

Abstract
Osteogenesis imperfecta (OI) is a family of heritable disorders of bone fragility. Most individuals with OI have mutations in the genes encoding type I collagen; at least 17 other genes have been associated with OI. Biallelic loss-of-function mutations in WNT1 cause severe OI. Heterozygous missense variants in WNT1 are responsible for early-onset osteoporosis with variable bone phenotypes. Herein we report a 3-generation family with four affected individuals, some presenting with multiple low-impact fractures in childhood and others presenting with early-onset osteoporosis without a striking fracture history. A WNT1 variant (c. 1051>C; p.Trp351Arg) was identified in the proband and segregated with a bone phenotype in three additional family members, consistent with autosomal dominant inheritance. In the proband, whole genome sequencing also revealed a de novo duplication (434 kb) of 22q11.2 that involves 25 genes, 4 of which are associated with human disease when haploinsufficient. Though smaller than the typical (1.5 Mb) 22q11.2 duplication, the duplication in the proband may be responsible for additional non-osseous aspects of his phenotype (hypotonia, developmental delay, small genitalia, strabismus, and depression in pre-adolescence). This case demonstrates the variability of bone phenotype conferred by a WNT1 variant and extends the spectrum of bone phenotypes associated with heterozygous WNT1 mutations.
Keywords: Osteogenesis imperfecta, WNT1 variant, Osteoporosis
INTRODUCTION
Osteogenesis imperfecta (OI) is a family of heritable disorders of connective tissue. The hallmark feature of OI is bone fragility, with susceptibility to fracture from minor trauma. Additional features include bone deformity, growth deficiency, hearing loss, blue sclerae, brittle teeth, kyphoscoliosis, short stature and easy bruising. It has been known for three decades that the majority of individuals with OI have mutations in COL1A1 or COL1A2, the two genes coding for collagen type I alpha chains, but in the past 10 years defects in at least 17 other genes have been linked to OI. WNT1 has been identified as a gene causing severe (progressively deforming) recessively-inherited OI [Trejo et al., 2016]. Homozygous or compound heterozygous mutations in WNT1 were recently described in 25 individuals from 16 families in a series of six publications [Kämpe et al., 2015; Fahiminiya et al., 2013; Faqeih et al. 2013; Keupp et al., 2013; Pyott et al., 2013; Stephen et al., 2015]. Heterozygous mutations in WNT1 are responsible for early onset osteoporosis [Keupp et al., 2013; Laine et al., 2013; Mäkitie et al., 2016; Palomo et al., 2014]. WNT1 is conserved back to at least Cnidaria and encodes a ligand for the WNT signaling pathway, which is a primary regulator of bone formation. WNT1 promotes bone formation by binding to the LRP5-FZD receptor complex and activating the canonical WNT signaling pathway. Heterozygous loss-of-function mutations in WNT1 result in less severe reductions in WNT signaling and milder bone disease (osteoporosis). WNT1 heterozygotes are characterized by early-onset, progressive osteoporosis with multiple fractures, affecting mainly the spine, but with normal growth and only slightly shortened stature due to kyphosis. Other extra-skeletal features, as well as abnormal biochemical findings (other than markers of bone turnover), are usually not present [Kämpe et al., 2015]. Herein we report a family with a novel WNT1 variant that segregates with a variable bone fragility phenotype.
CASE REPORT
An 11-year-old boy was evaluated for recurrent fractures. He was delivered vaginally at 39 weeks to a 36 year G7P1 SAB5, weighing 4.309 kg. The pregnancy was conceived by in vitro fertilization (IVF) because of endometriosis. The mother denied exposures during pregnancy. Fetal movement was vigorous. Maternal aneuploidy screening showed an elevated risk for trisomy 18, but fetal ultrasound was unremarkable and amniocentesis karyotype was 46,XY. Delivery was uncomplicated.
As an infant, the patient had hypotonia, severe gastroesophageal reflux, delayed gross motor skills (walked at age 2 years), a stiff waddling gait in early childhood (negative evaluation for hip dysplasia) and small penis. His gait improved with physical therapy and participation in sports. He also received occupational therapy for feeding difficulties. Strabismus required two surgeries. He has flat feet and hypermobile joints but no history of dislocations. He has no myopia or lens subluxation. Magnetic resonance imaging (MRI) of brain and electromyogram (EMG) were normal. He is currently experiencing psychiatric symptoms and is being treated for depression.
He has a lifetime history of eight fractures; all but one occurred in low impact settings (Table 1). A dual energy X-ray absorptiometry (DXA) scan showed bone mineral density greater than three standard deviations (SD) below the mean for his age. He was started on bisphosphonate infusions and has completed several rounds of infusions.
Table 1.
DXA measurements and fracture history of family members
| DXA Z-score | Age at DXA | WNT1 variant | Fracture history | Vertebral fracture | Other symptoms | |
|---|---|---|---|---|---|---|
| III-2 | −3.5 | 10 years | present | 8 (7 low-impact) | No | hypotonia, delayed gross motor skills, and stiff, waddling gait, pes planus, hypermobile joints |
| II-4 | −3 | 50 years | present | none | No | pes planus, scoliosis, shin splints with minimal running, osteonecrosis of the knee, femoral acetabular impingement |
| III-1 | −1.6 | 16 years | present | 3 (low-impact) | No | epilepsy, severe gluten intolerance, and two knee dislocations |
| I-2 | −2.2 | 80 years | present | 2 fractures (high impact) | No | – |
| II-1 | −1.2 | 54 years | absent | 3 fractures (2 toe and 1 high impact) | No | – |
| II-2 | −0.1 to −0.8 | 51 years | absent | none | No | – |
| II-3 | ND | NA | absent | none | No | – |
At age 11 years, he weighed 33.6 kg (24th centile), had a height of 154.6 cm (87th centile), and had a head circumference of 53.1 cm (42nd centile). He demonstrated increased range of motion in elbows and wrists. Tall forehead was noted, but there were no significant dysmorphic features. Mild left paraspinous elevation was noted on forward bending. He continues to exhibit poor coordination and suffers from back pain. The patient’s radiographs shown in Figure 1 demonstrate gracile fibulas, lack of vertebral fractures, and absences of obvious osteopenia.
Figure 1.
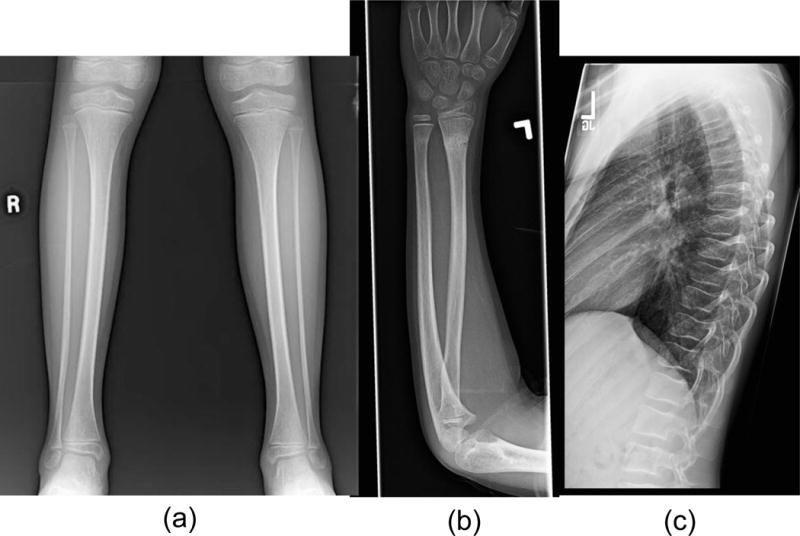
Proband radiographs showing (a) gracile fibulas (b) healing distal radius and ulna fractures and (c) thoracic spine with normal alignment, preservation of disc spaces and lack of fractures. The interpreting radiologist made no comments about bone density in each case. Radiographs all predated treatment with bisphosphonates.
The pedigree is shown in Figure 2. Family history was notable for father (II-4) having pes planus and scoliosis. As a young adult, he experienced debilitating shin splints and stress fractures with minimal running at 20 years of age. He had symptoms consistent with femoral acetabular impingement beginning at age 40 years, as well as osteonecrosis of the knee at 47 years of age (with no clear explanation or precipitating factors). Osteoporosis was documented at 50 years of age with a DXA scan showing bone mineral density was 3 SD below the mean for his age. The proband’s sister (III-1) has a history of 3 low-impact fractures, osteopenia, epilepsy, severe gluten intolerance, and two knee dislocations. At age 16 years, DXA scan showed her bone mineral density was 1.6 SD below the mean. The paternal grandmother (I-2) was diagnosed with osteoporosis at 69 years of age based on DXA scan. Her DXA scan done at age 80 showed her bone mineral density was 2.2 standard deviations below that of age matched controls (T-score is−3.0). She has a history of 2 fractures at age 56 and 60 years. She was treated with alendronate sodium for 15 years, then switched to denosumab recently. She reports having lost four inches of height in the last 5 years. None of the individuals in this family have experienced vertebral fractures.
Figure 2. Family Pedigree.
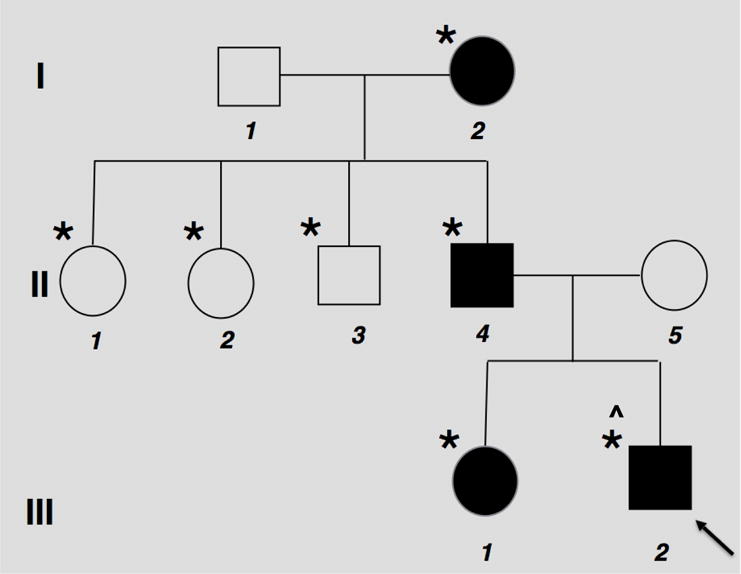
Squares represent male family members, circles female family members, black symbols affected family members, (*) means individual tested for WNT1 variant, (^) 22q11.2 duplication positive.
METHODS
Molecular clinical testing using a multi-gene next generation sequencing (NGS) panel for autosomal recessive osteogenesis imperfecta was performed on the proband at the University of Washington Collagen Diagnostics Laboratory. Concurrent clinical whole genome sequencing (WGS) of the proband, his sister, and both parents were undertaken through the philanthropic Rare and Undiagnosed Disease Program (iHOPE) in the Illumina Clinical Services Laboratory. Whole genome quad analysis was performed to identify variants that the affected individuals (III-1, III-2, II-4) shared. Additionally, copy number variation using WGS was simultaneously assessed in the quad analysis. Subsequent targeted variant analysis was performed on additional family members using Sanger sequencing at the University of Washington.
In Vitro Experiments
Plasmid construction
The coding region of WNT1 was cloned into pLenti CMV Puro DEST (w118-1) (Addgene plasmid #17452) vector named as WT vector. The c.1051T>C mutation was introduced in WNT1 by site-directed mutagenesis using QuikChange Lightning Site-Directed Mutagenesis kit (Agilent Technologies). The mutated vector was named WNT1 c.1051T>C.
Reporter Assays
Super TOPFLASH firefly luciferase reporter contains seven transcription factors T cell factor (TCF)/ lymphoid enhancer binding factor (LEF) binding sites driving the firefly luciferase gene. HEK293T cells were transfected 500ng of empty vector (pLenti CMV Puro DEST vector), WT vector plus empty vector (250ng each), or mutated vector (WNT1 c.1051T>C) plus empty vector with 400ng of Super TOPFLASH firefly luciferase reporter and 120ng of TK renilla luciferase reporter for transfection efficiency normalization per well in a 12-well plate using Superfect (Qiagen). To test dominant negative effect, we compared the WT plus empty vector with WT plus the mutated vector (WNT1 c.1051T>C) (250ng each). 24h post transfection, the firefly and TK renilla luciferase activity were measured using a dual luciferase assay kit from three parallel wells per condition (Dual-GloTM Luciferase Assay System, Promega). Super TOPFLASH firefly luciferase activity was normalized to TK renilla luciferase activity. The statistical difference between the WT and mutated WNT1 (WNT1 c.1051T>C) transfected cells was tested by Student’s t-test.
RESULTS
NGS panel testing revealed a missense variant (c.1051T>C; p.Trp351Arg) in exon 4 of the WNT1 gene in the proband that was classified as a variant of uncertain significance. WGS results also identified the p.Trp351Arg variant in the proband, and additionally found the variant in the affected sibling and father. Although the variant is unreported in the literature, multiple publications have observed autosomal dominant pathogenic missense variants in exon 4 in WNT1 in patients with bone-related phenotypes [Kämpe et al., 2015; Keupp et al., 2013; Laine et al., 2013; Mäkitie et al., 2016]. Additionally, amino acid position 351 (Figure 3) is highly conserved, and the c.1051T>C variant is not reported in any allele frequency population databases. Based on the combined evidence of segregation of the variant and bone abnormality phenotypes, the conservation of the amino acid position, and rarity of the variant in the general population, the variant was classified as a variant of unknown significance but suspicious for pathogenicity.
Figure 3. Conservation of WNT1 c.1051T>C (p.Trp351Arg).

The c.1051T>C (p.Trp351Arg) mutation identified in this pedigree affects an amino acid that is conserved across all vertebrate lineages and sits within a region of predicted disulfide bonds (UniProt: P04628).
Copy number analysis revealed a de novo copy number duplication of 434 kB on 22q11.21 (chr22: 21052009- 21484438) (Figure 4). This duplication encompassed 25 genes, four of which are associated with human disease when haploinsufficient.
Figure 4. 22q11 duplication in proband.

Whole genome sequencing in proband revealed an ~438kb duplication (hg19 chr22:21052009-21484438) that encompasses 11 canonical RefSeq genes, four of which have an established disease association. Three reported pathogenic duplications of similar size have been previously reported and are shown here, including work from Fernandez et al. (2009).
Sanger sequencing revealed the c.1051T>C variant in an additional family member (I-2) with an osteoporosis phenotype. Four additional unaffected family members did not carry the variant. The limitation of having only one extended family for the analysis may explain the lack of statistical significance.
After 14 months of treatment (3 infusions of zolendronic acid), the proband’s BMD by DXA improved from a z-score of −2.2 to −1.6 (subtotal whole body) and −3.2 to −2.5 (lumbar spine).
To test the functional consequence of the WNT1 variant, we assessed the induction of canonical WNT signaling. When canonical WNT signaling is activated, active β-catenin will accumulate in the nucleus, where β-catenin binds to the transcription factors T cell factor (TCF)/ lymphoid enhancer binding factor (LEF) and activates target gene transcription [Behrens, von Kries et al. 1996, van de Wetering, Cavallo et al. 1997]. Super TOPFLASH-luciferase reporter can be used to detect β-catenin-mediated transcription activation [Veeman, Slusarski et al. 2003]. We found that the mutant WNT1 c.1051T>C had significantly reduced capacity to activate canonical WNT signaling compared with wild type WNT1 (Figure 5). Moreover, co-transfection of mutated WNT1 c.1051T>C with wild type WNT1 did not interfere with the induction of the luciferase activity, which suggests that WNT1 c.1051T>C does not function in a dominant negative manner. Overall, the in vitro experiment suggests that WNT1 c.1051T>C mutant impairs the normal activity of WNT ligand which can induce canonical WNT signaling.
Figure 5.
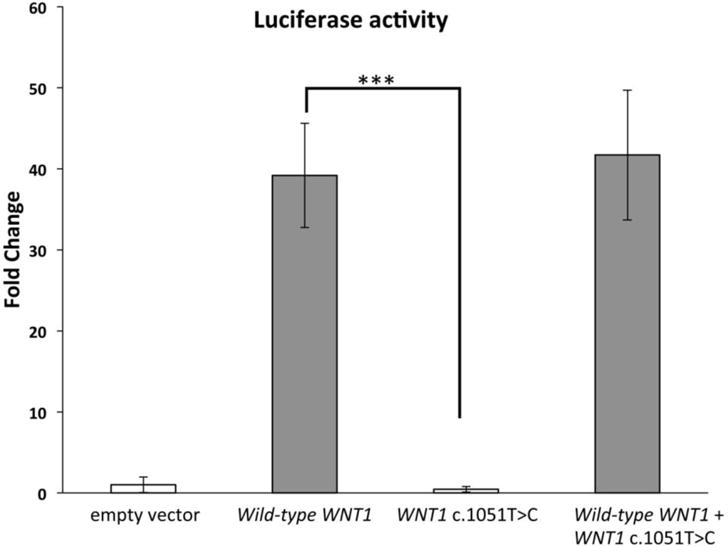
Super TOPFLASH luciferase reporter assay for canonical WNT signaling for the identified WNT1 mutation - The expression of mutant WNT1 (c.1051T>C) could not induce canonical WNT signaling compared to the wild-type WNT1 in the Super TOPFLASH luciferase reporter assay. Co-expressing wild-type and mutated WNT1 (c.1051T>C) did not reduce the induced luciferase activity by wild-type WNT1. *** p<0.0005, error bars represent S.D., N=3.
DISCUSSION
Homozygous or compound heterozygous WNT1 mutations cause severe (progressively deforming) OI, while heterozygous mutations cause early-onset osteoporosis. Here we report on a family with a WNT1 variant (c.1051T>C; p.Trp351Arg) that segregates with osteopenia on DXA scan and fractures. Two family members experienced frequent low-impact fractures, and all members who tested positive for WNT1 mutation had low BMD z-scores, ranging from −3 to −1.6, on DXA scan.
In addition to the bone phenotype, the proband has some other clinical features: strabismus, infantile hypotonia, delayed walking and talking, small genitalia, and recently psychological problems (suicidal ideation). In addition to his single nucleotide variant in WNT1, WGS identified a de novo duplication (434 kb) of 22q11.2 (involving 25 genes, 4 of which are associated with human disease when haploinsufficient). The 22q11.2 duplication syndrome usually involves 1.5-3 Mb and is associated with a variable phenotype ranging from apparently normal to intellectual disability/learning disability, delayed psychomotor development, growth retardation, and/or hypotonia. Though smaller than typical, the 22q11.2 duplication likely accounts for the non-osseous aspects of the proband’s phenotype.
This family demonstrates that heterozygotes with WNT1 mutations can present with a milder OI phenotype with childhood fractures. This confirms the observations of Laine et al. (2013), who described dominantly inherited early-onset osteoporosis caused by heterozygous mutations in WNT1 in 10 affected members of a Finnish family, 6 of whom had multiple vertebral fractures, and 8 of whom had low-impact peripheral fractures; one affected individual fractured as early as infancy. An extension of this family study was reported in 2016 [Makitie et al., 2016], detailing the features in 8 individuals ages 10-30 years. Height z-scores were −1.9 to +0.9, with only one individual having short stature (−2.4 at 18 years). Three individuals had mild scoliosis, and 3 had mild joint hypermobility without dislocations. All eight individuals had a bone density z-score below 0 and four had a bone density z score below −2.0. The epilepsy found in one girl in the Finnish family and individual III-1 of the family reported herein is likely coincidental given the lack of a neurological phenotype found in those with WNT1 mutations generally. However the osteonecrosis experienced by individual II-4 and the avascular necrosis of the femoral head reported in a 23-year-old female member of the Finnish family may indeed relate to a susceptibility conferred by WNT1 haploinsufficiency.
We believe the preponderance of the evidence from the literature and from this family’s presentation supports the WNT1 variant as causative for the bone phenotype in this family. Functional studies support pathogenicity of this variant. However, curiously, mutation carriers in this family do not exhibit the vertebral fractures that are prevalent in WNT1-associated osteoporosis.
Acknowledgments
This work was supported by the NIH P01 HD070394 (BL), U54 AR068069 (BL) and K01 AR069002 (KSJ). Health Sciences International, University of California San Diego and Ministry of Higher Education, Saudi Arabia.
Footnotes
There are no conflicts of interest to declare.
References
- Behrens J, Kries JP, Kühl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. Functional interaction of β-catenin with the transcription factor LEF-1. Nature. 1996;382(6592):638–642. doi: 10.1038/382638a0. [DOI] [PubMed] [Google Scholar]
- Fahiminiya S, Majewski J, Mort J, Moffatt P, Glorieux FH, Rauch F. Mutations in WNT1 are a cause of osteogenesis imperfecta. Journal of Medical Genetics. 2013;50(5):345–348. doi: 10.1136/jmedgenet-2013-101567. [DOI] [PubMed] [Google Scholar]
- Faqeih E, Shaheen R, Alkuraya FS. WNT1 mutation with recessive osteogenesis imperfecta and profound neurological phenotype. Journal of Medical Genetics. 2013;50(7):491–492. doi: 10.1136/jmedgenet-2013-101750. [DOI] [PubMed] [Google Scholar]
- Fernández L, Nevado J, Santos F, Heine-Suñer D, Martinez-Glez V, García-Miñaur S, Palomo R, Delicado A, López Pajares I, Palomares M, García-Guereta L, Valverde E, Hawkins F, Lapunzina P. A deletion and a duplication in distal 22q11.2 deletion syndrome region. Clinical implications and review. BMC Medical Genetics. 2009;10(1) doi: 10.1186/1471-2350-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämpe AJ, Mäkitie RE, Mäkitie O. New Genetic Forms of Childhood-Onset Primary Osteoporosis. Hormone Research in Paediatrics. 2015;84(6):361–369. doi: 10.1159/000439566. [DOI] [PubMed] [Google Scholar]
- Keupp K, Beleggia F, Kayserili H, Barnes AM, Steiner M, Semler O, Fischer B, Yigit G, Janda CY, Becker J, Breer S, Altunoglu, Grunhagen J, Krawitz P, Hecht J, Schinke T, Makareeva E, Lausch E, Cankaya T, Caparros-Martin JA, Lapunzina P, Teammate S, Aglan M, Zabel B, Eysel P, Koerber F, Leikin S, Garcia KC, Netzer C, Schonau E, Ruiz-Perez VL, Mundlos S, Amling M, Kornak U, Marini J, Wollnik B. Mutations in WNT1 cause different forms of bone fragility. The American Journal of Human Genetics. 2013;92(4):565–574. doi: 10.1016/j.ajhg.2013.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine CM, Joeng KS, Campeau PM, Kiviranta R, Tarkkonen K, Grover M, Lu JT, Pekkinen M, Wessman M, Heino TJ, Nieminen-Pihala V, Aronen M, Laine T, Kroger H, Cole WG, Lehesjoki AE, Nevarez L, Krakow D, Curry CJ, Cohn DH, Gibbs RA, Lee BH, Makitie O. WNT1 mutations in early-onset osteoporosis and osteogenesis imperfecta. N Engl J Med. 2013;368:1809–1816. doi: 10.1056/NEJMoa1215458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkitie RE, Haanpää M, Valta H, Pekkinen M, Laine CM, Lehesjoki AE, Schalin-Jäntti C, Mäkitie O. Skeletal Characteristics of WNT1 Osteoporosis in Children and Young Adults. Journal of Bone and Mineral Research. 2016;31(9):1734–1742. doi: 10.1002/jbmr.2841. [DOI] [PubMed] [Google Scholar]
- Palomo T, Al-Jallad H, Moffatt P, Glorieux FH, Lentle B, Roschger P, Klaushofer K, Rauch F. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone. 2014;67:63–70. doi: 10.1016/j.bone.2014.06.041. [DOI] [PubMed] [Google Scholar]
- Pyott SM, Tran TT, Leistritz DF, Pepin MG, Mendelsohn NJ, Temme RT, Fernandez BA, Elsayed SM, Elsobky E, Verma I, Nair S, Turner EH, Smith JD, Jarvik GP, Byers PH. WNT1 mutations in families affected by moderately severe and progressive recessive osteogenesis imperfecta. The American Journal of Human Genetics. 2013;92(4):590–597. doi: 10.1016/j.ajhg.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephen J, Girisha KM, Dalal A, Shukla A, Shah H, Srivastava P, Kornak U, Phadke SR. Mutations in patients with osteogenesis imperfecta from consanguineous Indian families. European Journal of Medical Genetics. 2015;58(1):21–27. doi: 10.1016/j.ejmg.2014.10.001. [DOI] [PubMed] [Google Scholar]
- Trejo P, Rauch F. Osteogenesis imperfecta in children and adolescents—new developments in diagnosis and treatment. Osteoporosis International. 2016;27(12):3427–3437. doi: 10.1007/s00198-016-3723-3. [DOI] [PubMed] [Google Scholar]
- Van De Wetering M, Cavallo R, Dooijes D, Van Beest M, Van Es J, Loureiro J, Ypama A, Hursh D, Jones T, Bejsovec A, Peifer M, Mortin M, Clevers H. Armadillo coactivates transcription driven by the product of the Drosophila segment polarity gene dTCF. Cell. 1997;88(6):789–799. doi: 10.1016/s0092-8674(00)81925-x. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical Wnt/Fz signaling, regulates gastrulation movements. Current Biology. 2003;13(8):680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]