S., Laval G., Barreiro L.B., Manry J., Vasseur E., Quintana-Murci L.. 2011, Evolution of the TIR domain-containing adaptors in humans: swinging between constraint and adaptation, Mol. Biol. Evol., 28, 3087–97. [DOI] [PubMed] [Google Scholar]
- 50. Gerdol M., Venier P., Edomi P., Pallavicini A.. 2017, Diversity and evolution of TIR-domain-containing proteins in bivalves and Metazoa: new insights from comparative genomics, Dev. Comp. Immunol., 70, 145–64. [DOI] [PubMed] [Google Scholar]
- 51. Zhang Y., He X., Yu F., et al. 2013, Characteristic and functional analysis of Toll-like receptors (TLRs) in the lophotrocozoan, Crassostrea gigas, reveals ancient origin of TLR-mediated innate immunity, PLoS One, 8, e76464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leulier F., Lemaitre B.. 2008, Toll-like receptors—taking an evolutionary approach, Nat. Rev. Genet., 9, 165–78. [DOI] [PubMed] [Google Scholar]
- 53. Girling J.E., Hedger M.P.. 2007, Toll-like receptors in the gonads and reproductive tract: emerging roles in reproductive physiology and pathology, Immunol. Cell Biol., 85, 481–9. [DOI] [PubMed] [Google Scholar]
- 54. Saeidi S., Shapouri F., Amirchaghmaghi E., et al. 2014, Sperm protection in the male reproductive tract by Toll-like receptors, Andrologia, 46, 784–90. [DOI] [PubMed] [Google Scholar]
- 55. Allam B., Raftos D.. 2015, Immune responses to infectious diseases in bivalves, J. Invertebr. Pathol., 131, 121–36. [DOI] [PubMed] [Google Scholar]
- 56. Hanington P.C., Zhang S.-M.. 2011, The primary role of fibrinogen-related proteins in invertebrates is defense, not coagulation, J. Innate Immun., 3, 17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Le Clec’h W., Anderson T.J.C., Chevalier F.D.. 2016, Characterization of hemolymph phenoloxidase activity in two Biomphalaria snail species and impact of Schistosoma mansoni infection, Parasites Vectors, 9, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Cerenius L., Lee B.L., Söderhäll K.. 2008, The proPO-system: pros and cons for its role in invertebrate immunity, Trends Immunol., 29, 263–71. [DOI] [PubMed] [Google Scholar]
- 59. Green T.J., Raftos D., O'Connor W., Adlard R.D., Barnes A.C.. 2011, Disease prevention strategies for QX disease (Marteilia sydneyi) of Sydney Rock Oysters (Saccostrea glomerata), J. Shellfish Res., 30, 47–53. [Google Scholar]
- 60. Luna-Acosta A., Rosenfeld E., Amari M., Fruitier-Arnaudin I., Bustamante P., Thomas-Guyon H.. 2010, First evidence of laccase activity in the Pacific oyster Crassostrea gigas, Fish Shellfish Immunol., 28, 719–26. [DOI] [PubMed] [Google Scholar]
- 61. Ameisen J.C. 2002, On the origin, evolution, and nature of programmed cell death: a timeline of four billion years, Cell Death Differ., 9, 367–93. [DOI] [PubMed] [Google Scholar]
- 62. Qu T., Zhang L., Wang W., et al. 2015, Characterization of an inhibitor of apoptosis protein in Crassostrea gigas clarifies its role in apoptosis and immune defense, Dev. Comp. Immunol., 51, 74–8. [DOI] [PubMed] [Google Scholar]
- 63. Chieffi G. 1984, Control of reproductive cycles in metazoa: exogenous and endogenous factors, Bolletino Zool., 51, 205–22. [Google Scholar]
- 64. Uchida K., Moriyama S., Chiba H., et al. 2010, Evolutionary origin of a functional gonadotropin in the pituitary of the most primitive vertebrate, hagfish, Proc. Natl. Acad. Sci., 107, 15832–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Stout E.P., La Clair J.J., Snell T.W., Shearer T.L., Kubanek J.. 2010, Conservation of progesterone hormone function in invertebrate reproduction, Proc. Natl. Acad. Sci. USA., 107, 11859–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Smith J.T., Clifton D.K., Steiner R.A.. 2006, Regulation of the neuroendocrine reproductive axis by kisspeptin-GPR54 signaling, Reproduction, 131, 623–30. [DOI] [PubMed] [Google Scholar]
- 67. Portet A., Pinaud S., Tetreau G., et al. 2017, Integrated multi-omic analyses in Biomphalaria-Schistosoma dialogue reveal the immunobiological significance of FREP-Sm PoMuc interaction, Dev. Comp. Immunol., 75, 16–27. [DOI] [PubMed] [Google Scholar]
- 68. Fernández Robledo J.A., Vasta G.R., Record N.R.. 2014, Protozoan parasites of bivalve molluscs: literature follows culture, PLoS One, 9, e100872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Dove M.C., Nell J.A., Mcorrie S., O’connor W.A.. 2013, Assessment of Qx and winter mortality disease resistance of mass selected Sydney Rock Oysters, Saccostrea glomerata (Gould, 1850), in the Hawkesbury River and Merimbula Lake, NSW Australia, J. Shellfish Res., 32, 681–7. [Google Scholar]
- 70. Raftos D.A., Kuchel R., Aladaileh S., Butt D.. 2014, Infectious microbial diseases and host defense responses in Sydney rock oysters, Front. Microbiol., 5, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Nell J.A., Cox E., Smith I.R., Maguire G.B.. 1994, Studies on triploid oysters in Australia. I. The farming potential of triploid Sydney rock oysters Saccostrea commercialis (Iredale and Roughley), Aquaculture, 126, 243–55. [Google Scholar]
- 72. Padilla D.K. 2010, Context-dependent impacts of a non-native ecosystem engineer, the Pacific Oyster Crassostrea gigas, Integr. Comp. Biol., 50, 213–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.